

Abstract
A model‐informed drug discovery and development strategy played a key role in the novel glucose‐responsive insulin MK‐2640’s early clinical development strategy and supported a novel clinical trial paradigm to assess glucose responsiveness. The development and application of in silico modeling approaches by leveraging substantial published clinical insulin pharmacokinetic–pharmacodynamic (PKPD) data and emerging preclinical and clinical data enabled rapid quantitative decision making. Learnings can be applied to define PKPD properties of novel insulins that could become therapeutically meaningful for diabetic patients.
Background
The use of insulin is hampered by its narrow therapeutic index and substantial day‐to‐day variability.1 Consequently, differences are small between a dose that achieves optimal control of blood glucose and a dose that triggers hypoglycemia. Two major classes of insulin analogs—mealtime and basal insulins—have progressively been developed over the last few decades with pharmacokinetic (PK) and pharmacodynamic (PD) profiles to more closely match the profiles of endogenous insulin secretion during prandial and postabsorptive conditions, respectively.2, 3, 4 Mealtime (prandial) insulins have rapid onset of action and intend to enable disposal of glucose ingested during meals.3, 5 In contrast, basal insulins are intended to sustain insulin action by aiming at a constant delivery of insulin over a 24‐hour period.3, 4, 6 Still, the risk of insulin‐induced hypoglycemia is a serious, frequent, and persisting complication of insulin therapy despite drug developments to create analogs with more predictable and desired PK and PD characteristics.1, 7, 8, 9
To improve insulin therapy, there is an aspiration to create a modified insulin with an improved therapeutic index that enables patients to achieve good glycemic control with a reduced risk for hypoglycemia. Ideally, an innovative insulin analog would modulate its action in coordination with changes in blood glucose, being fully functional under hyperglycemic conditions, but have scalable attenuation of insulin action as blood glucose approaches euglycemia, and as such would address patients’ needs for safer insulins.1, 10, 11 Forty years ago, Brownlee and Cerami12, 13 first proposed a glucose‐responsive insulin (GRI) using the concept of modifying insulin to render its release from an injection depot in response to a rise of glucose. Diverse approaches to the creation of a GRI using this concept have continued,14, 15, 16, 17, 18 including several that have been tested in preclinical studies, but none has progressed into clinical testing.1, 17 Recently, an alternative approach was proposed, wherein the elimination of the insulin, rather than the absorption, was altered in a glucose‐responsive manner resulting in the novel insulin analog MK‐2640.19, 20
MK‐2640 is a novel insulin oligosaccharide conjugate which binds to, activates, and is cleared via the insulin receptor. In addition, MK‐2640 has a second clearance pathway through binding to the lectin receptor mannose receptor C‐type 1 (MRC1) in competition with glucose, making it a GRI.19 Glucose‐dependent modulation of MK‐2640 binding to MRC1 has been demonstrated in the physiological glucose concentration range in vitro and in vivo.19, 20 In vivo, in both minipigs and dogs, at high ambient glucose, MK‐2640 clearance by the MRC1 pathway is reduced, and oppositely, at normal and low ambient glucose, MK‐2640 systemic clearance is increased by ~ 30%.20 MK‐2640 has been the first GRI that has advanced to the clinic to examine clinical translation of its glucose responsiveness.21 The MK‐2640 first‐in‐human study (ClinicalTrials.gov Identifier: NCT02269735) aimed to examine the safety, tolerability, and pharmacokinetic–pharmacodynamic (PKPD) relationship of intravenous (i.v.) administered MK‐2640 in healthy nondiabetic subjects under euglycemic clamp conditions, and to examine proof of mechanism (PoM) in type 1 diabetes mellitus (T1DM) subjects by demonstrating a (glucose‐responsive) change in clearance between euglycemic and hyperglycemic conditions. MK‐2640 was generally well tolerated and was found to be 25‐fold less potent than regular human insulin (RHI) at euglycemia, confirming the preclinical findings. However, MK‐2640 did not display a glucose‐dependent change in MK‐2640 systemic clearance in T1DM subjects between euglycemic and hyperglycemic conditions as was observed preclinically, although an increased glucose utilization was observed in clinical clamp setting at hyperglycemia compared with RHI. While MK‐2640 was subsequently discontinued for further clinical development, reverse translation of clinical PKPD data can provide crucial insights into next steps for developing an insulin oligosaccharide conjugate as a clinically effective glucose‐responsive insulin analog.
The central challenges at the time of advancing MK‐2640 to the clinic were (i) designing informative clinical trials to demonstrate proof of the glucose‐responsive clearance mechanism, (ii) predicting MK‐2640 human PK and PD properties through interspecies scaling of minipig and dog data that demonstrated both insulin action and the glucose‐responsive mechanism, (iii) understanding what a therapeutically meaningful reduction in hypoglycemia would entail that could provide differentiation from standard‐of‐care insulins, and (iv) illustrating the optimal properties of GRI that are critical for improved therapeutic index, thereby meeting the needs of diabetic patients. These key development questions in pursuit of an insulin with an improved therapeutic index are summarized in Table 1. To this end, we developed a model‐informed drug discovery and development (MID3) strategy to inform MK‐2640’s early clinical strategy and to quantify the target product profile requirements for a GRI. Employing an MID3 strategy aims to improve the quality, efficiency, and cost‐effectiveness of decision making by building a quantitative framework through integrated models of compound, mechanism, and disease level data to address key questions that arise in the discovery and development of novel therapies.22 Moreover, a fully integrated MID3 strategy intends to make optimal use of the interdependence between experimental conditions, data generation, and evidence generation based on modeling and simulations in a learning‐confirming paradigm.22
Table 1.
Tabular summary of the MID3 strategy for each pertinent question around the early clinical strategy for MK‐2640 and the target product profile requirements for a GRI using MID3 elements described in Marshall et al., 2016
| MID3 strategy element | Early clinical strategy MK‐2640 | Target product profile requirements for GRI | ||
|---|---|---|---|---|
| MK‐2640 PoM trial design | PKPD translation in support of first‐in‐human study | Differentiation potential | Compound properties | |
| Key Question | What is an appropriate and feasible clinical trial design to demonstrate glucose‐responsive clearance mechanism (proof of mechanism (PoM))? | What is the predicted human PKPD profile for MK‐2640 in nondiabetic healthy subjects (Dose escalation, Part 1) and T1DM subjects (PoM, Part 2)? | How much reduction in hypoglycemia event rate would be required to make a meaningful therapeutic difference and provide improvement over standard‐of‐care insulins in T1DM and T2DM patients, respectively? | What are the required PKPD properties for a glucose responsive insulin to be therapeutically relevant in diabetic patients in prandial and basal use, respectively? |
| Key Theme | Study Design | PK and PD | Medical need and Commercial viability | Efficacy and Safety |
| Activity Level | Mechanism | Compound | Disease | Mechanism & Compound |
| Data Step (Data & prior models) |
Clinical RHI clamp PKPD database in healthy nondiabetic and T1DM subjects Univ. of Virginia/Padova T1DM metabolic simulation platform (T1DM simulator)36 Emerging clinical data from clinical RHI pilot study (MK‐0000‐339) |
Preclinical MK‐2640 and RHI data in dog and minipig Emerging clinical data MK‐2640 (ClinicalTrials.gov Identifier: NCT02269735) |
A database of study‐level aggregate data from published clinical trials for RHI and lispro in T1DM subjects A database of study‐level aggregate data from published clinical trials reporting HbA1c for basal insulins and GLP1 agonists in T2DM subjects |
T1DM simulator36 MSD T2DM QSP simulator45 Subcutaneous comparator clamp PKPD database Basal insulin studies from comparator outcome database |
| Sequential Modeling Approach |
Development of steady‐state clinical RHI clamp PKPD model 23, 24 Exploration of use of T1DM simulator to predict multiglycemic clamp study for RHI25 Qualification of simulations with pilot RHI clinical study results25, 26 Modifying T1DM simulator to implement GRI mechanism of action 42 Perform trial design scenario simulations for MK‐2640 PoM study42 |
Development of translational PKPD model based on integrated glucose insulin model 27, 28, 40 Exposure‐response modeling of emerging healthy volunteer PKPD steady‐state data relative to RHI literature model to assess potency difference and allow dose setting for PoM study 21, 23, 24 |
Model‐based meta‐analysis was performed on outcome databases for prandial and basal insulins in T1DM and T2DM population, respectively 41, 43 |
T1DM simulator was modified to implement a glucose responsive insulin while reflecting key observations from preclinical and clinical MK‐2640 data42 T1DM simulator was used to simulate clamp trials and explore therapeutic relevance of hypothetical prandial GRIs and impact of absorption rate42 T2DM simulator was built in house based on literature data, s.c. PKPD information on SoC insulins and calibrated against outcome studies45, 46, 47 T2DM simulator explored therapeutic relevance for basal GRIs against basal SoC47 |
| Key Assumptions |
Implementation of GRI was done adequately (i.e., dog predicts human glucose‐responsiveness) T1DM simulator can be used for clamp study predictions |
Insulin action and GRI mechanism are translatable from animal to human on basis of bodyweight Insulin potency difference between healthy and T1DM subjects is also applicable to insulin action of a GRI |
Populations and inclusion criteria and study designs for insulin trials used in comparator databases are relevant for GRI program |
Implementation of GRI was done adequately (i.e., dog predicts human glucose‐responsiveness) Qualification for RHI and glargine give confidence that QSP model can be used to simulate GRI outcomes |
| Inference | Understanding of feasibility of steady‐state conditions and decisions around dose setting in MK‐2640 PoM trial |
Understanding of translatability of insulin action for RHI and MK‐2640 from animal to man and from healthy volunteers to patients Revealing gaps in understanding interspecies differences in glucose‐responsive mechanism |
Understanding what magnitude of reduction in hypoglycemia event rate that constitutes a meaningful therapeutic difference and provides improvement over standard‐of‐care insulins | Understanding the range of parameters that provide desired PKPD properties for a glucose responsive insulin to be therapeutically relevant in diabetic patients in prandial and basal use |
| Decisions Impacted |
Design & analysis of first‐in‐human study for MK‐2640 and RHI pilot study in T1DM subjects Supporting dose rationale for MK‐2640 first‐in‐human study Dose setting MK‐2640 PoM clamp study |
Shift from prandial to basal paradigm for GRI development Informing backup molecule properties Support of diabetes portfolio prioritizations |
||
GLP1, glucagon‐like peptide‐1; GRI, glucose‐responsive insulin; HbA1c, glycated hemoglobin A1c; MID3, model‐informed drug discovery and development; PD, pharmacodynamics; PK, pharmacokinetics; PKPD, pharmacokinetic–pharmacodynamic; PoM, proof of mechanism; QSP, quantitative systems pharmacology; RHI, regular human insulin; SoC, standard of care; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus.
