

Abstract
PURPOSE
Lifirafenib is an investigational, reversible inhibitor of B-RAFV600E, wild-type A-RAF, B-RAF, C-RAF, and EGFR. This first-in-human, phase I, dose-escalation/dose-expansion study evaluated the safety, tolerability, and efficacy of lifirafenib in patients with B-RAF– or K-RAS/N-RAS–mutated solid tumors.
METHODS
During dose escalation, adult patients with histologically/cytologically confirmed advanced solid tumors received escalating doses of lifirafenib. Primary end points were safety/tolerability during dose escalation and objective response rate in preselected patients with B-RAF and K-RAS/N-RAS mutations during dose expansion.
RESULTS
The maximum tolerated dose was established as 40 mg/d; dose-limiting toxicities included reversible thrombocytopenia and nonhematologic toxicity. Across the entire study, the most common grade ≥ 3 treatment-emergent adverse events were hypertension (n = 23; 17.6%) and fatigue (n = 13; 9.9%). One patient with B-RAF–mutated melanoma achieved complete response, and 8 patients with B-RAF mutations had confirmed objective responses: B-RAFV600E/K melanoma (n = 5, including 1 patient treated with prior B-RAF/MEK inhibitor therapy), B-RAFV600E thyroid cancer/papillary thyroid cancer (PTC; n = 2), and B-RAFV600E low-grade serous ovarian cancer (LGSOC; n = 1). One patient with B-RAF–mutated non–small-cell lung cancer (NSCLC) had unconfirmed partial response (PR). Patients with K-RAS–mutated endometrial cancer and K-RAS codon 12–mutated NSCLC had confirmed PR (n = 1 each). No responses were seen in patients with K-RAS/N-RAS–mutated colorectal cancer (n = 20).
CONCLUSION
Lifirafenib is a novel inhibitor of key RAF family kinases and EGFR, with an acceptable risk-benefit profile and antitumor activity in patients with B-RAFV600–mutated solid tumors, including melanoma, PTC, and LGSOC, as well as K-RAS–mutated NSCLC and endometrial carcinoma. Future comparisons with first-generation B-RAF inhibitors and exploration of lifirafenib alone or as combination therapy in patients with selected RAS mutations who are resistant/refractory to first-generation B-RAF inhibitors are warranted.
INTRODUCTION
The RAS-RAF-mitogen-activated protein kinase (MAPK) pathway plays a prominent role in tumorigenesis.1 K-RAS mutations are common in pancreatic, colorectal, lung, and biliary tract tumors and occur in 15%–30% of endometrioid carcinomas.1,2 H-RAS mutations are common in salivary gland tumors1; N-RAS mutations are frequent in melanomas. Among RAF serine/threonine kinases, B-RAF has the greatest inherent kinase activity.1,3 B-RAF mutations occur in approximately 50% of malignant melanomas,4 40% of thyroid carcinomas,3 between 2% and 38% of low-grade serous ovarian cancers (LGSOCs),5-8 and approximately 10% of metastatic colorectal cancers (CRCs).9 Despite this evident role in tumor biology, pursuit of RAS as a therapeutic target has proven difficult.10
First-generation B-RAF inhibitors have demonstrated clinical benefit in B-RAFV600E metastatic melanoma11,12 but not as monotherapy in B-RAFV600E CRC.13,14 Cutaneous squamous cell carcinoma (SCC) develops in 10%-26% of patients who receive B-RAF inhibitors, including treatment-related keratoacanthomas that result from paradoxical MAPK signal activation.15,16 Most patients who receive these therapies develop resistance and ultimately experience relapse.17
In melanoma, resistance to first-generation B-RAF inhibitors stems from several mechanisms, including epidermal growth factor receptor (EGFR)–mediated reactivation of MAPK signaling,18 RAS-independent RAF kinase dimerization induced by B-RAFV600E splice variants,19,20 AKT-dependent signaling pathways,17,21 and MAPK-redundant signaling pathways.17,22 In patients with B-RAF–mutated CRC, nonresponsiveness to first-generation B-RAF inhibitors is associated with EGFR-mediated reactivation of the MAPK pathway.14
Lifirafenib (BGB-283) is a novel, first-in-class, investigational RAF dimer inhibitor with potent, reversible inhibition of wild-type A-RAF, B-RAF, C-RAF, and B-RAFV600E as well as EGFR15,23 and K-RAS.24,25 Preclinical studies have suggested that lifirafenib leads to a greater number of responses in B-RAF–mutated cancers than first-generation B-RAF inhibitors, including vemurafenib and dabrafenib.15 This study was designed to investigate safety/tolerability, examine pharmacokinetics (PK), establish the maximum tolerated dose (MTD) and recommended phase II dose (RP2D) during dose escalation, and evaluate preliminary antitumor activity of lifirafenib in patients with tumors that harbor B-RAF, N-RAS, or K-RAS mutations.
METHODS
Study Design and Participants
This first-in-human, dose-escalation/dose-expansion, phase I study was conducted from November 20, 2013, to October 19, 2017, across 20 sites in Australia and New Zealand. Adult patients with histologically/cytologically confirmed advanced/metastatic solid tumors and an Eastern Cooperative Oncology Group performance status ≤ 1 were eligible if no effective standard therapy was available and they had locally assessed B-RAF, N-RAS, or K-RAS mutation-positive solid tumors or pancreatic cancer with unknown mutation status. Exclusion criteria included untreated leptomeningeal or brain metastases, major surgery within 28 days or radiotherapy within 14 days of enrollment, or unresolved toxicity grade > 1 from prior cancer therapy. All inclusion/exclusion criteria are presented in the Data Supplement (online only).
During dose escalation, patients were enrolled in successive dose-escalation cohorts of 3-6 patients each. Lifirafenib was administered on day 1 followed by a 2-day treatment-free period to allow for single-dose PK sample collection; on days 4-24, patients received once-daily lifirafenib. After cycle 1, patients received continuous treatment with once-daily lifirafenib in 21-day cycles. The first patient received 15 mg lifirafenib twice daily, but the dosing regimen was changed to 5 mg once daily after reviewing the safety and PK profile in that patient. Subsequent dose escalation cohorts examined 5, 10, 20, 30, 40, 50, and 60 mg lifirafenib once daily. Because modeling and PK data suggested that discontinuous dosing regimens resulted in comparable or increased lifirafenib plasma concentrations versus continuous dosing schedules, an alternate 50-mg once-daily regimen on a week on/off cycle was examined (n = 3).
