

ABSTRACT
Retinal regeneration is robust in some cold-blooded vertebrates, but this process is ineffective in warm-blooded vertebrates. Understanding the mechanisms that suppress the reprogramming of Müller glia into neurogenic progenitors is key to harnessing the regenerative potential of the retina. Inflammation and reactive microglia are known to influence the formation of Müller glia-derived progenitor cells (MGPCs), but the mechanisms underlying this interaction are unknown. We used a chick in vivo model to investigate nuclear factor kappa B (NF-κB) signaling, a critical regulator of inflammation, during the reprogramming of Müller glia into proliferating progenitors. We find that components of the NF-κB pathway are dynamically regulated by Müller glia after neuronal damage or treatment with growth factors. Inhibition of NF-κB enhances, whereas activation suppresses, the formation of proliferating MGPCs. Following microglia ablation, the effects of NF-κB-agonists on MGPC-formation are reversed, suggesting that signals provided by reactive microglia influence how NF-κB impacts Müller glia reprogramming. We propose that NF-κB is an important signaling ‘hub’ that suppresses the reprogramming of Müller glia into proliferating MGPCs and this ‘hub’ coordinates signals provided by reactive microglia.
KEY WORDS: Retina, Glia, Müller glia, NF-κB, Regeneration
Summary: In vivo studies in chick reveal that NF-κB plays significant roles in regulating glial reactivity, neuronal survival and the reprogramming of Müller glia into proliferating Müller glia progenitor cells.
INTRODUCTION
Müller glia are the primary type of support cell of the retina and have the capacity to reprogram into proliferating neurogenic progenitor cells (Fischer and Reh, 2001). Although Müller glia have the potential to act as a source of retinal regeneration, their regenerative potential varies greatly across vertebrate species. In teleost fish, Müller glia readily undergo a neurogenic program to produce different types of retinal neurons and restore visual function (Bernardos et al., 2007; Fausett and Goldman, 2006; Hitchcock and Raymond, 1992; Lenkowski and Raymond, 2014). By contrast, mammalian Müller glia undergo a gliotic program after damage and fail to reprogram into proliferating progenitor-like cells (Bringmann et al., 2009; Dyer and Cepko, 2000). Interestingly, avian Müller glia have a regenerative potential that lies between that of Müller glia in fish and mammals (Fischer and Reh, 2001; Gallina et al., 2014a). In the chick retina, Müller glia can de-differentiate, acquire a progenitor phenotype and proliferate to produce numerous progeny; of these progeny, only a fraction differentiate into neurons (Fischer and Reh, 2001, 2002). The neurogenic potential of Müller glia-derived progenitor cells (MGPCs) can be enhanced by targeting cell-signaling pathways, including Notch (Ghai et al., 2010; Hayes et al., 2007), glucocorticoid (Gallina et al., 2014b), Jak/Stat (Todd et al., 2016b) and retinoic acid signaling (Todd et al., 2018). In damaged mouse retina, reprogramming of Müller glia can be driven by the forced expression of the Ascl1, a pro-neural bHLH transcription factor, and inhibition of histone de-acetylases (HDACs) (Jorstad et al., 2017; Ueki et al., 2015). Identifying pathways and factors that promote retinal regeneration in the zebrafish, but suppress regeneration in the chick and mouse, is expected to guide the development of novel therapeutic strategies that could restore vision in diseased retinas.
In damaged retinas, injured neurons and activation of immune cells are believed to initiate and guide the process of reprogramming of Müller glia into MGPCs. The immune system and pro-inflammatory signals regulate neurogenesis in zebrafish brain (Kyritsis et al., 2012). Both acute damage and chronic degeneration of the retina result in inflammation, characterized and mediated by the accumulation of reactive microglia (Graeber and Stre'rt, 1990; Karlstetter et al., 2010; Wang and Wong, 2014). In response to damage or pro-inflammatory signals, retinal microglia rapidly respond by migrating and upregulating cytokines (Fischer et al., 2014; Todd et al., 2019; Zelinka et al., 2012). In the chick retina, the ablation of microglia suppresses the formation of proliferating MGPCs (Fischer et al., 2014). Similarly, the ablation of microglia in the zebrafish retina impairs neuronal regeneration (Conedera et al., 2019; White et al., 2017). In addition, pro-inflammatory signals, such as IL6 or TNFα, promote the formation of MGPCs in the zebrafish retina (Conner et al., 2014; Zhao et al., 2014). These pro-inflammatory cytokines are known to signal through the NF-κB pathway (Osborn et al., 1989).
NF-κB mediates inflammation in response to injury and infection, but also regulates cell survival, apoptosis, proliferation and differentiation in various cellular contexts (Hayden and Ghosh, 2004). Currently, the role of NF-κB in the formation of MGPCs in the retina is not understood. Accordingly, we investigate how NF-κB influences the reprogramming of Müller glia into proliferating MGPCs in the avian retina, and whether microglia are involved in regulating NF-κB.
RESULTS
Expression patterns of NF-κB signaling components in damaged retinas
To provide a cellular context for NF-κB in the chick retina we queried single cell RNA-sequencing (scRNA-seq) databases generated from chick retinas at different times after treatment with 1000 nmol NMDA (Fig. 2A). NF-κB genes queried and corresponding protein names are listed in Fig. 1. UMAP plots of aggregate scRNA-seq databases revealed discrete clustering of Müller glia from control retinas and 24 h after NMDA treatment, whereas Müller glia from 48 and 72 h after treatment were clustered together (Fig. 2B, Fig. S1). Müller glia were identified based on expression of VIM, SLC3A1, GLUL and RLBP1, and MGPCs were identified based on downregulation of GLUL and RLBP1, and upregulation of PCNA, CDK1 and TOP2A (Fig. 2B, Fig. S1). Clusters of different types of retinal neurons were identified based on well-established markers (see Materials and Methods).
Fig. 1.

Schematic summary of NF-κB signaling, and the drugs and factors used in this study. Green text and lines indicate factors or drugs and sites of activation for NF-κB. Red text and lines indicate drugs and sites of inhibition for NF-κB. The list provides the proteins and corresponding genes that are involved in NF-κB signaling.
Fig. 2.

Expression patterns of NF-κB components in damaged retinas. Libraries for scRNA-seq were established for control retinas and retinas at 24, 48 and 72 h after NMDA treatment. (A,B) UMAP plots show distinct clustering of different retinal cell populations from control and NMDA-damaged retinas (at different times after treatment), and the numbers of cells surveyed within each cluster (in parentheses). Each dot represents one cell. Clusters of different types of retinal cells were identified based on collective expression of different cell-distinguishing markers as described in the Materials and Methods. (C,D,F) UMAP plots illustrate expression of NFKBIA, NFKBIB, NFKBIZ, CHUK, TNFSF10, TNFRSF1A and TNFRSF21. (E,G) Violin/scatter plots illustrate expression levels of NFKBIA, NFKBIB, NFKBIZ, CHUK, TNFSF10, TNFRSF1A and TNFRSF21 in Müller glia and MGPCs in controls and at different times after NMDA treatment. (H) Clusters identified as Müller glia and MGPCs were re-embedded for pseudotime analysis. Pseudotime states included: (1; peach) resting Müller glia, (2; olive) transitional MG, (3; green) transitional MG, (4; blue) MGPCs and (5; magenta) activated Müller glia. (I) Dimensional reduction of pseudotime on the x-axis placed the resting Müller glia to the far left and the MGPCs to the far right. Expression of GLUL and CDK1 are shown across stretched pseudotime in different branches, one branch ending in MGPCs and one branch ending in activated Müller glia. (J,K) Relative levels of expression among Müller glia and MGPCs was assessed across branched pseudotime (J) and across pseudotime states in violin plots (K). Expression levels were assessed for components of the NF-κB-pathway, NFKBIA, NFKBIB and NFKBIZ, and TNF-related factors and receptors, TNFSF10, TNFRSF1A and TNTRSF21. Significance of difference (**P<0.01, ***P<0.001) was determined by using a Wilcoxon rank sum with Bonferroni correction.
We probed for different components of the NF-κB pathway, including NFKBIA, NFKBIB, NFKBIZ and CHUK. NF-κB transcription factors include P65 (RELA), RelB (RELB), Rel (REL), P50 (NFKB1) and P52 (NFKB2) (Wang et al., 2017). NF-κB signaling is regulated by cytoplasmic inhibitor of kappa B (IκB), which comprises IkBα (NFKBIA), IκBβ (NFKBIB), IκBε (NFKBIE), IκBγ (IKBKG) and IkBζ (NFKBIZ), which mask the nuclear localization sequences of NF-κB transcription factors (Fig. 1) (Ghosh et al., 1998). Inhibitor of kappa B kinases (IKKs; CHUK and IKBKB) phosphorylate IkB, targeting it for degradation, thereby resulting in liberation of NF-κB transcription factors (Fig. 1) (Karin and Ben-Neriah, 2000; Zhang et al., 2017). NFKBIA was prominently expressed in microglia (Fig. S1), with scattered expression in bipolar cells, non-astrocytic inner retinal glial (NIRG) cells, Müller glia, MGPCs, amacrine cells and photoreceptors (Fig. 2C). Among Müller glia, levels of NFKBIA were significantly reduced at 24 h after treatment with 1000 nmol NMDA, but not significantly in Müller glia at 48 and 72 h after NMDA treatment or in MGPCs (Fig. 2E). By comparison, NFKBIB was expressed by most types of retinal cells and was prominent in resting Müller glia (Fig. 2C). NFKBIB was broadly detected, but at significantly reduced levels, in Müller glia at 24, 48 and 72 h after NMDA treatment, and further downregulated in MGPCs (Fig. 2C,E). Similarly, NFKBIZ was predominantly expressed by resting Müller glia, and levels were decreased in Müller glia at 24, 48 and 72 h after NMDA treatment, and further reduced in MGPCs (Fig. 2D,E). CHUK was expressed by scattered Müller glia, MGPCs, NIRG cells, oligodendrocytes, amacrine cells, bipolar cells and ganglion cells (Fig. 2D).
TNF-related ligands are known to activate NF-κB in different cellular contexts (Hayden and Ghosh, 2004, 2014; Osborn et al., 1989; Schütze et al., 1992, 1995). Accordingly, we probed for TNF-related ligands and receptors in our scRNA-seq libraries. TNFα has not been identified in the chick genome, but tumor necrosis factor super family 15 (TNFSF15)/TL1A may function in its place (Migone et al., 2002; Takimoto et al., 2005). We found that TNFSF15 was detected only in microglia (Fig. S1), consistent with scRNA-seq data from the mouse retina where microglia are the only source of IL1A, IL1B and TNF (Todd et al., 2019). By comparison, TNFSF10 was detected in relatively few Müller glia in control retinas but was expressed at significantly elevated levels in Müller glia at 48 and 72 h after NMDA, and was reduced in MGPCs (Fig. 2F,G). Other isoforms of TNF-related ligands, including TNFSF6, TNFSF8 and TNFSF11 were not expressed at significant levels (not shown). We detected expression of TNFSF receptors predominantly in Müller glia in control and damaged retinas. In control retinas, TNFRSF1A was detected at relatively high levels in scattered Müller glia (Fig. 2F,G). In damaged retinas, TNFRSF1A was detected in many Müller glia at 24 h, but levels were significantly reduced (Fig. 2F,G). TNFRSF1A levels were increased in Müller glia at 48 and 72 h after NMDA treatment, and levels were reduced in MGPCs (Fig. 2F,G). By comparison, the expression of TNFRSF21 was widespread in control Müller glia, in Müller glia from damaged retinas and in MGPCs (Fig. 2F,G). Levels of TNFRSF21 were significantly lower in MGPCs (Fig. 2F,G). TNFRSF21 is a receptor for TNFSF15/TL1a (Migone et al., 2002). Although orthologs for mammalian TNFα have not been identified in the chicken genome, chicken TNFSF15 is homologous to mammalian TL1A and likely acts as TNFα in the chicken (Takimoto et al., 2005).