To support this GRI program, a suite of in silico models were developed and applied in a learning and confirming paradigm including PKPD models (translational and clinical clamp data on RHI as active comparator), quantitative systems pharmacology models (T1DM and T2DM simulators) and comparator models (model‐based meta‐analysis of outcome data for standard‐of‐care insulins from randomized clinical trials in T1DM and T2DM subjects), that were adapted to include the glucose‐responsive mechanism and emerging (pre) clinical data for MK‐2640 (Figure 1). These models integrated the nonlinear PKPD relationship of glucose and insulin and their complex interplay. The combination of these modeling tools resulted in the comprehensive characterization of the preclinical and clinical pharmacology of MK‐2640 relative to standard‐of‐care insulins, demonstrated a robust interspecies translation of insulin action, supported the early clinical strategy, and explored the differential potential for MK‐2640 relative to standard‐of‐care insulins. Herein, we review the wide‐ranging results of these analyses and the benefits accrued from using an MID3 strategy for the novel glucose‐responsive insulin MK‐2640 during its early clinical development, and how these learnings can be used to further explore optimal PKPD properties for a glucose‐responsive insulin that could become therapeutically meaningful for diabetic patients.
Figure 1.
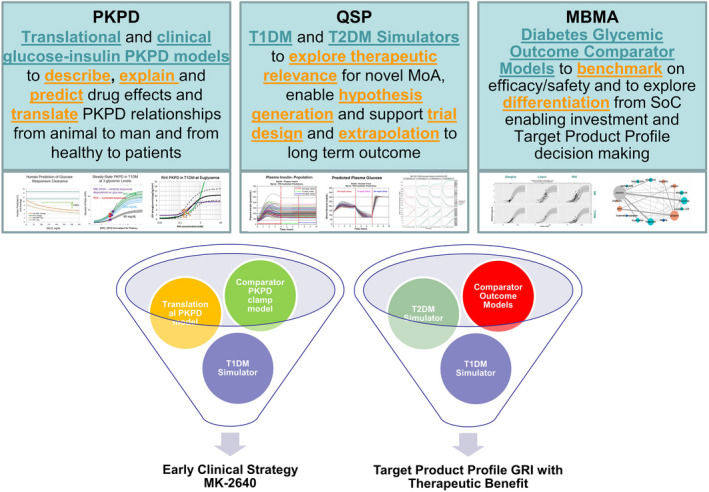
A diabetes modeling toolbox was developed in support of the MID3 GRI strategy. The in silico models developed were (left) PKPD models aiming at prediction and quantification of human insulin pharmacology: i.e., translational PKPD and clinical clamp PKPD models for MK‐2640 and RHI (regular human insulin) in healthy and diabetic subjects; (middle) QSP (quantitative systems pharmacology) simulation models for T1DM and T2DM to evaluate therapeutic relevance through in silico hypothesis testing; (right) diabetes comparator models based on MBMA (model‐based meta‐analysis) of outcome data from randomized clinical trials in T1DM and T2DM subjects to allow benchmarking to standard‐of‐care insulins. Each of these models can be applied or reapplied throughout the discovery and development of novel insulins. In various combinations, these models were used to address key questions in the early clinical strategy of MK‐2640 and the understanding of optimal PKPD properties for a therapeutically meaningful GRI. GRI, glucose‐responsive insulin; MID3, model‐informed drug discovery and development; PKPD, pharmacokinetic–pharmacodynamic; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus.
Early Clinical Strategy for MK‐2640
To progress MK‐2640 from discovery to first‐in‐human, the first question focused on establishing a relevant experimental medicine paradigm to test the glucose‐responsive clearance mechanism (PoM study). The second question focused on the predicted human PKPD profile for MK‐2640 based on preclinical data, in support of the dose rationale for the first‐in‐human study for healthy nondiabetic subjects (dose escalation, Part 1) and T1DM subjects (PoM, Part 2). To this end, a model‐based meta‐analysis of hyperinsulinemic glucose clamp clinical data was conducted to quantitatively characterize differences in RHI PKPD in T1DM compared with healthy nondiabetic subjects.23, 24 Characterization of the change in insulin action between healthy and diabetic subjects supported the dose setting for MK‐2640 in T1DM subjects based on PKPD observations in nondiabetics in Part 1. In addition, the University of Virginia / Padova University T1DM metabolic simulation platform (UVA/Padova T1DMS) was used, in a novel application of the platform, to explore the glucose dynamics of RHI under multiglycemic clamp conditions to support design of an operational feasibility study in preparation of the PoM clinical clamp study.25, 26 Furthermore, preclinical PKPD data in minipigs and dogs were analyzed through a translational PKPD model and yielded human PK and PD predictions for RHI and MK‐2640. All these analyses and predictions were included in the opening investigational new drug application in support of the dose rationale.27, 28
Developing an experimental medicine approach to test glucose‐responsive mechanism
As for any new insulin analog, first‐in‐human exploration of pharmacokinetics and pharmacodynamics, both with respect to steady‐state and time‐action, is performed in the hyperinsulinemic‐euglycemic glucose clamp procedure to lower the risk for subjects to experience hypoglycemia.29 The hyperinsulinemic clamp is an experimental platform for measuring insulin’s effect on whole body glucose disposal for characterizing a subject’s insulin resistance and insulin secretion both under euglycemic and hyperglycemic conditions.30, 31, 32, 33 The hyperinsulinemic clamp proceeds by infusing exogenous insulin and glucose at known rates into the systemic circulation to achieve target glucose concentrations. Endogenous insulin and glucose production are suppressed via either hyperinsulinemia, or somatostatin infusion, or otherwise can be accounted for through exogenous infusate labeling. For T1DM, endogenous insulin secretion is severely impaired due to the destruction of beta cells in the pancreas, and hence suppression is not required. At steady‐state clamp conditions, the total glucose input rate, which is the sum of the exogenous glucose infusion rate (GIR) plus the endogenous glucose production rate, equals the glucose output rate, called the whole‐body glucose disposal rate (GDR). At hyperinsulinemia, endogenous glucose production is often assumed to be suppressed, in which case GDR is approximately equal to GIR.34 Whole‐body GDR reflects both insulin and noninsulin mediated glucose disposal. Performing the hyperinsulinemic clamp at a range of insulin infusion rates establishes a concentration (dose)‐response curve between insulin concentration and glucose disposal, which can be used to characterize insulin action by its responsiveness and sensitivity. This is typically done at fixed normal glucose concentration (euglycemic conditions). Insulin responsiveness is measured as the maximal glucose uptake at a saturating insulin concentration, while insulin sensitivity is measured by the insulin concentration at half‐maximal response. Insulin resistance is characterized by a reduction in insulin responsiveness, sensitivity, or both.
To test MK‐2640’s glucose‐responsive clearance mechanism, the experimental clinical paradigm needs to include multiple glycemic levels (euglycemia and hyperglycemia) and be able to demonstrate glucose responsiveness when glucose changes in both directions. Therefore, quantitative understanding of the insulin‐glucose interaction at multiple glycemic levels was required. Although clamp studies have been conducted for more than 30 years, their key physiological results have not been aggregated or fully exploited to quantitatively understand the PKPD relationship and covariates such as patient population and characteristics. In a first step, we developed a joint PKPD model with the aim to describe and explain insulin PK and action at steady‐state based on a comprehensive study from Yki‐Jarvinnen.31 The developed model was physiologically plausible and could describe and explain insulin PK and action during the hyperinsulinemic clamp across a wide range of insulin infusion rates and glucose clamp levels (Figure 2, 23). Note that glucose disposal rate (glucose utilization) is dependent on both the glucose clamp level and the insulin infusion rate. This initial model described observations from a single study in T1DM subjects and hence did not allow for estimating population (nondiabetic vs. T1DM) differences. Therefore, we subsequently extended the analysis to a model‐based analysis utilizing 21 clinical study publications examining glucose disposal rates under different insulin infusion rates and steady‐state insulin and glucose concentrations to estimate population differences in insulin PK and resistance of subjects with T1DM compared with nondiabetic controls.24 In this analysis, the PK model’s insulin clearance was saturable, resulting in total clearance declining with increasing insulin concentrations. No population differences in insulin PK were identified, but a population difference was estimated for insulin responsiveness and sensitivity. T1DM subjects have a 1.5‐fold reduced insulin sensitivity and a 13% reduction in the insulin responsiveness compared with nondiabetic subjects for a given fixed glycemic target level (Figure 2, 24). This model was applied to calculate steady‐state infusion rates for both RHI and glucose for a proposed multiglycemic clamp study for RHI in T1DM subjects (Table 2).
Figure 2.