During dose expansion, patients with B-RAF and K-RAS/N-RAS mutations received lifirafenib 30 mg/d (RP2D) administered in 21-day cycles; patients could have melanoma (treatment naïve or resistant to B-RAF/MEK inhibitor therapy), CRC, non–small-cell lung cancer (NSCLC), thyroid cancer, endometrial cancer, pancreatic cancer, or other solid tumors. For both phases, patients continued treatment until unacceptable toxicity or patient withdrawal. Treatment beyond progression was permitted in patients who derived clinical benefit. Criteria for dose reductions/interruptions and treatment discontinuation are outlined in the Data Supplement.
End Points and Assessments
The primary end point during dose escalation was safety and tolerability, including dose-limiting toxicity (DLT) assessments and treatment-emergent adverse events (TEAEs). Adverse events (AEs) were graded by severity and evaluated by National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03). Secondary end points included PK parameters from blood plasma samples obtained at prespecified time points, objective response rates (ORRs; complete response [CR] + partial response [PR]) and stable disease (SD) on the basis of RECIST version 1.1, progression-free survival (PFS), duration of response (DoR), and duration of SD. Exploratory end points included 18F-fluorodeoxyglucose (FDG) uptake using FDG positron emission tomography-computed tomography (FDG-PET-CT) scans at screening and on cycle 2, day 1 (± 3 days).
The primary end point during dose expansion was investigator-assessed ORR. Secondary end points included PFS, DoR, duration of SD, disease control rate (DCR), clinical benefit rate (CR + PR + durable SD [SD ≥ 24 weeks]), safety/tolerability, and steady-state predose lifirafenib plasma concentration at prespecified time points.
Statistical Analysis
Dose escalation sample size was determined by dose levels and emerging toxicities; 30 patients were required to establish the RP2D and schedule. Expansion cohorts were to include ≤ 20 patients (total: up to 200 patients); enrollment in some arms stopped early because of difficulties in patient recruitment. Descriptive statistics summarized continuous and categorical variables; Kaplan-Meier estimators summarized time-to-event variables. The safety analysis set consisted of patients who received ≤ 1 dose of lifirafenib. The DLT analysis set included patients who experienced DLTs (or patients who received ≥ 80% of planned lifirafenib doses) during cycle 1.
Study Oversight
This study was conducted in accordance with the ethical principles of the Declaration of Helsinki, Good Clinical Practice guidelines, and the requirements of public registration of clinical trials. The protocol was approved by site-specific institutional review boards. Written informed consent was obtained from each patient at enrollment.
RESULTS
Patient Disposition
During dose escalation (n = 35), 31 patients received once-daily lifirafenib (5 mg, n = 3; 10 mg, n = 3; 20 mg, n = 3; 30 mg, n = 3; 40 mg, n = 10; 50 mg, n = 6; 60 mg, n = 3). Four patients received alternate lifirafenib dosing regimens (15 mg twice daily, n = 1; 50 mg/d on a week on/off schedule, n = 3). All 35 patients were included in the safety analysis set; 31 patients comprised the DLT analysis set. Reasons for treatment discontinuation included disease progression (n = 24), AEs (n = 5), investigator/sponsor decision (n = 1), and other (transition to lifirafenib compassionate use [thyroid cancer, n = 2; melanoma, n = 1]; clinical progression, n = 1; DLT, n = 1). During dose expansion (n = 96), reasons for treatment discontinuation included disease progression (n = 57), AEs (n = 19), withdrawal of consent (n = 4), investigator/sponsor decision (n = 3), death (n = 2), and other (transition to lifirafenib compassionate use [melanoma, n = 2; thyroid cancer, CRC, NSCLC, and LGSOC, n = 1 each], clinical progression, n = 5; noncompliance, n = 1). Across the entire study, 11 patients were treated > 2 weeks beyond progression (B-RAF, n = 7; K-RAS, n = 4); mean duration of treatment beyond progression was 157 days (range, 41-420 days) for patients with B-RAF mutations and 215 days (range, 100-256 days) for those with K-RAS mutations.
Demographic and Baseline Characteristics
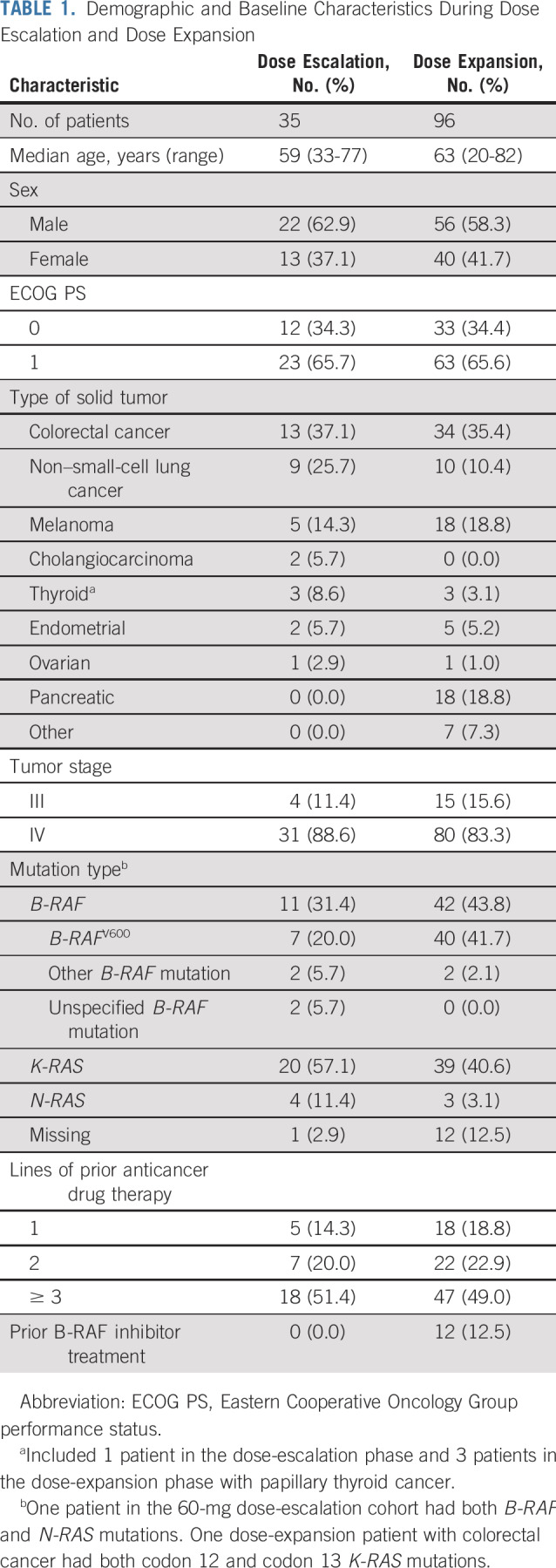
Demographic and baseline characteristics are listed in Table 1. Patients were heavily pretreated, with approximately 50% receiving ≥ 3 lines of anticancer therapy and ≥ 75% having prior anticancer surgery (dose escalation, 77.1%; dose expansion, 87.5%). Twelve patients with B-RAF mutations received prior B-RAF inhibitor treatment. During dose escalation, 31.4% of patients harbored B-RAF mutations, and 57.1% harbored K-RAS mutations; similar proportions of patients harbored B-RAF (43.8%) or K-RAS (40.6%) mutations during dose expansion.