To better understand the expression patterns of NF-κB-related genes in Müller glia and MGPCs, we assessed expression across pseudotime. Pseudotime states and trajectories were established by analyzing the highly variable genes among Müller glia and MGPCs. This analysis revealed five distinct pseudotime states and trajectories, including distinct branches for resting Müller glia, activated Müller glia, transitional Müller glia and MGPCs (Fig. 2H). In addition, we performed a branched pseudotime analysis with the first branch point segregating analyses from resting (state 1) to activated (state 5) Müller glia, and from resting Müller glia through transitional Müller glia (states 2 and 3) to MGPCs (state 4). Resting Müller glia (state 1) from control retinas were located to the far left of the pseudotime axis, and expressed high levels of mature glial markers, such as GLUL (Fig. 2I). MGPCs from 24, 48 and 72 h after NMDA treatment were located to the far right of the pseudotime axis (state 4) and expressed high levels of markers for proliferation and progenitor cells, such as CDK1, whereas non-proliferative activated Müller glia from 72 h after NMDA treatment were located in the descending pseudotime branch (state 5) (Fig. 2I).
Expression across pseudotime revealed small but significant decreases in relative levels of NFKBIB in MGPCs compared with activated Müller glia (Fig. 2J,K). By comparison, levels of NFKBIZ increased in MGPCs relative to levels in activated Müller glia (Fig. 2J,K). Expression of TNFRSF10 was significantly increased over pseudotime in MGPCs, but this trend was not significant in activated Müller glia (Fig. 2J,K). TNFRSF1A was decreased in MGPCs compared with resting, transitional and activated Müller glia (Fig. 2J,K). Expression of TNFRSF21 was relatively high in resting Müller glia, lower in transitional Müller glia, and further decreased in activated Müller glia and MGPCs (Fig. 2J,K). Taken together, these findings indicate that essential components of the NF-κB pathway are dynamically expressed in Müller glia after damage and during the process of reprogramming into MGPCs. The damage-induced changes in expression of NF-κB components and TNF-ligands/receptors in Müller glia implies that these signals are involved in the responses of Müller glia to retinal damage and may be involved in the reprogramming of Müller glia into MGPCs.
IκBα protein levels in normal, damaged and inhibitor-treated retinas
Previous studies have indicated that NMDA-induced damage in the mouse retina results in the activation of NF-κB in Müller glia (Lebrun-Julien et al., 2009). We failed to identify antibodies to components of the NF-κB pathway, including RelA/p65, phospho-p65, phospho-IKBα/β or RelB, that produced plausible and reproducible patterns of labeling in retinal sections or in western blot assays. As IκBα is targeted for degradation to liberate NF-κB transcription factors during active signaling (Fig. 1) (Karin and Ben-Neriah, 2000; Zhang et al., 2017), we used western blot analyses to probe for levels of IκBα. We found that levels of total IκBα were high in saline-treated retinas, and significantly decreased in NMDA-damaged retinas (Fig. 3A,B). By comparison, in NMDA-damaged retinas treated with sulfasalazine to inhibit NF-κB, levels of IκBα were significantly increased (Fig. 3A,B). Collectively, the findings of western blot analyses for IκBα indicate that NF-κB signaling is increased in damaged retinas, and the activity is effectively inhibited by sulfasalazine.
Fig. 3.
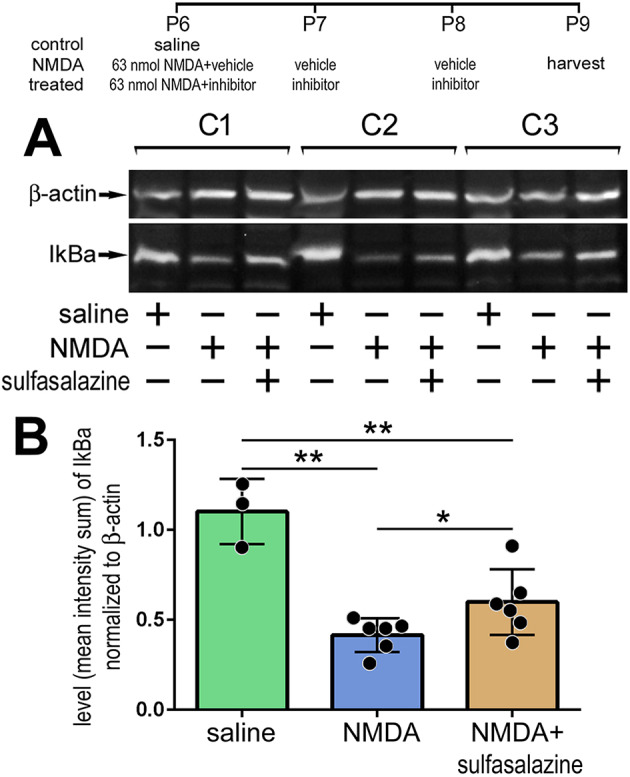
Western blot analysis of IκBα: NF-κB is activated after NMDA damage and treatment with sulfasalazine diminished signaling. (A) Representative images of chemiluminescent western blots for IκBα and β-actin for three individual retinas treated with saline, NMDA+vehicle or NMDA+sulfasalazine. (B) Histogram shows the mean (±s.d. and individual data points) of pixel intensity of IκBα above threshold and normalized to β-actin. Significance of difference (*P<0.05, **P<0.001) was determined by using ANOVA and Tukey's post-hoc test.
NF-κB regulates the formation of MGPCs in damaged retinas
To determine whether NF-κB influences the formation of MGPCs in the chick retina, we applied small molecule activators/inhibitors of NF-κB to NMDA-damaged retinas. We tested whether small molecule antagonists, i.e. sulfasalazine, 15-deoxy-delta-12,14-prostaglandin J2 (PGJ2) or SC75741 (SC757), that act at different levels of the NF-κB pathway (Fig. 1) influence the formation of proliferating MGPCs. Sulfasalazine is an anti-inflammatory agent that potently suppresses NF-κB activity by preventing IκBα phosphorylation and degradation, resulting in persistent sequestration of NF-κB transcription factors in the cytoplasm (Wahl et al., 1998). Sulfasalazine has also been shown to act at the cysteine/glutamate-antiporter (SLC7A11) (Gout et al., 2001), but this transporter is not expressed at detectable levels in scRNA-seq libraries (not shown). PGJ2 is a derivative of prostaglandin D2, and acts as a ligand for peroxisome proliferator-activated receptor γ (PPARγ) to regulate inflammation (Ricote et al., 1998; Straus et al., 2000), but also inhibits NF-κB in a PPARγ-independent manner (Lindström and Bennett, 2005). PGJ2 represses IKK activity, thus reducing IκBα phosphorylation and degradation (Lindström and Bennett, 2005; Straus et al., 2000). Additionally, PGJ2 covalently interacts with P50, an NF-κB transcription factor, to directly block its DNA-binding ability (Cernuda-Morollón et al., 2001; Straus et al., 2000). SC757 belongs to a novel class of NF-κB inhibitors (Leban et al., 2007) that acts by blocking the DNA binding of P65, without affecting nuclear translocation or IκBα (Ehrhardt et al., 2013).
Proliferation is considered to be a culminative read-out of reprogramming of Müller glia into MGPCs (Gallina et al., 2014a; Lamba et al., 2008). Furthermore, scRNA-seq data provide compelling data for a comprehensive correlation of progenitor-related transcription factors and markers of proliferation in MGPCs in both fish and chick model systems (Hoang et al., 2019 preprint). Accordingly, we used proliferation as a primary read-out of reprogramming of Müller glia into MGPCs. The proliferation of MGPCs in the chick retina is maximal with higher (≥1000 nmol) doses of NMDA, and numbers of proliferating MGPCs are diminished with lower (≤80 nmol) doses of NMDA (Fischer et al., 2004). Thus, to investigate whether drugs stimulate the formation of proliferating MGPCs we used treatment paradigms with a lower dose of NMDA, whereas to investigate whether drugs inhibit the formation of proliferating MGPCs, we used treatment paradigms with a high dose of NMDA, where numbers of proliferating MGPCs were maximal.
Application of NF-κB inhibitors after a low dose of NMDA (63 nmol) significantly increased numbers of proliferating MGPCs. Treatment with sulfasalazine, PGJ2 or SC757 following NMDA significantly increased numbers of Sox9+/EdU+ cells (Fig. 4A-D) and numbers of pHH3+/neurofilament+ mitotic cells in the INL and ONL (Fig. 4E-G). Neurofilament and phospho-histone H3 (pHH3) are known to be transiently expressed by proliferating MGPCs (Fischer and Reh, 2001; Zelinka et al., 2016). In addition, application of sulfasalazine following NMDA resulted in increased expression of stem cell-associated transcription factor Pax6 in Sox2-positive cells (Fig. 4H-J), suggesting that inhibition of NF-κB promotes the reprogramming of Müller glia into progenitor-like cells. Pax6 is expressed by retinal progenitor cells during development (Ashery-Padan and Gruss, 2001) and is expressed by resting Müller glia at low levels, but is upregulated during reprogramming into MGPCs (Fischer and Reh, 2001, 2002). Additionally, treatment of NMDA-damaged retinas with PGJ2 or SC757, but not sulfasalazine, decreased the proliferation of microglia (Fig. S2). Collectively, these findings suggest that inhibition of NF-κB at different levels of the pathway promotes the formation of proliferating MGPCs in damaged retinas; however, we cannot exclude the possibility that pharmacological agents may have off-target actions that could influence proliferation.
Fig. 4.

Inhibition of NF-κB promotes MGPC proliferation after NMDA damage. (A-J) NMDA-damaged retinas were treated with three consecutive daily doses of sulfasalazine, PGJ2 or SC757, or vehicle controls. Retinas were harvested 72 h after damage. (A,E,H,I) Retinal sections were labeled for Sox9 (green; A), EdU (red; A), neurofilament (green; E), Sox2 (red; E,H,I), pHistone H3 (PHH3) (blue; E) or Pax6 (green; H,I). (B-D,F,G,J) The histograms in B-D,F,G show the mean (±s.d. and individual data points) of proliferating cells; the histogram in J show mean (±s.d. and individual data points) pixel intensity above threshold for Pax6 immunofluorescence. Significance of difference (P<0.05) was determined by using a paired t-test. Arrows indicate proliferating MGPCs in A. Arrows in E represent mitotic figures. Arrows in H represent MGPCs expressing Pax6. Scale bars: 50 µm. ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
We next investigated whether activation of NF-κB following damage influenced the formation of MGPCs. Prostratin activates NF-κB by stimulating phosphorylation and degradation of IκBα in an IKK-dependent manner (Williams et al., 2004). Prostratin-induced NF-κB activation is likely mediated by PKC (Lin et al., 2000; Williams et al., 2004). Additionally, we applied TNFSF15 to control or NMDA-damaged retinas. TNF-ligands are known to activate NF-κB (Hayden and Ghosh, 2004; Schütze et al., 1995). TNFSF15/TL1a activates NF-κB through TNFRSF25/DR3 and also binds TNFRSF21/DR6 (Migone et al., 2002). Orthologues for mammalian TNFα and TNFRSF25 have not been identified in the chick genome. Chicken TNFSF15 is homologous to mammalian TL1a, and it is likely that TL1a/TNFSF15 acts as TNFα in the chick (Takimoto et al., 2005).