Understanding insulin/glucose nonlinearities in a clinical setting. Upper figure: We developed a joint PKPD model with the aim to describe and explain insulin PK (pharmacokinetics) and action based on a comprehensive study from Yki‐Jarvinnen,31 which studied 22 healthy male subjects at four porcine insulin infusion rates (0, 20, 60, 400 mU/minutes/m2) and four glucose clamp levels (90, 160, 250, 400 mg/dL). Details of model and results are provided in Fancourt et al.23 The PK of insulin is nonlinear and dependent on its concentration, but not on glucose concentration. The maximum effect on GDR (glucose disposal rate) but not potency of insulin is dependent on the glucose clamp concentration. PK (top panels) and PD (pharmacodynamics) (bottom panels) model fits to clinical clamp data. Open circles are data, closed circles are model predictions at measured values, and lines are model predictions at nominal values. Lower figure: VPC (visual predictive check) for final PD model from a model‐based meta‐analysis of 21 hyperinsulinemic glucose clamp clinical trials that was conducted to quantitatively characterize differences in standard insulin pharmacokinetics (PK) and glucose metabolism (PD) in T1DM (type 1 diabetes mellitus) patients compared with nondiabetics. Details of model and results for the RHI (regular human insulin) literature PKPD model are provided in Burroughs et al.24Conc, concentration; Glc, glucose; PKPD, pharmacokinetic–pharmacodynamic.
Table 2.
Summary of clinical predictions and observations for the multiglycemic clamp clinical study with RHI (MK‐0000‐339) in T1DM subjects based on translational PKPD, RHI meta‐analysis, and T1DMS modeling approaches25, 26, 27, 28
| Multiglycemic clamp study predictions | Steady‐state predictions | Predicted from minipig | Predicted from dog | Predicted from clinical clamp RHI meta‐analysis | Predicted from T1DMS | Observed in RHI multiglycemic clamp study (mean ± SD) |
|---|---|---|---|---|---|---|
| Interval 1: Glucose Infusion Rate is maintained at 5 mg/kg/minutes. A glucose clamp @ 200 mg/dL glucose is obtained by titrating Insulin infusion rate | Insulin conc (pM) | 170 | 112 | 195 (169–226) | 206 (109–529) | 220 ± 89 |
| Insulin clearance (mL/minutes/kg) | 11.1 | 19.9 | 15.1 | 16.5 | 15 ± 4 | |
| Interval 1 = Insulin infusion rate (pmol/kg/minutes) aiming @200 mg/dL glucose clamp | 1.9 | 2.2 | 2.9 (2.6–3.3) | 3.3 (1.1–7.4) | 3.4 ± 1.3 | |
| Interval 2 and 3: Insulin Infusion Rate is maintained from Interval 1. New glycemic levels are obtaind by titrating the glucose infusion rate | Interval 2 = Glucose infusion rate (mg/kg/minutes) aiming @75 mg/dL glucose clamp | 1.9 | 2.4 | 2.6 (2.2–3.0) | 2.1 (0–5.6) | 3.1 ± 1.3 |
| Interval 3 = Glucose infusion rate (mg/kg/minutes) aiming @300 mg/dL glucose clamp | 7.5 | 6.4 | 6.4 (5.5–7.4) | 6.6 (2.6–8.8) | 7.4 ± 1.9 |
RHI, regular human insulin; T1DMS, Type 1 Diabetes Simulator.
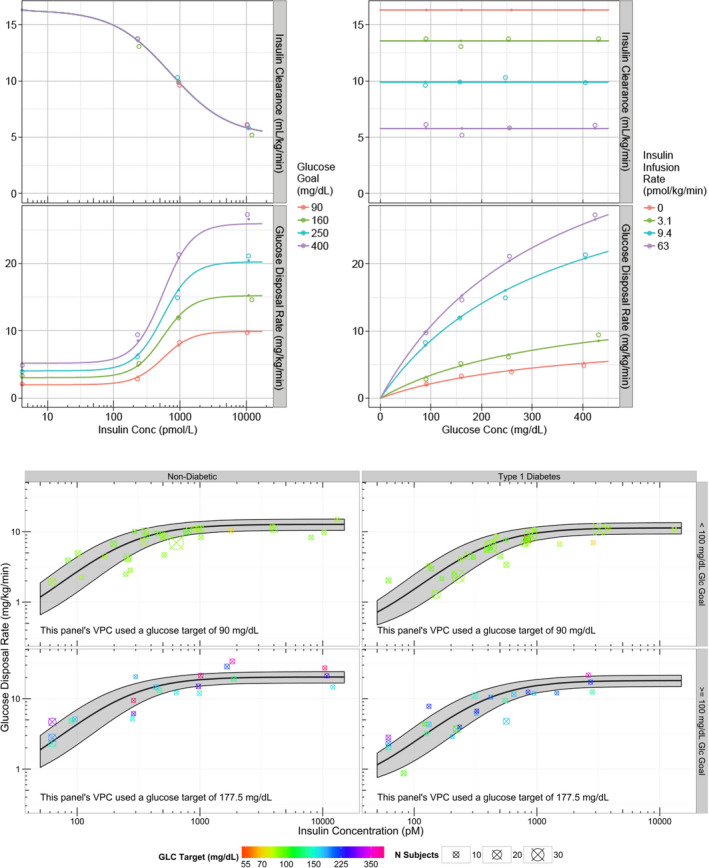
Before conducting the MK‐2640 PoM study, it was important to understand the operational feasibility of a multiglycemic clamp study that allows for reaching steady‐state within 3‐hour intervals at various glycemic levels and insulin infusion rates. Hence, a simulation tool was required that included temporal dynamics and could reflect clinical clamp controller procedures. To this end, the utility of the Uva/Padova Type 1 Diabetes Simulator (T1DMS, The Epsilon Group, Charlottesville, VA) was explored for designing clinical clamp studies through prediction of the glucose dynamics of RHI under multiglycemic clamp conditions. This T1DMS is a computer‐based model of human glucose–insulin dynamics.35, 36 This model was originally developed as a controller for subcutaneous insulin delivery via insulin pump, as part of closed‐loop control in T1DM. The model has been validated with observed results of clinical studies in T1DM subjects and has been accepted by the US Food and Drug Administration (FDA) as a substitute for preclinical trials to test the robustness of closed‐loop control algorithms for artificial pancreas systems.37 Here, it was explored for the first time whether this simulator could be useful for glucose clamp trial design and subsequently could serve as a benchmark for in silico comparison of a novel insulin against insulin. Through simulations, multiple variations of clamp durations and glycemic levels were explored, before finalization of the multiglycemic experimental medicine protocol for RHI (Figure 3).25 The experimental medicine clinical study (MK‐0000‐339) aimed to test the operational feasibility of multiglycemic clamp conditions. In this study, 12 T1DM subjects received infusions of RHI and glucose during a 9‐hour clamp study attaining three glucose levels (200, 75, and 300 mg/dL) each for a 3‐hour period. During period 1, the insulin infusion rate was varied, while the glucose infusion rate was fixed at 5 mg/kg/minutes, to attain a glucose clamp at 200 mg/dL. During periods 2 and 3, the insulin infusion rate was fixed at steady‐state obtained in period one, and subsequently the glucose infusion rate was varied to achieve the glucose targets of 75 and 300 mg/dL for period 2 and 3, respectively. Results of this study confirmed that both the T1DMS and the model‐based analysis could predict PK and PD steady‐state conditions for RHI (Table 2).25, 26 However, the study results also confirmed the simulation insights that it was more challenging to reach steady‐state within 3 hours when varying insulin infusion rate compared with changing glucose infusion rate. This led to the decision to simplify the clinical protocol for demonstrating PoM for MK‐2640 to attain only two glycemic levels with longer time to obtain steady‐state per level and removing the period in which insulin infusion rate was titrated.
Figure 3.
Establishing novel clinical experimental paradigm to demonstrate glucose responsive clearance mechanism. Upper figure: Simulations for insulin concentrations, insulin infusion rates, glucose concentrations, and glucose infusion rates for RHI study MK‐0000‐339 using the University of Virginia / Padova T1DM metabolic simulation platform.25 Lower figure: steady‐state observations from RHI study MK‐0000‐339 26 on model predictions from MBMA analysis,24 confirming that the model‐based analysis of steady‐state clamp data performed well in predicting steady‐state RHI PK (pharmacokinetics) and PD (pharmacodynamics). GIR, glucose infusion rate; MBMA, model‐based meta‐analysis; PID, proportional integral differential; RHI, regular human insulin; T1DM, type 1 diabetes mellitus.