TABLE 1.
Demographic and Baseline Characteristics During Dose Escalation and Dose Expansion
Safety and Tolerability
During dose escalation, the most frequent TEAEs were fatigue (n = 24; 68.6%) and dermatitis acneiform (n = 15; 42.9%; Table 2). Five patients (14.3%) experienced TEAEs that led to discontinuation. Treatment-related TEAEs were predominantly grades 1-2. The most common grade ≥ 3 treatment-related TEAEs during dose escalation were thrombocytopenia (14.3%), hypertension (11.4%), and fatigue (11.4%; Data Supplement). Among patients eligible for DLT assessment (n = 31), 6 experienced reversible DLTs, 5 of which occurred at doses ≥ 40 mg/d (Data Supplement). Observed DLTs included grade 3 increased ALT (n = 1) and grade 4 thrombocytopenia (n = 5); thrombocytopenia typically occurred within 2-3 weeks of initial dosing. Comprehensive investigations (including bone marrow biopsies) in the first 2 patients with thrombocytopenia revealed normal bone marrow morphology and reserve, which suggested a peripheral cause. Patients were treated with platelet transfusion (n = 1) or prednisolone administration (n = 4) and withholding of lifirafenib; platelet counts recovered within 6-20 days. Grade ≥ 3 TEAEs (including DLTs) occurred in 26 patients (74.3%), with more patients reporting grade ≥ 3 TEAEs in the 40, 50, and 60 mg/d cohorts (61.5%) versus lower-dose cohorts (38.5%). Grade ≥ 3 TEAEs that occurred in patients who received ≥ 40 mg/d are listed in the Data Supplement. The MTD was established at 40 mg/d. Eighty percent (4 of 5 patients) of dose-limiting thrombocytopenia occurred in patients who received 40 and 60 mg/d (n = 2 each), and 70% of patients treated with 40 mg/d had dose interruptions/reductions as a result of drug toxicity, typically between days 13 and 28 of cycle 1. On the basis of these data, the RP2D was established at 30 mg/d.
TABLE 2.
Incidence of Treatment-Emergent Adverse Events That Occurred in More Than 10% of Patients in Either Phase

Of 96 patients who received lifirafenib during dose expansion, the most commonly reported TEAEs were fatigue (n = 47; 49%) and decreased appetite (n = 35; 36.5%; Table 2). During the entire study, cutaneous SCC or keratoacanthoma was not reported. Grade ≥ 3 TEAEs occurred in 68 patients (70.8%), and serious TEAEs occurred in 56 patients (58.3%). TEAEs led to discontinuation in 19 patients (19.8%), most commonly fatigue (n = 5) and thrombocytopenia (n = 2; Data Supplement). Fifty patients (52%) experienced AEs that led to a dose adjustment, which resulted in a median relative dose intensity of 95.0%. Four patients (4.2%) experienced 6 TEAEs considered unrelated to treatment that led to death: pericardial effusion, sepsis, pleural effusion, intracranial hemorrhage, intestinal perforation as a result of disease progression, and small intestinal obstruction (n = 1 each). During dose expansion, the most common grade ≥ 3 treatment-related AEs were hypertension (8.3%) and fatigue (7.3%); 2 patients discontinued because of treatment-related grade ≥ 3 thrombocytopenia.
Antitumor Activity
Patients with B-RAF and K-RAS mutations from both phases had responses (Table 3). Among patients with B-RAF mutations, 8 (15.1%) of 53 achieved PR, including 1 patient with melanoma who received prior RAF inhibitor therapy, and 27 (50.9%) of 53 had SD; time to response and DoR of patients with select tumors are shown in Figure 1. During dose escalation, 1 patient with B-RAF–mutated melanoma (40 mg/d) achieved CR (Data Supplement). Across the entire study, 5 of 23 patients with B-RAFV600 melanoma achieved confirmed PR, including 4 treatment-naïve patients and 1 who progressed after 6 months of prior B-RAF inhibitor therapy (Fig 1; Data Supplement). Confirmed PRs occurred in patients with B-RAF–mutated thyroid cancer/PTC (n = 2) and B-RAFV600E LGSOC (n = 1). One patient with B-RAF–mutated NSCLC achieved unconfirmed PR.
TABLE 3.
