

Abstract
Antiretroviral therapy during pregnancy reduces the risk of vertical HIV‐1 transmission. However, drug dosing is challenging as pharmacokinetics (PK) may be altered during pregnancy. We combined a pregnancy physiologically‐based pharmacokinetic (PBPK) modeling approach with data on placental drug transfer to simulate maternal and fetal exposure to dolutegravir (DTG). First, a PBPK model for DTG exposure in healthy volunteers was established based on physiological and DTG PK data. Next, the model was extended with a fetoplacental unit using transplacental kinetics obtained by performing ex vivo dual‐side human cotyledon perfusion experiments. Simulations of fetal exposure after maternal dosing in the third trimester were in accordance with clinically observed DTG cord blood data. Furthermore, the predicted fetal trough plasma concentration (Ctrough) following 50 mg q.d. dosing remained above the concentration that results in 90% of viral inhibition. Our integrated approach enables simulation of maternal and fetal DTG exposure, illustrating this to be a promising way to assess DTG PK during pregnancy.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Pregnancy is associated with a variety of physiological and anatomic changes that can alter maternal antiretroviral pharmacokinetics (PK). Placental transfer of antiretrovirals may cause fetal toxicity or have beneficial effects as antenatal prophylaxis.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Can fetal exposure to dolutegravir (DTG) be simulated by incorporating ex vivo placental drug transfer data in a pregnancy physiologically‐based pharmacokinetic (PBPK) model?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Model simulations adequately captured maternal and fetal DTG exposure during late pregnancy. A dose of 50 mg DTG q.d. was predicted to result in sufficiently high maternal plasma levels during pregnancy to achieve viral suppression and provide fetal prophylaxis.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Simulation of maternal and fetal DTG PK by pregnancy‐PBPK modeling is a valuable tool to guide maternal dosing.
Dolutegravir (DTG) is being adopted as the preferred first‐line treatment for HIV infection.1, 2 DTG‐containing regimens have a favorable efficacy and tolerability benefit risk ratio, compared with a triple regimen consisting of either efavirenz or boosted protease inhibitors. Additionally, the low chance of resistance development and low costs make DTG superior to other antiretroviral therapy (ART) regimens. DTG is administered once daily either with emtricitabine and tenofovir disoproxil fumarate or as a fixed‐dose combination tablet with abacavir and lamivudine.3
As women of reproductive age represent a large proportion of all HIV‐positive patients, knowledge about the safety of DTG during pregnancy is required. In 2016, there were 17.8 million women living with HIV worldwide.4 They must be treated during pregnancy, not only to ensure their own health, but also because this reduces the risk of mother‐to‐child transmission (MTCT) of the virus from 20–45% to <1%.5 A systematic review reported that DTG use in pregnancy did not result in increased rates of stillbirth, preterm birth, small for gestational age, or congenital anomalies.6 This is in line with results from a large observational study that compared birth outcomes among women initiating either DTG‐based ART or efavirenz‐based ART (World Health Organization (WHO) first‐line recommended regimen until 2016) and found no statistically significant difference between the regimens when started during pregnancy.7 However, a preliminary analysis of an ongoing study in Botswana reported an increased incidence of neural tube defects associated with exposure around the time of conception (0.9% compared with a background incidence of 0.1%).8 The final data analysis—expected in 2019—and other ongoing randomized clinical trials will provide more information on the safety of fetal DTG exposure.9
With the gradual introduction of DTG in perinatal guidelines it is also necessary to evaluate the effect of pregnancy on maternal exposure, as physiological changes taking place in pregnancy may influence antiretroviral pharmacokinetics (PK). For some antiretroviral drugs, for instance raltegravir and elvitegravir, a reduction in maternal exposure is reported due to pregnancy‐induced changes that affect either the absorption, distribution, metabolism, or excretion of the drug.10, 11 Subtherapeutic maternal antiretroviral exposure may result in virologic breakthrough leading to MTCT and/or resistance.12 Because of the long period between US Food and Drug Administration (FDA) approval and the first published results on PK in pregnancy (median 6 years), prescription of antiretrovirals during pregnancy is often off‐label, without sufficient information on the dose‐exposure relationship in this specific population.13 To improve our understanding of the dose‐exposure relationship during this period, pregnancy physiologically‐based pharmacokinetic (p‐PBPK) models can be of great value. By combining data on human anatomy, physiology, and drug‐specific parameters in a p‐PBPK model, concentration‐time profiles in plasma and various other tissues can be simulated. This physiologically‐based approach may provide additional insights for selecting an adequate dosing regimen for HIV‐positive pregnant women.14, 15 It is equally important to predict fetal drug exposure following maternal dosing. Exposure of the developing unborn child could lead to adverse effects but it may also have beneficial effects from a perspective of fetal pre‐exposure prophylaxis. Next to the DTG‐driven reduction in maternal viral load, fetal plasma DTG levels above the concentration that results in 90% of viral inhibition (0.324 µg/mL) may contribute to a reduced chance of MTCT.16, 17
The delicate balance between possible fetal toxicity and potential beneficial effects of fetal exposure in terms of fetal pre‐exposure prophylaxis highlights that it is relevant to adequately describe fetal PK following maternal dosing. Although the umbilical cord blood‐to‐maternal blood (C:M) concentration ratio is used in clinical practice as an indicator of fetal exposure, its use has some drawbacks. The C:M concentration ratio provides only a very crude estimate of fetal drug exposure, as this single time‐point measurement is subject to a large interindividual variability and it does not provide any information on the fetal plasma concentration‐time profile. Moreover, the value of the C:M concentration ratio is dependent on the time of sampling relative to dosing, as transfer from maternal to fetal plasma may be delayed, which, in turn, depends on the rate of placental transfer. Simulating fetal exposure using a p‐PBPK model may account for this. However, to fulfill its potential with respect to predicting the placental passage of drugs, the model should allow a reliable description of placental disposition. It has been shown that ex vivo placenta perfusion is a valid method to study placental transfer of compounds and is predictive of placental transfer in vivo. 17, 18 We previously demonstrated that mechanistic data derived from ex vivo placental perfusion experiments can be used for parameterization of the fetal‐placental unit, allowing simulation of fetal darunavir exposure.19
In this study, placental transfer of DTG was assessed via ex vivo dual‐side cotyledon perfusion experiments. Results were subsequently incorporated into a p‐PBPK model that was used to investigate the ability to simulate maternal and fetal DTG PK simultaneously and to guide maternal dosing.
