

ABSTRACT
Heme is a cofactor and signaling molecule that is essential for much of aerobic life. All heme-dependent processes in eukaryotes require that heme is trafficked from its site of synthesis in the mitochondria to hemoproteins located throughout the cell. However, the mechanisms governing the mobilization of heme out of the mitochondria, and the spatio-temporal dynamics of these processes, are poorly understood. Here, using genetically encoded fluorescent heme sensors, we developed a live-cell assay to monitor heme distribution dynamics between the mitochondrial inner membrane, where heme is synthesized, and the mitochondrial matrix, cytosol and nucleus. Surprisingly, heme trafficking to the nucleus is ∼25% faster than to the cytosol or mitochondrial matrix, which have nearly identical heme trafficking dynamics, potentially supporting a role for heme as a mitochondrial–nuclear retrograde signal. Moreover, we discovered that the heme synthetic enzyme 5-aminolevulinic acid synthase (ALAS, also known as Hem1 in yeast), and GTPases in control of the mitochondrial dynamics machinery (Mgm1 and Dnm1) and ER contact sites (Gem1), regulate the flow of heme between the mitochondria and nucleus. Overall, our results indicate that there are parallel pathways for the distribution of bioavailable heme.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: Heme, Heme transport, Mitochondrial dynamics, Yeast
Highlighted Article: A live-cell assay to monitor heme distribution kinetics identifies GTPases in control of mitochondrial–ER contact sites and dynamics as being critical modulators of heme trafficking.
INTRODUCTION
Heme b is an essential but cytotoxic metallocofactor and signaling molecule. As a cofactor, heme facilitates diverse processes that span electron transfer, chemical catalysis and gas synthesis, storage and transport (Hanna et al., 2017; Reddi and Hamza, 2016). As a signaling molecule, heme-mediated regulation of transcription factors, kinases, ion channels and microRNA processing proteins collectively controls iron and redox homeostasis, oxygen sensing, mitochondrial metabolism, apoptosis, circadian rhythms, cell cycle progression, proliferation and proteostasis (Hanna et al., 2017; Reddi and Hamza, 2016).
Although essential for life, heme and its biosynthetic precursors can also act as toxins, necessitating careful handling of the synthesis and trafficking of this compound by cells. The hydrophobicity and redox activity of heme causes it to disrupt membrane and protein structure and deleteriously oxidize various biomolecules (Kumar and Bandyopadhyay, 2005; Sassa, 2004). Porphyrins and porphyrinogens, heme biosynthetic intermediates, are photosensitizers that can catalyze the formation of reactive oxygen species (Sachar et al., 2016). Many diseases are associated with defects in heme management, including certain cancers (Shen et al., 2014), cardiovascular disease (Wu et al., 2011), neurodegenerative diseases (Atamna and Frey, 2004; Atamna et al., 2002; Schipper et al., 2009), porphyrias (Puy et al., 2010) and anemias (Yang et al., 2016). Despite the tremendous importance of heme in physiology, the cellular and molecular mechanisms that govern the assimilation of heme into metabolism remain poorly understood.
In eukaryotes, heme is synthesized via a highly conserved eight-step process. The first and the last three reactions in metazoans (the first and last two in the yeast Saccharomyces cerevisiae) take place in the mitochondria and the remaining reactions occur in the cytosol (Piel et al., 2019). The first committed step of heme synthesis is the condensation of glycine with succinyl coenzyme A to form 5-aminolevulinic acid (ALA), which is catalyzed by ALA synthase (ALAS, also known as Hem1 in yeast). ALA is then exported from the mitochondria into the cytosol where it is converted to coproporphyrinogen III in four steps. Coproporphyrinogen III is then transported back into the mitochondria where it is converted to protoporphyrin IX (PPIX) in two steps by the enzymes coproporphyrinogen oxidase (CPOX) and protoporphyrinogen oxidase (PPOX). In the final step of heme synthesis, ferrochelatase (FECH) catalyzes the insertion of ferrous iron into PPIX to make heme.
Interestingly, it was recently proposed that the mitochondrial heme biosynthetic enzymes, ALAS, PPOX and FECH, form a super complex, or metabolon, that includes additional factors like iron and porphyrinogen transporters, and putative heme chaperones (Medlock et al., 2015; Piel et al., 2016). The rationale for a heme metabolon is that it would facilitate the channeling of potentially toxic reactive substrates, including porphyrinogen, porphyrin and iron, for efficient production and trafficking of heme, thereby mitigating their ability to diffuse freely throughout the cell in an unproductive and deleterious manner. However, given this rationale, it is unclear why ALAS would interact with FECH in the heme metabolon if ALA were not the immediate substrate for FECH. Rather, it is possible that ALAS may regulate FECH to affect heme synthesis or trafficking.
Once heme is synthesized by FECH on the matrix side of the mitochondrial inner membrane (IM), it must be mobilized to hemoproteins present in virtually every subcellular compartment. However, the specific factors that govern the trafficking of heme to target hemoproteins are poorly understood (Hanna et al., 2017; Reddi and Hamza, 2016). The current paradigm for subcellular heme trafficking is a sequential one in which mitochondrial heme transporters export heme into the cytosol, where it can then be distributed to other locales. This sequential paradigm for heme distribution was established with the discovery of feline leukemia virus subgroup C cellular receptor 1b (FLVCR1b), the only putative mitochondrial heme transporter identified to date (Chiabrando et al., 2012).
An alternative, but far less studied potential mechanism for heme distribution is through mitochondrial membrane contact sites (Hanna et al., 2017). Endoplasmic reticulum (ER)–mitochondrial encounter structures (ERMES), one type of mitochondrial–ER contact site (Elbaz-Alon et al., 2014), are highly dynamic tethers that physically link the mitochondrial and ER networks and have been proposed to play a role in the transfer of lipids and regulate iron homeostasis (Lackner, 2019; Murley and Nunnari, 2016; Xue et al., 2017). The frequency of ERMES is in part dependent on mitochondrial division (Elbaz-Alon et al., 2014). ER tubules facilitate mitochondrial fission by wrapping around and constricting mitochondria in an ERMES-dependent fashion, thereby ensuring that the GTPase Dnm1 can oligomerize around a compressed mitochondrial outer membrane (OM) to catalyze the scission of mitochondria (Friedman et al., 2011). Following fission, the GTPase Gem1 disengages ERMES to dissolve the ER–mitochondrial contact site (Murley et al., 2013). As a consequence, ERMES is dependent on mitochondrial fission and fusion dynamics (Elbaz-Alon et al., 2014). Indeed, mutants with defects in mitochondrial fission or fusion have increased or decreased numbers of ERMES, respectively (Elbaz-Alon et al., 2014).
Here, using genetically encoded ratiometric fluorescent heme sensors (HS1) targeted to the mitochondrial matrix, cytosol or nucleus (Hanna et al., 2016, 2018; Sweeny et al., 2018), we developed a live-cell assay in yeast to monitor heme distribution kinetics and probe the role of ALAS, ERMES and mitochondrial dynamics in subcellular heme trafficking. Surprisingly, we found that heme trafficking rates from the matrix side of the IM to the mitochondrial matrix and cytosol are similar, whereas trafficking to the nucleus is ∼25% faster. We propose that the increased rate of nuclear heme trafficking is required for heme-based mitochondrial–nuclear retrograde signaling. These data also indicate that heme is distributed from the mitochondrial IM to other locales simultaneously via parallel pathways rather than sequentially. Moreover, we discovered that ALAS negatively regulates mitochondrial–nuclear heme trafficking, highlighting the close coordination of heme synthesis and trafficking. In addition, we identified GTPases that directly (Gem1) and indirectly (Dnm1 and Mgm1) regulate ERMES as being modulators of nuclear heme transport. Based on our results, we propose a model in which heme is trafficked via ER–mitochondrial contact sites to other organelles such as the nucleus.
RESULTS
Inter-compartmental heme transport kinetics
In order to probe inter-compartmental heme trafficking in yeast, we developed an in vivo pulse-chase assay in which, upon the initiation of heme synthesis, heme mobilization into the mitochondrial matrix, cytosol and nucleus is monitored using a previously reported genetically encoded ratiometric fluorescent heme sensor (HS1) (Hanna et al., 2016). Heme binding to HS1 (Fig. 1A) results in a decrease in the eGFP to mKATE2 fluorescence ratio of the sensor. We targeted HS1 to the mitochondria and nucleus by appending N-terminal Cox4 mitochondrial matrix or C-terminal SV40 nuclear localization sequences, respectively, as previously demonstrated (Hanna et al., 2016) and confirmed the localization by microscopy (Fig. 1B) and organelle isolation and immunoblotting (Fig. 1C,D). HS1 lacking nuclear or mitochondrial targeting sequences had a diffuse pattern of expression throughout the cytosol (Fig. 1B) and was found primarily in the cytosolic fractions of mitochondrial and nuclear preparations (Fig. 1C,D). Nuclear- and mitochondrial-targeted HS1 colocalized with 4′,6-diamidino-2-phenylindole (DAPI) (Fig. 1B), which can be used to selectively mark nuclear or mitochondrial DNA depending on concentration or duration of exposure (Shadel and Seidel-Rogol, 2007; Williamson and Fennell, 1979), and was found primarily in nuclear and mitochondrial fractions, respectively (Fig. 1C,D).
Fig. 1.