Patient Responses by Mutation Status
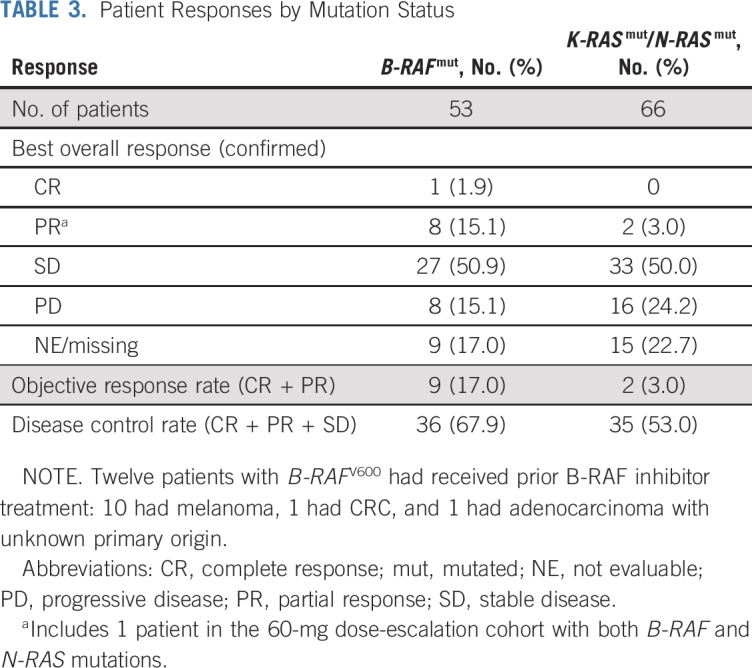
FIG 1.

(A and B) Responses in patients with B-RAF–mutated cancers (both phases). (>) Ongoing treatment indicates ongoing response at time of data cutoff. An additional 6 patients not included in this figure had prior B-RAF inhibitor treatment (4 had melanoma, 1 had colorectal cancer, and 1 had adenocarcinoma with unknown primary origin). (*) Patients who received prior B-RAF/MEK inhibitor treatment. CR, complete response; NSCLC, non–small-cell lung cancer; PD, progressive disease; PR, partial response; SD, stable disease.
Across the entire study, 2 patients with K-RAS mutations (endometrial cancer [20 mg/d] and codon 12–mutated NSCLC [30 mg/d], n = 1 each) had confirmed responses, which resulted in an ORR of 3.4%; 32 patients (54.2%) with K-RAS mutations had SD (Fig 2). Twelve patients with K-RAS– or N-RAS–mutated CRC had SD (Data Supplement); 5 patients with K-RAS–mutated endometrial cancer (codon 12 mutated, n = 4; codon 2 mutated, n = 1) and 2 patients with K-RAS–mutated NSCLC (codon 12 mutated and K-RAS G13D, n = 1 each) had SD (Data Supplement). Median DoR was 31.7 months (95% CI, 6.8 to 31.7 months) during dose escalation and 11.1 months (95% CI, 1.9 months to not reached) during dose expansion. Median duration of SD during dose expansion was 4.1 months (95% CI, 3.5 to 4.9 months). Duration of treatment was > 1 year for several patients with B-RAF (n = 10) and K-RAS mutations (n = 5).
FIG 2.

(A and B) Responses in patients with K-RAS–mutated cancers (excluding colorectal cancer; both phases). NE, not evaluable; NSCLC, non–small-cell lung cancer; PD, progressive disease; PR, partial response; SD, stable disease.
PK Outcomes
Systemic exposure increased from 5 to 50 mg on cycle 1, day 1, and cycle 2, day 1 (Data Supplement). Although not powered to assess proportionality, the log-log regression model accounted for > 80% of the observed variation from 10 to 60 mg for cycle 1, day 1. Lifirafenib was rapidly absorbed, with a median time to reach maximum plasma concentration of 3 hours. The accumulation ratio for maximum serum concentration (Cmax), area under the plasma concentration curve from 0-9 hours (AUC0-9), and AUC0-24 estimated from cycle 2, day 1/cycle 1, day 1, was similar from 10 to 50 mg (Data Supplement). Average accumulation ranged from 3.3- to 6.1-fold for Cmax and 3.6- to 7.6-fold for AUC0-9 and AUC0-24. Three patients had measurable terminal half-life (range, 15-59 hours); terminal half-life estimates should be interpreted with caution because samples were not collected beyond 72 hours after dosing.
FDG-PET-CT Scans
During dose escalation, 34 patients (including those who received lifirafenib 15 mg twice weekly or 50 mg once daily on a week on/off cycle) had tumor imaging on cycle 2, day 1. FDG-PET-CT scans showed no FDG uptake in 1 patient, low FDG uptake in 1 patient, moderate FDG uptake in 10 patients, and high FDG uptake in 15 patients (7 patients were missing postbaseline results). Complete metabolic reduction was observed in 1 patient with a B-RAF mutation who received 40 mg/d; 11 patients had partial metabolic reduction per European Organization for Research and Treatment of Cancer criteria.26 Decreases in maximum and average standardized uptake volumes, indicative of significant pharmacodynamic activity, were generally observed at lifirafenib doses ≥ 30 mg. At cycle 2, day 1, changes in FDG uptake from baseline were observed in 9 patients, including 7 (20.6%) who changed from high to moderate uptake, 1 (2.9%) who changed from high to low uptake, and 1 (2.9%) who changed from low to no uptake. All patients with confirmed clinical responses (n = 3; 8.6%) had FDG-PET-CT responses, but not all patients with FDG-PET-CT responses had clinical responses (6 had SD). Among patients who received once-daily lifirafenib (n = 31), FDG-PET-CT responses were observed in patients with B-RAF, K-RAS, and N-RAS mutations (Fig 3).
FIG 3.

Responses across lifirafenib dose cohorts by 18F-fluorodeoxyglucose positron emission tomography-computed tomography (FDG-PET-CT) analysis. Two patients were missing a postbaseline PET scan. (*) Patients who were treated beyond progression. CR, complete response; NE, not evaluable; PD, progressive disease; PR, partial response; SD, stable disease; SUVave, average standardized uptake volume.
DISCUSSION
This first-in-human dose-escalation/dose-expansion study evaluated lifirafenib, a novel investigational oral RAF family kinase inhibitor, in patients with B-RAF–, K-RAS–, or N-RAS–mutated solid tumors. The MTD was determined as 40 mg once daily; DLTs included reversible thrombocytopenia and increased ALT.