Methods
Placental transfer of DTG ex vivo
Dual‐side placental perfusion experiments were performed to study transfer of DTG ex vivo, using human, term placentas. A detailed description of the method can be found in File S1 and an illustration of the ex vivo dual‐side cotyledon perfusion model in Figure S1 .
Physiologically‐based pharmacokinetic modeling
A physiologically‐based pharmacokinetic (PBPK) model was developed for a healthy, nonpregnant population using Berkeley Madonna software (version 8.3.18). For model development, prediction, and verification, covariates were matched to the mean values in the clinical studies. In this way, PK of an average subject was simulated. Details on the modeling approach are described in File S2 and Table S1 . Drug physicochemical properties and parameters describing human physiology were derived from literature as well as from the virtual healthy population in Simcyp (version 17; Certara UK Limited, Simcyp Division, Sheffield, UK) and the model was optimized using in vivo PK data for DTG from nonpregnant patients. Subsequently, the model was used to predict maternal and fetal DTG exposure in pregnancy after incorporation of gestational age‐dependent changes in the maternal and fetal physiology and in vitro and ex vivo data obtained from human cotyledon perfusion experiments. The model performance of the p‐PBPK model was verified against observed maternal and fetal DTG concentration data.
Development of a PBPK model in healthy volunteers
The PBPK model resembling a healthy nonpregnant individual consisted of 13 compartments representing organs and a venous and arterial blood compartment. The change in DTG concentrations in these compartments was described using time‐based differential equations. For noneliminating tissues, the assumption has been made that at steady‐state an equilibrium is established between drug concentrations in the tissue compartments and the blood circulation. The tissue‐to‐plasma partition coefficients that describe the movement of DTG into tissues were predicted by the method described by the Rodgers and Rowland method available within the Simcyp Simulator.
For all nonelimination tissue compartments, perfusion rate‐limited equations were used:
| (1) |
where V, C, and Q denote the volume of the compartment, the drug concentration in the compartment, and blood flow in and out of the compartment, respectively. The parameters Kp and BP represent the tissue‐to‐plasma partition coefficient and blood‐to‐plasma ratio, respectively. C arterial refers to the arterial blood concentration.
Absorption of DTG was defined as a one‐compartmental process, with a first‐order absorption rate (k a) describing the uptake of the drug across the gut wall. Subsequently, the drug enters the liver via the portal vein, which was described with a well‐stirred model.
The well‐stirred model applied for the liver compartment:
| (2) |
where V, C, and Q denote the volume of the compartment, the drug concentration in the compartment, and blood flow in and out of the compartment, respectively. Tissue‐to‐plasma partition coefficients are indicated by Kp values and BP represents the tissue‐to‐plasma partition coefficient. The subscripts ha, arterial, spl, gut, and liv denote the hepatic artery, arterial blood, spleen, gut, and liver, respectively. Scaled liver clearance and the unbound concentration in the liver are represented by CLliv and Cu liv.
A well‐stirred liver model was used, and hepatic clearance was modeled based on in vitro‐in vivo extrapolation of reported recombinant CYP3A4 and UGT1A1 biotransformation data.20 This resulted in an underestimation of clearance and associated overestimation of DTG systemic exposure. Therefore, an empirical scaling factor was required to be able to capture clinically observed data on DTG exposure. The value of scaling factor was fitted by comparing simulations with observed clinical PK data published by Min et al.21 This resulted in a refined well‐stirred model for the liver compartment. The following equation was used to describe this process, under the assumption that the fraction unbound in the liver is similar to the fraction unbound in plasma (fuplasma):
| (3) |
| (4) |
where CLliv and CLint_total represent scaled liver clearance and intrinsic metabolic clearance in the liver, respectively. The scaling factor is represented by SF. The unbound and total concentrations in the liver are represented by Cu liv and C liv, respectively. Systemic clearance of DTG was considered to mainly occur via the liver via uridine diphosphate‐glucuronosyltransferase 1A1 (UGT1A1), therefore, renal clearance was not included in the model.22 The established PBPK model was used as a starting point for building the p‐PBPK model.
Development of a p‐PBPK model
In a next step, the PBPK model for healthy volunteers was extended with a fetal‐placental unit to simulate DTG PK in pregnant women and fetus. A schematic overview of the p‐PBPK model is given in Figure 1.
Figure 1.
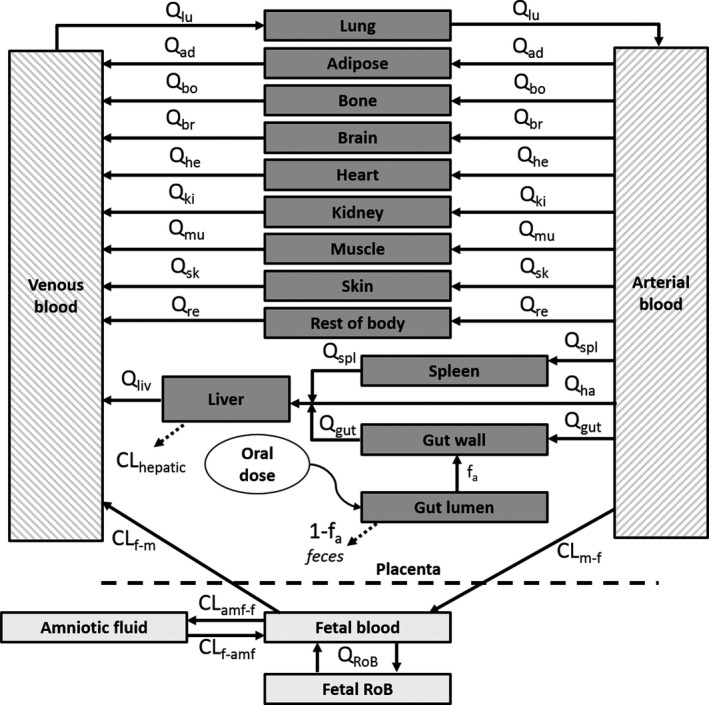
Schematic overview of the pregnancy physiologically‐based pharmacokinetic model. Time‐based changes in dolutegravir concentrations in tissues and blood are described with tissue‐specific blood flow (Q) and clearance values (CL). The subscripts denote the following tissues: ad, adipose; amf‐f, amniotic fluid‐to‐fetus; bo, bone; br, brain; f‐amf, fetus‐to‐amniotic fluid; f‐m, fetal‐to‐maternal; gut, gut wall; ha, hepatic artery; he, heart; ki, kidney; liv, liver; lu, lung; m‐f, maternal‐to‐fetal; mu, muscle; re, rest of body; RoB, rest of body fetus; sk, skin; spl, spleen.