Human dose predictions for MK‐2640 in healthy and T1DM subjects
To provide predictions for MK‐2640 PK and PD in humans, a translational PKPD modeling approach was developed that characterized the dynamic interaction of glucose and insulin following administration of MK‐2640 and RHI in minipigs and dogs with the ability to translate to humans. The model aimed to quantify the PK and PD parameters for RHI and MK‐2640, and their relative differences in diabetic (hyperglycemia) and nondiabetic (euglycemia) state in minipigs and under steady‐state conditions in dogs. Plasma exposure and blood glucose data were available from both diabetic (n = 90) and nondiabetic (n = 55) minipigs administered a single i.v. bolus injection of RHI or MK‐2640.20 In some of the MK‐2640 experimental arms, the MCR1 receptor was blocked using an infusion of α‐methyl mannose to investigate the influence of the MCR1 receptor on MK‐2640’s clearance.20 The dog data were generated in clamp‐studies in somatostatin‐infused healthy dogs (n = 48) at multiple glycemic clamp conditions.20 An integrated glucose‐insulin model, as first introduced by Silber,38 was modified to incorporate a glucose dependent MK‐2640 clearance mechanism and used to describe the minipig and dog PKPD data and allometrically scaled to man.27, 28, 39 This glucose‐dependent clearance mechanism was assumed to be a linear change in systemic clearance between low and high glucose concentrations. Both minipig and dog models predicted a ~ 30% lower MK‐2640 clearance at hyperglycemia (300 mg/dL) compared with euglycemia (75 mg/dL). In vitro, minipig, and dog predicted a similar (25, 17, and 40‐fold, respectively) decrease of MK‐2640 potency relative to RHI. The human RHI PKPD predictions from the translational PKPD model agreed with the pilot study data26 and the literature clinical RHI PKPD model,23, 24 indicating that insulin PK and PD was translated well from minipig and dog to man (Table 2, 27, 40) and yielded confidence for MK‐2640 clearance and PKPD relationship predictions.28
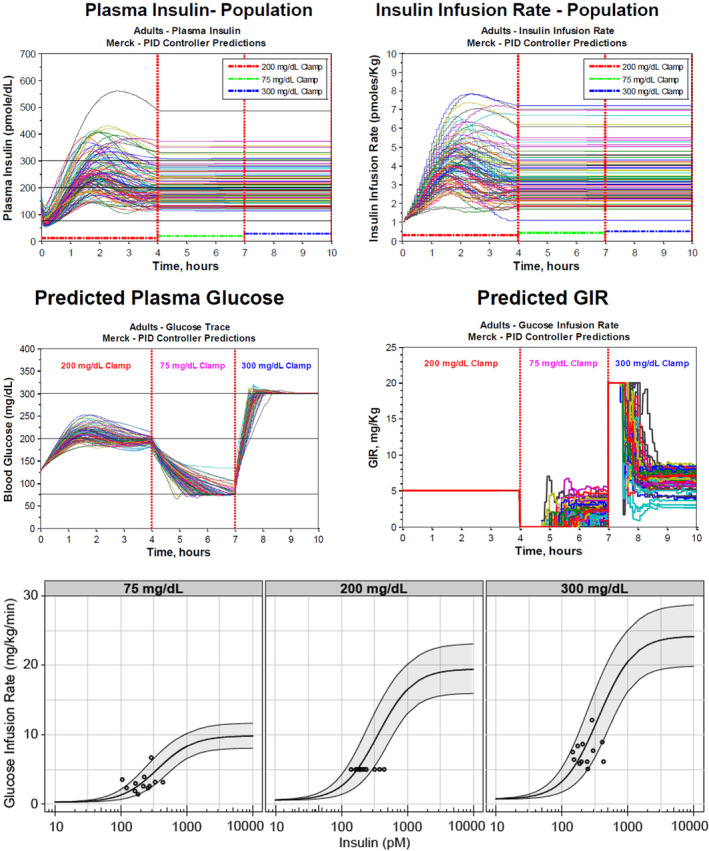
The MK‐2640 first‐in‐human study (ClinicalTrials.gov Identifier: NCT02269735) consisted of two parts: the first part was a rising dose study of i.v.‐administered MK‐2640 in healthy nondiabetic subjects, conducted under euglycemic clamp conditions, to evaluate safety and tolerability, PK, and PD. The goal of the second part was to demonstrate PoM for MK‐2640 in T1DM subjects. In this study, glucose‐responsiveness of i.v.‐administered MK‐2640, where MK‐2640 PK, measured as clearance, and PD, measured as GIR, was evaluated at euglycemia (90 mg/dL) vs. hyperglycemia (300 mg/dL) in comparison to RHI.21 During Part 1, the PK and PD for MK‐2640 was assessed at euglycemia. Real‐time modeling of the Part 1 data provided estimates of the MK‐2640 clearance and the relative potency difference against RHI in nondiabetics based on the literature clinical RHI PKPD model (Fancourt 2015). MK‐2640 clearance was found to be ~ twofold higher in humans than predicted from preclinical species. This finding is unexplained to date as we were not able to discriminate between experimental and mechanistic considerations without doing further studies. At higher MK‐2640 concentrations a saturation of MK‐2640 clearance was observed (Figure 3 in Krug et al.21), similar to RHI’s nonlinear clearance (Figure 2, left upper panel23) but right‐shifted by 25‐fold. This suggests that the saturation in MK‐2640 clearance, similar as for RHI, is driven by the insulin‐receptor mediated clearance.
MK‐2640 displayed similar maximal effect (insulin responsiveness) compared with RHI in nondiabetics. The relative potency (insulin sensitivity) difference between RHI and MK‐2640 was found to be ~ 25‐fold (Figure 4 upper panel), in similar range as estimated from preclinical species and in vitro measurements. For Part 2, the RHI infusion rate was set at 1.4 pmol/minutes/kg with a target GIR of 1.5 mg/kg/minutes to achieve a 90 mg/dL glucose concentration using the literature clinical RHI PKPD model. Based on the observed MK‐2640 clearance and potency difference in nondiabetics, the infusion rate for MK‐2640 was set in Part 2 in T1DM to 40 pmol/minutes/kg to reach equipotent steady‐state levels that can maintain a 90 mg/dL glucose clamp target. In this PoM assessment, MK‐2640 displayed a nonsignificant (6%) decrease in clearance at hyperglycemia vs. euglycemia,21 rather than the predicted 30% based on preclinical studies.28 Despite its PK limitations, MK‐2640 manifested at hyperglycemia vs. euglycemia a modest PD differentiation from regular human insulin, stimulating a greater increase in glucose infusion rate, reflecting that there may be a glucose‐dependent change in insulin action but that this is not reflected in systemic clearance change (Figure 4 lower panel). We found that insulin PK and insulin action scaled well between species (Table 2), confirming that preclinical animal models such as dogs and minipigs can be useful in the characterization of insulin action for novel insulins. In contrast, the interspecies differences for MK‐2640 with respect to glucose responsiveness and total clearance may be due to insufficient understanding of interspecies differences in the MRC1 receptor expression and function and the underlying tissue‐specific mechanism of action.21 Further experimental and integrative modeling work will be required to fully understand the mechanism of action that may explain the gap in interspecies translation.
Figure 4.
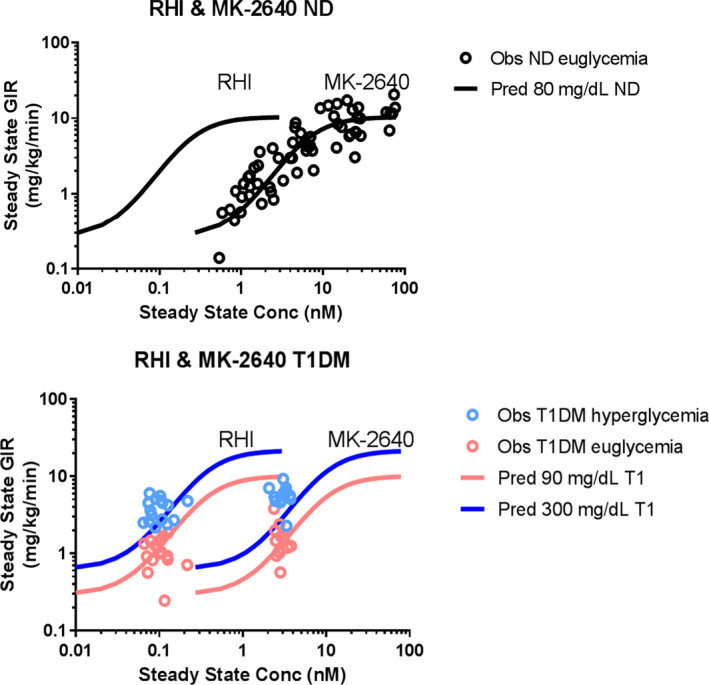
PKPD relationship for RHI and MK‐2640 in nondiabetics and T1DM subjects. Upper panel: Observed and predicted population relationship for concentration at steady‐state vs. glucose infusion rate (GIR) for RHI and MK‐2640 in healthy subjects. Lower panel: Observed and predicted population relationship for concentration at steady‐state vs. glucose infusion rate (GIR) for RHI and MK‐2640 in T1DM subjects for glucose target concentrations at euglycemia (90 mg/dL) and hyperglycemia (300 mg/dL). Observations for MK‐2640 and RHI are individual measurements in the first‐in‐human study.21 The predictions for RHI were derived from the RHI clinical literature model at euglycemia and hyperglycemia (Burroughs 2015) and for MK‐2640 with inclusion of a potency drop‐off of 25‐fold at a GIR of 5 mg/kg/minutes (projected half‐maximal effective concentration). The 25‐fold potency difference between RHI and MK‐2640 was initially based on in vitro estimates and later confirmed and estimated in the first‐in‐human study.21 All observations for RHI at both glycemic levels and the observations for MK‐2640 at euglycemia are predicted by the PKPD relationships. However, the observations for MK‐2640 for hyperglycemia are above the mean prediction line and illustrate that more insulin action is observed than what is predicted. These insulin effects would be expected at higher MK‐2640 concentrations (i.e., right‐shifted) by a systemic change in CL. The absence of a demonstrated systemic change in CL at hyperglycemia but with a larger observed insulin effect for the observed systemic concentration may suggest that tissue concentrations may be more relevant to study glucose responsiveness. CL, clearance; Conc, concentration; ND, nondiabetic; Obs, observed; PKPD, pharmacokinetic–pharmacodynamic; Pred, predicted; RHI, regular human insulin; T1DM, type 1 diabetes mellitus.
Understanding GRI compound requirements to offer therapeutic benefit for diabetic patients
In addition to the specific questions around MK‐2640 as described above, two more general questions focused on the target product profile for a GRI: i.e., what would be the required properties for a novel insulin to provide improved therapeutic benefit to patients over existing treatment options. The first question focused on the therapeutic relevance, i.e., how much reduction in hypoglycemia event rate would be required to make a meaningful therapeutic difference and allows for improvement over standard‐of‐care insulins? The second question entailed the compound requirements, i.e., what are the required PKPD properties for a GRI to be therapeutically relevant in diabetic patients for prandial and basal use? Through combination of model‐based meta‐analysis and quantitative systems pharmacology (QSP) modeling approaches, sets of properties were explored and benchmarked to standard‐of‐care insulins to understand what a transformational mealtime and basal insulin would be for both T1DM and T2DM subjects.