We found that three consecutively daily injections of prostratin or TNFSF15 did not stimulate the formation of proliferating MGPCs (Fig. S3). By comparison, we found that treatment with prostratin after a high dose of NMDA (1 µmol) resulted in a significant decrease in the numbers of Sox9+/EdU+ cells in the INL (Fig. 5A,B), whereas there was no significant change in the proliferation of microglia (Fig. 5D). Additionally, there was a significant decrease in the number of Sox2+/NF+/pHH3+ mitotic cells in the INL/ONL of NMDA damaged retinas treated with prostratin relative to NMDA controls, indicating a decrease in the formation of reprogrammed, proliferating MGPCs (Fig. 5E,F). Treatment with TNFSF15 following NMDA did not influence the proliferation of MGPCs; there was no significant difference in numbers of Sox9+/EdU+ cells between control and treated retinas (Fig. 5C). It is possible that TNF receptors on Müller glia were saturated in damaged retinas and, thus, addition of exogenous TNFSF15 had no significant effect upon the formation of MGPCs. However, TNFSF15 treatment recruited microglia to the vitreal surface of the retina in control and NMDA-damaged retinas (Fig. S4), suggesting a chemotactic influence upon the immune cells.
Fig. 5.

Stimulation of NF-κB suppresses MGPC proliferation in damaged retinas. (A-F) NMDA-damaged retinas were treated with three consecutive daily doses of prostratin or TNFSF15 or vehicle controls, and retinas were harvested 72 h after damage. (A,E) Retinal sections were labeled for Sox9 (green; A), EdU (red; A), neurofilament (green; E), Sox2 (red; E) or pHistone H3 (PHH3) (blue; E). (B-D,F) Histograms show the mean (±s.d. and individual data points) of proliferating cells. Significance of difference (P<0.05) was determined by using a paired t-test. Arrows indicate proliferating MGPCs in A. Scale bars: 50 µm.
Inhibition of NF-κB is neuroprotective against NMDA-induced retinal damage
Levels of retinal damage and cell death are known to positively correlate with the formation of MGPCs (Fischer and Reh, 2001, 2003), NF-κB is known to influence inflammation and neuronal survival (Hayden and Ghosh, 2008; Lanzillotta et al., 2015; Lebrun-Julien et al., 2009; Schneider et al., 1999). Thus, we investigated whether inhibition of NF-κB influenced cell death and neuronal survival, which may secondarily impact the formation of MGPCs. In the chick retina, NMDA induces cell death within 4 h, with numbers of dying cells peaking around 24 h, and continuing through to 72 h after treatment (Fischer et al., 1998, 2015). In retinas treated with sulfasalazine following NMDA (1 µmol or 63 nmol), we found significantly fewer TUNEL-positive cells at 4 h, 24 h and 72 h after damage relative to NMDA alone (Fig. 6A-D). Consistent with these findings, numbers of TUNEL-positive cells were significantly reduced 72 h after NMDA damage in retinas treated with PGJ2 compared with controls (Fig. 6E), suggesting that PGJ2 may be neuroprotective. By comparison, application of prostratin following a NMDA (1 µmol) had no effect upon numbers of dying cells (Fig. 6F), whereas application of TNFSF15 following NMDA resulted in a modest, but significant, increase in numbers of dying cells (Fig. 6G). The TNFSF15 likely activates multiple isoforms of TNF receptors and may activate signaling pathways in addition to NF-κB, and thereby elicit different effects on cell death when compared to prostratin treatment.
Fig. 6.

Inhibition of NF-κB is neuroprotective to different types of retinal neurons. NF-κB inhibitors (sulfasalazine or PGJ12) or activators (prostratin or TNFSF15) were applied after NMDA- or colchicine-induced damage. Retinas were harvested at different times after damage. (A-G) Retinas were harvested at 4 h (A,B), 24 h (A,C) or 72 h (A,D-G). In retinal sections, dying cells containing fragmented DNA were labeled using TUNEL (A). Histograms in B-G show the mean number (±s.d. and individual data points) of TUNEL-positive cells. (H-J) Colchicine-damaged retinas were treated with sulfasalazine or vehicle control, followed by six consecutive daily treatments of sulfasalazine or vehicle, and retinas were harvested 9 days after colchicine treatment. (H) Retinal whole-mounts were labeled using antibodies to Brn3. (I,J) Histograms show the mean number (±s.d. and individual data points) of ganglion cells in dorsal (I) and ventral (J) regions of the retina. Significance of difference (*P<0.05) was determined by using a paired t-test. Scale bars: 50 µm.
We next examined whether NF-κB influences the survival of retinal ganglion cells. NMDA damage primarily destroys amacrine cells, bipolar cells and horizontal cells in the chick retina, whereas colchicine treatment causes ganglion cell death within 3 days of treatment in newly hatched chicks (Fischer et al., 1998; Stanke and Fischer, 2010). Colchicine treatment of the chick retina results in cell death spread across the first 5 days following treatment (Stanke and Fischer, 2010). Thus, determination of end-point neuronal survival is more reliable and simpler than assaying for numbers of TUNEL+ cells collectively across the first 5 days following colchicine treatment. Treatment with colchicine significantly reduced the total number of surviving Brn3+ ganglion cells compared with undamaged controls at 9 days post-treatment (Fig. 6I,J). Application of sulfasalazine following colchicine treatment resulted in significantly greater numbers of surviving Brn3+ ganglion cells in both dorsal and ventral regions relative to colchicine treatment alone (Fig. 6H-J). However, sulfasalazine treatment did not restore the numbers of ganglion cells to those of undamaged retinas (Fig. 6I,J). Taken together, these data indicate that TNFSF15 and NF-κB promotes the death of retinal neurons, whereas inhibition of NF-κB promotes neuronal survival.
NF-κB promotes glial cell fate after damage
During neural development NF-κB acts as a pro-gliogenic pathway (Fujita et al., 2011; Keohane et al., 2010; Mondal et al., 2004). Thus, we tested whether inhibition of NF-κB with sulfasalazine following NMDA damage influenced the differentiation of newly generated cells. We found that inhibition of NF-κB signaling in damaged retinas resulted in an increased number of proliferating MGPCs at 7 days after damage (Fig. 7A,B), but did not influence the percentage of progeny that differentiated into neurons (EdU+/HuD+; Fig. 7C or EdU+/Otx2+; not shown) or the percentage of progeny that differentiated as glia (EdU+/GS+) (Fig. 7C). We next tested whether activation of NF-κB influenced cellular differentiation. The NF-κB inhibitor (sulfasalazine) was applied with NMDA at P6, and at P7 and P8, to increase numbers of proliferating MGPCs. Then, NF-κB activator (prostratin) was applied at P11 and P12, after MGPCs have undergone proliferation, and retinas were harvested at P15. We found no significant difference in the number of newly generated EdU+ cells between treated and control conditions (Fig. 7E). However, we found a significant twofold increase in the percentage of MGPC-progeny that differentiated into glia (EdU+/GS+) (Fig. 7D-F). There was no significant difference in the differentiation of newly generated neurons (EdU+/HuD+; Fig. 7F, or EdU+/Otx2+; not shown). These data indicate that activation of NF-κB promotes glial cell fate from the progeny of MGPCs.
Fig. 7.

NF-κB promotes glial cell-fate after damage. (A-C) Retinas were treated with NMDA and vehicle (control) or NMDA and sulfasalazine (treated) at P6, followed by vehicle or sulfasalazine at P7, P8, P9 and P10, and retinas harvested at P13. (D-F) Alternatively, retinas were treated with NMDA and vehicle (control) or NMDA and sulfasalazine (treated) at P6, followed by vehicle or sulfasalazine at P7 and P8, followed by vehicle or prostratin at P11 and P12, and retinas harvested at P15. (A,D) Retinal sections were labeled for EdU (A, grayscale; D, red) and GS (D, green). (B,E) Histograms show the mean number (±s.d. and individual data points) of proliferating MGPCs. (C,F) Histograms show the mean percentage change (±s.d. and individual data points) of EdU-positive cells that are co-labeled for GS (Müller glia) or HuD (neurons). Significance of difference (P<0.05) was determined by using a paired t-test. Scale bars: 50 µm. Arrowheads indicate proliferating MGPCs; arrows indicate newly generated GS-positive cells .
The impact of NF-κB on MGPC proliferation after damage is dependent on the presence of reactive microglia
Signals from reactive microglia are known to impact the reprogramming of Müller glia into neurogenic MGPCs (Conner et al., 2014; Fischer et al., 2014; Nelson et al., 2013; White et al., 2017; Zhao et al., 2014). We performed scRNA-seq on normal and damaged retinas with and without microglia. We ablated retinal microglia via an intraocular injection of clodronate liposomes, which ablates >99% of microglia within 2 days of treatment (Zelinka et al., 2012). We have reported previously that DiI-labeled clodronate liposomes accumulate at the vitreal surface of the retina, are only taken up by reactive microglia and do not deplete numbers of Müller glia (Fischer et al., 2014; Zelinka et al., 2012). Retinas were treated with clodronate liposomes at P6 and then treated with NMDA or saline controls at P9. Retinas (±microglia) were harvested for scRNA-seq at 24 h after treatment with saline or NMDA, objects identified as Müller glia were isolated, and analyses were performed. No microglia-specific genes were identifiable from scRNA-seq libraries of clodronate-treated retinas, indicating complete ablation of microglia. Regardless of the presence of microglia, unbiased tSNE plots revealed distinct clustering of Müller glia from saline-treated retinas and clustering of Müller glia from damaged retinas (Fig. 8A). Expression levels of VIM in Müller glia were significantly increased as a result of damage, regardless of the presence of microglia (Fig. 8B,C). VIM was significantly increased in Müller glia from damaged retinas without microglia compared with levels in Müller glia from damaged retinas with microglia; however, there was no significant difference between levels of VIM in Müller glia from saline-treated retinas with or without microglia (Fig. 8B,C). By comparison, damage-induced downregulation of RLBP1 was significant, but the levels of RLBP1 were significantly increased in NMDA-treated Müller glia when microglia were ablated (Fig. 8B,C). Although the absence of microglia had no effects upon the relative expression of NF-κB components in Müller glia in saline-treated retinas, levels of expression of NFKBIB and TNFRSF21 were reduced in NMDA-damaged retinas missing reactive microglia (Fig. 8C). Although damage induced a significant decrease in NFKBIA, the presence of microglia had no significant effect upon levels in normal and damaged retinas (Fig. 8C). These findings suggest that the NF-κB pathway may be diminished in Müller glia from damaged retinas devoid of reactive microglia.
Fig. 8.

The impact of NF-κB on MGPC proliferation depends on the presence of reactive microglia. (A) Libraries for scRNA-seq were established for cells from retinas 24 h after saline or NMDA treatment with (control) and without (clodronate liposomes) microglia. Müller glia were bioinformatically isolated from saline-treated retinas with and without microglia, and from NMDA-damaged retinas with and without microglia. tSNE plots show distinct clustering of Müller glia from saline and NMDA-damaged retinas. (B) Müller glia were identified based on collective expression of VIM, GLUL, RLBP1 and SLC1A3. (C) Violin/scatter plots illustrate expression levels of VIM, RLBP1, NFKBIA, NFKBIB and TNFRSF21 in Müller glia controls (±microglia) and at 24 h after NMDA-treatment (±microglia). Significance of difference (**P<0.01, ***P<0.001) was determined by using a Wilcoxon rank sum with Bonferroni correction. (D-H) Retinas were treated with clodronate liposomes at P6, NMDA plus vehicle or sulfasalazine at P9, vehicle or sulfasalazine plus EdU at P10 and P11, and retinas harvested at P12. (I-N) Alternatively, retinas were treated with clodronate liposomes at P6, NMDA plus vehicle or prostratin or TNFSF15 at P9, EdU plus vehicle or prostratin or TNFSF15 at P10 and P11, and retinas harvested at P12. (D,I,N) Retinal sections were labeled for EdU (red; D,I), Sox9 (green; D,I), CD45 (magenta; D,I) or TUNEL (N). Histograms in E,G,J and K show the mean number (±s.d. and individual data points) of proliferating MGPCs. Histograms in F,H,L and M show the mean number (±s.d. and individual data points) of TUNEL-positive cells. Arrows in D and I represent proliferating MGPCs. Significance of difference was determined by using a paired t-test. Scale bars: 50 µm.