P‐PBPK model structure
To simulate DTG PK in pregnant women, drug‐specific parameter values used to build the nonpregnancy model were kept constant while physiological parameters were adjusted to reflect maternal physiological changes during pregnancy. Published regression equations describing anatomic, physiological, and biological changes according to gestational age were incorporated in the p‐PBPK model.11 Parameter values were calculated based on a gestational age of 34 weeks to be able to compare simulations with clinical data (Table S2 ). Potential effects of pregnancy on DTG kinetics were incorporated in the p‐PBPK model as follows. DTG is highly bound (> 99%) to plasma proteins, especially albumin.23, 24 The reported drop in albumin concentration from 45.8 g/L pre‐pregnancy to 37.6 g/L at 34 weeks of pregnancy results in an increased fraction unbound, which is known from clinical studies.25 The effect of changing albumin concentrations during pregnancy on the fraction unbound was incorporated into the model, with the assumption that pregnancy did not influence protein affinity or configuration. In addition to the effect of pregnancy on plasma protein levels, an altered activity of key metabolizing enzymes is reported.26 DTG is extensively metabolized, primarily via UGT1A1 and to a lesser extent by cytochrome P450 (CYP)‐3A4.22 As DTG is partly converted by CYP3A4, the reported 16% increased enzyme activity was incorporated in the model.11 Pregnancy also leads to an upregulation of UGT1A1 activity. This is indicated by an increased clearance of labetalol that is seen during pregnancy, most likely due to a rise in progesterone. Although this enzyme plays a more dominant role in DTG metabolism, it is difficult to parameterize the increase in UGT1A1 activity in a quantitative manner due to the limited number of clinical studies, the use of nonspecific substrates, and high interindividual variability.27, 28 The assumption was made that pregnancy‐induced upregulation of UGT1A1 activity is proportional to that of CYP3A4. As maternal variations in UGT1A1 activity may affect fetal DTG exposure levels, expression levels and interindividual variability should be considered in future p‐PBPK modeling studies when these become available.
Incorporation of a fetoplacental unit
In this study, the placenta was considered as a barrier separating the maternal and fetal circulation and the fetal PBPK model comprised a fetal blood compartment, amniotic fluid, and a compartment representing the rest of the fetal body. Table 1 describes the parameter values used to establish the fetoplacental unit. Equations describing the gestational age‐dependent changes can be found in Table S2 .
Table 1.
Parameter values for incorporation of the fetoplacental unit
| Parameter | Value | References | |
|---|---|---|---|
| Fetal volume, L | VtotalF | 2.32 | 11 |
| Fetal blood, L | FbloodF | 0.171 | 48 |
| Fetal cardiac output, L/hour | CO_F | 76.9 | 49 |
| Fetal blood flow directed to placenta, fraction of fetal cardiac output | frQplaF | 0.23 | 50 |
| Amniotic fluid volume, L | Vamf | 0.906 | 11 |
| Maternal‐to‐fetal apparent ex vivo cotyledon clearance, mL/minutes, mean ± SD | CLappcotmf | 1.03 ± 0.06 | Determined |
| Fetal‐to‐maternal apparent ex vivo cotyledon clearance, mL/minutes, mean ± SD | CLappcotfm | 1.03 ± 0.23 | Determined |
| Fraction unbound in maternal perfusion buffer | fumf | 0.048 | Determined |
| Fraction unbound in fetal perfusion buffer | fufm | 0.043 | Determined |
| Number of cotyledons per placenta | Ncot | 30 | 51 |
To simulate fetal DTG exposure, placental transfer data obtained from ex vivo dual‐side cotyledon perfusion experiments was incorporated into the model. An elimination rate constant (k e) for DTG disappearance was calculated by linear regression analysis of the ln‐transformed DTG concentration in the closed reservoir. Bidirectional clearance values were determined using the following formula:
| (5) |
where CLappcot denotes the apparent intrinsic cotyledon clearance, ke is a placental elimination rate constant calculated from the slope of the ln‐transformed concentration‐time profile, and V represents the volume of the buffer in the closed circulation.
Because of the high degree of binding of DTG to plasma proteins, albumin was added to both circulations. A fetal‐to‐maternal albumin concentration ratio of 1.2 was applied to mimic physiological conditions. To determine whether this resulted in a similar unbound fraction as determined in vivo, the fraction unbound in maternal and fetal perfusion samples (both n = 3) was measured. A mean ± SD (%) fraction unbound of 4.8 ± 0.5% and 4.3 ± 0.4% was found in maternal and fetal perfusion samples, respectively (i.e., ~ 10‐fold higher than the fraction unbound observed in vivo).25 Based on the measured fraction unbound in perfusion samples, the unbound ex vivo intrinsic cotyledon clearance was calculated. This was subsequently scaled to an in vivo placental clearance using the reported fraction unbound in vivo and the number of cotyledons of a single placenta:
| (6) |
| (7) |
where CLplac and CLappcot represent the scaled in vivo placental blood clearance and ex vivo apparent intrinsic cotyledon clearance, respectively. The fraction unbound in the perfusion samples is denoted by fu and fub refers to the fraction unbound in blood found in vivo. The subscript mf denotes maternal‐to‐fetal transfer and fm denotes fetal‐to‐maternal transfer. The average number of cotyledons for scaling purposes is denoted by Ncot.
The fetoplacental unit is comprised of the fetal blood compartment, the compartment representing the rest of the fetal body, and the amniotic fluid compartment, and the following equations were used to describe DTG distribution:
Fetal blood compartment:
| (8) |
Compartment representing rest of fetal body:
| (9) |
Amniotic fluid compartment:
| (10) |
where V, C, and Q denote the volume of the compartment, the drug concentration in the compartment, and blood flow in and out of the compartment, respectively. The parameters Kp and BP represent the tissue‐to‐plasma partition coefficient and blood‐to‐plasma ratio, respectively. The subscripts blood_F, arterial, rob_F and amf denote the compartments fetal blood, maternal arterial blood, rest of fetal body, and amniotic fluid, respectively. The scaled placental clearance values are represented by CL, in either the maternal‐to‐fetal (mf) or fetal‐to‐maternal (fm) direction. The estimated clearance from the fetal blood to the amniotic fluid compartment and vice versa is represented by CLf‐amf and CLamf‐f, respectively. Drug movement to and from the amniotic fluid compartment is parameterized based on fluid exchange rates reported in literature, describing fetal‐to‐amniotic fluid flow (oral, nasal, tracheal, and pulmonary secretion) and amniotic fluid‐to‐fetus flow (swallowing and intramembraneous uptake of amniotic fluid).29 As < 1% of DTG is excreted in urine as unchanged compound,22 the renal elimination pathway is not included in the model.