Lifirafenib had an acceptable risk-benefit profile. The dose-escalation phase suggested that once-daily lifirafenib was better tolerated at doses ≤ 30 mg than ≥ 40 mg, irrespective of schedule. Treatment-related dermatologic toxicities were observed, but cutaneous SCCs and keratoacanthomas reported with other B-RAF inhibitor treatments27 were not. Treatment-related thrombocytopenia and hypertension were manageable and likely stemmed from off-target inhibition of kinases other than B-RAF, including EGFR and PDGF. A prior report demonstrated that lifirafenib inhibited additional kinases, including EGFR, DDR1, DDR2, EPHA3, FLT3, VEGFR2, ABL1, RET, EPHGA7, EPHB2, MNK2, and ZAK, with half-maximal inhibitory concentrations (IC50s) within 10-fold of that for B-RAFV600E.15 With an IC50 of 108 nM, inhibition of VEGFR2 by lifirafenib could potentially account for the clinically observed hypertension in our study and was similar to off-target effects of other kinase inhibitor therapies.15,28 The precise mechanism of thrombocytopenia remains unclear but seems dose related because a higher proportion of patients experienced thrombocytopenia at doses ≥ 40 versus ≤ 30 mg/d. Thrombocytopenia resolved rapidly with drug interruption, and although steroids were initiated on the basis of an assumed peripheral (possibly immune-mediated) mechanism, the specific impact this intervention had remains unclear. A more direct kinase-mediated effect, particularly given lifirafenib’s novel kinase inhibition profile, remains possible and warrants additional exploration. Kinases, including EGFR and PDGF, have individually, but rarely, been implicated in platelet dysfunction,29-31 and the effect of collectively inhibiting these kinases is unclear. Of note, thrombocytopenia was manageable at or below the RP2D and did not recur with ongoing dosing in patients who continued treatment. Additional exploration of alternate dose schedules and underlying causative mechanisms remains an important priority in the ongoing development of lifirafenib.
Lifirafenib was rapidly absorbed, and systemic exposure increased during cycles 1 and 2 from 10 to 50 mg/d. During dose escalation, FDG-PET-CT scans were used as a surrogate marker of MAPK pathway inhibition to define optimal biologic dosing.32 Partial/complete metabolic reductions by FDG-PET-CT scan were observed in 12 of 34 patients, including 11 who received ≥ 30 mg/d lifirafenib, which suggests significant pharmacodynamic activity at doses ≥ 30 mg/d. The accumulation ratio was similar from 10-50 mg/d. On the basis of the safety/tolerability, antitumor activity, PK profile, and pharmacodynamic activity, the RP2D was established at 30 mg/d.
While the overall antitumor activity of lifirafenib was modest, antitumor activity was observed in patients with B-RAF–mutated solid tumors, including melanoma, thyroid cancer, and LGSOC. Limited information with regard to pharmacodynamic markers of response and/or resistance were collected; detailed genetic information for responders/nonresponders (beyond underlying B-RAF and K-RAS mutations) was unavailable for most patients. Additional investigation may be needed to understand the underlying mechanism of response and resistance to this novel agent and to inform future treatment decisions and/or trial design. During dose expansion, 4 of 7 patients with B-RAFV600–mutated melanoma who had not received prior B-RAF/MEK inhibitor therapy had confirmed PR (ORR, 57.1%; DCR, 85.7%). Because 2 patients with responses and no PD transferred to a compassionate use program, DoR was not reached. One of 3 patients with B-RAFV600–mutated thyroid cancer had confirmed PR (ORR, 33.3%; DCR, 100%; Data Supplement), comparable to reported clinical activities for first-generation B-RAF inhibitors, such as vemurafenib and dabrafenib, which demonstrated an ORR of 48% and 50% (DCR, 91%), respectively, in patients with B-RAF–mutated melanoma.12,33 Importantly, antitumor activity was observed in K-RAS–mutant cancers, including NSCLC and endometrial carcinoma (Data Supplement), with prolonged disease control. In contrast, lifirafenib had limited clinical activity in patients with K-RAS–mutated CRC or pancreatic cancer. This observation was unlikely due to differences in K-RAS mutations between NSCLC and CRC or pancreatic cancer because these results were similar to phase I results of a novel RAF/MEK inhibitor, RO5126766, that showed promise in KRAS-mutated NSCLC and endometrial/ovarian cancers but not in K-RAS–mutated CRC.34,35 This suggests that oncogenic K-RAS may be context dependent, potentially requiring different approaches on the basis of tumor types. Two novel investigational K-RASG12C covalent inhibitors, AMG 510 and MRTX849, recently demonstrated preliminary antitumor activity in K-RAS–mutant tumors.36,37 Albeit early, the data showed promising efficacy signals and may represent a breakthrough in treatment of K-RAS–mutant tumors. However, targeting only K-RASG12C mutations limits the utility of these new therapies, resulting in a continued unmet medical need for tumors with oncogenic K-RAS mutations beyond K-RASG12C.
In summary, lifirafenib demonstrated an acceptable risk-benefit profile given the safety results and responses in patients with B-RAF–mutated melanoma, thyroid cancer, and LGSOC and K-RAS–mutated NSCLC and endometrial cancer. Our findings suggest that lifirafenib could potentially benefit patients with MAPK pathway–associated kinase alterations beyond B-RAFV600 mutations, including activated K-RAS. Additional investigation of the safety/efficacy of lifirafenib as monotherapy or in combination is warranted, especially in K-RAS–mutated solid tumor malignancies other than CRC; a phase I/II trial of lifirafenib in combination with a MEK inhibitor in patents with B-RAF– and RAS-mutant tumors (ClinicalTrials.gov identifier: NCT03905148) has recently commenced enrollment.
ACKNOWLEDGMENT
We acknowledge the investigative center study staff, the study patients, and their families. Financial support for the development of this article, including medical writing and editorial assistance, under the authors’ guidance, provided by Stephan Lindsey, PhD, and Elizabeth Hermans, PhD, of OPEN Health Medical Communications (Chicago, IL), was provided by BeiGene.
PRIOR PRESENTATION
Presented at the American Association for Cancer Research Annual Meeting 2017, Washington, DC, April 1-5, 2017, and American Association for Cancer Research Annual Meeting 2018, Chicago, IL, April 14-18, 2018.