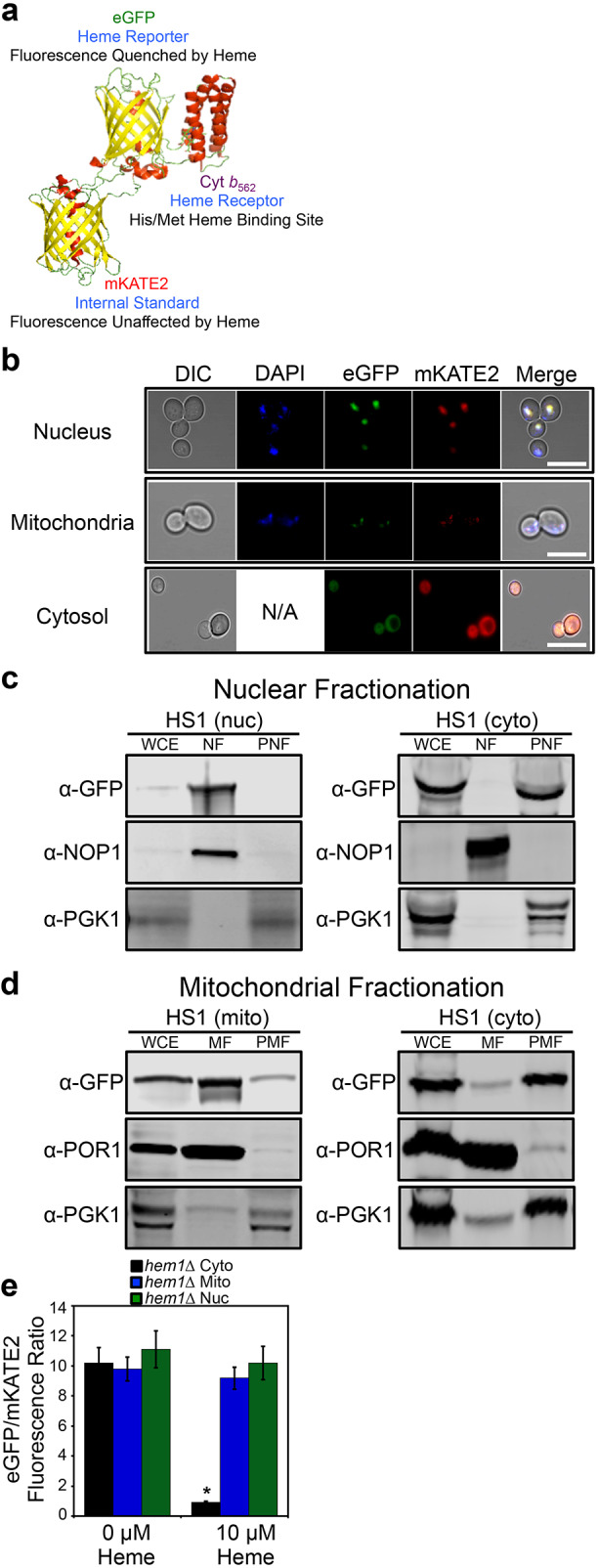
Molecular model and subcellular targeting of the heme sensor HS1. (A) Model of HS1 derived from the X-ray structures of mKATE (PDB: 3BXB) and CG6 (PDB: 3U8P). (B) The subcellular localization of HS1 was assessed by laser-scanning confocal microscopy. Cells expressing nuclear- or mitochondrial-targeted HS1 were stained with DAPI to label nuclei or mitochondria and live cells were imaged at a magnification of 63×. Images shown are representative of n=3 experiments. ‘Merge’ indicates the merged images of fluorescence from the DAPI, EGFP and mKATE2 fluorescence channels. N/A, not applicable. Scale bar: 5 μm. (C,D) Confirmation of mitochondrial (mito), nuclear (nuc), and cytosolic (cyto) HS1 localization as assessed by cell fractionation and immunoblotting. Nuclei (C) and mitochondria (D) were isolated and expression of HS1 was probed using anti-GFP antibodies. The indicated fractions were confirmed by probing the expression of PGK1, NOP1, and POR1, which are cytosolic, nuclear, and mitochondrial marker proteins, respectively. Whole cell extract (WCE), derived from the spheroplast fraction (5% of input loaded), nuclear fraction (NF), mitochondrial fraction (MF), post-mitochondrial fraction (PMF), or post-nuclear fraction (PNF) were electrophoresed on a 14% Tris-glycine SDS–PAGE gel. Blots shown are representative of n=2 experiments. (E) Validation that mitochondrial- and nuclear-targeted HS1 do not respond to cytosolic heme. hem1Δ cells expressing cytosolic, nuclear, or mitochondrial HS1 were permeabilized with digitonin, a plasma-membrane-specific permeabilizing agent, and incubated with the indicated concentration of heme. Fluorimetry data represent mean±s.d. of three biological replicates. *P<0.00001 (one-way ANOVA with Dunnett's post-hoc test).
It is possible that a small fraction of mitochondrial- or nuclear-targeted sensor might be present in the cytosol and confound analysis of sub-compartmental heme. In order to assess whether this were the case, we permeabilized heme-deficient cells lacking HEM1, which encodes ALAS, with digitonin, which selectively permeabilizes the plasma membrane, and treated cells with heme. As indicated in Fig. 1E, only cells expressing cytosolic HS1 exhibited a significant heme-dependent reduction in eGFP to mKATE2 fluorescence ratio, with little perturbation to the fluorescence of mitochondrial- and nuclear-targeted HS1. These data indicate that mitochondrial- and nuclear-targeted HS1 is unresponsive to cytosolic heme.
Inter-compartmental heme trafficking rates were monitored by inhibiting heme synthesis with 500 μM succinylacetone (SA), an inhibitor of the second enzyme in the heme biosynthetic pathway, porphobilinogen synthase (PBGS; also known as ALAD), then removing the block in heme synthesis by resuspending cells into medium lacking SA and monitoring the subsequent time-dependent change in the eGFP to mKATE2 ratio (R) of HS1 localized to different locations upon the re-initiation of heme synthesis (Fig. 2A–D). As described by Eqn 1, the fractional saturation of HS1 (% Bound) is calculated by considering the R-value of the sensor relative to that when it is 0% (Rmin) or 100% (Rmax) bound (Fig. 2E), which are parameters derived from parallel cultures of cells continuously maintained in medium with and without SA (Hanna et al., 2016, 2018) (Fig. 2A–C).
Fig. 2.
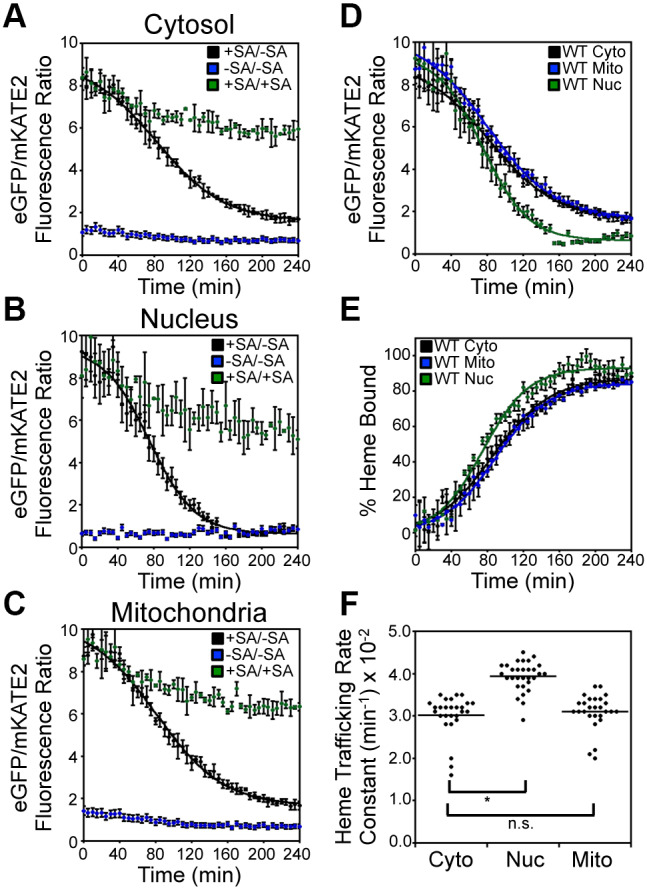
Inter-compartmental heme trafficking dynamics as measured by the heme sensor HS1. WT cells expressing HS1 in the cytosol (A), nucleus (B), or mitochondria (C) were either depleted of heme by addition of 500 μM succinylacetone (SA; heme biosynthetic inhibitor) before removal of SA to allow re-initiation of heme synthesis (+SA/−SA), or continually treated (+SA/+SA) or untreated (−SA/−SA) with SA. Upon re-initiation of heme synthesis in +SA/−SA samples, the rates of heme trafficking to the indicated subcellular locations were monitored by measuring the time-dependent change in sensor eGFP/mKATE2 fluorescence ratio (D) or the fractional saturation of HS1 (E), which is determined using Eqn 1 and the ‘raw’ fluorescence ratio values shown in A–C. The data in D and E are fitted to Eqn 2. (F) Rate constants for cytosolic, nuclear, and mitochondrial heme trafficking, which were extracted from fits to the data represented in E, for triplicate cultures in nine independent experimental trials, are shown. *P<0.0001; ns, not significant (one-way ANOVA with Dunnett's post-hoc test). Heme trafficking kinetics data shown in A–E represent mean±s.d. of independent triplicate cultures.
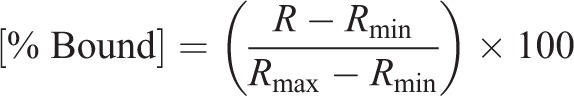 |
(1) |
To confirm the SA-mediated block in heme synthesis, we determined that SA-conditioned wild-type (WT) cells exhibited intracellular heme concentrations (Michener et al., 2012) (Fig. S1A) and HS1 R-values (Hanna et al., 2016) (Fig. S1B) similar to hem1Δ cells, which cannot make heme due to the deletion of ALAS (Hanna et al., 2016). SA pre-conditioned cells shifted to medium lacking SA exhibited a time-dependent decrease in HS1 R-values (Fig. 2A–C) in the cytosol, nucleus, and mitochondria, which is characteristic of increased heme loading of the sensor due to binding of newly synthesized heme (Fig. 2E). By contrast, the HS1 R-values in cells continuously maintained with or without SA remained characteristically high or low, respectively (Fig. 2A–C), albeit with some modest time-dependent changes.
The change in sensor heme saturation following the re-initiation of heme synthesis revealed three distinct phases: a lag phase, an exponential phase and a stationary phase (Fig. 2E). The lag phase is interpreted to be the time required to alleviate the SA-mediated block in heme synthesis and re-initiate heme production. The exponential phase represents the rate of heme binding to HS1, which is governed by the relative rates of heme synthesis and trafficking to the different subcellular locations. However, when comparing HS1 heme saturation kinetics between different compartments within a given strain, the data strictly represent the inter-compartmental heme trafficking rates since the contribution from heme synthesis is a constant. Indeed, expression of cytosolic, nuclear or mitochondrial HS1 did not perturb heme synthesis (Fig. S2A). Since the final step of heme synthesis occurs with the insertion of ferrous iron into PPIX, a reaction catalyzed by FECH at the matrix side of the mitochondrial IM, the heme trafficking rates reflect heme mobilization from the mitochondrial IM to the matrix, cytosol, or nucleus, as well as equilibration of heme between the sensor and endogenous hemoproteins within a given compartment. Given the high affinity of HS1 for both ferric and ferrous heme, 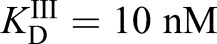 and
and 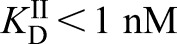 (calculated to be 1 pM based on a thermodynamic cycle) at pH 7.0 (Hanna et al., 2018), which is amongst the tightest measured for an intracellular soluble hemoprotein (Hanna et al., 2016), we assume the HS1 reporter acts as a heme sink that does not exchange with other endogenous hemoproteins due to their presumably weaker heme affinities. If this assumption is valid, the exponential phase of sensor heme saturation would only reflect the inter-compartmental transport rates. Additionally, due to the high affinities of ferric and ferrous heme and their ability to similarly affect HS1 fluorescence (Hanna et al., 2016), we cannot resolve the two oxidation states of heme. The stationary phase represents the maximum limiting heme saturation of the sensor 4 h after alleviating the SA-mediated block in heme synthesis and typically spans ∼70–100% saturation.
(calculated to be 1 pM based on a thermodynamic cycle) at pH 7.0 (Hanna et al., 2018), which is amongst the tightest measured for an intracellular soluble hemoprotein (Hanna et al., 2016), we assume the HS1 reporter acts as a heme sink that does not exchange with other endogenous hemoproteins due to their presumably weaker heme affinities. If this assumption is valid, the exponential phase of sensor heme saturation would only reflect the inter-compartmental transport rates. Additionally, due to the high affinities of ferric and ferrous heme and their ability to similarly affect HS1 fluorescence (Hanna et al., 2016), we cannot resolve the two oxidation states of heme. The stationary phase represents the maximum limiting heme saturation of the sensor 4 h after alleviating the SA-mediated block in heme synthesis and typically spans ∼70–100% saturation.