DTG is mainly metabolized by UGT1A1, an enzyme known to be expressed in third trimester placental tissue as well and hypothesized to contribute to decreased fetal exposure to xenobiotics like bisphenol‐A.30, 31 However, the interindividual variability of its activity is high and the contribution of placental metabolism to total metabolic DTG clearance could not be quantified.32 Therefore, placental metabolism was not considered in this model and the organ was considered to be a barrier rather than a compartmental structure. Hence, a partitioning coefficient and tissue volume were not included for this organ. Consequently, CLplac values were considered to reflect both active transport and passive diffusion processes.
Simulations of both maternal and fetal plasma concentration‐time profiles as well as amniotic fluid concentrations were performed following oral administration of 50 mg DTG q.d. to a virtual pregnant woman at 34 weeks of gestation. The simulated maternal plasma concentration‐time profile at steady‐state was compared with available clinical data from the PANNA network and the IMPAACT group.33, 34 In addition, simulated fetal exposure was compared to in vivo fetal exposure, as umbilical cord blood‐to‐maternal blood concentration ratios are reported.34, 36 To study the impact of modeling assumptions and uncertainty around some parameter values on simulated concentration‐time profiles, sensitivity analyses were conducted.
Results
Ex vivo dual‐side cotyledon perfusion experiments
Six successful ex vivo placental perfusion experiments were performed, n = 3 in either direction. As can be seen in Figure 2a, DTG concentration in the closed maternal circulation decreased during 180 min of perfusion with a constant fraction ending up in the fetal outflow. A similar pattern of DTG transport from the closed fetal circulation to the maternal side can be seen (Figure 2b). A detailed description of the results can be found in File S3 and Figure S3 .
Figure 2.
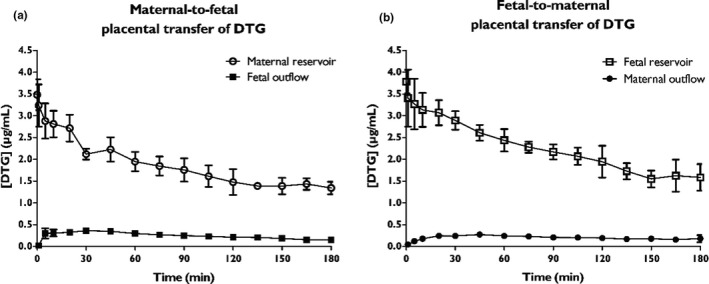
Bidirectional transfer of dolutegravir (DTG) in ex vivo dual‐side cotyledon perfusion experiments. (a) Concentration‐time profiles of DTG upon administration to the closed maternal circulation (open circles). (b) Concentration‐time profiles of DTG upon administration to the closed fetal circulation (open squares). Results are depicted as mean ± SD, n = 3 for each direction.
PBPK model in healthy volunteers
A PBPK model for DTG exposure in healthy volunteers was established and model performance was verified against clinical PK datasets of several DTG dosing regimens.21 Simulated profiles slightly overestimated the observed total plasma exposure, whereas peak exposure (Cmax) was slightly underestimated (Figure 3).
Figure 3.
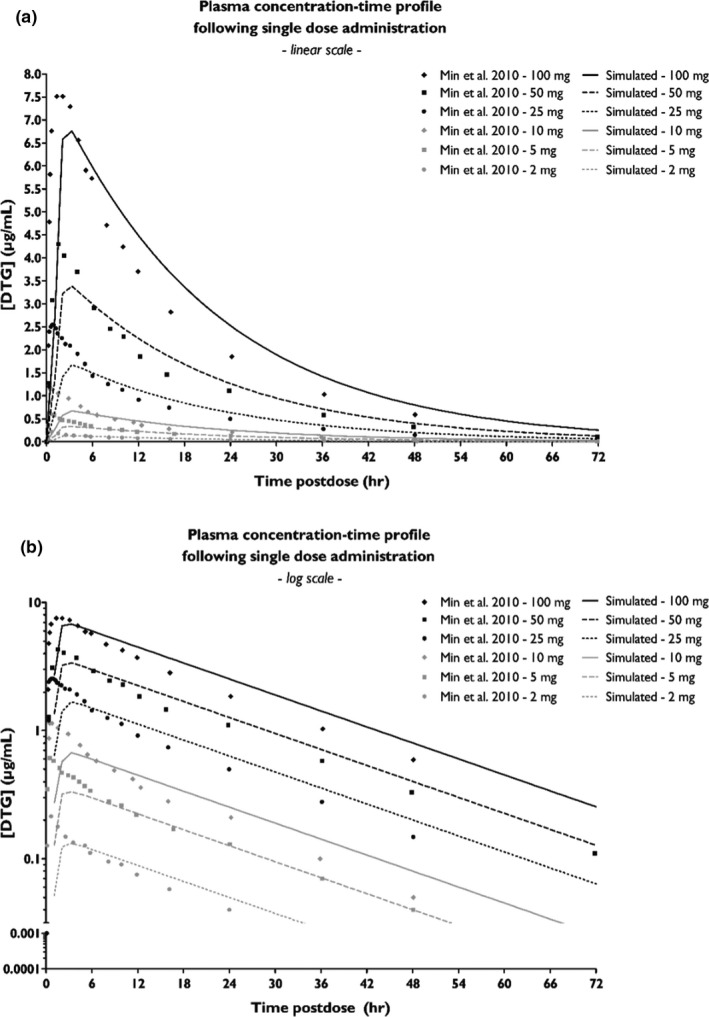
Concentration‐time profile of dolutegravir (DTG) upon administration of different dosing regimen. The simulated plasma concentration‐time profiles (lines) upon administration of a single dose of 2, 5, 10, 25, 50, or 100 mg DTG are plotted against observed in vivo clinical data (dots) as reported by Min et al.,21 either plotted on a linear scale (a) or on a log‐scale (b).