Demonstrating therapeutic benefit to T1DM subjects with an insulin with a glucose‐responsive clearance mechanism
In a first step, an exploratory meta‐analysis of efficacy (glycated hemoglobin A1c (HbA1c)) and safety (hypoglycemia) data for mealtime (prandial) insulins in T1DM subjects was conducted to quantify effect sizes for lispro and RHI as standard‐of‐care insulins.41 HbA1c and hypoglycemic event rates were quantified as change from baseline for RHI and lispro individually, and relative to each other while understanding the impact of design characteristics (covariates) such as fasting plasma glucose target, HbA1c baseline, run‐in period duration, and titration strategy. The meta‐analysis provided predictions for HbA1c and hypoglycemic event rates for RHI and lispro for a chosen trial design, such as a 13‐week trial with T1DM subjects, in which lispro demonstrated increased reduction in HbA1c and lower major hypoglycemic event rate compared with RHI, albeit not statistically significant.41 In silico predictions of HbA1c and hypoglycemic event rates for novel insulins, such as through the T1DMS as described below, can be benchmarked against these results for standard‐of‐care insulins. Such an approach can guide a proof of concept (PoC) trial design aiming for demonstration of superiority of novel glucose‐responsive mealtime insulins against standard of care.
An alternative approach to understanding therapeutic benefit was to modify the T1DMS.25, 28, 42 Although not previously used in the context of drug development, the T1DMS was modified through inclusion of a glucose‐responsive clearance that linearly decreased with plasma glucose over the range of 300–75 mg/dL, based on dog and minipig observations.20 First, to understand therapeutic index, continuous glucose profiles (N = 100) were predicted over 24 hours with meals at 7:00 am (50 g carbohydrates), noon (80 g), and 6:00 pm (80 g). Optimal dosing per virtual subject was provided for the first two meals and 125% of optimal dose was given with the third meal such that 50% of the virtual subjects experienced hypoglycemia (plasma glucose < 70 mg/dL). Combinations of glucose‐dependent clearance (0%, 15%, 30%, 50%, 70%), insulin potency, and absorption were simulated, predicting that T1DM virtual patients would experience less hypoglycemia with a glucose‐responsive insulin; i.e., glycemic control could be improved relative to RHI under conditions of increased dose requirements, and under the assumption that absorption rate is not altered (Figure 5, 42). Generally, the insulin apparent elimination after s.c. injection is absorption driven, even for fast‐acting mealtime insulins. Simulations demonstrated that improving the absorption rate for RHI into a faster acting insulin, such as lispro, was more impactful on improving glycemic control in mealtime settings than solely adding glucose‐responsive elimination to an RHI‐like insulin. Hence, we believe that a prandial GRI effect will allow for an improved therapeutic index from a non‐glucose‐responsive insulin if it comes with rapid absorption characteristics similar to or better than fast acting insulins (unpublished simulations). As described above, the T1DMS was used to accurately predict PKPD of RHI in multilevel glycemic clamps after i.v. administration (Table 2, 23). A similar simulation approach was used for the MK‐2640 PoM trial using the simulator with the implemented glucose‐responsive clearance. When comparing the emerging observations for MK‐2640 from the PoM study to T1DMS simulations, the predicted PD of MK‐2640 was consistent with observed for euglycemia.42 However, it was clear that predicting optimal dosing would require additional considerations for the mechanism of action to explain the presence of a PD effect in absence of clinical glucose‐responsive PK.
Figure 5.
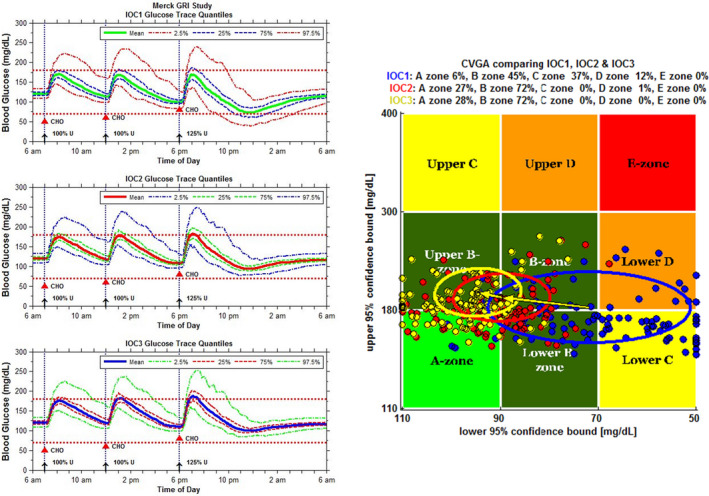
The University of Virginia / Padova University Type 1 diabetes mellitus human metabolic simulation platform (T1DMS) was used to predict therapeutic index of GRI.42 GRI action was simulated as glucose‐dependent CL decreasing linearly by 0% (IOC1, RHI), 30% (IOC2) and 50% (IOC3) over a plasma glucose range from 75 to 300 mg/dL. Virtual patients received s.c. bolus insulin unit doses optimized per virtual patient and 3 meals. In order to evaluate GRI therapeutic index relative to RHI, a 125% dose of regular human insulin in conjunction with the third meal was chosen to have 50% of individuals experience hypoglycemia (left, top graph). The mean glucose (green line) remains above 70 mg/dL plasma glucose with 95% confidence interval (red dashed curve) dropping below. The same regimen with a 30% glucose‐dependent CL GRI (left, middle graph) is predicted to prevent hypoglycemic excursions for all subjects. CVGA (right) for the simulation shows individuals treated with RHI (blue) experience overcorrection or failure to deal with hypoglycemia (zones lower C and D, respectively) whereas individuals treated with a 30% glucose‐dependent CL GRI (red) or 50% glucose‐dependent CL GRI (yellow) stay within accurate control (zone A) or benign deviations of control (zone B). GRI‐treated individuals trend to somewhat higher glycemia than RHI‐treated individuals. CHO, carbohydrate; CL, clearance; CVGA, control variability grid analysis; GRI, glucose‐responsive insulin; IOC1, insulin oligosasccharide conjugate 1; RHI, regular human insulin; s.c., subcutaneous; U, unit.
Demonstrating therapeutic benefit to T2DM subjects with an insulin with a glucose‐responsive clearance mechanism
A model‐based meta‐analysis of basal insulin effects in randomized clinical trials in T2DM subjects was conducted to quantify glycemic efficacy and safety of standard‐of‐care insulins, and to be able to benchmark predictions on long‐term glycemic control of a GRI to understand differentiation potential.43 To this end, a database of study‐level aggregate data from published clinical trials for basal insulins and GLP‐1 agonists in T2DM subjects was constructed. The hypoglycemic event rates were classified as minor events (symptomatic and nonsymptomatic: < 70 mg/dL), major events (requiring third party assistance), and nocturnal events (occurred between 0:00 and 6:00 am). Data for glucagon‐like peptide‐1 agonists were included in the analysis to add a greater volume of placebo reference data. A longitudinal model was used to describe HbA1c over time as function of dose and baseline (similar to Vaddady et al.44). Mean drug effect models for minor, nocturnal, and major hypoglycemia were developed and included exploration of a dose–response model. Inclusion of glucagon‐like peptide‐1 data enhanced stability of HbA1c and hypoglycemia model and precision of parameter estimates. HbA1c change data were well described by a longitudinal dose–response model with covariate effects for baseline status, background therapy, body weight, and Japanese race. Hypoglycemia rate data were described equally well by a mean drug effect model and dose–response model using glargine as reference treatment. The final models were simulated independently by sampling 1000 parameter sets from the final parameter estimates with uncertainties. This analysis demonstrated that at the same HbA1c response, degludec has slightly lower, detemir equal, and NPH higher hypoglycemia rates than glargine (Figure 6). This analysis enhanced understanding of the differentiation potential of novel basal insulin treatment options.
Figure 6.
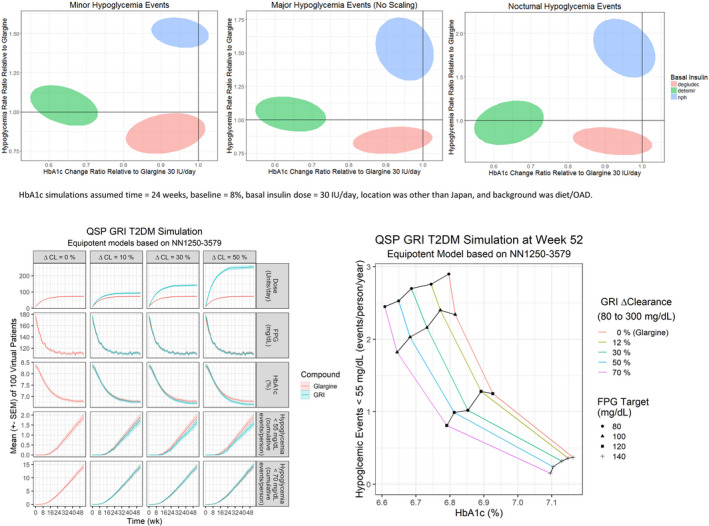
Differentiation potential for GRI from standard‐of‐care basal insulins. Upper panels: Basal insulin relationship between HBA1c change and minor, major, and nocturnal hypoglycemia events in randomized clinical trials in T2DM subjects.43 Bottom panels: Exploration of GRI through QSP simulations.47 Left: Performance of three GRIs with varying glucose responsiveness in comparison with glargine during a 52‐week simulated trial in T2DM subjects. Right: Performance of four GRIs with varying glucose responsiveness in T2DM subjects at week 52 for various FPG targets. CL, clearance; FPG, fasting plasma glucose; GRI, glucose‐responsive insulin; HBA1c, glycated hemoglobin A1c; IU, international units; NN1250‐3579, clinical trial comparing insulin degludec vs. insulin glargine (NCT00982644); OAD, oral antidiabetic; QSP, quantitative systems pharmacology; T2DM, type 2 diabetes mellitus.