SUPPORT
Supported by BeiGene.
CLINICAL TRIAL INFORMATION
DATA SHARING STATEMENT
Upon request, and subject to certain criteria, conditions, and exceptions, BeiGene will provide access to individual de-identified participant data from BeiGene-sponsored global interventional clinical studies conducted for medicines (1) for indications that have been approved or (2) in programs that have been terminated. BeiGene will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data requests may be submitted to medicalinformation@beigene.com.
See accompanying article on page 2197
AUTHOR CONTRIBUTIONS
Conception and design: Jayesh Desai, Hui Gan, Michael Millward, Gary Richardson, Phillip Parente, Lai Wang, Lusong Luo, Benjamin Solomon
Provision of study material or patients: Catherine Barrow, Michael Jameson, Victoria Atkinson, Andrew Haydon, Michael Millward, Stephen Begbie, Michael Brown, Ben Markman, William Patterson, Andrew Hill, Adnan Nagrial, Christopher Jackson, Michael Friedlander, Ben Tran, Lusong Luo, Benjamin Solomon
Collection and assembly of data: Jayesh Desai, Hui Gan, Catherine Barrow, Michael Jameson, Victoria Atkinson, Andrew Haydon, Michael Millward, Stephen Begbie, Michael Brown, Ben Markman, William Patterson, Andrew Hill, Lisa Horvath, Adnan Nagrial, Gary Richardson, Christopher Jackson, Michael Friedlander, Phillip Parente, Ben Tran, Yunxin Chen, Dewan Zeng, Benjamin Solomon
Data analysis and interpretation: Jayesh Desai, Hui Gan, Catherine Barrow, Michael Jameson, Victoria Atkinson, Andrew Haydon, Michael Millward, Stephen Begbie, Ben Markman, Gary Richardson, Christopher Jackson, Phillip Parente, Ben Tran, Zhiyu Tang, Wendy Huang, John Wu, Dewan Zeng, Lusong Luo, Benjamin Solomon
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Phase I, Open-Label, Dose-Escalation/Dose-Expansion Study of Lifirafenib (BGB-283), an RAF Family Kinase Inhibitor, in Patients With Solid Tumors
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Jayesh Desai
Consulting or Advisory Role: Eli Lilly, Eisai, BeiGene, Biocon, Amgen (Inst)
Research Funding: Roche (Inst), GlaxoSmithKline (Inst), Novartis (Inst), Bionomics (Inst), BeiGene (Inst), Eli Lilly (Inst), Bristol-Myers Squibb (Inst), AstraZeneca (Inst), MedImmune (Inst)
Hui Gan
Consulting or Advisory Role: Bristol-Myers Squibb
Speakers’ Bureau: Eisai, Merck Serono
Research Funding: AbbVie
Catherine Barrow
Travel, Accommodations, Expenses: Merck
Michael Jameson
Research Funding: Merck (Inst), Pfizer (Inst), BeiGene (Inst), Dynavax (Inst), Sabinsa (Inst), Bristol-Myers Squibb (Inst), Eli Lilly (Inst)
Victoria Atkinson
Honoraria: Bristol-Myers Squibb, Novartis, Merck Sharp & Dohme, Pierre Fabre, Roche, Genentech, Merck Serono
Consulting or Advisory Role: Bristol-Myers Squibb, Merck Sharp & Dohme, Novartis, Merck Serono, Pierre Fabre, Roche
Speakers’ Bureau: Roche, Genentech, Bristol-Myers Squibb, Novartis, Merck Sharp & Dohme, Merck Serono
Travel, Accommodations, Expenses: Bristol-Myers Squibb, OncoSec, Merck Sharp & Dohme, Pierre Fabre
Andrew Haydon
Honoraria: Novartis, Merck
Consulting or Advisory Role: Novartis, Pierre Fabre
Speakers’ Bureau: Novartis, Merck
Michael Millward
Consulting or Advisory Role: Roche, Bristol-Myers Squibb, AstraZeneca, Merck Sharp & Dohme, Novartis, Pfizer, EMD Serono
Research Funding: Bristol-Myers Squibb (Inst), Novartis (Inst), Roche (Inst), AstraZeneca (Inst), Takeda Pharmaceuticals (Inst), GlaxoSmithKline (Inst), BeiGene (Inst), Eli Lilly (Inst), Apollomics (Inst), PIN Pharma (Inst), Albion (Inst), AkesioBio (Inst), AbbVie (Inst), C-Stone Pharmaceuticals (Inst), Therapim (Inst), Five Prime Therapeutics (Inst), Dizal (Inst), Maxinovel (Inst)
Travel, Accommodations, Expenses: Roche, Bristol-Myers Squibb, AstraZeneca
Stephen Begbie
Consulting or Advisory Role: AstraZeneca, Janssen Oncology, Roche, Astellas Pharma, MSD Oncology, Noxopharm,
Research Funding: Astellas Pharma (Inst), Medivation (Inst), Merck Serono (Inst), Janssen Oncology (Inst), Roche (Inst), Boston Biomedical (Inst), Merck (Inst), Pfizer (Inst), EMD Serono (Inst), Bristol-Myers Squibb (Inst)