PBPK model in pregnant subjects
Based on six successful perfusions, DTG cotyledon clearances were calculated. The clearance values (mean ± SD) were 1.03 ± 0.06 and 1.03 ± 0.23 mL/minutes in maternal‐to‐fetal and fetal‐to‐maternal direction, respectively. After incorporation of scaled placental clearance values into the p‐PBPK model, as described in the Method section, the DTG concentration in maternal plasma, fetal plasma, and amniotic fluid was simulated.
As can be seen in Figure 4, maternal plasma concentrations were simulated upon administration of 50 mg q.d. until a steady‐state was reached after ~ 4 days. Simulations were compared with clinical PK data of third trimester HIV‐1‐infected women. Mean plasma concentrations of PK curves from the PANNA network (n = 15) and the IMPAACT group (n = 28) are shown. Simulations indicate that administration of 50 mg DTG q.d. would result in a maternal C0 of 0.99 µg/mL, which is in line with the reported C0 of 1.00 and 0.96 µg/mL, by the PANNA network and IMPAACT group, respectively.33, 34 This regimen resulted in sufficient high maternal plasma levels during pregnancy to achieve viral suppression.
Figure 4.
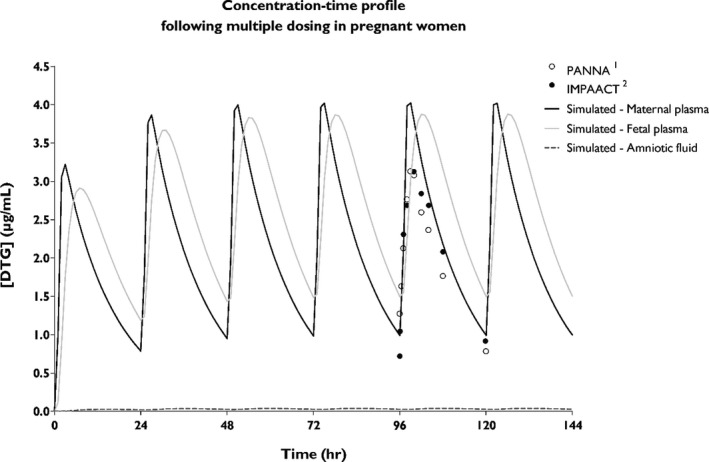
Concentration‐time profile of dolutegravir (DTG) in pregnancy. The simulated concentration‐time profiles of maternal plasma (black line), fetal plasma (gray line), and in the amniotic fluid (dotted line) are shown upon multiple dosing of 50 mg DTG q.d. Clinical maternal plasma concentrations are indicated with dots, representing mean values reported by the PANNA network (n = 15) and the IMPAACT group (n = 28).33, 34
Simulations of fetal DTG exposure were compared with in vivo data from the PANNA network, reporting umbilical cord plasma‐to‐maternal plasma concentrations of 10 mother‐infant pairs. The simulated cord blood‐to‐maternal blood concentration ratios range between 0.57 and 1.51 over an entire dosing interval and are in line with the range of reported ratios in vivo by the PANNA network (0.64–1.81, n = 10) and Rimawi et al. (0.68–1.68, n = 3) and the mean (interquartile range) cord blood‐to‐maternal blood concentration ratio of 1.25 (1.07–1.40, n = 23) reported by the IMPAACT group.34, 35 The predicted fetal trough plasma concentration (Ctrough) of 1.50 µg/mL is above the concentration that results in 90% of viral inhibition (0.324 µg/mL), indicating potential use of DTG for fetal pre‐exposure prophylaxis at this dose level.16
An amniotic fluid compartment was included in the p‐PBPK model, allowing simulation of DTG concentration in this compartment over time. A low amniotic fluid concentration (0.03 µg/mL) was reported in the literature,37 and simulations were similar to this value (range 0.025–0.039 µg/mL).
Sensitivity analyses
Sensitivity analyses on maternal and fetoplacental input parameters showed that DTG fraction unbound and placental clearance are the most important parameters influencing DTG plasma concentrations. Results can be found in File S4 and Figures S4 and S5 .
Discussion
The integration of data derived from ex vivo experiments into in silico models provides an ethically acceptable framework to study maternal and fetal exposure to drugs used during pregnancy. To fulfill its potential with respect to predicting the placental passage of drugs, accurate parameterization based on human physiology and in vitro and ex vivo data describing (placental) disposition is warranted. The current work focused on parameterizing the model with ex vivo cotyledon perfusion data. Although this model is, to our knowledge, the first to describe DTG PK in pregnancy, p‐PBPK models have been used to predict maternal and fetal exposure to other antivirals (e.g., darunavir,19 nevirapine,38 tenofovir, and emtricitabine).39 A key question that remains is whether the approach based on parameterization with ex vivo placenta perfusion data is reproducible, also for other drugs with different physicochemical properties, and of which the placental disposition may be driven by other mechanisms. In this respect, DTG has a logP of 2.2 compared with darunavir (logP = 2.8), nevirapine (logP = 2.5), tenofovir (logP = −3.4), and emtricitabine (logP = −0.9). Moreover, DTG is mainly metabolized via glucuronidation, whereas the previously studied drugs are not subject to biotransformation.
As a first step toward simulating DTG disposition in pregnancy, we established a PBPK model reflecting nonpregnant healthy volunteers. Exposure to DTG was overestimated using a well‐stirred liver model based on recombinant CYP and UGT clearance data alone. The introduction of an empirically determined scaling factor was required to capture the clinical plasma concentration‐time profiles following single ascending doses in the nonpregnant population. The necessity of introducing a scaling factor indicates that yet unknown (hepatic) clearance mechanisms may play a role in vivo and would need to be included in the PBPK model. On one hand, the underestimation of clearance may be related to the intrinsic activity of UGT1A1 and CYP3A4 enzymes or the role of extrahepatic clearance. Although the PBPK model in the current work did not incorporate any metabolism apart from the liver, and also because the effect of pregnancy on extrahepatic clearance is unknown, it is expected that accounting for metabolism in the kidneys and gut could improve oral clearance predictions.40 On the other hand, the amount of DTG available for metabolism may yet be underestimated. Active basolateral uptake of the drug into hepatocytes via yet unknown routes or an underestimation of the fraction unbound present within the hepatocytes may be a cause for the underestimation of clearance in our model. Given the lack of experimental data on actual intracellular unbound DTG concentrations, we now used the fraction unbound in plasma as an indicator of the fraction unbound in the liver with an additional scaling factor. This needs further experimental corroboration.