As components of the NF-κB pathway are expressed by both Müller glia and microglia (Fig. 2), pharmacological manipulations of the pathway are expected to directly influence both cell types. Thus, we investigated how NF-κB influences Müller glia reprogramming in the absence of microglia. In the absence of microglia, inhibition of NF-κB with sulfasalazine or PGJ2 following NMDA treatment had no significant effect upon numbers of proliferating MGPCs (Fig. 8D,E,G), or on total numbers of TUNEL+ dying cells (Fig. 8F,H). Treatment with SC757 produced the same outcomes as sulfasalazine and PGJ2 treatment (not shown). Taken together, these findings suggest that, in the absence of microglia, NF-κB-inhibitors have no effect, likely because there was little or no active signaling to inhibit. It is possible that activated microglia provide the signals required to activate NF-κB in Müller glia after damage.
We next investigated whether activation of NF-κB in the absence of microglia influenced the formation of MGPCs. In retinas treated with clodronate liposomes 2 days prior, a high dose (1 µmol) NMDA was applied in combination with prostratin, TNFSF15 or vehicle controls. Interestingly, treatment with prostratin or TNFSF15 caused significant increases in numbers Sox9+/EdU+ cells in microglia-depleted, NMDA-damaged retinas (Fig. 8I-K). These data suggest that, in the absence of microglia, activation of NF-κB promotes MGPC proliferation, which is opposite to the effects of NF-κB activation on MGPC proliferation in damaged retinas with reactive microglia (Fig. 5). Additionally, in the absence of microglia, treatment of damaged retinas with TNFSF15 significantly increased numbers of TUNEL+ cells (Fig. 8M,N), whereas prostratin had no effect (Fig. 8L). Collectively, these findings suggest that reactive microglia are required to induce NF-κB activation in Müller glia and initiate a gliotic response that progresses into reprogramming; however, sustained activation of NF-κB suppresses proliferation of MGPCs.
Expression patterns of NF-κB-related genes in retinal cells after treatment with insulin and FGF2
In the chick retina, MGPCs are known to form in the absence of retinal damage in response to three consecutive daily injections of insulin and FGF2 (Fischer and Reh, 2002). Accordingly, we establish scRNA-seq libraries for retinas treated with insulin and FGF2 (Fig. 9A). Müller glia were identified based on expression of VIM, SOX2 and SOX9, and MGPCs were identified based on downregulation of GLUL and RLBP1, and upregulation of PCNA, CDK1 and NES (Fig. 9B). We analyzed expression of NF-κB components in Müller glia and MGPCs, as treatment with insulin and FGF2 was not expected to have significant effects upon retinal neurons. NFKBIA had scattered expression in Müller glia and MGPCs in saline and growth factor-treated retinas, without significant change (Fig. 9C). By comparison, NFKBIB was expressed by many resting Müller glia in control retinas and by MGPCs in treated retinas, but levels were significantly decreased in non-proliferative Müller glia treated with insulin and FGF2 (Fig. 9C). By comparison, expression of TNFRSF21 was widespread in control Müller glia and was significantly downregulated in Müller glia from growth factor-treated retinas but was not decreased in MGPCs compared with resting Müller glia (Fig. 9C). TNFSF10 and TNFRSF1a were detected in scattered Müller glia treated with saline or insulin and FGF2 without significant change between treatments (not shown).
Fig. 9.

Expression patterns of NF-κB-related genes in retinal cells after treatment with insulin and FGF2. (A) Libraries for scRNA-seq were established for cells from control retinas and from retinas treated with two or three consecutive daily doses of insulin and FGF2. tSNE plots show distinct clustering of different retinal cell types and numbers of cells surveyed within each cluster (in parentheses). (B) Each individual point represents one cell. Müller glia were identified based on expression of SOX2, SOX9 and VIM. Progenitors were identified based on expression of CDK1, ASCL1, PCNA and NES. (C) Violin/scatter plots for expression levels of NFKBIA, NFKBIB and TNFRSF21 in Müller glia in control retinas, 2× insulin and FGF2, 3× insulin and FGF2, and MGPCs. (D) Clusters identified as Müller glia and MGPCs were re-embedded for pseudotime analysis. Pseudotime states included: (1; peach) resting Müller glia, (2; green) activated Müller glia and (3; blue) MGPCs. Dimensional reduction of trajectories to stretched pseudotime to the x-axis placed the resting Müller glia to the far left, and activated Müller glia and MGPCs to the far right. (E,F) Relative levels of gene expression were assessed among Müller glia and MGPCs across branched pseudotime (E) and across different pseudotime states in violin plots (F). Relative levels were assessed for components of the NF-κB -pathway, NFKBIA, NFKBIB and NFKBIZ; and TNF-related factors and receptors, TNFSF10, TNFRSF1A and TNFRSF21. Significance of difference (**P<0.01, ***P<0.001) was determined by using a Wilcoxon rank sum with Bonferroni correction.
Pseudotime analysis revealed three distinct states and a trajectory that included branches for resting Müller glia (state 1), two doses insulin+FGF2 (IF; state 2) and three doses IF (state 3; Fig. 9D). Resting Müller glia were located in the left branch of the pseudotime trajectory (state 1), and expressed at high levels of mature glial markers, such as GLUL (Fig. 9D). Activated Müller glia, from retinas treated with two doses of insulin and FGF2, were located in the upper-right branch of the pseudotime trajectory (state 2), and uniquely expressed high levels of factors such as TGFB2, WNT4 and WNT6 (not shown). MGPCs, mostly from retinas treated with three doses of insulin and FGF2, were located to the lower-right branch of the pseudotime trajectory (state 3), and expressed markers for proliferation and progenitors, such as CDK1 (Fig. 9D). Levels of expression of NFKBIA, NFKBIZ, TNFSF10 and TNFRSF1A were relatively low and did not change significantly across pseudotime (Fig. 9E,F). Relative levels of NFKBIB were high in resting Müller glia, and significantly decreased in both activated Müller glia and MGPCs (Fig. 9E,F). Relative expression level of TNFRSF21 was high in resting Müller glia and MGPCs but was decreased in activated Müller glia (Fig. 9E,F). Taken together, these findings indicate that components of the NF-κB pathway and TNF receptors are differentially expressed by Müller glia in retinas treated with insulin and FGF2, thereby suggesting that NF-κB and TNF signaling may influence the formation of MGPCs in the absence of neuronal damage.
NF-κB influences MGPC formation in the absence of retinal damage
Treatment FGF2 is known to stimulate the formation of MGPCs in the absence of damage (Fischer et al., 2014) and activate a network of cell-signaling pathways, including MAPK, Wnt/β-catenin, BMP/Smad and Jak/Stat, while modestly activating microglia, but not incurring neuronal damage (Fischer et al., 2009b, 2014; Gallina et al., 2015; Todd et al., 2016b, 2017). Thus, we investigated whether NF-kB suppresses the formation of MGPCs in FGF2-treated retinas. We found that consecutive daily injections of sulfasalazine, prostratin or TNFSF15 alone did not induce the proliferation of MGPCs (Fig. S3) or induce cell death (not shown). By comparison, four consecutive daily doses of NF-κB inhibitors (sulfasalazine or PGJ2) with FGF2 significantly increased the numbers of EdU+/Sox9+ proliferating MGPCs (Fig. 10A-C), and decreased microglia reactivity, indicated by decreased levels CD45-immunofluorescence (Fig. 10D,E). Application of the NF-κB activator prostratin with FGF2 significantly decreased the numbers of EdU+/Sox2+ proliferating MGPCs (Fig. 10F,G), but did not influence numbers of proliferating microglia (Fig. 10H). Similarly, treatment with TNFSF15 and FGF2 significantly decreased MGPC proliferation compared with treatment with FGF2 alone, while there was no change in microglial proliferation (Fig. 10G,H). Thus, in the absence of retinal damage, NF-κB signaling acts to suppress the formation of MGPCs in retinas treated with FGF2.
Fig. 10.

Inhibition of NF-κB promotes MGPC proliferation in the absence of damage. Retinas were treated with FGF2 plus NF-κB inhibitors (sulfasalazine or PGJ2), NF-κB activators (prostratin or TNFSF15) or vehicle at P6-P9. EdU was added to injections at P8-P10 and retinas harvested at P11. (A,D,F) Retinal sections were labeled for EdU (red; A,F) or using antibodies to Sox9 (green; A), CD45 (green; D) or Sox2 (green; F). (B,C,G) Histograms show mean number (±s.d. and individual data points) of proliferating MGPCs. (E,H) Histograms show mean number (±s.d. and individual data points) of proliferating microglia. Arrows represent proliferating MGPCs (A,F) or microglia (D). Scale bars: 50 µm.
DISCUSSION
Our findings indicate that components of the NF-κB pathway are dynamically expressed in Müller glia during the process of reprogramming into MGPCs. Furthermore, our findings indicate that active NF-κB in damaged retinas acts to suppress the formation of proliferating MGPCs and may contribute to detrimental inflammation that exacerbates cell death. Interestingly, the effects of activation of NF-κB on the formation of MGPCs in damaged retinas are reversed when the microglia are absent. We propose that pro-inflammatory signals from microglia are required to induce NF-κB activation and initiate a gliotic response that progresses into reprogramming, but this reprogramming is suppressed by sustained activation of NF-κB. Finally, we find that NF-κB is part of the network of pathways activated by FGF2 treatment, and in the absence of neuronal damage NF-κB activation suppresses the formation of MGPCs. Our findings do not exclude the possibility that NF-kB signaling in other retinal cells, in addition to Müller glia, impacts neuronal survival, microglial reactivity and the formation of proliferating MGPCs. These findings are summarized in Fig. 11.
Fig. 11.

Schematic summary of findings. (A) After NMDA, microglia are required for activation of NF-κB. Inhibition of NF-κB promotes Müller glia reprogramming into proliferating MGPCs, while sustained activation suppresses the formation of MGPCs. NF-κB activation in the progeny of MGPCs promotes glial fate. (B) In the absence of microglia, NF-kB signaling is diminished in Müller glia and MGPC formation is not initiated. Stimulation of NF-κB after damage promotes Müller glia reactivity to initiate reprogramming and promote the formation of proliferating MGPCs.
NF-κB agonists and antagonists failed to induce the formation of MGPCs unless combined with damage or FGF2 treatment. This ‘gating’ effect is similar to that seen with the effects of EGF on mouse Müller glia, wherein damage was required for Müller glia to upregulate the EGF receptor and render these glia responsive to EGF (Close et al., 2006). Similarly, activation of BMP/Smad-, Jak/Stat, Wnt/β-catenin and retinoic acid signaling must be combined with retinal damage or FGF2 treatment to drive the proliferation of MGPCs in the chick retina (Gallina et al., 2015; Todd et al., 2016b, 2017, 2018). Our findings are consistent with the notion that pro-inflammatory signals from reactive microglia activate NF-κB in Müller glia in damaged retinas. Microglia become highly reactive in NMDA-damaged retinas (Fischer et al., 1998, 2014; Todd et al., 2019; Wada et al., 2013), and participate in bi-directional communication with Müller glia (Wang et al., 2011, 2014). Ablation of microglia prior to retinal damage suppresses the formation of MGPCs in the chick retina (Fischer et al., 2014). In NMDA-damaged retinas, microglia rapidly and transiently upregulate IL-1α, IL-1β and TNFα (Todd et al., 2019), and these cytokines are known to activate NF-κB in different cells and contexts (Hayden and Ghosh, 2004; Osborn et al., 1989). In most instances, the formation of MGPCs requires neuronal damage, and levels of neuronal damaged are positively correlated to numbers of proliferating MGPCs (Gallina et al., 2014b; Todd et al., 2017). However, MGPCs can form in the absence of neuronal death in chick and fish model systems (Fischer et al., 2002, 2009a; Gallina et al., 2014a; Wan et al., 2012, 2014). Despite decreased cell death in NMDA-damaged retinas treated with NF-κB-inhibitors, we find increased MGPC proliferation with reactive microglia present, whereas there was no change in MGPC proliferation in damaged retinas treated with NF-κB inhibitors when microglia were absent. Our findings suggest that the relationship between levels of damage and formation of MGPCs can be uncoupled by the inhibition of NF-κB.