The kinetics of heme binding to HS1 (Fig. 2E) can be fitted to the logistic function described in Eqn 2, where [A] is the maximal value of HS1 fractional saturation (amplitude), k is the first order rate constant (min−1), t is the time (min), and t0.5 is the midpoint of the transition. The lag time can be defined as t0.5–2/k (Arosio et al., 2015).
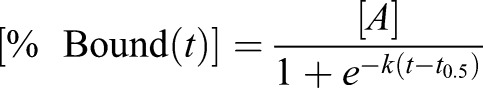 |
(2) |
Quite surprisingly, the kinetics of HS1 heme saturation indicated that heme trafficking to locations as disparate as the mitochondrial matrix, cytosol, and nucleus is similar, but with heme transport to the nucleus exhibiting an ∼25% increase in rate constant relative to heme transport to the cytosol and mitochondrial matrix (Fig. 2E,F). This observation was highly reproducible and the rate constants for heme trafficking to the cytosol (kCYTO), nucleus (kNUC), and mitochondrial matrix (kMITO) were found to be 0.030±0.005 min−1, 0.039±0.003 min−1 and 0.031±0.004 min−1, respectively (mean±s.d.; Fig. 2F). Importantly, in every trial conducted, even if the absolute rates were different, the relative differences always followed the same trend, with kNUC>kCYTO∼kMITO. Fitting of the ‘raw’ unprocessed sensor ratios, R, revealed a similar trend in the relative rates of heme binding to HS1, with kNUC (0.039±0.003 min−1)>kCYTO (0.026±0.001 min−1)=kMITO (0.027±0.002 min−1) (Fig. 2D), albeit with different absolute rate constants to those determined through fitting processed ‘% Bound’ traces. This is due to the fact that, for reasons not completely understood, there are fluctuations in Rmin and Rmax over the course of a heme trafficking experiment (Fig. 2A–C). The ‘% Bound’ is our preferred metric because it accounts for non-heme dependent changes in fluorescence ratios.
The similarity in the observed rates of heme trafficking to different locales might suggest that heme binding to the sensor is rate limiting. However, measurements of the rate of heme binding to HS1 in cell lysates of WT cells depleted of heme indicated that heme binding to the cytosolic, mitochondrial, and nuclear sensors occurs in less than 2 min (Fig. S3), which is much faster than the ∼90–120 min it takes 50% of total cellular heme to be synthesized (Fig. S2A) or 50% of HS1 to become heme saturated in the SA pulse-chase assays (Fig. 2).
In order to rule out any potential artefacts associated with heme sensor expression itself perturbing the observed heme trafficking rates, we expressed cytosolic HS1 under the control of weak (pADH1), medium (pTEF1), or strong (pGPD) promoters and found that the observed heme trafficking rates were not affected (Fig. S2B) despite a nearly 10-fold range in sensor expression (Fig. S2C). These results are consistent with our previous findings that sensor expression does not perturb heme homeostasis or otherwise affect cell viability (Hanna et al., 2016). Analogous experiments titrating expression with mitochondrial and nuclear sensors could not be completed due to low sensor expression and correspondingly low sensor signal-to-noise ratios associated with pTEF1- or pADH1-driven expression. Taken together, our data indicate that the SA pulse-chase assays can be used to measure the rates of heme trafficking to multiple compartments and that the rates of mitochondrial–nuclear heme trafficking are ∼25% faster relative to heme trafficking to the cytosol and mitochondria.
In order to rule out that the observed rates of heme trafficking are due to artefacts associated with SA treatment, we recapitulated the in vivo heme transport assay in hem1Δ cells pulsed with a bolus of ALA to initiate heme synthesis. As shown in Fig. 3A, heme trafficking kinetics to the matrix, cytosol, and nucleus in hem1Δ cells were qualitatively similar to the results obtained from the SA pulse-chase assay using WT cells (Fig. 2E,F), with transport to the nucleus (kNUC=0.059±0.005 min−1) being faster than to the cytosol (kCYTO=0.030±0.003 min−1) and mitochondrial matrix (kMITO=0.028±0.004 min−1) (Table S1). Notably, the rate enhancement for heme trafficking to the nucleus was greater in magnitude in hem1Δ cells pulsed with ALA (kNUC/kCYTO=2.0±0.1; Fig. 3A) than in WT cells that have the SA-mediated block in heme synthesis alleviated (kNUC/kCYTO=1.3±0.2; P<0.01, one-way ANOVA; n=3) (Fig. 2E,F). This observation suggests that either the use of SA suppresses nuclear heme trafficking or that the first enzyme in the heme biosynthetic pathway, ALAS, is a negative regulator of mitochondrial–nuclear heme trafficking.
Fig. 3.
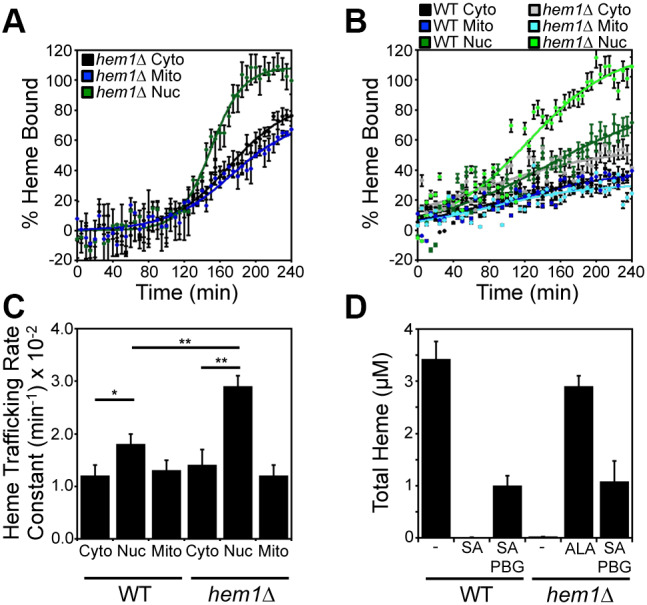
ALA synthase (Hem1) negatively regulates mitochondrial–nuclear heme trafficking. (A) hem1Δ cells expressing HS1 in the cytosol (black), nucleus (green), or mitochondria (blue) were pulsed with a bolus of ALA to initiate heme synthesis and the rates of heme trafficking to the indicated subcellular locations were monitored by measuring the fractional saturation of HS1 over time, as in Fig. 2. (B) WT and hem1Δ cells expressing the HS1 sensor were pre-cultured with SA to deplete heme and pulsed with a bolus of PBG to re-initiate heme synthesis. The rates of heme trafficking to the indicated subcellular locations were monitored by measuring the fractional saturation of HS1 over time, as in Fig. 2. Data shown in A and B are mean±s.d. of n=3 cultures (C) Heme trafficking rate constants for the indicated compartments of WT and hem1Δ strains, based on data represented in B. Data are mean±s.d. of triplicate cultures. *P<0.05, **P<0.001 (one-way ANOVA with Dunnett's post-hoc test). (D) Analysis of total heme in WT and hem1Δ strains treated with SA, ALA or PBG. Data represent the mean±s.d. of triplicate cultures.
ALAS regulates mitochondrial–nuclear heme trafficking
In order to determine if ALAS plays a role in regulating heme distribution kinetics, we directly compared inter-compartmental heme trafficking kinetics in WT and hem1Δ cells. To this end, we monitored heme distribution dynamics in WT and hem1Δ cells with SA-inactivated PBGS that were pulsed with 500 μM porphobilinogen (PBG), the product of the reaction catalyzed by PBGS, to re-initiate heme biosynthesis. As with the SA pulse-chase experiment in WT cells (Fig. 2E,F), the rates of heme trafficking to the nucleus were greater than rates of trafficking to the cytosol and mitochondrial matrix in SA-treated WT cells pulsed with PBG (Fig. 3B,C): kNUC/kCYTO=1.5±0.2 versus kMITO/kCYTO=1.1±0.2 (P=0.05, one-way ANOVA; n=3). Strikingly, in SA-treated hem1Δ cells pulsed with PBG, heme trafficking to the nucleus was significantly augmented relative to that in WT cells (Fig. 3B,C): kNUC/kCYTO=2.0±0.1 in hem1Δ cells versus kNUC/kCYTO=1.5±0.2 in WT cells (P=0.05, one-way ANOVA; n=3). The relatively low final fractional saturation of the sensor using PBG can be attributed to the fact that heme-deprived cells were less efficient at using PBG to make heme (Fig. 3D). WT and hem1Δ cells treated with SA and PBG made ∼33% of the heme that WT or hem1Δ cells treated with ALA made. Thus, it is all the more impressive that in hem1Δ cells pulsed with PBG, the nuclear sensor was completely saturated with heme, unlike in WT cells. Importantly, WT and hem1Δ cells synthesized similar levels of heme when supplied with PBG (Fig. 3D). In total, our results demonstrate that ALAS negatively regulates mitochondrial–nuclear heme trafficking.
Gem1, a GTPase that regulate ERMES, negatively modulates mitochondrial–nuclear heme trafficking
Since mitochondria form dynamic physical contacts with other organelles such as the ER (Elbaz-Alon et al., 2014; Murley and Nunnari, 2016), we surmised that one potential route for the distribution of mitochondrial heme to other compartments, such as the nucleus, could be through membrane contact sites. In this scenario, heme may be trafficked to the nucleus via the ER network, bypassing the cytosol, thereby accounting for the increased rate of nuclear heme trafficking. To test this idea, we probed heme distribution rates in deletion mutants with increased or decreased ER–mitochondrial contact sites.
The ER–mitochondrial encounter structure (ERMES), which constitutes one type of physical interface between these organelles, is a complex that consists of four proteins, Mmm1, Mdm10, Mdm12 and Mdm34 (Boldogh et al., 2003; Kornmann et al., 2009; Sogo and Yaffe, 1994; Youngman et al., 2004). Mmm1 and Mdm10 are anchored in the ER and outer mitochondrial membranes, respectively, while the cytosolic protein Mdm12 and the mitochondrial protein Mdm34 help to bridge the interaction between Mmm1 and Mdm10 in the ERMES complex (Boldogh et al., 2003; Kornmann et al., 2009; Sogo and Yaffe, 1994; Youngman et al., 2004). The absence of any single ERMES protein results in dissociation of the entire complex, compromising the formation of ER–mitochondrial contact sites, mitochondrial division and morphology (Boldogh et al., 2003; Burgess et al., 1994; Kornmann et al., 2009; Sogo and Yaffe, 1994; Youngman et al., 2004). ERMES is negatively regulated by Gem1, which is a mitochondrial outer-membrane localized GTPase that disengages ERMES after mitochondrial division (Kornmann et al., 2011; Murley et al., 2013).