Travel, Accommodations, Expenses: Astellas Pharma
Michael Brown
Honoraria: Bristol-Myers Squibb Australia (Inst), MSD Oncology (Inst), Novartis (Inst)
Consulting or Advisory Role: Amgen (Inst), Bristol-Myers Squibb (Inst), Merck Sharp & Dohme (Inst), Novartis (Inst), GlaxoSmithKline (Inst), Roche (Inst), Cartherics
Research Funding: Bristol-Myers Squibb (Inst), GlaxoSmithKline (Inst), Merck Sharp & Dohme (Inst), Roche (Inst), Novartis (Inst), Pharmaust (Inst), Zucero (Inst), C-Stone Pharmaceuticals (Inst)
Ben Markman
Travel, Accommodations, Expenses: Akeso Biopharma
Andrew Hill
Employment: Tasman Oncology
Stock and Other Ownership Interests: Tasman Health Care and Tasman Oncology Research
Honoraria: Bristol-Myers Squibb
Travel, Accommodations, Expenses: Bristol-Myers Squibb, Merck
Lisa Horvath
Employment: Connected Medical Solutions (I)
Leadership: Connected Medical Solutions (I)
Stock and Other Ownership Interests: Connected Medical Solutions (I), Imagion Biosystems
Consulting or Advisory Role: Imagion Biosystems
Research Funding: Astellas Pharma
Travel, Accommodations, Expenses: Astellas Pharma, Janssen-Cilag, Pfizer
Adnan Nagrial
Consulting or Advisory Role: Bristol-Myers Squibb, MSD Oncology, AstraZeneca, Roche
Research Funding: Bristol-Myers Squibb (Inst), MSD Oncology (Inst), AstraZeneca (Inst), MedImmune (Inst), Amgen (Inst), Akeso Biopharma (Inst)
Gary Richardson
Research Funding: Bristol-Myers Squibb (Inst), Roche (Inst), Genentech (Inst), AstraZeneca (Inst), Merck (Inst), Takeda Pharmaceuticals (Inst), BeiGene (Inst), Pfizer (Inst), CBT Pharmaceuticals (Inst), Corvus Pharmaceuticals (Inst), Novotech (Inst), Shanghai Fosun Pharmaceutical Development Co (Inst), Shanghai Henlius Biotech (Inst), Five Prime Therapeutics (Inst), Suzhou Alphamab Co (Inst)
Christopher Jackson
Honoraria: MSD Oncology, Athenex
Research Funding: Athenex (Inst)
Patents, Royalties, Other Intellectual Property: Patent pending Oraxol, Athenex, no financial interest
Travel, Accommodations, Expenses: Athenex
Michael Friedlander
Honoraria: AstraZeneca, MSD, Eli Lilly, Takeda Pharmaceuticals
Consulting or Advisory Role: AstraZeneca, MSD, AbbVie, Eli Lilly, Takeda Pharmaceuticals
Speakers’ Bureau: AstraZeneca
Research Funding: BeiGene (Inst)
Ben Tran
Honoraria: Astellas Pharma, Janssen-Cilag, Sanofi, Tolmar, Amgen
Consulting or Advisory Role: Amgen, Astellas Pharma, Bayer AG, Sanofi, Tolmar, Janssen-Cilag, Bristol-Myers Squibb, Ipsen, MSD Oncology
Research Funding: Astellas Pharma (Inst), Janssen-Cilag (Inst), Amgen (Inst), Pfizer (Inst), Genentech (Inst), AstraZeneca (Inst), Bayer AG (Inst), Pfizer (Inst), Bristol-Myers Squibb, Merck Sharp & Dohme
Travel, Accommodations, Expenses: Amgen, Astellas Pharma
Lai Wang
Employment: BeiGene
Stock and Other Ownership Interests: BeiGene
Yunxin Chen
Employment: BeiGene (Beijing) Co, Bayer China
Stock and Other Ownership Interests: BeiGene (Beijing) Co
Zhiyu Tang
Employment: BeiGene
Stock and Other Ownership Interests: BeiGene
Wendy Huang
Employment: Rakuten Medical, BeiGene
John Wu
Employment: BeiGene
Stock and Other Ownership Interests: BeiGene
Dewan Zeng
Employment: BeiGene
Stock and Other Ownership Interests: BeiGene
Travel, Accommodations, Expenses: BeiGene
Lusong Luo
Employment: BeiGene, BeiGene (I)
Leadership: MapKure
Stock and Other Ownership Interests: BeiGene, Bristol-Myers Squibb, GlaxoSmithKline, Gilead, Regeneron Pharmaceuticals, Incyte, Merck, Clovis Oncology, Adaptimmune, Rigel, Arrowhead Pharmaceuticals, AbbVie, AstraZeneca
Patents, Royalties, Other Intellectual Property: Multiple patents obtained as BeiGene employee, multiple patents awarded as BeiGene employee (I)
Travel, Accommodations, Expenses: BeiGene, BeiGene (I)
Benjamin Solomon
Honoraria: Bristol-Myers Squibb, AstraZeneca, Merck Sharp & Dohme, Roche, Genentech, Pfizer
Consulting or Advisory Role: Bristol-Myers Squibb, Merck Sharp & Dohme, AstraZeneca, Pfizer (Inst), Roche, Genentech, Loxo Oncology
Research Funding: Pfizer (Inst)
Patents, Royalties, Other Intellectual Property: Royalties from Veristrat (Biodesix), UpToDate
No other potential conflicts of interest were reported.