NF-κB is coordinated with a network of cell-signaling pathways that regulate the reprogramming of Müller glia into MGPCs. In the chick, the formation of MGPCs is known to be influenced by many different cell-signaling pathways, including BMP/TGFβ/Smad and Wnt/β-catenin (Gallina et al., 2014a, 2015; Todd and Fischer, 2015; Todd et al., 2016b, 2017, 2018). There is evidence that NF-κB- and Wnt/β-catenin signaling mutually inhibit each other in different cellular contexts (reviewed by Ma and Hottiger, 2016). Wnt signaling promotes the formation of proliferating MGPCs in the chick, fish and mouse model systems (Gallina et al., 2015; Meyers et al., 2012; Osakada et al., 2007; Ramachandran et al., 2011; Yao et al., 2016, 2018). It is possible that the reprogramming-suppressing effects of NF-κB are mediated, in part, by Smad2/3. NF-κB can induce cell cycle arrest and terminal differentiation via IKKα-dependent regulation of Smad2/3 target genes (Descargues et al., 2008). TGFβ/Smad2/3-signaling inhibits the formation of MGPCs in the chick retina (Todd et al., 2017), and suppression of TGFβ-signaling is required for the formation of MGPCs during retinal regeneration in zebrafish (Lenkowski et al., 2013). Further studies are required to better establish how NF-κB fits into the hierarchy of cell-signaling pathways that regulate the formation of MGPCs.
Differences in activation of NF-κB may underlie differences in the reprogramming potential of MGPCs in different vertebrates. In the mouse retina, it has been shown that NMDA-induced excitotoxic damage activates NF-κB in Müller glia (Lebrun-Julien et al., 2009). In zebrafish retina, TNFα is required for MGPC proliferation via activation of Stat3 (Nelson et al., 2013). A recent comparative transcriptomic and epigenomic study has indicated that following NMDA-induced damage, components of the NF-κB pathway are predominantly expressed, and their regulatory elements are accessible for transcription, in mouse Müller glia. This is seen to a lesser degree in chick and fish Müller glia (Hoang et al., 2019 preprint). Rodent Müller glia rapidly transition into an activated state and then revert back to a resting state (Hoang et al., 2019 preprint); this may be driven by NF-kB signaling. Collectively, these data suggest the following model for the role of NF-κB in reprogramming of Müller glia into MGPCs (Fig. 11): (1) neuronal damage rapidly induces microglial reactivity; (2) increased production of pro-inflammatory cytokines from microglia occurs; (3) a rapid initial activation of NF-κB in Müller glia, and perhaps other retinal cell types, initiates gliosis; (4) transition to reprogramming (downregulation of glial genes and upregulation of progenitor genes) occurs; but (5) sustained NF-κB maintains reactive phenotype and pushes glia back to a resting phenotype (mouse) or suppresses reprogramming of Müller glia into proliferating MGPCs (chick). In the absence of reactive microglia, our data suggest that the process of reprogramming requires initiation of glial reactivity and de-differentiation, which can be provided by exogenous TNFSF15 or NF-κB-agonist.
We found that activation of NF-κB promoted glial differentiation from the progeny of proliferating MGPCs. Our findings are consistent with reports that pro-inflammatory cytokines and NF-κB promote gliogenesis during neural development (Bonni et al., 1997; Deverman and Patterson, 2009; Mondal et al., 2004). For example, IL-1 promotes acquisition of astrocyte cell fate, with peak expression concurrent with the generation of astrocytes in development (Giulian et al., 1988). Furthermore, IL-1 is known to activate NF-κB and there is evidence that NF-κB promotes specification of astrocytes (Mondal et al., 2004). Additionally, TNFα stimulates hippocampal neural precursors to promote astrocyte formation, and this occurs via upregulation of the pro-glial/progenitor gene bHLH transcription factor Hes1 (Keohane et al., 2010). Collectively, these findings suggest that NF-κB acts to promote glial differentiation from MGPCs.
NF-κB influences cell death in a context-specific manner. NF-κB has been shown to promote neuron death (Schneider et al., 1999), but has also been shown to support neuronal survival (Bhakar et al., 2002). NF-κB is activated by a variety of factors, including TNFα, reactive oxygen species, lipopolysaccharide and various growth factors (Schreck et al., 1991). In the mouse retina, NF-κB has been shown to be involved in NMDA-induced retinal neuron death, and inhibition of NF-κB prevents retinal neuron death (Lebrun-Julien et al., 2009). Consistent with these findings, we found that inhibition of NF-κB reduced numbers of dying amacrine and bipolar neurons in NMDA-damaged retinas and promoted the survival of ganglion cells in colchicine-damaged retinas. This may have resulted from suppressed production of TNF-ligands from reactive microglia.
Conclusions
Our findings suggest that NF-κB plays significant roles in regulating glial reactivity, neuronal survival and the reprogramming of Müller glia into proliferating MGPCs. NF-κB activity suppresses the formation of proliferating MGPCs, and the influence of NF-κB depends on reactive microglia. Despite reducing levels of cell death, NF-κB-inhibitors also stimulate the formation of MGPCs. In the absence of damage, in retinas treated with FGF2, NF-κB is recruited into a network of cell-signaling pathways that regulate the reprogramming of Müller glia into proliferating MGPCs. We propose that reactive microglia may provide signals to activate Müller glia via NF-κB to initiate the process of reprogramming.
MATERIALS AND METHODS
Animals
The use of animals in these experiments was in accordance with the guidelines established by the National Institutes of Health and the Ohio State University. Newly hatched wild-type leghorn chickens (Gallus gallus domesticus) were obtained from Meyer Hatchery (Polk, OH, USA). Postnatal chicks were kept on a cycle of 12 h light, 12 h dark (lights on at 8:00 AM). Chicks were housed in a stainless steel brooder at 25°C and received water and Purina chick starter ad libitum. All experiments were started when the chicks were 6-7 days post-hatch (P6-P7), with the exception of experiments involving colchicine, where the experiments began at P2.
Intraocular injections
Chickens were anesthetized via inhalation of 2.5% isoflurane in oxygen and intraocular injections performed as described previously (Fischer et al., 1998). For all experiments, the vitreous chamber of right eyes of chicks were injected with the experimental compound and the contralateral left eyes were injected with a control vehicle. Compounds were injected into chick eyes in 20 μl sterile saline with up to 30% (v/v) di-methyl-sulfoxide (DMSO) and 0.05 mg/ml bovine serum albumin added as a carrier. Compounds used in these studies included N-methyl-D-aspartate (NMDA) (9.2 or 147 μg/dose; M3262; Sigma-Aldrich), FGF2 (250 ng/dose; 233-FB; R&D systems), sulfasalazine (5.0 μg/dose; S0883: Sigma-Aldrich), 15-Deoxy-delta12,14-prostaglandin J2 (PGJ2; 5.0 μg/dose; J67427; Alfa Aesar), prostratin (5.0 μg/dose; Sigma-Aldrich), TNFSF15 (250 ng/dose; RP0116C; KingFisher Biotech) and SC75741 (SC757; 2.0 μg/dose; s7273; Selleck Chem). EdU (2.0 μg; 5-ethynyl-2′-deoxyuridine; 900584; ThermoFisher Scientific) was injected to label proliferating cells. Injection paradigms are included in each figure.
Fixation, sectioning and immunocytochemistry
Tissues were fixed, sectioned and immunolabeled as described previously (Fischer et al., 2008, 2009b). Working dilutions and sources of antibodies used in this study are listed in Table S1. Antibody labeling was validated by comparison with patterns of labeling observed in previous reports (Fischer et al., 2014; Gallina et al., 2014b; Todd and Fischer, 2015; Todd et al., 2018; Zelinka et al., 2016) and/or referencing the Journal of Comparative Neurology antibody database (onlinelibrary.wiley.com/page/journal/10969861/homepage/other_resources.htm#AntibodyDatabase).
None of the observed labeling was due to non-specific labeling of secondary antibodies or autofluorescence because sections labeled with secondary antibodies alone were devoid of fluorescence. Secondary antibodies included donkey-anti-goat-Alexa488/568, goat-anti-rabbit-Alexa488/568/647, goat-anti-mouse-Alexa488/568/647, goat anti-rat-Alexa488 (Life Technologies) diluted to 1:1000 in PBS plus 0.2% Triton X-100.
Western blotting
Retinas were harvested in ice-cold Hank's Balanced Salt Solution and sonicated in ice-cold RIPA-buffer (Bio-Rad) with protease inhibitor cocktail (Roche). After incubating the suspension on ice for 5 min, supernatant lysates were collected following centrifugation at 5000 g for 20 min at 4°C. Protein concentration was determined using a BCA Protein Assay (Thermo Scientific) in accordance with manufacturer's guidelines. Protein was loaded into 4-15% polyacrylamide gels (Bio-Rad) with Precision Plus Protein Dual Color Standards (Bio-Rad) for electrophoresis and then transferred onto nitrocellulose membranes (Thermo Fisher). Membranes were blocked with Tris-buffered saline+0.1% Tween (TBS-T) containing 5% (w/v) nonfat dry milk for 1 h and incubated in primary antibodies against P65, phospho-P65 and β-actin (Table S1) diluted in TBS-T+5% milk over night at 4°C. Membranes were washed in TBS-T prior to incubation in horseradish-peroxidase-conjugated secondary antibodies (diluted 1:2000 in TBS-T+0.5% milk) for 1 h at room temperature. Proteins were visualized using chemiluminescence detection (Thermo Scientific; SuperSignal West Pico Plus) and imaged on BioRad Chemidoc XRS+ system, and pixel intensity was quantified using ImageJ.
Labeling for EdU
Immunolabeled tissue sections were fixed in 4% formaldehyde in PBS for 5 min at room temperature, washed for 5 min with PBS, permeabilized with 0.5% Triton X-100 in PBS for 1 min at room temperature and washed twice for 5 min in PBS. Sections were incubated for 30 min at room temperature in 2 M Tris, 50 mM CuSO4, Alexa Fluor 568 or 647 Azide (ThermoFisher Scientific), and 0.5 M ascorbic acid in distilled H2O. Sections were washed with PBS for 5 min and further processed for immunofluorescence as required.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)
To identify dying cells that contained fragmented DNA, the TUNEL method was used. We used an In Situ Cell Death Kit (TMR red; 12156792910; Roche Applied Science), as per the manufacturer's instructions.