As shown in Fig. 4A,B, gem1Δ cells, which have increased ERMES, exhibited a significantly increased rate of mitochondrial–nuclear heme trafficking, with minimal impact on heme trafficking to the cytosol and mitochondrial matrix: kNUC/kCYTO=1.6±0.1 in gem1Δ cells versus kNUC/kCYTO=1.2±0.1 in WT cells (P<0.01, one-way ANOVA: n=3). These data suggest that more stable ER–mitochondrial contacts increase the flow of heme to the nucleus. However, in ERMES-compromised mdm12Δ and mdm34Δ strains, heme trafficking rates were unaffected (Fig. 4C–F).
Fig. 4.
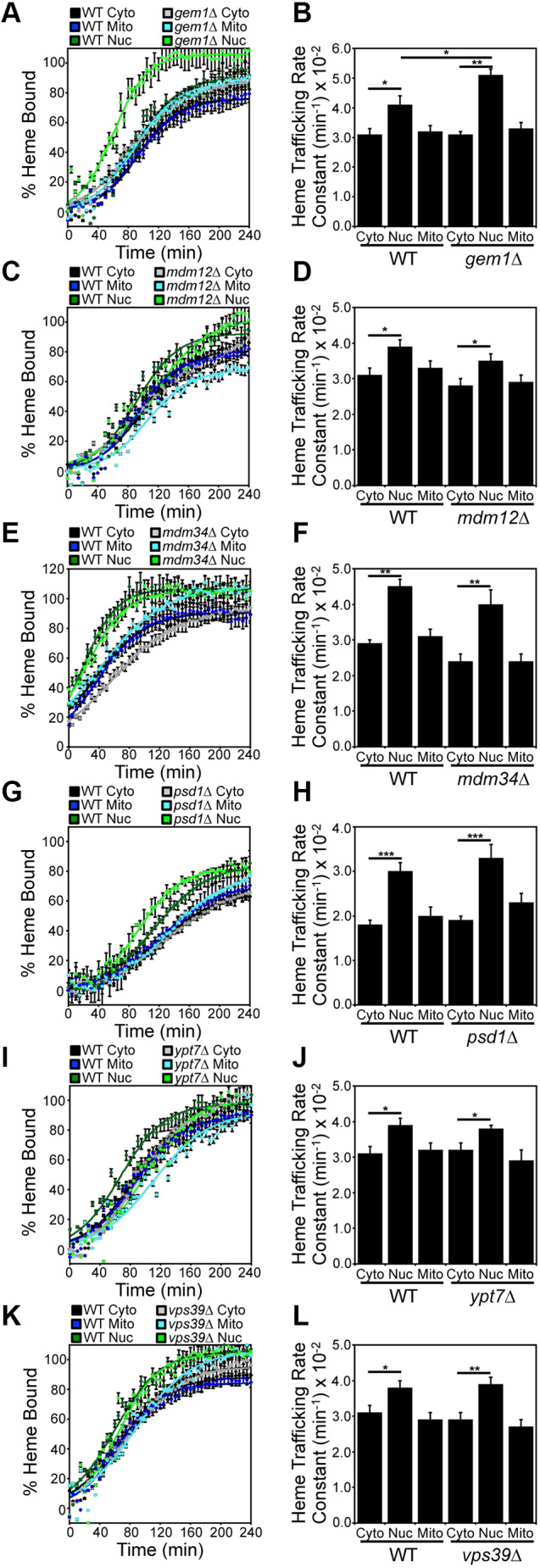
Gem1 is a negative regulator of mitochondrial–nuclear heme trafficking. Heme trafficking rates were monitored using the SA pulse-chase assay and rate constants calculated, as described in Fig. 2, for (A,B) gem1Δ, (C,D) mdm12Δ, (E,F) mdm34Δ, (G,H) psd1Δ, (I,J) ypt7Δ, and (K,L) vps39Δ cells. Data represent the mean±s.d. of independent triplicate cultures. *P<0.05, **P<0.01, ***P<0.001 (one-way ANOVA with Dunnett's post-hoc test).
ERMES has been implicated in the transfer of lipids, for example phosphatidylserine (PS), between the ER and mitochondria (Kawano et al., 2018; Kornmann et al., 2009; Nguyen et al., 2012). PS is required for the synthesis of phosphatidylethanolamine (PE) and phosphatidylcholine (PC) (Schuler et al., 2016). In order to rule out confounding contributions that may arise due to general defects in PE or PC synthesis in ERMES mutants, we tested heme trafficking dynamics in psd1Δ cells, which lack PS decarboxylase, the enzyme that catalyzes the formation of PE. As shown in Fig. 4G,H, there were no defects in heme trafficking dynamics in psd1Δ cells.
ERMES are highly dynamic and sensitive to the presence of mitochondrial–vacuolar contact sites, termed vCLAMPs for vacuolar and mitochondrial patches. Like ERMES, vCLAMPs are important for metabolite trafficking between mitochondria and the endomembrane system (Elbaz-Alon et al., 2014). The absence of one causes expansion of the other, and ablation of both ERMES and vCLAMPs is lethal (Elbaz-Alon et al., 2014). We therefore sought to determine if a defect in vCLAMPs also increased mitochondrial–nuclear heme trafficking rates as in gem1Δ cells, which have an increased number of ERMES foci as found in vCLAMP mutants. However, neither vps39Δ nor ypt7Δ cells (Fig. 4I–L), which lack a component of the HOPS (homotypic fusion and vacuole protein sorting) tethering complex (Vps39) and a Rab GTPase (Ypt7), both core components of vCLAMPs (Elbaz-Alon et al., 2014; Hönscher et al., 2014; Murley and Nunnari, 2016), exhibited defects in heme distribution kinetics. We conclude that Gem1, but not the core components of ERMES and vCLAMP machineries, regulates mitochondrial–nuclear heme trafficking.
Mgm1 and Dnm1 are positive and negative regulators of mitochondrial–nuclear heme trafficking, respectively
The mitochondrial network is highly dynamic and is constantly remodeled by fusion and fission events. The dynamic behavior of the mitochondrial network is thought to be responsible for the proper cellular distribution and trafficking of a number of mitochondrial-derived metabolites, including various lipids (Chan, 2012; Tatsuta et al., 2014). Given that we identified that Gem1, an ERMES-regulating GTPase, modulates mitochondrial–nuclear heme trafficking, and because ERMES are associated with sites of mitochondrial division and the frequency of their formation can be impacted by mitochondrial fission and fusion (Elbaz-Alon et al., 2014; Friedman et al., 2011), we reasoned that other GTPases that regulate mitochondrial dynamics might regulate mitochondrial–nuclear heme trafficking.
Mitochondrial fusion
In yeast, a pair of dynamin-like GTPases, Fzo1 and Mgm1, drive OM and IM fusion, respectively (Westermann, 2008). In order to coordinate double membrane fusion, a protein spanning the mitochondrial intermembrane space, Ugo1, physically tethers Fzo1 and Mgm1. Mgm1 is proteolytically processed by the rhomboid protease Pcp1 (DeVay et al., 2009; Zick et al., 2009) into long (l-Mgm1) and short (s-Mgm1) isoforms. It is thought that l-Mgm1, which has an inactive GTPase domain, acts as an anchor in the IM and interacts with and activates the GTPase activity of s-Mgm1 in the intermembrane space to promote IM fusion (DeVay et al., 2009; Zick et al., 2009).
The mgm1Δ strain exhibited a marked defect in nuclear heme trafficking relative to that in WT cells, with a greater than 50% reduction in HS1 heme loading (Fig. 5A). The modest changes in sensor loading and variability between biological replicates resulted in a poor goodness-of-fit (R2=0.5293) and large error in the rate constant when fitting the mitochondrial–nuclear heme trafficking data to Eqn 2 (Fig. 5B, Table S1). Conversely, heme trafficking to the mitochondrial matrix and cytosol was not significantly impacted in mgm1Δ cells (Fig. 5A,B). Interestingly, heme availability to HS1 in the nucleus was similar between WT and mgm1Δ cells after a prolonged growth period of ∼16 h, suggesting that there are compensatory mechanisms that overcome the defects in nuclear heme trafficking in mgm1Δ cells (Fig. S4A). The rate of heme synthesis was unperturbed by loss of Mgm1, indicating that the nuclear trafficking defect is not related to general defects in heme synthesis (Fig. S4B). The effects of the mgm1Δ mutant on nuclear heme trafficking is not due to the loss of mitochondrial DNA, which occurs with high frequency in mgm1Δ cells; rho0 cells, which lack mitochondrial DNA, did not exhibit an appreciable defect in nuclear heme trafficking (Fig. 5C,D). Surprisingly, ablation of other components of the mitochondrial fusion machinery, including Fzo1 and Ugo1, did not affect heme trafficking rates (Fig. 5E–H). Because the regulation of nuclear heme transport is specific to Mgm1, we tested whether proteolytic processing of Mgm1 was important for heme mobilization. As seen in Fig. 5I,J, pcp1Δ cells, which cannot convert l-Mgm1 to s-Mgm1, did not exhibit a defect in mitochondrial–nuclear heme trafficking.
Fig. 5.
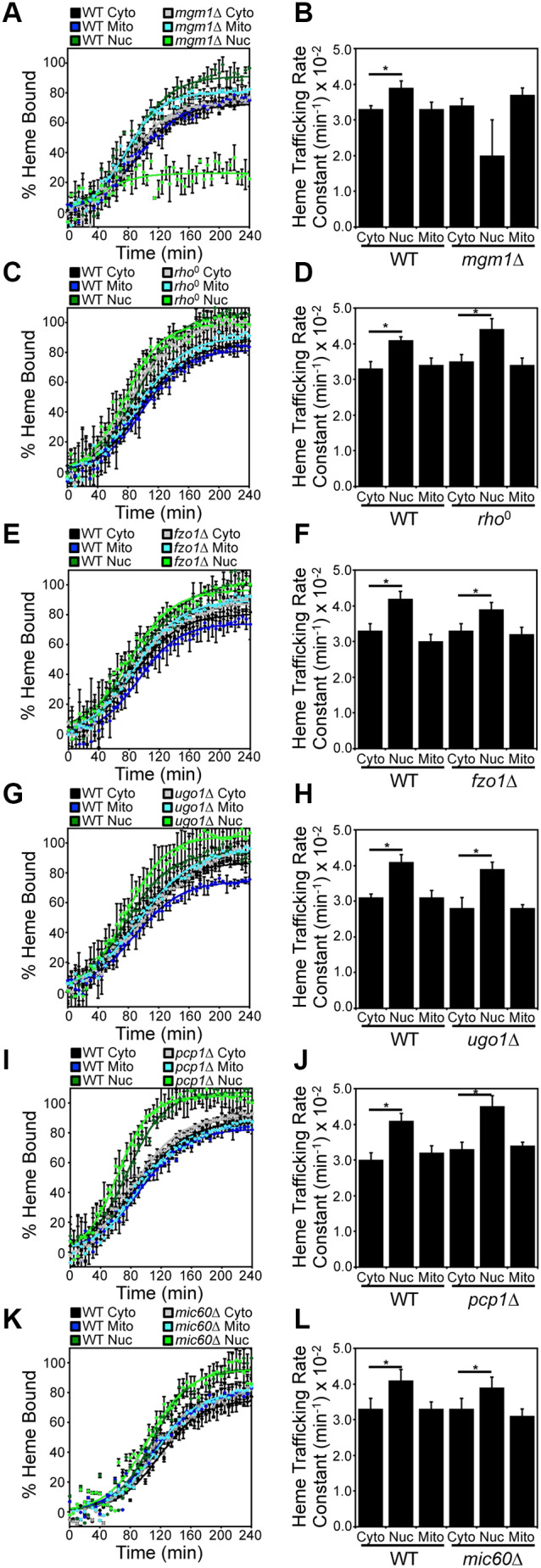
Mgm1 is a positive regulator of mitochondrial–nuclear heme trafficking. Heme trafficking rates were monitored using the SA pulse-chase assay and rate constants calculated, as described in Fig. 2, for (A,B) mgm1Δ, (C,D) rho0, (E,F) fzo1Δ, (G,H) ugo1Δ, (I,J) pcp1Δ cells and (K,L) mic60Δ cells. Data represent the mean±s.d. of independent triplicate cultures. *P<0.05 (one-way ANOVA with Dunnett's post-hoc test).