Where for T1DM an existing simulation platform was used,36 for T2DM a simulator was developed within MSD. The MSD Diabetes QSP model represents important processes in glucose homeostasis using 11 physiological modules, six dynamic state variables, and 35 parameters.45 A curated database of PKPD data for subcutaneously administered insulins in clinical glucose clamp studies was modeled, affirming that lispro, RHI, and glargine time‐action profiles can be explained by the same structural PKPD model that was derived from intravenous clamp studies at steady‐state, and that these insulins only differ in their bioavailability and absorption rate.46 The subcutaneous PKPD insulin models were useful for the implementation of standard‐of‐care insulins into the T2DM simulator. The T2DM simulator was calibrated to the glargine arm of a 52‐week study in T2DM subjects using fasting plasma glucose, HbA1c, and confirmed hypoglycemia event rate (model glucose < 55 mg/dL) for 100 random virtual patients.47 A GRI was implemented with assumed equipotency to glargine, and a clearance with competitive glucose inhibition. The GRI glucose responsive clearance was changing 0% (glargine), 12%, 30%, 50%, or 70% between glucose of 80–300 mg/dL. In simulations, the GRI and glargine were titrated to a fasting plasma glucose target of 80, 100, 120, or 140 mg/dL in the same 100 virtual patients for 52 weeks. These simulations show that GRI outperformed glargine on both HbA1c and hypoglycemia (Figure 6): i.e., a GRI with a 50% glucose responsive clearance and a 120 mg/dL fasting plasma glucose achieved similar HbA1c (~ 6.8%) as glargine targeting a 80 mg/dL fasting plasma glucose, but had 1/3 lower hypoglycemia rate. Alternatively, GRI with a 70% glucose‐responsive clearance targeting a 80 mg/dL fasting plasma glucose achieved similar hypoglycemia (~ 2.5 events/person/year) as glargine with target 100 mg/dL fasting plasma glucose, but had 0.2% lower HbA1c. However, when average glucose levels drop, then higher doses of a GRI are required to maintain glucose homeostasis due to the increased clearance (Figure 6, lower left panel). In summary, these simulations demonstrated that a basal GRI with large changes in glucose‐responsive clearance has the potential to outperform glargine on both glucose lowering and reducing hypoglycemia, albeit at the expense of higher cost‐of‐goods.
Inference, Decisions, and Learnings
What have we learned through this MID3 approach and what decisions were impacted? As described above, we have developed a suite of models (PKPD, QSP, and MBMA) based on a wealth of available literature and in‐house–generated data, including insulin pharmacology, preclinical and clinical data on RHI as active comparator, and long‐term outcome data for standard‐of‐care insulins in T1DM and T2DM from randomized clinical trials, and emerging (pre) clinical data for MK‐2640. These models allowed us to simulate and explore the clinical trial paradigm, predict and set doses for first‐in‐human MK‐2640 trial, and understand the critical compound properties relevant for therapeutic benefit and to allow differentiation from standard‐of‐care treatment. These areas of inference and decisions will be further discussed below.
The first set of inferences is centered around quantifying insulin PKPD behavior at steady‐state and in dynamic conditions to design a trial paradigm to study the glucose‐responsive clearance mechanism. To our knowledge, a detailed quantitative characterization of insulin resistance, in terms of both responsiveness and sensitivity in clamp studies, has been lacking. Kaul et al.48 summarized the literature on insulin resistance in T1DM and concluded that insulin resistance is an integral feature of T1DM but did not estimate a numerical value. Donga et al.49 published a meta‐analysis of insulin sensitivity in clamp studies of T1DM subjects vs. healthy controls and found that insulin resistance was increased in T1DM subjects. However, their approach did not differentiate between insulin responsiveness and sensitivity, nor did it consider the differences in insulin infusion rate or glucose clamp levels in the studies, limiting the interpretation and preventing any predictions. Results of our model‐based analysis of clinical insulin steady‐state clamp data24 agree with other reports using tracer data that were published after our analysis.50 The initial analysis on the Yki Yarvinnen study31 is within the prediction range for the T1DM population.23, 24 This work provided a quantitative framework around RHI PK and PD and allowed for the prediction of insulin effects at various glycemic clamp targets and under various insulin infusion regimens and translation between healthy and T1DM subjects. As such, it can be used as reference to understand potential for differentiation based on differences in PK and/or PD for novel insulins and for design of clinical hyperinsulinemic clamp studies. In fact, the steady‐state RHI clamp model has been used to compare the translational predictions based on RHI PKPD in minipig in acute bolus injection setting and in dog under steady‐state clamp conditions (Table 2, 25) and played a role in the subsequent optimization of the translational predictions from minipigs.40 The work has enabled the design and analysis of a clinical RHI pilot study to assess the operational feasibility of a three‐period clamp study.26 More importantly, it was used to quantify the differences for a novel glucose responsive insulin and dose setting for the PoM part of the first‐in‐human study21 in T1DM subjects, based on results in nondiabetic subjects. Finally, the model was implemented in a QSP model for diabetes.46, 47 As limitations, one could consider that the model was built on a selection of references from the literature with a focus on reports that included both T1DM subjects and healthy subjects and multiple glucose and insulin infusion rates. Many more reports exist on hyperinsulinemic glucose clamp data, therefore the between‐study variability might be larger than estimated here. Also, the model is built on meta‐level steady‐state data, and thus cannot predict glucose/insulin over time on an individual basis. For that latter reason, the utility of the T1DMS was explored. The application of the T1DMS for prospective simulation of clamp studies was new and untested. However, the robustness of the predictions of RHI and glucose time‐course under various perturbations compared with the pilot study results was encouraging,25 thereby laying the foundation for model‐informed testing of modified insulins or insulin mimetic therapeutic agents and mechanistic, hypothesis‐generating interpretation of their clinical effects.42 Also, this quantitative simulation framework aids the understanding of opportunities and challenges for various prandial and basal PoM clinical trial designs and enables extrapolation from steady‐state (i.v. infusions) clamp conditions to dynamic (s.c. injections) designs. While this extrapolation from clamp to s.c. setting was demonstrated for regular insulins, we could not confirm this for MK‐2640 for the reason that a clinical s.c. study was not feasible due to high dose (injection volume) and slow absorption characteristics of the clinical formulation.
The second set of inferences is around translatability of insulin and GRI effects across species to guide dose selection. Translational PKPD models for insulin action and glucose‐responsive mechanism were developed based on animal data to predict the human PKPD profile of MK‐2640 and the comparator RHI.26, 27, 40 Emerging results from the first‐in‐human study with MK‐2640 demonstrated that the dog and minipig translational PKPD models accurately predicted the potency difference between MK‐2640 and RHI as well as RHI’s PK and PD properties. However, disappointingly, no significant glucose‐dependent change in clearance was demonstrated in humans, though a glucose‐dependent augmentation of the glucose infusion rate as PD marker was observed.21 This finding of MK‐2640 resonates with the recently reported two‐stage (euglycemia and hyperglycemia) clamp study in dogs that used portal venous, hepatic vein, and hepatic arterial catheterization to ascertain PKPD across the hepatic bed of a similar GRI analog to MK‐2640.51 Moore51 delineated that hepatic actions were the predominant contributors to glucose‐responsive PD effects of the GRI. These findings warrant further development of more mechanistic translational and physiologically‐based pharmacokinetic models that include the MRC1 biology, the tissue‐specific (hepatic) effects, and any differences between dog and man as can be studied in tracer studies.52 Expanding the T1DM simulator with more physiologically‐based pharmacokinetic and mechanistic tissue information on MRC1 expression may be a suitable option.
The third domain of inference comprises the learnings on the PKPD and long‐term glycemic efficacy and safety of standard‐of‐care insulins. For oral antidiabetic treatments, model‐based meta‐analysis of outcome data from randomized clinical trials have been reported that quantify the differentiation between antidiabetic drug classes at their therapeutic dose and to predict outcomes from early markers, such as fasting plasma glucose.44, 53, 54 Here, comparator models, based on model‐based meta‐analyses of randomized clinical trials in T1DM and T2DM, were developed to quantify glycemic efficacy and safety of standard‐of‐care insulins, and to be able to benchmark predictions on long term glycemic control of a GRI to understand its differentiation potential.41, 43 To our knowledge this is the first time that such analysis was applied to insulins in mealtime and basal setting. These analyses provided insights that the incremental benefit of optimizing PK and PD properties indeed is accompanied with incremental benefit in long‐term glycemic control and reductions in hypoglycemic events. However, demonstration of superiority requires substantial improvements in PKPD properties of novel insulins combined with large sample sizes to demonstrate this in clinical trials.
The fourth inference was around the compound properties of a GRI that would lead to clinically meaningful differences compared with existing treatment. Two quantitative systems pharmacology models—T1DMS and MSD T2DM Diabetes simulator—were used to explore the therapeutic relevance of GRI PKPD properties through prediction of long‐term outcome in both T1DM and T2DM population and to support early clinical strategy through trial design explorations.25, 42 In addition, meta PKPD analyses of intravenous and subcutaneous clinical clamp studies of the comparator insulins were performed to inform the standard‐of‐care insulins implementation in the T2DM QSP model, and to bridge from i.v. to s.c. dosing.23, 24, 46 It was shown that for RHI, lispro, and glargine, the PKPD can be explained by the same structural model with only differences in bioavailability and absorption properties. Simulations demonstrated that for both mealtime and basal insulins, improvements in glycemic control can be derived when a substantial (more than 50% change of clearance over diabetic glucose range) glucose‐responsive clearance change is present. However, this will come at an increased dose demand (increase in cost of goods) as the GRI is cleared faster at hyperglycemia. Embedding a T1DMS in‐house was important to provide a quick turnaround between trial design ideas in the team and in silico assessment of the feasibility of these ideas. One key learning was that in the prandial setting, improving absorption rate for mealtime insulins is more impactful than adding a glucose‐responsive clearance mechanism without the disadvantage of increasing the dose demand. Still, in clinical practice, demonstrating true clinical benefit for incrementally improved absorption rate (for example, comparing incremental improvements in absorption rate for rapid acting and inhaled insulins) is challenging.41, 55, 56 Comparing T2DM simulations with the information from the T2DM comparator model enabled a more refined understanding of the properties required for backup molecules to demonstrate therapeutically relevant improvements in the basal setting, thereby helping in framing a clinical strategy for the development of basal GRIs. Moreover, the developed QSP models can be further used to generate hypothesis for improved mechanistic understanding of the clinical findings. This will require the implementation of more physiologically‐based pharmacokinetic models that allow for expression of tissue‐specific MRC1 effects. Overall, the combination of MBMA results and the QSP simulators enabled our understanding of the required reduction in hypoglycemia event rate that would constitute a meaningful therapeutic improvement over standard‐of‐care insulins in both prandial and basal setting. In addition, the models allowed for simulations of PoC trial designs that would demonstrate superiority in both T1DM and T2DM patient populations. This begs the question of how to develop a transformational insulin that meets these criteria. Development of such a novel insulin presents a drug discovery challenge that we believe should be solved or tackled with an iterative learning‐confirming modeling and simulation approach.