Preparation of clodronate liposomes
The preparation of clodronate liposomes was similar to previous descriptions (Van Rooijen, 1989; Zelinka et al., 2012). 50 ng cholesterol and 8 mg egg lecithin [L-α-Phosphatidyl-DL-glycerol sodium salt (Sigma P8318)] were dissolved in chloroform in a round-bottom flask. The solution was evaporated under nitrogen until a white liposome residue remained. Dichloro-methylene diphosphonate (158 mg; clodronate; Sigma-Aldrich) in sterile PBS was added and mixed. Clodronate encapsulation and vesicle size normalization were facilitated by sonication at 42,000 Hz for 5 min. The liposomes were centrifuged at 10,000× g for 15 min and re-suspended in 150 ml sterile PBS. We are unable to determine the exact concentration of clodronate because of the stochastic nature of the clodronate combining with the liposomes. We titered doses to levels where >99% of microglia were ablated at 2 days after treatment.
scRNA-seq
Retinas were acutely dissociated via papain digestion and mild trituration. Dissociated cells were loaded onto the 10X Chromium Controller using Chromium Single Cell 3′ v2 reagents. Sequencing libraries were prepared following the manufacturer's instructions (10X Genomics), with 10 cycles used for cDNA amplification and 12 cycles for library amplification. The resulting sequencing libraries were sequenced with Paired End reads, with Read 1 (26 base pairs) and Read 2 (98 base pairs), on a Nextseq500 at the Genomics Resources Core Facility (High Throughput Center) at Johns Hopkins University. Raw sequence data was processed with Cell Ranger software (10X Genomics) to align sequences, de-multiplex, annotate to ENSMBL databases, count reads, assess levels of expression and construct gene-cell matrices. t-distributed stochastic neighbor embedding (tSNE) plots were generated and probed using Cell Ranger and Cell Browser software (10X Genomics) using the following parameters: perplexity=30, float=0.5, max dimensions=2 and max iterations=1000. Uniform manifold approximation and projection (UMAP) plots were generated using Seurat with the following parameters: dims=1:10, spread=2, n.neighbors=30L, min.dist=0.1. Plots were generated via aggregate cluster analysis of nine separate cDNA libraries, including two replicates of control undamaged retinas, and retinas at different times after NMDA treatment. The identity of clustered cells was established using known cell type-specific markers. Seurat was used to generate violin/scatter plots for candidate genes in identified clusters of cells (Powers and Satija, 2015; Satija et al., 2015). The violin/scatter plots provide the probability density distribution and relative expression level of genes within an identified cluster of cells. The width of the violins provides a measure of the relative abundance of cells within a cluster that are present at a relative expression level. Wilcoxon rank sum with Bonferroni correction was used to determine whether there are significant differences in relative expression levels. Relative expression level refers to Log Transcripts Per Million (TPM) normalized across scRNA-seq libraries.
scRNA-seq libraries and numbers of cells were as follows: for Fig. 2, (1) control saline-treated retinas (16,381 cells), (2) NMDA-treated retinas at 24 h (14,298 cells), (3) NMDA-treated retinas at 48 h (8421 cells) and (4) NMDA-treated retinas at 72 h (18,130 cells) after NMDA-treatment; for Fig. 8, (5) Müller glia from saline-treated with microglia (1040 cells), (6) Müller glia from saline-treated without microglia (1459 cells), (7) Müller glia from 24 h NMDA-treated retina with microglia (954) and (8) Müller glia from 24 h NMDA-treated retina without microglia (1007 cells); for Fig. 9, (9) control saline-treated retinas (16,913 cells), (10) two consecutive daily doses of insulin and FGF2 (7568 cells); and (11) three consecutive daily doses of insulin and FGF2 (9653 cells).
Markers that were used to identify different types of retina cell that were clustered in tSNE/UMAP plots included the following: (1) Müller glia: GLUL, VIM, SLC13A and RLBP1; (2) microglia: C1QA, C1QB, CCL4, CSF1R and TMEM22; (3) ganglion cells: THY1, POU4F2, RBPMS2, NEFL and NEFM; (4) amacrine cells: GAD67, CALB2 and TFAP2A; (5) horizontal cells: PROX1, CALB2 and NTRK1; (6) bipolar cells: VSX1, OTX2, GRIK1 and GABRA1; (7) cone photoreceptors: CALB1, GNAT2 and OPN1LW; and (8) rod photoreceptors: RHO, NR2E3 and ARR3. Monocle 2.1 was used to establish pseudotime trajectories and levels of gene expression across pseudotime for cells identified as Müller glia or MGPCs (Trapnell et al., 2014). Pseudotime trajectories were generated in an unbiased manner. The Müller glia have an overabundant representation in the scRNA-seq databases. This likely results from fortuitous capture-bias and/or tolerance of the of the Müller glia to the dissociation process. scRNA-seq libraries are available at proteinpaint.stjude.org/F/2019.retina.scRNA.html (Hoang et al., 2019 preprint).
Photography, measurements, cell counts and statistics
Wide-field photomicroscopy was performed using a Leica DM5000B microscope equipped with epifluorescence and Leica DC500 digital camera or a Zeiss AxioImager M2 equipped with epifluorescence and a Zeiss AxioCam MRc. Confocal images were obtained using a Leica SP8 imaging system at the Department of Neuroscience Imaging Facility at The Ohio State University. Images were optimized for color, brightness and contrast, multiple channels overlaid, and figures constructed using Adobe Photoshop. Cell counts were performed on representative images. To avoid the possibility of region-specific differences within the retina, cell counts were consistently made from the same region of the retina for each dataset.
Similar to previous reports (Fischer et al., 2009a,b; Ghai et al., 2009), immunofluorescence was quantified by using ImagePro6.2 (Media Cybernetics). Identical illumination, microscope and camera settings were used to obtain images for quantification. Retinal areas were sampled from 5.4 MP digital images. These areas were randomly sampled over the inner nuclear layer (INL) where the nuclei of the bipolar and amacrine neurons were observed. Measurements of immunofluorescence were performed using ImagePro 6.2 as described previously (Ghai et al., 2009; Stanke et al., 2010; Todd and Fischer, 2015). The density sum was calculated as the total of pixel values for all pixels within thresholded regions. The mean density sum was calculated for the pixels within threshold regions from at least five retinas for each experimental condition. GraphPad Prism 6 was used for statistical analyses.
Measurements for expression of Pax6 in the nuclei of Müller glia/MGPCs were made from single optical confocal sections by selecting the total area of pixel values above threshold (≥70) for Sox2 or Sox9 immunofluorescence (in the red channel) and copying nuclear Pax6 from only Müller glia (in the green channel). The Müller glia-specific Pax6 was quantified (as described below or copied onto a 70% grayscale background for figures). Measurements were made for regions containing pixels with intensity values of 70 or greater (0=black and 255=saturated). The total area was calculated for regions with pixel intensities above threshold. The intensity sum was calculated as the total of pixel values for all pixels within threshold regions. The mean intensity sum was calculated for the pixels within threshold regions from at least eight retinas for each experimental condition.
The identity of EdU-labeled cells was determined based on previous findings that 100% of the proliferating cells in the chick retina comprise Sox2/9+ Müller glia in the INL/ONL, Sox2/9/Nkx2.2+ non-astrocytic inner retinal glial (NIRG) cells in the IPL, GCL and NFL, and CD45+ microglia. The NIRG cells are a unique type of retinal glia that has been described in the retinas of birds and some reptiles (Rompani and Cepko, 2010; Todd et al., 2016a; Zelinka et al., 2012). Sox2+ nuclei in the INL were identified as Müller glia based on their large size and fusiform shape that was distinctly different from the Sox2+ nuclei of cholinergic amacrine cells, which are small and round (Fischer et al., 2010).
GraphPad Prism 6 was used for statistical analyses and generation of histograms and bar graphs. Where statistical significance of difference was determined between treatment groups accounting for intra-individual variability within a biological sample, we performed a two-tailed, paired t-test. Where significance of difference was determined between two treatment groups comparing inter-individual variability we performed two-tailed, unpaired t-tests. When evaluating significance in difference between multiple groups we performed ANOVA followed by Tukey's test.
Supplementary Material
Acknowledgements
Thanks to Alex Campbell and Levi Todd for comments and discussions that shaped the final form of the paper.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: I.P., A.J.F.; Methodology: I.P., T.V.H., S.B., A.J.F.; Investigation: I.P., K.D., T.V.H.; Resources: I.P., T.V.H., S.B., A.J.F.; Data curation: I.P., K.D., T.V.H., A.J.F.; Writing - original draft: I.P., A.J.F.; Writing - review & editing: I.P., K.D., T.V.H., S.B.; Visualization: I.P., A.J.F.; Supervision: S.B., A.J.F.; Funding acquisition: S.B., A.J.F.
Funding
This work was supported by grants from National Institutes of Health (National Eye Institute; RO1 EY022030-06 to A.J.F.; UO1 EY027267-03 to A.J.F. and S.B.). Deposited in PMC for release after 12 months.
Data availability
RNA-seq data can be accessed via GitHub https://github.com/jiewwwang/Single-cell-retinal-regeneration. scRNA-seq data can be queried at https://proteinpaint.stjude.org/F/2019.retina.scRNA.html (Hoang et al., 2019 preprint).