In order to determine whether the defect in mitochondrial–nuclear heme trafficking in mgm1Δ cells is related to the role of Mgm1 in maintaining the proper folding and structure of cristae in the IM (Meeusen et al., 2006), we determined whether heme trafficking was altered in a mutant that lacks the mitochondrial contact site and cristae organizing system (MICOS). As seen in Fig. 5K,L, mic60Δ cells, which are missing a core component of MICOS that is required for proper IM folding (Hessenberger et al., 2017), did not exhibit a defect in mitochondrial–nuclear heme trafficking. Taken together, our data strongly suggest that full length Mgm1, but not mitochondrial fusion or IM architecture per se, positively regulates mitochondrial–nuclear heme trafficking (Fig. 5).
Mitochondrial fission
In yeast, mitochondrial fission involves recruitment of the GTPase Dnm1 to its receptor Fis1 on the mitochondrial OM, which is dependent on the paralogous adapter proteins Mdv1 and Caf4 (Westermann, 2008). Once assembled and oligomerized around the OM, Dnm1 drives the GTP-dependent constriction and scission of mitochondrial tubules.
Relative to WT cells, the dnm1Δ strain exhibited statistically significant increases in heme trafficking rates to all three compartments tested, including the matrix, cytosol and nucleus, but with the most pronounced effect on nuclear heme trafficking: kNUC/kCYTO=1.5±0.1 in dnm1Δ cells versus kNUC/kCYTO=1.2±0.1 in WT cells (P<0.01, one-way ANOVA; n=3) (Fig. 6A,B). After prolonged growth for 16 h, heme availability to HS1 in all three compartments was similar in WT and dnm1Δ cells (Fig. S4A). Additionally, the rate of heme synthesis was unperturbed by loss of Dnm1, indicating that the increase in the rate of heme trafficking is not due to an increase in the rate of heme synthesis (Fig. S4C). However, loss of other essential components of the mitochondrial fission machinery, including Fis1 (Fig. 6C,D) and Caf4/Mdv1 (Fig. 6E,F), did not affect heme trafficking. Taken together, our data indicate that Dnm1, but not mitochondrial fission per se, negatively regulates mitochondrial–nuclear heme trafficking (Fig. 6).
Fig. 6.
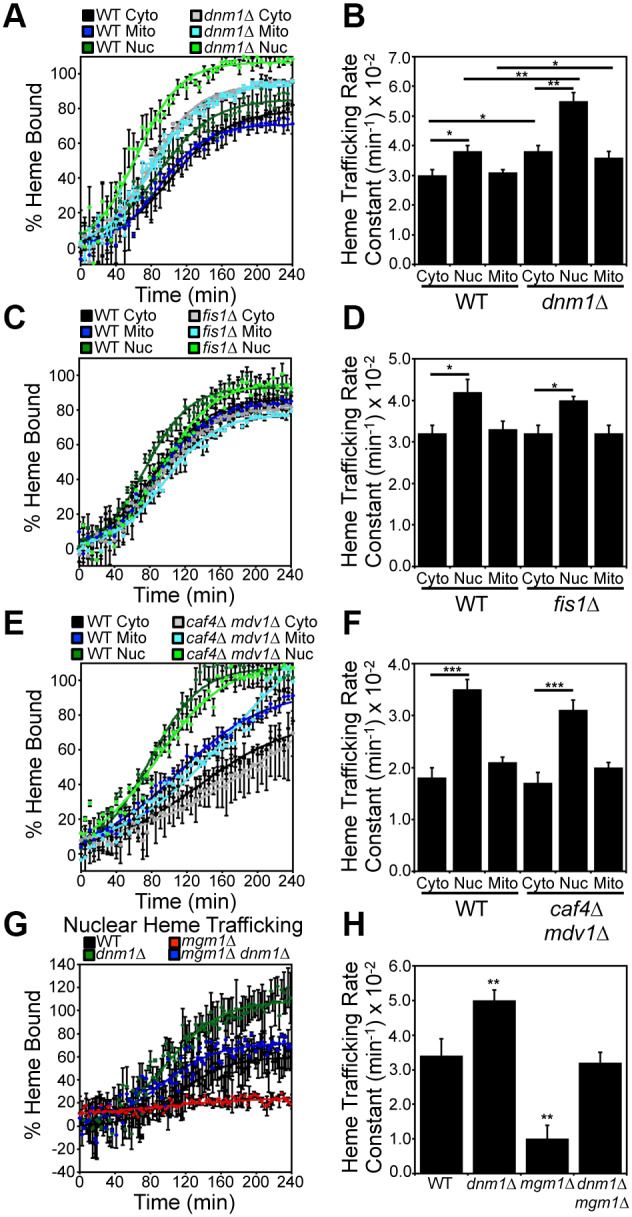
Dnm1 is a negative regulator of mitochondrial–nuclear heme trafficking. Heme trafficking rates were monitored using the SA pulse-chase assay and rate constants calculated, as described in Fig. 2, for (A,B) dnm1Δ, (C,D) fis1Δ, (E,F) caf4Δ mdv1Δ cells and (G,H) mgm1Δ dnm1Δ cells. Fluorimetry data represent the mean±s.d. of three independent cultures. *P<0.05, **P<0.01, ***P<0.001 (one-way ANOVA with Dunnett's post-hoc test).
We next sought to determine the consequence of ablating both Mgm1 and Dnm1 on mitochondrial–nuclear heme trafficking. Interestingly, mgm1Δ dnm1Δ double mutants exhibited WT rates of mitochondrial–nuclear heme trafficking (Fig. 6G,H). Therefore, Mgm1 and Dnm1 regulate heme trafficking in opposing directions with similar magnitude.
Mgm1 and Dnm1 regulate the activation of the nuclear heme-regulated transcription factor Hap1
Given that Mgm1 and Dnm1 are positive and negative regulators of mitochondrial–nuclear heme trafficking, we sought to determine their impact on the activation of the heme-regulated transcription factor Hap1. Heme binding to Hap1 alters the ability of the protein to promote or repress transcription of a number of target genes, including CYC1, which Hap1 positively regulates (Hanna et al., 2016; Pfeifer et al., 1989; Zhang et al., 1993, 1998; Zhang and Hach, 1999). In order to probe Hap1 activity, we used a transcriptional reporter that employs the promoter of a Hap1 target gene, pCYC1, to drive the expression of enhanced green fluorescent protein (eGFP) (Hanna et al., 2016). As demonstrated in Fig. 7A, not only was steady-state Hap1 activity greatly diminished in the heme-deficient hem1Δ mutant and in response to SA treatment, as expected, it was also reduced nearly 3-fold as a result of loss of Mgm1. Conversely, loss of Dnm1 did not affect basal Hap1 activity (Fig. 7A, –).
Fig. 7.
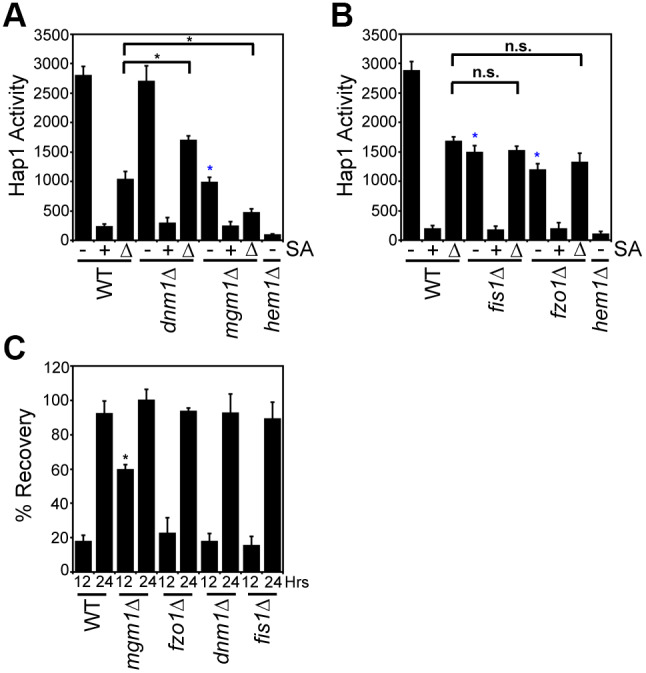
Mitochondrial–nuclear heme trafficking regulates Hap1 activity and sensitivity to new heme synthesis. (A,B) Hap1 activity in the indicated strains as measured by a transcriptional reporter that uses eGFP driven by the CYC1 promoter, a Hap1 target gene. Hap1 activity in cells that were untreated with SA (−), treated with 500 μM SA (+), or treated with 500 μM SA followed by shifting to medium lacking SA for 4 h (Δ) is shown. Fluorimetry data represent the mean±s.d. of three biological replicates. *P<0.001, asterisks not associated with an indicated pair-wise comparison (blue) were compared to WT untreated samples; ns, not significant (one-way ANOVA with Bonferroni's post-hoc test). (C) Cellular tolerance to new heme synthesis. Cells were pre-cultured overnight with or without 500 μM SA then allowed to recover on medium without SA. Sensitivity to new heme synthesis was scored as ‘% Recovery’, as described by Eqn 3, by comparing growth at the indicated times to growth of cells continually maintained in medium without SA and with SA. Growth data represent the mean±s.d. of three biological replicates. *P<0.001 (one-way ANOVA with Dunnett's post-hoc test relative to WT cells).
Because basal Hap1 activity in WT cells might already reflect heme saturation of Hap1, it is possible that the effects of the loss of Dnm1 are masked. To address this, we also measured Hap1 activity under non-heme saturating conditions in which Hap1 activity was probed just 4 h after alleviation of the SA-mediated block in heme synthesis. Under these conditions, dnm1Δ cells had increased Hap1 activity, and mgm1Δ cells exhibited diminished Hap1 activity relative to that in WT cells (Fig. 7A, Δ).