Decisions that were made during the early development of MK‐2640 based on the inferences discussed above were as follows: the first decision was to simplify clinical protocol for GRI from three to two glycemic states with confidence that simulations could predict the RHI arm and explore effect sizes for MK‐2640 based on its predicted properties. The second decision was around the dose setting for both RHI and MK‐2640 for the PoM part of the first‐in‐human trial. An equivalent dose was required to maintain glucose at euglycemia, which could not have been done without the modeling as no insulin titration was allowed. The third decision was to discontinue MK‐2640 without further clinical exploration and shift focus to renewed discovery efforts. The limited glucose responsiveness, low potency (which led to a large dose and volume for prandial use), and the inferior absorption rate for the clinical formulation for such high dose, were all predicted to lead to inferiority in a PoC trial against a fast‐acting mealtime insulin. The final decision was to shift the project strategy from prandial to basal application of future GRI candidates, as this shift was considered to faster lead to a proof‐of‐concept molecule.
Beyond these inferences and decisions, some additional learnings can be noted. It was critical to make investments in developing these tools proactively and in time to allow for maximal impact on learnings and decisions. In early drug development when the program direction shifts rapidly as soon as in vitro and preclinical data are generated, this may not be easy. Hence, it is important to anticipate a program’s needs and to develop a quantitative development plan, detailing how these models can provide inferences and support decisions.22 Also, investments in model and platform development such as comparator and QSP models should be assessed at the disease level, rather than being compound specific, due to the opportunity to create incremental benefit and return on investment when applying to more programs.57 While these models require a substantial investment up front, the impact on improved trial design and program probability of success is warranted. Moreover, these models can be used across novel insulin programs with adaptations that would be specific for the underlying mechanisms. Here, the comparator and QSP models not only provided value for the GRI program but also were used for assessing therapeutic relevance assessment for other modified insulin programs with other mechanisms of action and allowed for program prioritization in the diabetes disease area portfolio. Importantly, these models can be useful for a rapid quantitative assessment of novel molecules (such as assessing in‐licensing opportunities) or to generate hypotheses on what could become a transformational insulin treatment. Moreover, these models could be used for exploring titration schemes and dose‐to‐unit conversions as is important in the insulin space. It is imperative, however, that these models are further developed with respect to emerging mechanistic understanding and emerging clinical data. Hence, maintaining proper documentation and ensuring operational knowledge of these models are prerequisites for further value.
As noted elsewhere, successful MID3 can only happen when there is full support within the project team, combined with a pull from decision makers.58 This requires building confidence and trust with respect to what the models can do and cannot do. With respect to stakeholder management, building trust was achieved through incremental model qualification with emerging data and across models. For example, the clinical PKPD and outcome data of standard‐of‐care insulins like RHI and glargine in the literature were compared against various model‐based predictions using the translational model, PKPD comparator models, and QSP simulators and were shown to be very consistent. The value of a comprehensive quantitative understanding and availability of various modeling tools were recognized by other novel insulin development teams. Other novel insulin project teams, who were familiar with the modeling approaches described here, embraced these tools and thus could make decisions in a more informed and efficient manner. An observed benefit was that the models enabled translation of information between the preclinical and clinical project teams, visualized gaps in understanding, and enabled the organization to focus on strategy that could bring differentiated therapies to diabetes patients.
Conclusions
The MID3 strategy using a suite of modeling tools informed the early clinical strategy for the glucose‐responsive insulin program, supported a novel clinical trial design to assess glucose responsiveness, and allowed prioritization against other modified insulin approaches. Moreover, it allowed for rapid, quantitative decision making by leveraging rich published data with emerging clinical data. Finally, the MID3 approach identified critical gaps in translational understanding of the glucose‐responsive mechanism. We envision that this case study can serve as a blueprint for implementation of MID3 in early clinical development, and that these developed models and the derived insulin knowledge base could be used or reused in support of discovering and developing novel insulin treatment options.
Funding
This research was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
Conflict of Interest
All authors are or were full‐time employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA at the time of the research and may own stock/stock options in Merck & Co., Inc., Kenilworth, NJ, USA.
Acknowledgments
We sincerely thank our (former) colleagues in the Discovery GRI team, the clinical MK‐2640 team, and the GRI M&S team for all discussions and contributions over the years. Also, we gratefully acknowledge the contributions from many collaborators at The Epsilon Group, Pharmetheus, qPharmetra, LAP&P, and Quantitative Solutions to the various modeling projects. A fair representation of individual contributions can be derived from the abstract publication references (as can be found in the supplemental material). Finally, we would like to express our gratitude to our management and the MSD decision makers who enabled this work through both supporting investments and recognition of its value.
References
Note to the reader: references 23, 24, 25, 26, 27, 28, 40, 41, 42, 43, 46, 47 are abstracts for scientific conferences such as ACOP, PAGE and ASCPT. Pdf copies of the associated posters presented at those meetings can be obtained from the corresponding author.
- 1. Zaykov, A.N. , Mayer, J.P. & DiMarchi, R.D. Pursuit of a perfect insulin. Nat. Rev. Drug Discov. 15, 425–439 (2016). [DOI] [PubMed] [Google Scholar]
- 2. Home, P.D. The pharmacokinetics and pharmacodynamics of rapid‐acting insulin analogues and their clinical consequences. Diabetes Obes. Metab. 14, 780–788 (2012). [DOI] [PubMed] [Google Scholar]
- 3. Cahn, A. , Miccoli, R. , Dardano, A. & Del Prato, S. New forms of insulin and insulin therapies for the treatment of type 2 diabetes. Lancet Diabetes Endocrinol. 3, 638–652 (2015). [DOI] [PubMed] [Google Scholar]
- 4. Heise, T. , Zijlstra, E. , Nosek, L. , Heckermann, S. , Plum‐Mörschel, L. & Forst, T. Euglycaemic glucose clamp: what it can and cannot do, and how to do it. Diabetes Obes. Metab. 18, 962–972 (2016). [DOI] [PubMed] [Google Scholar]
- 5. Anderson, J.H. Jr , Brunelle, R.L. , Koivisto, V.A. , Trautmann, M.E. , Vignati, L. & DiMarchi, R. Improved mealtime treatment of diabetes mellitus using an insulin analogue. Multicenter Insulin Lispro Study Group. Clin. Ther. 19, 62–72 (1997). [DOI] [PubMed] [Google Scholar]
- 6. Owens, D.R. , Matfin, G. & Monnier, L. Basal insulin analogues in the management of diabetes mellitus: what progress have we made? Diabetes Metab. Res. Rev. 30, 104–119 (2014). [DOI] [PubMed] [Google Scholar]
- 7. Frier, B.M. Hypoglycaemia in diabetes mellitus: epidemiology and clinical implications. Nat. Rev. Endocrinol. 10, 711–722 (2014). [DOI] [PubMed] [Google Scholar]
- 8. Cryer, P.E. Severe hypoglycemia predicts mortality in diabetes. Diabetes Care 35, 1814–1816 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seaquist, E.R. et al Hypoglycemia and diabetes: a report of a workgroup of the American Diabetes Association and the Endocrine Society. Diabetes Care 36, 1384–1395 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Veiseh, O. , Tang, B.C. , Whitehead, K.A. , Anderson, D.G. & Langer, R. Managing diabetes with nanomedicine: challenges and opportunities. Nat. Rev. Drug Discov. 14, 45–57 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang, R. et al A glucose‐responsive insulin therapy protects animals against hypoglycemia. JCI Insight 3, 97476 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brownlee, M. & Cerami, A. A glucose‐controlled insulin‐delivery system: semisynthetic insulin bound to lectin. Science 206, 1190–1191 (1979). [DOI] [PubMed] [Google Scholar]
- 13. Brownlee, M. & Cerami, A. Glycosylated insulin complexed to concanavalin A. Biochemical basis for a closed‐loop insulin delivery system. Diabetes 32, 499–504 (1983). [DOI] [PubMed] [Google Scholar]
- 14. Chou, D.H. et al Glucose‐responsive insulin activity by covalent modification with aliphatic phenylboronic acid conjugates. Proc. Natl. Acad. Sci. USA 112, 2401–2406 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Traitel, T. , Cohen, Y. & Kost, J. Characterization of glucose‐sensitive insulin release systems in simulated in vivo conditions. Biomaterials 21, 1679–1687 (2000). [DOI] [PubMed] [Google Scholar]
- 16. Webber, M.J. & Anderson, D.G. Smart approaches to glucose‐responsive drug delivery. J. Drug Target. 23, 651–655 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu, Q. , Wang, L. , Yu, H. , Wang, J. & Chen, Z. Organization of glucose‐responsive systems and their properties. Chem. Rev. 111, 7855–7875 (2011). [DOI] [PubMed] [Google Scholar]
- 18. Yu, J. et al Microneedle‐array patches loaded with hypoxia‐sensitive vesicles provide fast glucose‐responsive insulin delivery. Proc. Natl. Acad. Sci. USA 112, 8260–8265 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang, R. et al A glucose‐responsive insulin therapy protects animals against hypoglycemia. JCI Insight 3, e97476 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaarsholm, N.C. et al Engineering Glucose Responsiveness Into Insulin. Diabetes 67, 299–308 (2018). [DOI] [PubMed] [Google Scholar]
- 21. Krug, A.W. et al Clinical evaluation of MK‐2640: an insulin analog with glucose‐responsive properties. Clin. Pharmacol. Ther. 105, 417–425 (2019). [DOI] [PubMed] [Google Scholar]
- 22. EFPIA MID3 Workgroup et al Good practices in model‐informed drug discovery and development: practice, application, and documentation. CPT Pharmacometrics Syst. Pharmacol. 5, 93–122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fancourt, C. , Valiathan, C. , Tatosian, D. , Cho, C. & Visser, S.A.G. Development of a joint PKPD model of the hyperinsulinemic glucose clamp. J. Pharmacokinet. Pharmacodyn. 42, S11–S107 (2015). [Google Scholar]
- 24. Burroughs, E. , Fancourt, C. , Dykstra, K. & Visser, S.A.G. A model‐based meta‐analysis of insulin PK‐PD in glucose clamp studies of diabetes mellitus type 1 and non‐diabetic human subjects. J. Pharmacokinet. Pharmacodyn. 42, S79–S80 (2015). [Google Scholar]
- 25. Fancourt, C. et al Using T1DMS simulation for the conceptualization and design of a clinical clamp studies in the development of modified insulin therapeutic agents. Part 1 17th Annual Diabetes Technology Meeting, Rockville, MD, November 2–4, 2017. 10.1177/1932296818761742 [DOI]
- 26. Kandala, B. et al A model‐based analysis of a multi‐level glycemic clamp regular human insulin study in T1DM patients. J. Pharmacokinet. Pharmacodyn. 42, S11–S107 (2015). [Google Scholar]
- 27. Visser , S.A.G. et al. Translational modelling of regular human insulin pharmacokinetics and glucose dynamics in minipig and dog. Twenty‐fourth meeting, Population Approach Group Europe, Hersonissos, Greece, June 2–5, 2015. Abstract 3490 <www.page-meeting.org/?abstract=3490 >.