Next, physiological parameters were adjusted to reflect pregnant women. We found a small discrepancy between the predicted and observed maternal concentration‐time profiles, which may be explained by missing information on quantitative data describing pregnancy or HIV‐related changes in some physiological parameters required to parameterize the model. Parameters describing body composition and organ blood flows were based on healthy rather than HIV‐infected women. Due to a lack of quantitative information, we did not incorporate any effect of HIV condition on drug disposition that can possibly result from hematological abnormalities or chronic liver disease.41 Furthermore, as renal clearance of DTG is very low, we did not take it into account in the models. Including renal clearance will likely lead to a slightly improved accuracy of maternal PK, as the pregnancy‐related decrease in albumin and increase in renal blood flow could result in an increased renal clearance of the free fraction of the drug. However, because no clinical renal clearance data are available for validation and the free concentration is reported not to be altered during pregnancy,25 further optimization was not conducted here.
Considering the physiology of the fetoplacental unit of the model, the placenta was incorporated as a barrier, rather than an organ compartment, with DTG clearances values derived from ex vivo cotyledon perfusion experiments. It should be noted that the use of healthy term placentas is a limitation of the study, as results of our simulations were compared with in vivo PK data of HIV‐infected women, collected at 34 weeks of gestation. For example, malperfusion of the placental bed seems to occur more frequently among HIV‐infected women.42 For safety and availability reasons, these placentas were not studied and potential changes in placental physiology taking place between 34 weeks of gestation and term were not considered.
In our sensitivity analyses, DTG fraction unbound in plasma is the most important parameter influencing both maternal and fetal steady‐state concentration‐time profiles. A high interindividual variability in fraction unbound was reported in this patient population,24 highlighting the importance of measuring the fraction unbound in clinical practice and including variability in in silico prediction models. In addition, ex vivo protein binding is relevant to consider. In our studies, albumin was added to both circulations during the perfusion experiment, with a 20% higher albumin concentration in the fetal circulation. Although the applied albumin concentration ratio was similar to the value reported in pregnancy43 and no statistically significant effect of HIV on albumin concentrations was found at term,44 the fraction unbound in perfusion samples of both circulations was found to be higher than those measured in vivo, indicating that binding kinetics may be different in vivo or that other binding proteins than albumin are involved. To circumvent problems associated with this, we measured and calculated unbound clearance values and incorporated these in the model to accurately extrapolate ex vivo cotyledon clearance to in vivo expected placental clearance. No additional empirical scaling factor for placental DTG transport seemed to be necessary for fitting purposes. This illustrates that our approach to incorporate ex vivo placental perfusion data in PBPK models is not only suitable for darunavir, as demonstrated earlier,19 but also for DTG. As the placenta is incorporated as a barrier in this model, possible retention of DTG within the placenta could not be simulated. This could, however, be of relevance as placental retention is reported to occur in vivo 36 and we were also able to measure DTG in placental tissue samples upon perfusion. However, concentrations did remain relatively low, and no relevant placental accumulation was observed as found for several other drugs.45, 46 An additional aspect that requires further research is that the fetal part of the model was composed of a blood compartment with the different organs lumped together in a “rest of body”‐compartment. This may be refined to be able to better estimate to which tissues DTG distributes to in the fetus and to better understand the possible adverse effects observed.8 Fetal hepatic metabolism of DTG was also not taken into account in the current model as fetal expression of UGT1A1 is very low compared with maternal levels.47 As more knowledge on fetal physiology emerges, the simulation of drug distribution using a full body fetal PBPK model becomes feasible. Simulation of DTG concentrations in amniotic fluid were performed by applying an empirical scaling factor on amniotic fluid transfer and associated drug clearance and fitting against one published measurement. However, sensitivity analysis showed no profound contribution of using different magnitudes of amniotic fluid clearance pathways to fetal or maternal DTG exposure in plasma.
In summary, simulations were able to adequately capture maternal DTG exposure during late pregnancy, with a slight overprediction of peak concentrations. Simulated cord blood plasma‐to‐maternal blood plasma concentration ratios were also comparable with those reported in literature. Individual cord blood DTG concentrations, rather than ratios, could be of value to draw a conclusion on the shape of the simulated fetal concentration‐time profile, but were unavailable at this time to use for validation purposes.
This study showed that fetal DTG exposure can be predicted using a p‐PBPK model established using placental transfer data obtained ex vivo. Based on these simulations, a dose of 50 mg DTG q.d. should result in sufficiently high maternal plasma levels during pregnancy to achieve viral suppression and may provide fetal pre‐exposure prophylaxis. Although both maternal and fetal exposure to DTG may reduce the chance of MTCT, recent concerns about fetal toxicity with maternal DTG use in the periconception period highlight the importance of monitoring birth outcomes and follow‐up of intrauterine‐exposed children.
Funding
This work was funded by Simcyp Grant and Partnership Scheme 2015/16 (Certara).
Conflict of Interest
S.S. is an employee of AstraZeneca and may own stock or stock options. A.P.C. has received research grants to her institution from PENTA, ViiV, Janssen Research, Gilead, and Merck. K.A. is an employee of Certara UK Limited, Simcyp Division. D.M.B. has received research grants to his institution from Merck, BMS, Janssen/Tibotec, ViiV Healthcare, and Gilead; has received educational grants from Merck and was speaker at a symposia for Merck; and is a member of the advisory board of ViiV Healthcare and Merck. All other authors declared no competing interests for this work.
Author Contributions
All authors wrote the manuscript. J.J.M.F., F.G.M.R., D.M.B., and R.G. designed the research. J.J.M.F, S.S., and R.G. performed the research. J.J.M.F. and R.G. analyzed the data.
Supporting information
File S1. Method: Placental transfer of DTG ex vivo.
Figure S1. Illustration of the ex vivo dual‐side cotyledon perfusion model.
File S2. Berkeley Madonna modeling script.
Table S1. Physicochemical and pharmacokinetic parameters for dolutegravir.
Table S2. Pregnancy‐related changes with gestational age.
Figure S2. Placental transfer of antipyrine.
File S3. Results: Placental transfer of DTG ex vivo.
Figure S3. DTG levels in perfused placental tissue.
File S4. Sensitivity analyses.
Figure S4. Sensitivity analyses for maternal input parameters.
Figure S5. Sensitivity analyses for fetoplacental input parameters.