REFERENCES
- 1.Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:103–119. doi: 10.1517/14728222.2011.645805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okuda T, Sekizawa A, Purwosunu Y, et al. Genetics of endometrial cancers. Obstet Gynecol Int. 2010;2010:984013. doi: 10.1155/2010/984013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Durrant DE, Morrison DK. Targeting the Raf kinases in human cancer: The Raf dimer dilemma. Br J Cancer. 2018;118:3–8. doi: 10.1038/bjc.2017.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239–1246. doi: 10.1200/JCO.2010.32.4327. [DOI] [PubMed] [Google Scholar]
- 5.Bösmüller H, Fischer A, Pham DL, et al. Detection of the BRAF V600E mutation in serous ovarian tumors: A comparative analysis of immunohistochemistry with a mutation-specific monoclonal antibody and allele-specific PCR. Hum Pathol. 2013;44:329–335. doi: 10.1016/j.humpath.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 6.Singer G, Oldt R, III, Cohen Y, et al. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst. 2003;95:484–486. doi: 10.1093/jnci/95.6.484. [DOI] [PubMed] [Google Scholar]
- 7.Jones S, Wang TL, Kurman RJ, et al. Low-grade serous carcinomas of the ovary contain very few point mutations. J Pathol. 2012;226:413–420. doi: 10.1002/path.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong KK, Tsang YT, Deavers MT, et al. BRAF mutation is rare in advanced-stage low-grade ovarian serous carcinomas. Am J Pathol. 2010;177:1611–1617. doi: 10.2353/ajpath.2010.100212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strickler JH, Wu C, Bekaii-Saab T. Targeting BRAF in metastatic colorectal cancer: Maximizing molecular approaches. Cancer Treat Rev. 2017;60:109–119. doi: 10.1016/j.ctrv.2017.08.006. [DOI] [PubMed] [Google Scholar]
- 10.Stephen AG, Esposito D, Bagni RK, et al. Dragging ras back in the ring. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 11.Chapman PB, Robert C, Larkin J, et al. Vemurafenib in patients with BRAFV600 mutation-positive metastatic melanoma: Final overall survival results of the randomized BRIM-3 study. Ann Oncol. 2017;28:2581–2587. doi: 10.1093/annonc/mdx339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 13. Kopetz S, Desai J, Chan E, et al: PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J Clin Oncol 28, 2010 (suppl; abstr 3534)
- 14.Corcoran RB, Ebi H, Turke AB, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang Z, Yuan X, Du R, et al. BGB-283, a novel RAF kinase and EGFR inhibitor, displays potent antitumor activity in BRAF-mutated colorectal cancers. Mol Cancer Ther. 2015;14:2187–2197. doi: 10.1158/1535-7163.MCT-15-0262. [DOI] [PubMed] [Google Scholar]
- 16. Anforth R, Menzies A, Byth K, et al: Factors influencing the development of cutaneous squamous cell carcinoma in patients on BRAF inhibitor therapy. J Am Acad Dermatol 72:809-815.e1, 2015. [DOI] [PubMed]
- 17.Shi H, Moriceau G, Kong X, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. doi: 10.1038/ncomms1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Girotti MR, Pedersen M, Sanchez-Laorden B, et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013;3:158–167. doi: 10.1158/2159-8290.CD-12-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rizos H, Menzies AM, Pupo GM, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin Cancer Res. 2014;20:1965–1977. doi: 10.1158/1078-0432.CCR-13-3122. [DOI] [PubMed] [Google Scholar]
- 21.Mao M, Tian F, Mariadason JM, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res. 2013;19:657–667. doi: 10.1158/1078-0432.CCR-11-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Temraz S, Mukherji D, Shamseddine A. Dual inhibition of MEK and PI3K pathway in KRAS and BRAF mutated colorectal cancers. Int J Mol Sci. 2015;16:22976–22988. doi: 10.3390/ijms160922976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tang Z, Liu Y, Jiang B, et al: BGB-283: A novel RAF dimer inhibitor, displays potent antitumor activity in HCC patient derived xenograft models. Cancer Res 76, 2016 (suppl; abstr 1230)
- 24. Desai J, Markman B, Sandhu SK, et al: Updated safety, efficacy, and pharmacokinetics (PK) results from the phase I study of BGB-A317, an antiprogrammed death-1 (PD-1) mAb in patients (pts) with advanced solid tumors. Presented at 31st Ann Meet Soc Immunother Cancer, National Harbor, MD, November 9−13, 2016.
- 25.Hao H, Muniz-Medina VM, Mehta H, et al. Context-dependent roles of mutant B-Raf signaling in melanoma and colorectal carcinoma cell growth. Mol Cancer Ther. 2007;6:2220–2229. doi: 10.1158/1535-7163.MCT-06-0728. [DOI] [PubMed] [Google Scholar]
- 26.Young H, Baum R, Cremerius U, et al. Measurement of clinical and subclinical tumour response using [18F]-fluorodeoxyglucose and positron emission tomography: Review and 1999 EORTC recommendations. Eur J Cancer. 1999;35:1773–1782. doi: 10.1016/s0959-8049(99)00229-4. [DOI] [PubMed] [Google Scholar]
- 27.Wu JH, Cohen DN, Rady PL, et al. BRAF inhibitor-associated cutaneous squamous cell carcinoma: New mechanistic insight, emerging evidence for viral involvement and perspectives on clinical management. Br J Dermatol. 2017;177:914–923. doi: 10.1111/bjd.15348. [DOI] [PubMed] [Google Scholar]
- 28.Robinson ES, Khankin EV, Karumanchi SA, et al. Hypertension induced by vascular endothelial growth factor signaling pathway inhibition: Mechanisms and potential use as a biomarker. Semin Nephrol. 2010;30:591–601. doi: 10.1016/j.semnephrol.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao H, Bhuyan AAM, Umbach AT, et al. Inhibitory effect of afatinib on platelet activation and apoptosis. Cell Physiol Biochem. 2017;43:2264–2276. doi: 10.1159/000484377. [DOI] [PubMed] [Google Scholar]
- 30.Ohnishi H, Yamaguchi K, Shimada S, et al. A new approach to the treatment of atherosclerosis and trapidil as an antagonist to platelet-derived growth factor. Life Sci. 1981;28:1641–1646. doi: 10.1016/0024-3205(81)90320-9. [DOI] [PubMed] [Google Scholar]
- 31.Ohnishi H, Kosuzume H, Hayashi Y, et al. Effects of trapidil on thromboxane A2-induced aggregation of platelets, ischemic changes in heart and biosynthesis of thromboxane A2. Prostaglandins Med. 1981;6:269–281. doi: 10.1016/0161-4630(81)90151-8. [DOI] [PubMed] [Google Scholar]
- 32.McArthur GA, Puzanov I, Amaravadi R, et al. Marked, homogeneous, and early [18F]fluorodeoxyglucose-positron emission tomography responses to vemurafenib in BRAF-mutant advanced melanoma. J Clin Oncol. 2012;30:1628–1634. doi: 10.1200/JCO.2011.39.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harris SJ, Luken MJ, Perez DR, et al: Updated efficacy and safety results from the phase I study of intermittent dosing of the dual MEK/RAF inhibitor, RO5126766 in patients (pts) with RAS/RAF mutated advanced solid tumours. J Clin Oncol 34, 2016 (suppl; abstr 2582)
- 35. Chenard-Poirier M, Kaiser M, Boyd K, et al: Results from the biomarker-driven basket trial of RO5126766 (CH5127566), a potent RAF/MEK inhibitor, in RAS- or RAF-mutated malignancies including multiple myeloma. J Clin Oncol 35, 2017 (suppl; abstr 2506)
- 36.Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–223. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 37. Hallin J, Engstrom LD, Hargis L, et al: The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov 10:54-71, 2020. [DOI] [PMC free article] [PubMed]