- 28. Kandala, B. et al. Translational modeling of minipig and dog glucose and insulin data accurately predicts human insulin action for MK2640 but not its glucose‐responsive pharmacokinetics. Clin. Pharmacol. Ther. 103, S71 (2018). [Google Scholar]
- 29. Heise, T. & Mathieu, C. Impact of the mode of protraction of basal insulin therapies on their pharmacokinetic and pharmacodynamic properties and resulting clinical outcomes. Diabetes Obes. Metab. 19, 3–12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. DeFronzo, R.A. , Tobin, J.D. & Andres, R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am. J. Physiol. 237, E214–E223 (1979). [DOI] [PubMed] [Google Scholar]
- 31. Yki‐Järvinen, H. , Young, A.A. , Lamkin, C. & Foley, J.E. Kinetics of glucose disposal in whole body and across the forearm in man. J. Clin. Invest. 79, 1713–1719 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tam, C.S. , Xie, W. , Johnson, W.D. , Cefalu, W.T. , Redman, L.M. & Ravussin, E. Defining insulin resistance from hyperinsulinemic‐euglycemic clamps. Diabetes Care. 35, 1605–1610 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shankar, S.S. , Shankar, R.R. , Railkar, R.A. , Beals, C.R. , Steinberg, H.O. &Kelley D.E. Early Clinical detection of pharmacologic response in insulin action in a nondiabetic insulin‐resistant population. Curr. Ther. Res. Clin. Exp. 14, 83–89 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rizza, R.A. , Mandarino, L.J. & Gerich, J.E. Dose‐response characteristics for effects of insulin on production and utilization of glucose in man. Am. J. Physiol. 240, E630–E639 (1981). [DOI] [PubMed] [Google Scholar]
- 35. Dalla Man, C.D. , Rizza, R.A. & Cobelli, C. Meal simulation model of the glucose‐insulin system. IEEE Trans. Biomed. Eng. 54, 1740–1749 (2007). [DOI] [PubMed] [Google Scholar]
- 36. Dalla Man, C. , Micheletto, F. , Lv, D. , Breton, M. , Kovatchev, B. & Cobelli, C. The UVA/PADOVA Type 1 diabetes simulator new features. J. Diabetes Sci. Technol. 8, 26–34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Visentin, R. , Dalla Man, C. , Kovatchev, B. & Cobelli, C. The university of Virginia/Padova type 1 diabetes simulator matches the glucose traces of a clinical trial. Diabetes Technol. Ther. 16, 428–434 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Silber, H.E ., Jauslin, P.M. , Frey, N. , Gieschke, R. , Simonsson, U.S. & Karlsson, M.O. An integrated model for glucose and insulin regulation in healthy volunteers and type 2 diabetic patients following intravenous glucose provocations. J. Clin. Pharmacol. 47, 1159–1171 (2007). [DOI] [PubMed] [Google Scholar]
- 39. Alskär, O. , Karlsson, M.O. & Kjellsson, M.C. Model‐based interspecies scaling of glucose homeostasis. CPT Pharmacometrics Syst. Pharmacol. 6, 778–786 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lu, Y. et al. Translational modeling of regular human insulin pharmacokinetics and glucose dynamics in minipigs. J. Pharmacokinet. Pharmacodyn. 42, S28 (2015). [Google Scholar]
- 41. Kandala, B. et al. Meta‐analysis of HbA1c and hypoglycemic event rates of meal‐time insulins in randomized clinical trials in T1DM subjects: exploration of POC trial design options. J. Pharmacokinet. Pharmacodyn. 44, S11–S143 (2017). [Google Scholar]
- 42. Cho, C.R. et al. Using T1DMS simulation for the conceptualization and design of clinical clamp studies in the development of modified insulin therapeutic agents. Part II – MK‐2640. Clin. Pharmacol. Ther. 105, S90 (2019). [Google Scholar]
- 43. Vaddady, P. et al. Differentiation of basal insulins in randomized clinical trials in T2DM subjects: a model‐based meta‐analysis of HbA1c and hypoglycemic event rates. J. Pharmacokinet. Pharmacodyn. 44, S11–S143 (2017). [Google Scholar]
- 44. Vaddady, P. et al A comprehensive model‐based meta‐analysis (MBMA) of diabetes studies in type 2 diabetes mellitus patients to quantify the relationship between HbA1c and fasting plasma glucose. Twenty‐sixth meeting, Population Approach Group Europe, Budapest, Hungary, June 6–9, 2017. Abstract 7301 <www.page-meeting.org/?abstract=7301 >.
- 45. Hagen, D. , Jajamovich, G.H. , Fancourt, C. , Topp, B. & Truijllo, M.E. Predicting phase III efficacy and safety in insulin response using QSP modeling. J. Pharmacokinet. Pharmacodyn. 44, S11–S143 (2017). [Google Scholar]
- 46. Fancourt, C. , Lommerse, J. , Kandala, B. , Kerbusch, T. & Visser, S.A.G. A PKPD model‐based meta‐analysis of subcutaneously administered insulins in clinical glucose clamp studies. J. Pharmacokinet. Pharmacodyn. 43, S1–122 (2016). [Google Scholar]
- 47. Fancourt, C. , Visser, S.A.G. , Trujillo, M. & Topp, B. A quantitative systems pharmacology model of glucose responsive insulin. ACOP 2017. J. Pharmacokinet. Pharmacodyn. 44, S11–S143 (2017). [Google Scholar]
- 48. Kaul, K. , Apostolopoulou, M. & Roden, M. Insulin resistance in type 1 diabetes mellitus. Metabolism 64, 1629–1639 (2015). [DOI] [PubMed] [Google Scholar]
- 49. Donga, E. , Dekkers, O.M. , Corssmit, E.P. & Romijn, J.A. Insulin resistance in patients with type 1 diabetes assessed by glucose clamp studies: systematic review and meta‐analysis. Eur. J. Endocrinol. 173, 101–109 (2015). [DOI] [PubMed] [Google Scholar]
- 50. Bizzotto, R. et al. Glucose uptake saturation explains glucose kinetics profiles measured by different tests. Am. J. Physiol. Endocrinol. Metab. 311, E346–E357 (2016). [DOI] [PubMed] [Google Scholar]
- 51. Moore, M.C. et al Superior glycemic control with a glucose‐responsive insulin analog: hepatic and nonhepatic impacts. Diabetes 67, 1173–1181 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Trujillo, M.E. et al. Semi‐mechanistic modeling of dog tracer study data to quantify insulin action on glucose disposal rate and inhibition of lipolysis. ACOP 2017. J. Pharmacokinet. Pharmacodyn. 44, S11–S143 (2017). [Google Scholar]
- 53. Liu, J. et al Prediction of long‐term efficacy of antidiabetic drugs on HbA1c using longitudinal model‐based meta‐analysis (MBMA) of literature data. Clin. Pharmacol. Ther. 99, 49 (2016).26509246 [Google Scholar]
- 54. Maloney, A. , Rosenstock, J. & Fonseca, V. A model‐based meta‐analysis of 24 antihyperglycemic drugs for type 2 diabetes: comparison of treatment effects at therapeutic doses. Clin. Pharmacol. Ther. 105, 1213–1223 (2019). [DOI] [PubMed] [Google Scholar]
- 55. van Bon, A.C. , Bode, B.W. , Sert‐Langeron, C. , DeVries, J.H. & Charpentier, G. Insulin glulisine compared to insulin aspart and to insulin lispro administered by continuous subcutaneous insulin infusion in patients with type 1 diabetes: a randomized controlled trial. Diabetes Technol. Ther. 13, 607–614 (2011). [DOI] [PubMed] [Google Scholar]
- 56. Klonoff, D.C. Afrezza inhaled insulin: the fastest‐acting FDA‐approved insulin on the market has favorable properties. J. Diabetes Sci. Technol. 8, 1071–1073 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Visser, S.A.G. , De Alwis, D.P. , Kerbusch, T. , Stone, J.A. & Allerheiligen, S.R. Implementation of quantitative and systems pharmacology in large pharma. CPT Pharmacometrics Syst. Pharmacol. 3, e142 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nayak, S. et al Getting innovative therapies faster to patients at the right dose: impact of quantitative pharmacology towards first registration and expanding therapeutic use. Clin. Pharmacol. Ther. 103, 378–383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]