The Dnm1 and Mgm1 dependent regulation of Hap1 is not due to general defects in fusion or fission since fis1Δ and fzo1Δ cells exhibited WT levels of Hap1 activity when probed 4 h after alleviation of the SA-mediated block in heme synthesis (Fig. 7B, Δ). However, for reasons unclear at this time, steady-state Hap1 activity was diminished in both fis1Δ and fzo1Δ strains (Fig. 7B, –). Taken together, the respective positive and negative effects of Mgm1 and Dnm1 on nuclear heme trafficking correlate with their effects on heme activation of Hap1 and appear to be distinct from their roles in fusion and fission dynamics (Fig. 7A,B, Δ).
We next addressed whether Mgm1- and Dnm1-regulated nuclear heme trafficking affected heme-dependent growth of cells. To this end, we determined the degree to which heme-depleted cells pre-cultured with SA were able to recover in medium lacking SA (GΔSA) relative to cells continually maintained with (G+SA) or without SA (G−SA). As such, the ‘% Recovery’ can be calculated using Eqn 3:
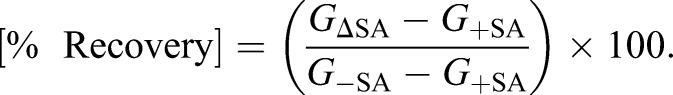 |
(3) |
Interestingly, as seen in Fig. 7C, after 12 h of growth, mgm1Δ cells were better able to tolerate the re-synthesis of heme compared to WT cells. The superior ability of mgm1Δ cells grown with SA to recover in medium lacking SA was not phenocopied by another fusion mutant, fzo1Δ, suggesting that Mgm1-mediated nuclear heme trafficking may be inhibitory to growth. Conversely, the fission mutants dnm1Δ and fis1Δ had WT levels of recovery from the alleviation of the SA-mediated block in heme synthesis, presumably because sufficient heme was accessing the nucleus to control growth in a WT manner. After 24 h, all cells achieved maximal growth recovery after re-initiation of heme synthesis (Fig. 7C). In total, our data suggest that Mgm1 is not only a positive regulator of mitochondrial–nuclear heme trafficking, but that this trafficking is negatively correlated with tolerance to the re-initiation of heme synthesis.
Heme regulates mitochondrial dynamics
Heme and targets of heme signaling, such as the heme activator proteins Hap1 and the HAP complex (Hap2/3/4/5), collectively transcriptionally regulate genes involved in mitochondrial metabolism, including respiration and fission-fusion dynamics (Buschlen et al., 2003; Hon et al., 2005; Knorre et al., 2016; Zhang et al., 2017). We therefore sought to determine whether heme itself could regulate mitochondrial fragmentation, a proxy for fission and fusion dynamics. To this end, by inhibiting heme synthesis using a 500 μM dose of SA for 4–6 h, we found that there was a nearly 2-fold increase in mitochondrial fragmentation in SA-treated cells relative to control cells as early as 4 h post-treatment (Fig. 8A,B). The number of cells with fragmented mitochondria further increased upon prolonged incubation with SA. The 500 μM dose of SA resulted in an ∼60–80% decrease in intracellular heme (Fig. 8C) and alterations in mitochondrial network preceded any appreciable changes in mitochondrial membrane potential (Fig. 8D). Hence, the SA-dependent increase in mitochondrial fragmentation is consistent with a role for heme in positively influencing mitochondrial fusion and/or negatively regulating mitochondrial fission.
Fig. 8.
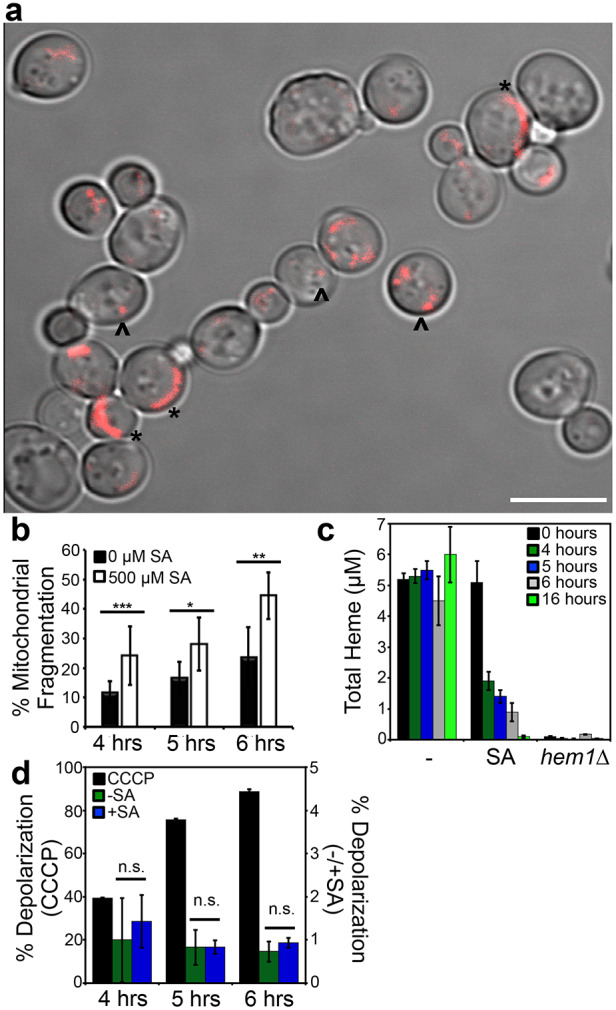
Heme regulates mitochondrial morphology. (A) Representative image of the mitochondrial network in cells conditioned with 500 μM SA, showing a mix of tubular (*) and punctate (^) morphology. Scale bar: 5 μm. (B) Quantitative analysis of the mitochondrial network in Su9–RFP-expressing WT cells incubated with or without 500 μM SA for varying lengths of time. Mitochondrial fragmentation was quantified as the ratio of the number of cells displaying punctate morphology versus the number of cells displaying tubular morphology. Data show mean±s.d. of three biological replicates, with 300–400 cells per biological replicate. *P<0.05, **P<0.01, ***P<0.001 (Student's t-test). (C) Heme levels in WT or hem1Δ cells treated or untreated with 500 μM SA for the indicated periods of time. Data represent the mean±s.d. of three biological replicates. (D) Changes in mitochondrial membrane potential in WT cells treated with or without 500 μM SA or 45 μM of the mitochondrial uncoupler CCCP for varying time periods. Cells were stained with 200 nM membrane-potential-sensitive dye DiOC6 and analyzed by flow cytometry using the FITC channel. Data show the mean±s.d. of three independent experiments. ns, not significant (t-test).
DISCUSSION
We developed a live-cell assay in yeast to monitor heme transport dynamics between the mitochondrial IM and the mitochondrial matrix, cytosol, and nucleus in order to identify new aspects of heme trafficking and distribution. Saccharomyces cerevisiae is an unparalleled model system to probe heme distribution dynamics because, unlike many eukaryotes including mammalian cells, yeast lacking heme can be made viable if supplemented with ergosterol and oleic acid. Thus, heme trafficking and transport dynamics can be monitored without a background associated with pre-existing heme pools. By integrating the heme trafficking dynamics assay with yeast molecular genetics approaches, we have, for the first time, probed the biodistribution of heme while it is being synthesized in live cells, and identified new factors that regulate heme trafficking, including the heme biosynthetic enzyme ALAS (also known as Hem1) (Fig. 3), and GTPases that regulate ERMES (Gem1; Fig. 4A) and mitochondrial fusion (Mgm1; Fig. 5A) and fission (Dnm1; Fig. 6). As discussed below, our results provide fundamental new insights into heme trafficking and signaling.
The heme trafficking kinetics data challenge the current conceptual paradigm for the cellular distribution of mitochondrial heme. The discovery of the first putative mitochondrial heme exporter, FLVCR1b, the only mitochondrial heme transporter identified to date (Chiabrando et al., 2012), has often been taken to imply that heme distribution is sequential, with heme first transported into the cytosol followed by its mobilization to other organelles (Hanna et al., 2017; Reddi and Hamza, 2016; Sweeny et al., 2018). However, we found that heme distribution to the mitochondrial matrix, cytosol, and nucleus occurs virtually simultaneously, suggesting the existence of parallel pathways for heme mobilization to different cellular locales (Figs 2 and 3). Thus, there must be additional factors and mechanisms that distribute heme, in addition to mitochondrial heme transporters that export heme into the cytosol. Our data suggest that GTPases that regulate mitochondrial dynamics and ER contact sites constitute novel components of a newly defined heme distribution network.
We propose that the ∼25% faster rate of heme trafficking to the nucleus relative to the cytosol or mitochondrial matrix (Fig. 2F) is suggestive of heme acting as a mitochondrial–nuclear retrograde signal that enables cells to adapt their metabolism and physiology for life with heme and properly functioning mitochondria. Indeed, the activity of the heme-regulated nuclear transcription factor, Hap1, correlated with perturbations to mitochondrial–nuclear heme trafficking caused by deletion of Mgm1 or Dnm1, which are positive and negative regulators of heme trafficking, respectively (Fig. 7A). Rather surprisingly, as part of this heme-based retrograde signal, diminished mitochondrial–nuclear heme trafficking in mgm1Δ cells correlated with enhanced cellular growth in response to the re-initiation of heme synthesis (Fig. 7C). This may be due to the detrimental effects of potentially cytotoxic levels of heme accumulation in the nucleus. Indeed, as a redox active molecule, heme may promote oxidative damage in the nucleus (Hanna et al., 2017; Reddi and Hamza, 2016). Alternatively, heme may act as an anti-proliferation signal through its ability to act as a transcriptional regulator by binding G-quadruplexes in DNA (Gray et al., 2019) or by controlling transcription via Hap1 and the HAP complex (Buschlen et al., 2003; Hon et al., 2005; Zhang et al., 1993, 1998; Zhang and Hach, 1999). Taken together, our results indicate that the relatively fast rate of nuclear heme acquisition may play an important role in mitochondrial–nuclear retrograde signaling.
Interestingly, we found that ALAS negatively regulated mitochondrial–nuclear heme trafficking (Fig. 3). This result may explain the functional role of ALAS and FECH interactions in a proposed IM heme biosynthetic supercomplex (Medlock et al., 2015; Piel et al., 2016). The rationale for protein-protein interactions in a biosynthetic pathway usually invokes the requirement for substrate channeling in order to mitigate the dissociation of potentially toxic substrates and ensure rapid flux. However, the biochemical necessity for an interaction between ALAS and FECH, the first and last enzymes in the heme synthesis pathway, is not clear in the context of substrate channeling. Our results suggest that interactions between ALAS and FECH may be regulatory because we find that ALAS decreases heme trafficking from FECH in the mitochondrial IM to the nucleus. Because a number of factors are known to regulate ALAS expression and mitochondrial import, including glucose, iron and heme (Keng and Guarente, 1987; Kubota et al., 2016), we propose that the regulation of mitochondrial–nuclear heme trafficking by ALAS might be part of a complex circuit that links nutrient status to heme-based retrograde signaling. However, the precise physiological context and mechanism of ALAS-mediated regulation of mitochondrial–nuclear heme trafficking needs to be further explored.