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.183418.supplemental
Peer review history
The peer review history is available online at https://dev.biologists.org/lookup/doi/10.1242/dev.183418.reviewer-comments.pdf
References
- Ashery-Padan R. and Gruss P. (2001). Pax6 lights-up the way for eye development. Curr. Opin. Cell Biol. 13, 706-714. 10.1016/S0955-0674(00)00274-X [DOI] [PubMed] [Google Scholar]
- Bernardos R. L., Barthel L. K., Meyers J. R. and Raymond P. A. (2007). Late-stage neuronal progenitors in the retina are radial Muller glia that function as retinal stem cells. J. Neurosci. 27, 7028-7040. 10.1523/JNEUROSCI.1624-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakar A. L., Tannis L.-L., Zeindler C., Russo M. P., Jobin C., Park D. S., MacPherson S. and Barker P. A. (2002). Constitutive nuclear factor-κB activity is required for central neuron survival. J. Neurosci. 22, 8466-8475. 10.1523/JNEUROSCI.22-19-08466.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonni A., Sun Y., Nadal-Vicens M., Bhatt A., Frank D. A., Rozovsky I., Stahl N., Yancopoulos G. D. and Greenberg M. E. (1997). Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science 278, 477-483. 10.1126/science.278.5337.477 [DOI] [PubMed] [Google Scholar]
- Bringmann A., Iandiev I., Pannicke T., Wurm A., Hollborn M., Wiedemann P., Osborne N. N. and Reichenbach A. (2009). Cellular signaling and factors involved in Müller cell gliosis: neuroprotective and detrimental effects. Prog. Retin. Eye Res. 28, 423-451. 10.1016/j.preteyeres.2009.07.001 [DOI] [PubMed] [Google Scholar]
- Cernuda-Morollón E., Pineda-Molina E., Cañada F. J. and Pérez-Sala D. (2001). 15-Deoxy-Delta 12,14-prostaglandin J2 inhibition of NF-kappaB-DNA binding through covalent modification of the p50 subunit. J. Biol. Chem. 276, 35530-35536. 10.1074/jbc.M104518200 [DOI] [PubMed] [Google Scholar]
- Close J. L., Liu J., Gumuscu B. and Reh T. A. (2006). Epidermal growth factor receptor expression regulates proliferation in the postnatal rat retina. Glia 54, 94-104. 10.1002/glia.20361 [DOI] [PubMed] [Google Scholar]
- Conedera F. M., Pousa A. M. Q., Mercader N., Tschopp M. and Enzmann V. (2019). Retinal microglia signaling affects Müller cell behavior in the zebrafish following laser injury induction. Glia 67, 1150-1166. 10.1002/glia.23601 [DOI] [PubMed] [Google Scholar]
- Conner C., Ackerman K. M., Lahne M., Hobgood J. S. and Hyde D. R. (2014). Repressing Notch signaling and expressing TNFalpha are sufficient to mimic retinal regeneration by inducing Muller glial proliferation to generate committed progenitor cells. J. Neurosci. 34, 14403-14419. 10.1523/JNEUROSCI.0498-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descargues P., Sil A. K., Sano Y., Korchynskyi O., Han G., Owens P., Wang X.-J. and Karin M. (2008). IKKalpha is a critical coregulator of a Smad4-independent TGFbeta-Smad2/3 signaling pathway that controls keratinocyte differentiation. Proc. Natl. Acad. Sci. USA 105, 2487-2492. 10.1073/pnas.0712044105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deverman B. E. and Patterson P. H. (2009). Cytokines and CNS Development. Neuron 64, 61-78. 10.1016/j.neuron.2009.09.002 [DOI] [PubMed] [Google Scholar]
- Dyer M. A. and Cepko C. L. (2000). p57(Kip2) regulates progenitor cell proliferation and amacrine interneuron development in the mouse retina. Development 127, 3593-3605. [DOI] [PubMed] [Google Scholar]
- Ehrhardt C., Rückle A., Hrincius E. R., Haasbach E., Anhlan D., Ahmann K., Banning C., Reiling S. J., Kühn J., Strobl S. et al. (2013). The NF-κB inhibitor SC75741 efficiently blocks influenza virus propagation and confers a high barrier for development of viral resistance. Cell. Microbiol. 15, 1198-1211. 10.1111/cmi.12108 [DOI] [PubMed] [Google Scholar]
- Fausett B. V. and Goldman D. (2006). A role for alpha1 tubulin-expressing Muller glia in regeneration of the injured zebrafish retina. J. Neurosci. 26, 6303-6313. 10.1523/JNEUROSCI.0332-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. J. and Reh T. A. (2001). Müller glia are a potential source of neural regeneration in the postnatal chicken retina. Nat. Neurosci. 4, 247-252. 10.1038/85090 [DOI] [PubMed] [Google Scholar]
- Fischer A. J. and Reh T. A. (2002). Exogenous growth factors stimulate the regeneration of ganglion cells in the chicken retina. Dev. Biol. 251, 367-379. 10.1006/dbio.2002.0813 [DOI] [PubMed] [Google Scholar]
- Fischer A. J. and Reh T. A. (2003). Potential of Müller glia to become neurogenic retinal progenitor cells. Glia 43, 70-76. 10.1002/glia.10218 [DOI] [PubMed] [Google Scholar]
- Fischer A. J., Seltner R. L., Poon J. and Stell W. K. (1998). Immunocytochemical characterization of quisqualic acid- and N-methyl-D-aspartate-induced excitotoxicity in the retina of chicks. J. Comp. Neurol. 393, 1-15. [DOI] [PubMed] [Google Scholar]
- Fischer A. J., McGuire C. R., Dierks B. D. and Reh T. A. (2002). Insulin and fibroblast growth factor 2 activate a neurogenic program in Müller glia of the chicken retina. J. Neurosci. 22, 9387-9398. 10.1523/JNEUROSCI.22-21-09387.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. J., Schmidt M., Omar G. and Reh T. A. (2004). BMP4 and CNTF are neuroprotective and suppress damage-induced proliferation of Müller glia in the retina. Mol. Cell. Neurosci. 27, 531-542. 10.1016/j.mcn.2004.08.007 [DOI] [PubMed] [Google Scholar]
- Fischer A. J., Ritchey E. R., Scott M. A. and Wynne A. (2008). Bullwhip neurons in the retina regulate the size and shape of the eye. Dev. Biol. 317, 196-212. 10.1016/j.ydbio.2008.02.023 [DOI] [PubMed] [Google Scholar]
- Fischer A. J., Scott M. A., Ritchey E. R. and Sherwood P. (2009a). Mitogen-activated protein kinase-signaling regulates the ability of Müller glia to proliferate and protect retinal neurons against excitotoxicity. Glia 57, 1538-1552. 10.1002/glia.20868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. J., Scott M. A. and Tuten W. (2009b). Mitogen-activated protein kinase-signaling stimulates Müller glia to proliferate in acutely damaged chicken retina. Glia 57, 166-181. 10.1002/glia.20743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. J., Zelinka C. and Scott M. A. (2010). Heterogeneity of glia in the retina and optic nerve of birds and mammals. PLoS ONE 5, e10774 10.1371/journal.pone.0010774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. J., Zelinka C., Gallina D., Scott M. A. and Todd L. (2014). Reactive microglia and macrophage facilitate the formation of Müller glia-derived retinal progenitors. Glia 62, 1608-1628. 10.1002/glia.22703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. J., Zelinka C. and Milani-Nejad N. (2015). Reactive retinal microglia, neuronal survival, and the formation of retinal folds and detachments. Glia 63, 313-327. 10.1002/glia.22752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita K., Yasui S., Shinohara T. and Ito K. (2011). Interaction between NF-κB signaling and Notch signaling in gliogenesis of mouse mesencephalic neural crest cells. Mech. Dev. 128, 496-509. 10.1016/j.mod.2011.09.003 [DOI] [PubMed] [Google Scholar]
- Gallina D., Todd L. and Fischer A. J. (2014a). A comparative analysis of Müller glia-mediated regeneration in the vertebrate retina. Exp. Eye Res. 123, 121-130. 10.1016/j.exer.2013.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallina D., Zelinka C. and Fischer A. J. (2014b). Glucocorticoid receptors in the retina, Muller glia and the formation of Muller glia-derived progenitors. Development 141, 3340-3351. 10.1242/dev.109835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallina D., Palazzo I., Steffenson L., Todd L. and Fischer A. J. (2015). Wnt/β-catenin-signaling and the formation of Müller glia-derived progenitors in the chick retina. Dev. Neurobiol. 76, 983-1002. 10.1002/dneu.22370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghai K., Zelinka C. and Fischer A. J. (2009). Serotonin released from amacrine neurons is scavenged and degraded in bipolar neurons in the retina. J. Neurochem. 111, 1-14. 10.1111/j.1471-4159.2009.06270.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghai K., Zelinka C. and Fischer A. J. (2010). Notch signaling influences neuroprotective and proliferative properties of mature Muller glia. J. Neurosci. 30, 3101-3112. 10.1523/JNEUROSCI.4919-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S., May M. J. and Kopp E. B. (1998). NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 16, 225-260. 10.1146/annurev.immunol.16.1.225 [DOI] [PubMed] [Google Scholar]
- Giulian D., Young D. G., Woodward J., Brown D. C. and Lachman L. B. (1988). Interleukin-1 is an astroglial growth factor in the developing brain. J. Neurosci. 8, 709-714. 10.1523/JNEUROSCI.08-02-00709.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout P. W., Buckley A. R., Simms C. R. and Bruchovsky N. (2001). Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia 15, 1633-1640. 10.1038/sj.leu.2402238 [DOI] [PubMed] [Google Scholar]
- Graeber M. B. and Stre'rt W. J. (1990). Microglia: immune network in the CNS. Brain Pathol. 1, 2-5. 10.1111/j.1750-3639.1990.tb00630.x [DOI] [PubMed] [Google Scholar]
- Hayden M. S. and Ghosh S. (2004). Signaling to NF-κB. Genes Dev. 18, 2195-2224. 10.1101/gad.1228704 [DOI] [PubMed] [Google Scholar]
- Hayden M. S. and Ghosh S. (2008). Shared principles in NF-κB signaling. Cell 132, 344-362. 10.1016/j.cell.2008.01.020 [DOI] [PubMed] [Google Scholar]
- Hayden M. S. and Ghosh S. (2014). Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 26, 253-266. 10.1016/j.smim.2014.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes S., Nelson B. R., Buckingham B. and Reh T. A. (2007). Notch signaling regulates regeneration in the avian retina. Dev. Biol. 312, 300-311. 10.1016/j.ydbio.2007.09.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchcock P. F. and Raymond P. A. (1992). Retinal regeneration. Trends Neurosci. 15, 103-108. 10.1016/0166-2236(92)90020-9 [DOI] [PubMed] [Google Scholar]
- Hoang T., Wang J., Boyd P., Wang F., Santiago C., Jiang L., Lahne M., Yoo S., Todd L. J., Saez C. et al. (2019). Cross-species transcriptomic and epigenomic analysis reveals key regulators of injury response and neuronal regeneration in vertebrate retinas. bioRxiv, 717876 10.1101/717876 [DOI] [Google Scholar]
- Jorstad N. L., Wilken M. S., Grimes W. N., Wohl S. G., VandenBosch L. S., Yoshimatsu T., Wong R. O., Rieke F. and Reh T. A. (2017). Stimulation of functional neuronal regeneration from Müller glia in adult mice. Nature 548, 103-107. 10.1038/nature23283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. and Ben-Neriah Y. (2000). Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol. 18, 621-663. 10.1146/annurev.immunol.18.1.621 [DOI] [PubMed] [Google Scholar]
- Karlstetter M., Ebert S. and Langmann T. (2010). Microglia in the healthy and degenerating retina: insights from novel mouse models. Immunobiology 215, 685-691. 10.1016/j.imbio.2010.05.010 [DOI] [PubMed] [Google Scholar]
- Keohane A., Ryan S., Maloney E., Sullivan A. M. and Nolan Y. M. (2010). Tumour necrosis factor-α impairs neuronal differentiation but not proliferation of hippocampal neural precursor cells: role of Hes1. Mol. Cell. Neurosci. 43, 127-135. 10.1016/j.mcn.2009.10.003 [DOI] [PubMed] [Google Scholar]
- Kyritsis N., Kizil C., Zocher S., Kroehne V., Kaslin J., Freudenreich D., Iltzsche A. and Brand M. (2012). Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science 338, 1353-1356. 10.1126/science.1228773 [DOI] [PubMed] [Google Scholar]
- Lamba D., Karl M. and Reh T. (2008). Neural regeneration and cell replacement: a view from the eye. Cell Stem Cell 2, 538-549. 10.1016/j.stem.2008.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzillotta A., Porrini V., Bellucci A., Benarese M., Branca C., Parrella E., Spano P. F. and Pizzi M. (2015). NF-κB in innate neuroprotection and age-related neurodegenerative diseases. Front. Neurol. 6, 98 10.3389/fneur.2015.00098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leban J., Baierl M., Mies J., Trentinaglia V., Rath S., Kronthaler K., Wolf K., Gotschlich A. and Seifert M. H. J. (2007). A novel class of potent NF-κB signaling inhibitors. Bioorg. Med. Chem. Lett. 17, 5858-5862. 10.1016/j.bmcl.2007.08.022 [DOI] [PubMed] [Google Scholar]