Acknowledgments
The authors thank Gerard Zijderveld for inclusion of participants, all women who donated placentas, and the caretakers who collected them. We are grateful to Certara for the Simcyp Grant and Partnership Scheme grant allowing research on this topic. The Simcyp Simulator is freely available to academic institutions and other non‐for‐profit organizations for research and teaching purposes. We thank Joyce van der Heijden for her assistance with the experiments, Marga Teulen for her help with the bioanalyses, and Laurens Verscheijden for providing valuable feedback on the modeling script.
References
- 1.World Health Organization (WHO) . Updated recommendations on first‐line and second‐line antiretroviral regimens and post‐exposure prophylaxis and recommendations on early infant diagnosis of HIV: interim guidelines <https://apps.who.int/iris/bitstream/handle/10665/277395/WHO-CDS-HIV-18.51-eng.pdf?ua=1> (2018). Accessed February 2019.
- 2. Dorward, J. et al Dolutegravir for first‐line antiretroviral therapy in low‐income and middle‐income countries: uncertainties and opportunities for implementation and research. Lancet HIV 5, e400–e404 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kanters, S. et al Comparative efficacy and safety of first‐line antiretroviral therapy for the treatment of HIV infection: a systematic review and network meta‐analysis. Lancet HIV 3, e510–e520 (2016). [DOI] [PubMed] [Google Scholar]
- 4. UNAIDS DATA 2017 <http://www.unaids.org/en/resources/documents/2017/2017_data_book>. Accessed February 2019.
- 5. Townsend, C.L. et al Earlier initiation of ART and further decline in mother‐to‐child HIV transmission rates, 2000–2011. AIDS 28, 1049–1057 (2014). [DOI] [PubMed] [Google Scholar]
- 6. Hill, A. , Clayden, P. , Thorne, C. , Christie, R. & Zash, R. Safety and pharmacokinetics of dolutegravir in HIV‐positive pregnant women: a systematic review. J. Virus Erad. 4, 66–71 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zash, R. et al Comparative safety of dolutegravir‐based or efavirenz‐based antiretroviral treatment started during pregnancy in Botswana: an observational study. Lancet Glob. Health. 6, e804–e810 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zash, R. , Makhema, J. & Shapiro, R.L. Neural‐tube defects with dolutegravir treatment from the time of conception. N. Engl. J. Med. 379, 979–981 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Transition to new antiretrovirals in HIV programmes . Policy brief. 2017. Contract No.: WHO/HIV/2017.20.
- 10. van der Galien, R. et al Pharmacokinetics of HIV‐integrase inhibitors during pregnancy: mechanisms, clinical implications and knowledge gaps. Clin. Pharmacokinet. 58, 309–323 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abduljalil, K. , Furness, P. , Johnson, T.N. , Rostami‐Hodjegan, A. & Soltani, H. Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: a database for parameters required in physiologically based pharmacokinetic modelling. Clin. Pharmacokinet. 51, 365–396 (2012). [DOI] [PubMed] [Google Scholar]
- 12. Siegfried, N. , van der Merwe, L., Brocklehurst, P. & Sint, TT Antiretrovirals for reducing the risk of mother‐to‐child transmission of HIV infection. Cochrane Database Syst. Rev. 7, CD003510 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Colbers, A. , Mirochnick, M. , Schalkwijk, S. , Penazzato, M. , Townsend, C. & Burger, D. Importance of prospective studies in pregnant and breastfeeding women living with HIV. Clin. Infect. Dis. 69, 1254–1258 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu, G. , Abduljalil, K. , Jamei, M. , Johnson, T.N. , Soltani, H. & Rostami‐Hodjegan, A. Physiologically‐based pharmacokinetic (PBPK) models for assessing the kinetics of xenobiotics during pregnancy: achievements and shortcomings. Curr. Drug Metab. 13, 695–720 (2012). [DOI] [PubMed] [Google Scholar]
- 15. Ke, A.B. , Greupink, R. & Abduljalil, K. Drug dosing in pregnant women: challenges and opportunities in using physiologically based pharmacokinetic modeling and simulations. CPT Pharmacometrics Syst. Pharmacol. 7, 103–110 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cottrell, M.L. , Hadzic, T. & Kashuba, A.D. Clinical pharmacokinetic, pharmacodynamic and drug‐interaction profile of the integrase inhibitor dolutegravir. Clin. Pharmacokinet. 52, 981–994 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McCormack, S.A. & Best, B.M. Protecting the fetus against HIV infection: a systematic review of placental transfer of antiretrovirals. Clin. Pharmacokinet. 53, 989–1004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hutson, J.R. , Garcia‐Bournissen, F. , Davis, A. & Koren, G. The human placental perfusion model: a systematic review and development of a model to predict in vivo transfer of therapeutic drugs. Clin. Pharmacol. Ther. 90, 67–76 (2011). [DOI] [PubMed] [Google Scholar]
- 19. Schalkwijk, S. et al Prediction of fetal darunavir exposure by integrating human ex‐vivo placental transfer and physiologically based pharmacokinetic modeling. Clin. Pharmacokinet. 57, 705–716 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reese, M.J. et al In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a HIV integrase inhibitor. Drug Metab. Dispos. 41, 353–361 (2013). [DOI] [PubMed] [Google Scholar]
- 21. Min, S. et al Pharmacokinetics and safety of S/GSK1349572, a next‐generation HIV integrase inhibitor, in healthy volunteers. Antimicrob. Agents Chemother. 54, 254–258 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Castellino, S. et al Metabolism, excretion, and mass balance of the HIV‐1 integrase inhibitor dolutegravir in humans. Antimicrob. Agents Chemother. 57, 3536–3546 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Song, I.H. et al Pharmacokinetics of single‐dose dolutegravir in HIV‐seronegative subjects with moderate hepatic impairment compared to healthy matched controls. Clin. Pharmacol. Drug Dev. 2, 342–348 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Metsu, D. et al Determination of dolutegravir's unbound fraction in human plasma using validated equilibrium dialysis and LC‐MS/MS methods. Clin. Chim. Acta 479, 56–65 (2018). [DOI] [PubMed] [Google Scholar]
- 25. Bollen, P. et al . First report of dolutegravir unbound plasma concentrations during pregnancy in HIV‐positive women. Nineteenth International Workshop on Clinical Pharmacology of Antiviral Therapy, Baltimore, MD, May 22–24, 2018.