What is the mechanism of mitochondrial–nuclear heme trafficking? We identified three conserved GTPases in control of mitochondrial fusion (Mgm1) and fission (Dnm1), and ER–mitochondrial contact sites (Gem1) as regulators of mitochondrial–nuclear heme trafficking. One unifying model consistent with our data is that mitochondrial–ER contact sites facilitate the distribution of heme to the nucleus and other extra-mitochondrial locales. Indeed, both dnm1Δ and gem1Δ cells, which exhibit more stable mitochondrial–ER contact sites (Elbaz-Alon et al., 2014; Murley et al., 2013), also exhibit increased rates of nuclear heme trafficking (Figs 4A, 6A).
In accordance with this model, we suggest that Mgm1-mediated regulation of mitochondrial–nuclear heme trafficking occurs in a Dnm1 and ERMES-dependent manner. In such a model, Mgm1 promotes mitochondrial–nuclear heme trafficking because of its ability to drive mitochondrial fusion and provide a balancing force to oppose Dnm1-dependent mitochondrial fission. As a consequence, mgm1Δ cells would be expected to – and do – exhibit diminished mitochondrial–nuclear heme trafficking because there is no Mgm1 present to counteract Dnm1-mediated fission (Fig. 5A). Consistent with this model, we observed a restoration of WT levels of mitochondrial–nuclear heme trafficking in mgm1Δ dnm1Δ cells (Fig. 6D).
However, a number of issues must be resolved regarding the ERMES-dependent model for heme trafficking. First, we do not currently have a means to monitor heme at ER membranes to more directly assess the impact of ERMES and ER–mitochondrial contact sites on heme trafficking. Second, it is unclear at this time why other fission and fusion mutants did not exhibit defects in heme trafficking (Figs 5, 6), which suggests additional functions of Mgm1 and Dnm1 beyond mitochondrial dynamics. Third, we did not observe that mutants with defects in essential components of the ERMES complex, including mdm12Δ and mdm34Δ, exhibit altered mitochondrial–nuclear heme trafficking rates. One explanation is that there are multiple types of ER–mitochondrial contact sites in addition to ERMES that exist, including the conserved ER membrane protein complex (EMC) (Lahiri et al., 2014). It is also possible that mitochondrial-derived vesicles, which are responsible for the trafficking of cargo to vacuoles and lysosomes, offer alternative routes of mitochondrial heme efflux (Reddi and Hamza, 2016). Thus, a heme distribution phenotype may only be observed in multigenic knockouts defective in many parallel routes for heme trafficking.
An alternative to the ERMES-dependent model for heme trafficking is that each GTPase identified may play distinct and unrelated roles in heme trafficking. For instance, the human homolog of Mgm1, OPA1, was previously found to physically associate with FECH in the heme metabolon (Piel et al., 2016). As such, it is possible that Mgm1/OPA1 regulates the heme metabolon, and its interactions with putative heme trafficking factors, to effect mitochondrial–nuclear heme trafficking. Given the lipid-like properties of heme (Reddi and Hamza, 2016) and the fact that Dnm1 and Gem1 are lipid-interacting OM-associated GTPases (Macdonald et al., 2014), we speculate that heme may be sequestered by Dnm1 or Gem1 in the OM to suppress heme trafficking. Gem1 also has roles in mitochondrial positioning and cytoskeleton anchoring that may contribute to heme distribution dynamics (Koshiba et al., 2011). We are currently exploring the molecular mechanisms underlying Mgm1-, Dnm1- and Gem1-mediated heme trafficking and whether they cooperate through a common mechanism or act independently of each other. Since ablation of both Mgm1 and Dnm1 restored heme trafficking to WT levels, these factors may positively and negatively regulate a common downstream target, respectively, and are likely to play indirect roles in regulating heme trafficking. It is also possible that the mutants identified to have altered heme trafficking rates also have changes in expression of endogenous hemoproteins that have the capacity to compete with HS1 for heme.
Interestingly, our data suggest that heme may impact mitochondrial dynamics (Fig. 8). We speculate that there may be feedback mechanisms by which heme itself, possibly acting through Mgm1 or Dnm1, can regulate heme distribution dynamics between the mitochondrial network and the nucleus. Notably, mitochondrial fragmentation alone does not impact heme trafficking dynamics since certain fission and fusion mutants with hyper-fused or hyper-fragmented mitochondria (i.e. fis1Δ and fzo1Δ cells, respectively) exhibited WT rates of heme trafficking. We are currently probing the underlying mechanisms that govern heme regulation of mitochondrial fragmentation.
The in vivo assay to monitor real-time dynamics of inter-compartmental heme trafficking has revealed new insights into mobilization. We expect that the tools and approaches presented here could be used to probe a number of human diseases, including certain cancers, neurodegenerative disorders and blood diseases, that are associated with defects in heme homeostasis, mitochondrial dynamics and contact sites (Atamna and Frey, 2004; Atamna et al., 2002; Wang et al., 2008). Indeed, given that mitochondrial dynamics and contact sites with the endomembrane system are conserved between yeast and humans, and the factors that regulate them have homologs or functional analogs between lower and higher eukaryotes (Abrams et al., 2015; Cipolat et al., 2004; MacVicar and Langer, 2016; Schrepfer and Scorrano, 2016; Westermann, 2008), our studies in yeast may be of broad applicability to better understand how membrane and organelle dynamics impact heme transport and trafficking.
MATERIALS AND METHODS
Yeast strains, transformations and growth conditions
S. cerevisiae strains used in this study were derived from BY4741 (MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0). fis1Δ::KanMX4, dnm1Δ::KanMX4, mgm1Δ::KanMX4, pcp1Δ::KanMX4, ugo1Δ::KanMX4, caf4Δ::KanMX4, mdv1Δ::KanMX4 strains were obtained from the yeast gene deletion collection (Thermo Fisher Scientific). We also used the previously reported strains LJ109 (rho0) (Reddi and Culotta, 2013) and DH001b-3 (hem1Δ::HIS3) (Hanna et al., 2016). AR1029-3 (mdv1Δ::KanMX4 caf4Δ::HIS3) was generated by deleting CAF4 with pAR1047 in mdv1Δ::KanMX4 cells. OM232 (mgm1Δ::HIS3) and OM233 (dnm1Δ::KanMX4 mgm1Δ::HIS3) were generated by deleting MGM1 with pAR1051 in WT and dnm1Δ::KanMX4 cells, respectively. All strains were confirmed by PCR, mitochondrial morphology (Fig. S5), and, if derived from the yeast deletion collection, sequencing the unique barcodes flanking the KanMX4 deletion cassette.
Yeast transformations were performed by the lithium acetate procedure (Gietz and Schiestl, 1991). Strains were maintained at 30°C on either enriched yeast extract (1%) and peptone (2%) based medium supplemented with 2% glucose (YPD), or synthetic complete medium (SC; Sunrise Science Products) supplemented with 2% glucose and the appropriate amino acids to maintain selection (Hanna et al., 2016). Cells cultured on solid medium plates were done so with YPD or SC media supplemented with 2% agar (Hanna et al., 2016). Selection for yeast strains containing the KanMX4 marker was done on YPD agar plates supplemented with G418 (200 μg/ml) (Hanna et al., 2016). WT cells treated with the heme synthesis inhibitor, SA, and hem1Δ cells were cultured in YPD or SC media supplemented with 50 μg/ml of 5-aminolevulinic acid (ALA) or 15 mg/ml of ergosterol and 0.5% TWEEN 80 (YPDE or SCE, respectively) (Hanna et al., 2016; Ness et al., 1998). All liquid cultures were maintained at 30°C and shaken at 220 rpm.
Plasmids
The caf4::HIS3 disruption plasmid pAR1047 was generated by first PCR amplifying the upstream (−650 to −129) and downstream (+2110 to +2479) sequences relative to the CAF4 translational start site, introducing 5′ BamHI and 3′ XhoI, and 5′ XbaI and 3′ BamHI restriction sites, respectively. The CAF4 PCR products were digested with the enzymes indicated and ligated in a trimolecular reaction into the HIS3 integrating plasmid pRS403 (Sikorski and Hieter, 1989) digested with XhoI and XbaI, resulting in pAR1047. Transformation of yeast strains with pAR1047 linearized with BamHI resulted in deletion of CAF4 sequences from −128 to +2109.
The mgm1::HIS3 disruption plasmid, pAR1051, was generated by first PCR amplifying the upstream (−532 to −5) and downstream (+2736 to +3023) sequences relative to the MGM1 translational start site, introducing 5′ BamHI and 3′ XhoI, and 5′ XbaI and 3′ BamHI restriction sites, respectively. The MGM1 PCR products were digested with the enzymes indicated and ligated in a trimolecular reaction into the HIS3 integrating plasmid pRS403 (Sikorski and Hieter, 1989) digested with XhoI and XbaI, resulting in pAR1051. Transformation of yeast strains with pAR1051 linearized with BamHI resulted in deletion of MGM1 sequences from −4 to +2735.
Cytosolic, mitochondrial- and nuclear-targeted heme sensors, HS1, were sub-cloned into pRS415 [American Type Culture Collection (ATCC)] and driven by ADH, TEF, or GPD promoters as previously described (Hanna et al., 2016). The Hap1 reporter plasmid in which eGFP is driven by the CYC1 promoter was also as previously described (Hanna et al., 2016).
Heme trafficking dynamics assay
Overview
Inter-compartmental heme trafficking rates were monitored by inhibiting heme synthesis with SA in sensor-expressing cells, then removing the block in heme synthesis by resuspending cells into medium lacking SA and monitoring the time-dependent change in the percentage of heme bound to heme sensor 1 (HS1) localized to the cytosol, nucleus, and mitochondrial matrix upon the re-initiation of heme synthesis. The fractional heme saturation of the sensor was determined using previously established sensor calibration protocols (Hanna et al., 2016). The percentage of the sensor bound to heme, % Bound, was calculated by determining the sensor eGFP/mKATE2 fluorescence ratio (R) under a given test condition relative to the eGFP/mKATE2 fluorescence ratio when the sensor was 100% (Rmax) or 0% (Rmin) bound to heme as described in Eqn 1 (Hanna et al., 2016).
Rmin was determined by measuring the HS1 eGFP/mKATE2 ratio in parallel cultures that were conditioned with SA, which inhibits the second enzyme in the heme biosynthetic pathway, ALA dehydratase (ALAD) (Ebert et al., 1979), and Rmax was determined by permeabilizing cells and adding an excess of heme to saturate the sensor (Hanna et al., 2016). Given HS1 is quantitatively saturated with heme in the cytosol, nucleus, and mitochondria of WT yeast, we typically determined Rmax by measuring the HS1 eGFP/mKATE2 ratio in parallel WT cultures grown without SA (Hanna et al., 2016).