- Lebrun-Julien F., Duplan L., Pernet V., Osswald I., Sapieha P., Bourgeois P., Dickson K., Bowie D., Barker P. A. and Di Polo A. (2009). Excitotoxic death of retinal neurons in vivo occurs via a non-cell-autonomous mechanism. J. Neurosci. 29, 5536-5545. 10.1523/JNEUROSCI.0831-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenkowski J. R. and Raymond P. A. (2014). Müller glia: stem cells for generation and regeneration of retinal neurons in teleost fish. Prog. Retin. Eye Res. 40, 94-123. 10.1016/j.preteyeres.2013.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenkowski J. R., Qin Z., Sifuentes C. J., Thummel R., Soto C. M., Moens C. B. and Raymond P. A. (2013). Retinal regeneration in adult zebrafish requires regulation of TGFβ signaling. Glia 61, 1687-1697. 10.1002/glia.22549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X., O'Mahony A., Mu Y., Geleziunas R. and Greene W. C. (2000). Protein kinase C-theta participates in NF-kappaB activation induced by CD3-CD28 costimulation through selective activation of Ikappa B kinase beta. Mol. Cell. Biol. 20, 2933-2940. 10.1128/MCB.20.8.2933-2940.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindström T. M. and Bennett P. R. (2005). 15-Deoxy-Δ12,14-prostaglandin j2 inhibits interleukin-1β-induced nuclear factor-κB in human amnion and myometrial cells: mechanisms and implications. J. Clin. Endocrinol. Metab. 90, 3534-3543. 10.1210/jc.2005-0055 [DOI] [PubMed] [Google Scholar]
- Ma B. and Hottiger M. O. (2016). Crosstalk between Wnt/β-catenin and NF-κB signaling pathway during inflammation. Front. Immunol. 7, 378 10.3389/fimmu.2016.00378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers J. R., Hu L., Moses A., Kaboli K., Papandrea A. and Raymond P. A. (2012). β-catenin/Wnt signaling controls progenitor fate in the developing and regenerating zebrafish retina. Neural Dev. 7, 30 10.1186/1749-8104-7-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migone T.-S., Zhang J., Luo X., Zhuang L., Chen C., Hu B., Hong J. S., Perry J. W., Chen S.-F., Zhou J. X. H. et al. (2002). TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity 16, 479-492. 10.1016/S1074-7613(02)00283-2 [DOI] [PubMed] [Google Scholar]
- Mondal D., Pradhan L. and LaRussa V. F. (2004). Signal transduction pathways involved in the lineage-differentiation of NSCs: can the knowledge gained from blood be used in the brain? Cancer Invest. 22, 925-943. 10.1081/CNV-200039679 [DOI] [PubMed] [Google Scholar]
- Nelson C. M., Ackerman K. M., O'Hayer P., Bailey T. J., Gorsuch R. A. and Hyde D. R. (2013). Tumor necrosis factor-alpha is produced by dying retinal neurons and is required for Muller glia proliferation during zebrafish retinal regeneration. J. Neurosci. 33, 6524-6539. 10.1523/JNEUROSCI.3838-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osakada F., Ooto S., Akagi T., Mandai M., Akaike A. and Takahashi M. (2007). Wnt signaling promotes regeneration in the retina of adult mammals. J. Neurosci. 27, 4210-4219. 10.1523/JNEUROSCI.4193-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn L., Kunkel S. and Nabel G. J. (1989). Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc. Natl. Acad. Sci. USA 86, 2336-2340. 10.1073/pnas.86.7.2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers A. N. and Satija R. (2015). Single-cell analysis reveals key roles for Bcl11a in regulating stem cell fate decisions. Genome Biol. 16, 199 10.1186/s13059-015-0778-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R., Zhao X.-F. and Goldman D. (2011). Ascl1a/Dkk/beta-catenin signaling pathway is necessary and glycogen synthase kinase-3beta inhibition is sufficient for zebrafish retina regeneration. Proc. Natl. Acad. Sci. USA 108, 15858-15863. 10.1073/pnas.1107220108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricote M., Li A. C., Willson T. M., Kelly C. J. and Glass C. K. (1998). The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 391, 79-82. 10.1038/34178 [DOI] [PubMed] [Google Scholar]
- Rompani S. B. and Cepko C. L. (2010). A common progenitor for retinal astrocytes and oligodendrocytes. J. Neurosci. 30, 4970-4980. 10.1523/JNEUROSCI.3456-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R., Farrell J. A., Gennert D., Schier A. F. and Regev A. (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495-502. 10.1038/nbt.3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A., Martin-Villalba A., Weih F., Vogel J., Wirth T. and Schwaninger M. (1999). NF-κB is activated and promotes cell death in focal cerebral ischemia. Nat. Med. 5, 554-559. 10.1038/8432 [DOI] [PubMed] [Google Scholar]
- Schreck R., Rieber P. and Baeuerle P. A. (1991). Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 10, 2247-2258. 10.1002/j.1460-2075.1991.tb07761.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütze S., Potthoff K., Machleidt T., Berkovic D., Wiegmann K. and Krönke M. (1992). TNF activates NF-κB by phosphatidylcholine-specific phospholipase C-induced “acidic” sphingomyelin breakdown. Cell 71, 765-776. 10.1016/0092-8674(92)90553-O [DOI] [PubMed] [Google Scholar]
- Schütze S., Wiegmann K., Machleidt T. and Krönke M. (1995). TNF-induced activation of NF-κB. Immunobiology 193, 193-203. 10.1016/S0171-2985(11)80543-7 [DOI] [PubMed] [Google Scholar]
- Stanke J. J. and Fischer A. J. (2010). Embryonic retinal cells and support to mature retinal neurons. Invest. Ophthalmol. Vis. Sci. 51, 2208-2218. 10.1167/iovs.09-4447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke J., Moose H. E., El-Hodiri H. M. and Fischer A. J. (2010). Comparative study of Pax2 expression in glial cells in the retina and optic nerve of birds and mammals. J. Comp. Neurol. 518, 2316-2333. 10.1002/cne.22335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus D. S., Pascual G., Li M., Welch J. S., Ricote M., Hsiang C.-H., Sengchanthalangsy L. L., Ghosh G. and Glass C. K. (2000). 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc. Natl. Acad. Sci. USA 97, 4844-4849. 10.1073/pnas.97.9.4844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto T., Takahashi K., Sato K. and Akiba Y. (2005). Molecular cloning and functional characterizations of chicken TL1A. Dev. Comp. Immunol. 29, 895-905. 10.1016/j.dci.2005.03.002 [DOI] [PubMed] [Google Scholar]
- Todd L. and Fischer A. J. (2015). Hedgehog signaling stimulates the formation of proliferating Müller glia-derived progenitor cells in the chick retina. Development 142, 2610-2622. 10.1242/dev.121616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd L., Suarez L., Squires N., Zelinka C. P., Gribbins K. and Fischer A. J. (2016a). Comparative analysis of glucagonergic cells, glia, and the circumferential marginal zone in the reptilian retina. J. Comp. Neurol. 524, 74-89. 10.1002/cne.23823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd L., Squires N., Suarez L. and Fischer A. J. (2016b). Jak/Stat signaling regulates the proliferation and neurogenic potential of Müller glia-derived progenitor cells in the avian retina. Sci. Rep. 6, 35703 10.1038/srep35703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd L., Palazzo I., Squires N., Mendonca N. and Fischer A. J. (2017). BMP- and TGFβ-signaling regulate the formation of Müller glia-derived progenitor cells in the avian retina. Glia 65, 1640-1655. 10.1002/glia.23185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd L., Suarez L., Quinn C. and Fischer A. J. (2018). Retinoic acid-signaling regulates the proliferative and neurogenic capacity of Müller glia-derived progenitor cells in the avian retina. Stem Cells 36, 392-405. 10.1002/stem.2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd L., Palazzo I., Suarez L., Liu X., Volkov L., Hoang T. V., Campbell W. A., Blackshaw S., Quan N. and Fischer A. J. (2019). Reactive microglia and IL1β/IL-1R1-signaling mediate neuroprotection in excitotoxin-damaged mouse retina. J. Neuroinflammation 16, 118 10.1186/s12974-019-1505-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N. J., Livak K. J., Mikkelsen T. S. and Rinn J. L. (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381-386. 10.1038/nbt.2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki Y., Wilken M. S., Cox K. E., Chipman L., Jorstad N., Sternhagen K., Simic M., Ullom K., Nakafuku M. and Reh T. A. (2015). Transgenic expression of the proneural transcription factor Ascl1 in Müller glia stimulates retinal regeneration in young mice. Proc. Natl. Acad. Sci. USA 112, 13717-13722. 10.1073/pnas.1510595112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rooijen N. (1989). The liposome-mediated macrophage ‘suicide’ technique. J. Immunol. Methods 124, 1-6. 10.1016/0022-1759(89)90178-6 [DOI] [PubMed] [Google Scholar]
- Wada Y., Nakamachi T., Endo K., Seki T., Ohtaki H., Tsuchikawa D., Hori M., Tsuchida M., Yoshikawa A., Matkovits A. et al. (2013). PACAP attenuates NMDA-induced retinal damage in association with modulation of the microglia/macrophage status into an acquired deactivation subtype. J. Mol. Neurosci. 51, 493-502. 10.1007/s12031-013-0017-5 [DOI] [PubMed] [Google Scholar]
- Wahl C., Liptay S., Adler G. and Schmid R. M. (1998). Sulfasalazine: a potent and specific inhibitor of nuclear factor kappa B. J. Clin. Invest. 101, 1163-1174. 10.1172/JCI992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J., Ramachandran R. and Goldman D. (2012). HB-EGF is necessary and sufficient for Müller glia dedifferentiation and retina regeneration. Dev. Cell 22, 334-347. 10.1016/j.devcel.2011.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J., Zhao X.-F., Vojtek A. and Goldman D. (2014). Retinal injury, growth factors, and cytokines converge on β-catenin and pStat3 signaling to stimulate retina regeneration. Cell Rep. 9, 285-297. 10.1016/j.celrep.2014.08.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. and Wong W. T. (2014). Microglia-Müller cell interactions in the retina. Adv. Exp. Med. Biol. 801, 333-338. 10.1007/978-1-4614-3209-8_42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Ma W., Zhao L., Fariss R. N. and Wong W. T. (2011). Adaptive Müller cell responses to microglial activation mediate neuroprotection and coordinate inflammation in the retina. J. Neuroinflammation 8, 173 10.1186/1742-2094-8-173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Wang X., Zhao L., Ma W., Rodriguez I. R., Fariss R. N. and Wong W. T. (2014). Macroglia-microglia interactions via TSPO signaling regulates microglial activation in the mouse retina. J. Neurosci. 34, 3793-3806. 10.1523/JNEUROSCI.3153-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Zhao L., Zhang Y., Ma W., Gonzalez S. R., Fan J., Kretschmer F., Badea T. C., Qian H.-H. and Wong W. T. (2017). Tamoxifen provides structural and functional rescue in murine models of photoreceptor degeneration. J. Neurosci. 37, 3294-3310. 10.1523/JNEUROSCI.2717-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White D. T., Sengupta S., Saxena M. T., Xu Q., Hanes J., Ding D., Ji H. and Mumm J. S. (2017). Immunomodulation-accelerated neuronal regeneration following selective rod photoreceptor cell ablation in the zebrafish retina. Proc. Natl. Acad. Sci. USA 114, E3719-E3728. 10.1073/pnas.1617721114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S. A., Chen L.-F., Kwon H., Fenard D., Bisgrove D., Verdin E. and Greene W. C. (2004). Prostratin antagonizes HIV latency by activating NF-κB. J. Biol. Chem. 279, 42008-42017. 10.1074/jbc.M402124200 [DOI] [PubMed] [Google Scholar]
- Yao K., Qiu S., Tian L., Snider W. D., Flannery J. G., Schaffer D. V. and Chen B. (2016). Wnt regulates proliferation and neurogenic potential of Müller glial cells via a Lin28/let-7 miRNA-dependent pathway in adult mammalian retinas. Cell Rep. 17, 165-178. 10.1016/j.celrep.2016.08.078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao K., Qiu S., Wang Y. V., Park S. J. H., Mohns E. J., Mehta B., Liu X., Chang B., Zenisek D., Crair M. C. et al. (2018). Restoration of vision after de novo genesis of rod photoreceptors in mammalian retinas. Nature 560, 484-488. 10.1038/s41586-018-0425-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelinka C. P., Scott M. A., Volkov L. and Fischer A. J. (2012). The reactivity, distribution and abundance of Non-Astrocytic Inner Retinal Glial (NIRG) cells are regulated by microglia, acute damage, and IGF1. PLoS ONE 7, e44477 10.1371/journal.pone.0044477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelinka C. P., Volkov L., Goodman Z. A., Todd L., Palazzo I., Bishop W. A. and Fischer A. J. (2016). mTor signaling is required for the formation of proliferating Müller glia-derived progenitor cells in the chick retina. Development 143, 1859-1873. 10.1242/dev.133215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Lenardo M. J. and Baltimore D. (2017). 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell 168, 37-57. 10.1016/j.cell.2016.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.-F., Wan J., Powell C., Ramachandran R., Myers M. G. Jr and Goldman D. (2014). Leptin and IL-6 family cytokines synergize to stimulate Müller glia reprogramming and retina regeneration. Cell Rep. 9, 272-284. 10.1016/j.celrep.2014.08.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.