- 26. Hodge, L.S. & Tracy, T.S. Alterations in drug disposition during pregnancy: implications for drug therapy. Expert Opin. Drug Metab. Toxicol. 3, 557–571 (2007). [DOI] [PubMed] [Google Scholar]
- 27. Jeong, H. , Choi, S. , Song, J.W. , Chen, H. & Fischer, J.H. Regulation of UDP‐glucuronosyltransferase (UGT) 1A1 by progesterone and its impact on labetalol elimination. Xenobiotica 38, 62–75 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fischer, J.H. et al Influence of gestational age and body weight on the pharmacokinetics of labetalol in pregnancy. Clin. Pharmacokinet. 53, 373–383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Underwood, M.A. , Gilbert, W.M. & Sherman, M.P. Amniotic fluid: not just fetal urine anymore. J. Perinatol. 25, 341–348 (2005). [DOI] [PubMed] [Google Scholar]
- 30. Collier, A.C. , Thevenon, A.D. , Goh, W. , Hiraoka, M. & Kendal‐Wright, C.E. Placental profiling of UGT1A enzyme expression and activity and interactions with preeclampsia at term. Eur. J. Drug Metab. Pharmacokinet. 40, 471–480 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ginsberg, G. & Rice, D.C. Does rapid metabolism ensure negligible risk from bisphenol A? Environ. Health Perspect. 117, 1639–1643 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Peterkin, V.C. et al Limited influence of UGT1A1*28 and no effect of UGT2B7*2 polymorphisms on UGT1A1 or UGT2B7 activities and protein expression in human liver microsomes. Br. J. Clin. Pharmacol. 64, 458–468 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Colbers, A. et al Dolutegravir plasma concentrations during pregnancy and postpartum. Conference on Retroviruses and Opportunistic Infections, Seattle, WA, March 4–7, 2019. Poster 758.
- 34. Mulligan, N. et al Dolutegravir pharmacokinetics in pregnant and postpartum women living with HIV. AIDS 32, 729–737 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Colbers, A. et al Dolutegravir pharmacokinetics during pregnancy and postpartum. Ninth International Workshop on HIV & Women, Seattle, WA, March 2‐3, 2019.
- 36. Rimawi, B.H. et al Pharmacokinetics and placental transfer of elvitegravir, dolutegravir, and other antiretrovirals during pregnancy. Antimicrob. Agents Chemother. 61, pii: e02213–02216 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schalkwijk, S. et al Substantially lowered dolutegravir exposure in a treatment‐experienced perinatally HIV‐1‐infected pregnant woman. AIDS 30, 1999–2001 (2016). [DOI] [PubMed] [Google Scholar]
- 38. De Sousa Mendes, M. et al A physiologically‐based pharmacokinetic model to predict human fetal exposure for a drug metabolized by several CYP450 pathways. Clin. Pharmacokinet. 56, 537–550 (2017). [DOI] [PubMed] [Google Scholar]
- 39. De Sousa Mendes M. et al Prediction of human fetal pharmacokinetics using ex vivo human placenta perfusion studies and physiologically based models. Br. J. Clin. Pharmacol. 81, 646–657 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu, S.N. , Lu, J.B.L. , Watson, C.J. , Lazarus, P. , Desta, Z. & Gufford, B.T. Mechanistic assessment of extrahepatic contributions to glucuronidation of integrase strand transfer inhibitors. Drug Metab. Dispos. 47, 535–544 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. De Santis, G.C. et al Hematological abnormalities in HIV‐infected patients. Int. J. Infect. Dis. 15, e808–e811 (2011). [DOI] [PubMed] [Google Scholar]
- 42. Kalk, E. et al Placental pathology in HIV infection at term: a comparison with HIV‐uninfected women. Trop. Med. Int. Health 22, 604–613 (2017). [DOI] [PubMed] [Google Scholar]
- 43. Hill, M.D. & Abramson, F.P. The significance of plasma protein binding on the fetal/maternal distribution of drugs at steady‐state. Clin. Pharmacokinet. 14, 156–170 (1988). [DOI] [PubMed] [Google Scholar]
- 44. Colbers, A. , Greupink, R. , Litjens, C. , Burger, D. & Russel, F.G. Physiologically based modelling of darunavir/ritonavir pharmacokinetics during pregnancy. Clin. Pharmacokinet. 55, 381–396 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Eliesen, G.A.M. et al Editor's highlight: Placental disposition and effects of crizotinib: an ex vivo study in the isolated dual‐side perfused human cotyledon. Toxicol. Sci. 157, 500–509 (2017). [DOI] [PubMed] [Google Scholar]
- 46. Freriksen, J.J.M. et al Placental disposition of the immunosuppressive drug tacrolimus in renal transplant recipients and in ex vivo perfused placental tissue. Eur. J. Pharm. Sci. 119, 244–248 (2018). [DOI] [PubMed] [Google Scholar]
- 47. Miyagi, S.J. & Collier, A.C. The development of UDP‐glucuronosyltransferases 1A1 and 1A6 in the pediatric liver. Drug Metab. Dispos. 39, 912–919 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smith, G.C. & Cameron, A.D. Estimating human fetal blood volume on the basis of gestational age and fetal abdominal circumference. BJOG 109, 721–722 (2002). [DOI] [PubMed] [Google Scholar]
- 49. De Smedt, M.C. , Visser, G.H. & Meijboom, E.J. Fetal cardiac output estimated by Doppler echocardiography during mid‐ and late gestation. Am. J. Cardiol. 60, 338–342 (1987). [DOI] [PubMed] [Google Scholar]
- 50. Kiserud, T. , Ebbing, C. , Kessler, J. & Rasmussen, S. Fetal cardiac output, distribution to the placenta and impact of placental compromise. Ultrasound Obstet. Gynecol. 28, 126–136 (2006). [DOI] [PubMed] [Google Scholar]
- 51. Syme, M.R. , Paxton, J.W. & Keelan, J.A. Drug transfer and metabolism by the human placenta. Clin. Pharmacokinet. 43, 487–514 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File S1. Method: Placental transfer of DTG ex vivo.
Figure S1. Illustration of the ex vivo dual‐side cotyledon perfusion model.
File S2. Berkeley Madonna modeling script.
Table S1. Physicochemical and pharmacokinetic parameters for dolutegravir.
Table S2. Pregnancy‐related changes with gestational age.
Figure S2. Placental transfer of antipyrine.
File S3. Results: Placental transfer of DTG ex vivo.
Figure S3. DTG levels in perfused placental tissue.
File S4. Sensitivity analyses.
Figure S4. Sensitivity analyses for maternal input parameters.
Figure S5. Sensitivity analyses for fetoplacental input parameters.