Growth for SA pulse-chase assay
HS1-expressing cells were cultured with or without 500 μM SA (Sigma-Aldrich) in SCE-LEU medium. Triplicate 5 ml cultures were seeded at an initial optical density of OD600=0.01–0.02 (2–4×105 cells/ml) and grown for 14–16 h at 30°C and shaking at 220 rpm until cells reached a final density of OD600nm∼1.0 (2×107 cells/ml). After culturing, 1 OD or 2×107 cells were harvested, washed twice with 1 ml of ultrapure water, and resuspended in 1 ml of fresh SC-LEU medium. The cells that were pre-cultured without SA provided HS1 Rmax values. The SA-conditioned cells were split into two 500 μl fractions. One fraction was treated with 500 μM SA to give HS1 Rmin values. The other fraction was not treated with SA so that heme synthesis could be re-initiated to give compartment-specific heme trafficking rates. HS1 fluorescence was monitored using 200 μl of a 1 OD/ml (2×107 cells/ml) cell suspension in black Greiner Bio-one flat bottom fluorescence plates with a Synergy Mx multi-modal plate reader. eGFP (excitation 488 nm, emission 510 nm) and mKATE2 (excitation 588 nm, emission 620 nm) fluorescence was recorded every 5 min for 4 h, with the plate being shaken at medium strength for 30 s prior to each read. Background fluorescence of cells not expressing the heme sensors was recorded and subtracted from the eGFP and mKATE2 fluorescence values.
Growth for ALA pulse-chase assay
HS1-expressing hem1Δ cells were cultured with or without 400 μM 5-aminolevulinic acid (ALA) (Sigma-Aldrich) in SCE-LEU medium. Triplicate 5 ml cultures were seeded at an initial optical density of OD600nm=0.01–0.02 (2–4×105 cells/ml) and grown for 14–16 h at 30°C and shaking at 220 rpm until cells reached a final density of OD600∼1.0 (2×107 cells/ml). After culturing, 1 OD or 2×107 cells were harvested, washed twice with 1 ml of ultrapure water, and resuspended in 1 ml of fresh SC-LEU medium. The cells that were pre-cultured with ALA provided HS1 Rmax values. The cells that were cultured without ALA were split into two 500 μl fractions. One fraction was used to give HS1 Rmin values. The other fraction was treated with 400 μM ALA so that heme synthesis could be initiated to give compartment-specific heme trafficking rates. HS1 fluorescence was monitored on 200 μl of a 1 OD/ml (2×107 cells/ml) cell suspension using black Greiner Bio-one flat bottom fluorescence plates and a Synergy Mx multi-modal plate reader. eGFP (excitation 488 nm, emission 510 nm) and mKATE2 (excitation 588 nm, emission 620 nm) fluorescence was recorded every 5 min for 4 h, with the plate being shaken at medium-strength for 30 s prior to each read. Background fluorescence of cells not expressing the heme sensors was recorded and subtracted from the eGFP and mKATE2 fluorescence values.
It should be noted that due to evaporation and drying of cell cultures in the plate reader, reliable measurements could not be achieved after 4 h of continuous monitoring.
Total heme quantification
Measurements of total heme were accomplished using a fluorimetric assay designed to measure the fluorescence of protoporphyrin IX upon the release of iron from heme as previously described (Michener et al., 2012). For all total heme measurements, ∼1×108 cells were harvested, washed in sterile ultrapure water, resuspended in 500 μl of 20 mM oxalic acid and stored in a closed box at 4°C overnight (16–18 h). Next, an equal volume (500 μl) of 2 M oxalic acid was added to the cell suspensions. The samples were split, with half the cell suspension transferred to a heat block set at 95°C and heated for 30 min and the other half of the cell suspension kept at room temperature (∼25°C) for 30 min. All suspensions were centrifuged for 2 min on a table-top microfuge at 21,000 g and the porphyrin fluorescence (excitation 400 nm, emission 620 nm) of 200 μl of each sample was recorded on a Synergy Mx multi-modal plate reader using black Greiner Bio-one flat bottom fluorescence plates. Heme concentrations were calculated from a standard curve prepared by diluting 500–1500 μM hemin chloride stock solutions in 0.1 M NaOH into ultrapure water, which was then added back to extra cell samples as prepared above. In order to calculate heme concentrations, the fluorescence of the unboiled sample (taken to be the background level of protoporphyrin IX) was subtracted from the fluorescence of the boiled sample (taken to be the free base porphyrin generated upon the release of heme iron). The cellular concentration of heme was determined by dividing the moles of heme determined in this fluorescence assay by the number of cells analyzed, giving moles of heme per cell, and then converting to a cellular concentration by dividing by the volume of a yeast cell, taken to be 50 fl (Hanna et al., 2016).
In order to measure heme synthesis rates, exponential phase cells were pre-conditioned with 500 μM SA for 14–16 h in SCE-LEU medium as described above for the heme trafficking dynamics assay. Following this, cells were washed and resuspended in medium lacking SA and aliquots of cells were harvested and washed for total heme quantification as described above.
Hap1 activity
Cells expressing p415-CYC1-eGFP, where eGFP expression is driven by the Hap1 regulated CYC1 promoter, were cultured in 50 ml of SCE-LEU in 250 ml Erlenmeyer flasks, with or without 500 μM SA, for 14–16 h to an optical density of OD600∼1.0 (2×107 cells/ml). Cells were washed with sterile ultrapure water and diluted into fresh SC-LEU medium at an optical density of OD600=0.25 and allowed to grow for an additional 4 h. The culture that was initially not conditioned with SA remained untreated during the 4 h growth phase and these cultures represented basal Hap1 activity. The culture that was pre-conditioned with SA was split into two fractions. One fraction was treated with 500 μM SA for the 4 h growth phase and these cultures served as a negative control, representing minimal Hap1 mediated activation of CYC1. The other fraction was not treated with SA for the 4 h growth phase and these cultures represented Hap1 activity under conditions in which Hap1 was not saturated with heme. After growth for 4 h, cells were washed in sterile ultrapure water and resuspended in PBS to a concentration of 1×108 cells/ml and 100 μl was used to measure eGFP fluorescence (excitation 488 nm, emission 510 nm). Background auto-fluorescence of cells not expressing eGFP was recorded and subtracted from the p415-CYC1-eGFP expressing strains.
Isolation of mitochondria and nuclei to confirm heme sensor localization
Mitochondria and nuclei were isolated using yeast mitochondrial (Cat # K259-50) and nuclear (Cat # K289-50) isolation kits from BioVision according to the manufacturer's instructions. All reagents and buffers were supplied by the isolation kits. For isolation of mitochondria, 50 ml of sensor expressing cells were cultured in SC-LEU in 250 ml Erlenmeyer flasks to a final density of OD600=1.0 (2×107 cells/ml). A total of 4×108 cells were harvested, washed in ultrapure water, and resuspended in ‘Buffer A’ containing 10 mM DTT for 30 min at 30°C with occasional gentle agitation. Cells were then harvested by centrifugation at 1500 g for 5 min at room temperature. Cells were then resuspended in 1 ml of ‘Buffer B’ containing manufacturer-provided zymolyase and incubated at 30°C with occasional gentle inversion. Spheroplast formation was monitored by diluting 10 μl of the cell suspension into ultrapure water and monitoring the decrease in OD600 such that it was >80% less than the initial value, which typically took 30–60 min. Spheroplasts were harvested by centrifugation at 1500 g for 5 min, resuspended in ‘Homogenization Buffer’ containing a protease inhibitor cocktail, and lysed with 15 strokes of a Dounce homogenizer on ice. The lysate was centrifuged at 600 g for 5 min at 4°C to remove cell debris. The supernatant, which contained the mitochondria, was again centrifuged for 5 min at 600 g to remove additional debris. Finally, mitochondria were harvested by centrifugation at 12,000 g for 10 min at 4°C. The mitochondrial pellet was resuspended in 50 μl of ‘Storage Buffer’. In order to validate sensor mitochondrial localization, 5% of the total spheroplast fraction, mitochondrial fraction, and the post-mitochondrial fraction (supernatant from the 12,000 g centrifugation step) were electrophoresed on a 14% Tris-glycine SDS–PAGE gel and immunoblotted using anti-PGK1 (Life Technologies; Cat # PA528612; 1:5000 dilution) to detect the cytosolic marker protein PGK1, anti-porin (Life Technologies; Cat # 459500; 1:5000 dilution) to detect the mitochondrial marker protein porin, and anti-GFP (Genetex; Cat # GTX30738; 1:5000 dilution) to probe HS1 expression. A goat anti-rabbit secondary antibody conjugated to a 680 nm emitting fluorophore (VWR/Biotium, Cat # 89138-520; 1:10,000 dilution) was used to probe for PGK1 and GFP. A goat anti-mouse secondary antibody conjugated to a 680 nm emitting fluorophore (VWR/Biotium, Cat # 89138-516; 1:10,000 dilution) was used to probe for Porin. All gels were imaged on a LiCOR Odyssey Infrared imager (Hanna et al., 2016; Reddi and Culotta, 2013).
For isolation of nuclei, 50 ml of sensor-expressing cells were cultured in SC-LEU in 250 ml Erlenmeyer flasks to a final density of OD600=1.0 (2×107 cells/ml). A total of 4×108 cells were harvested, washed in ultrapure water, and resuspended in ‘Buffer A’ containing 10 mM DTT for 30 min at 30°C with occasional gentle agitation. Cells were then harvested by centrifugation at 1500 g for 5 min at room temperature. Cells were then resuspended in 1 ml of ‘Buffer B’ containing manufacturer provided zymolyase and incubated at 30°C with occasional gentle inversion. Spheroplast formation was monitored by diluting 10 μl of the cell suspension into ultrapure water and monitoring the decrease in OD600 such that it was >80% less than the initial value, which typically took 30–60 min. Spheroplasts were harvested by centrifugation at 1500 g for 5 min, resuspended in 1 ml of ‘Buffer N’ containing a protease inhibitor cocktail, and lysed with 5 strokes of a Dounce homogenizer on ice. The suspension was incubated for 30 min at room temperature with gentle agitation every 3–5 min. The lysate was centrifuged at 1500 g for 5 min at 4°C to remove cell debris. Finally, nuclei were harvested by centrifugation at 20,000 g for 10 min at 4°C. The nuclear pellet was resuspended in 100 μl of ‘Buffer N’. In order to validate sensor nuclear localization, 5% of the total spheroplast fraction, nuclear fraction, and the post-nuclear fraction (supernatant from the 20,000 g centrifugation step) were electrophoresed on a 14% Tris-glycine SDS–PAGE gel and immunoblotted using anti-PGK1 (Life Technologies; Cat # PA528612; 1:5000 dilution) to detect the cytosolic marker protein PGK1, anti-NOP1 (Life Technologies; Cat # MA110025; 1:10,000 dilution) to detect the nuclear marker protein NOP1, and anti-GFP (Genetex; Cat # GTX30738; 1:5000 dilution) to probe HS1 expression. A goat anti-rabbit secondary antibody conjugated to a 680 nm emitting fluorophore (VWR/Biotium; Cat # 89138-520; 1:10,000 dilution) was used to probe for PGK1 and GFP. A goat anti-mouse secondary antibody conjugated to a 680 nm emitting fluorophore (VWR/Biotium; Cat # 89138-516; 1:10,000 dilution) was used to probe for NOP1. All gels were imaged on a LiCOR Odyssey Infrared imager (Hanna et al., 2016; Reddi and Culotta, 2013).
Confirmation of heme sensor localization by microscopy
To confirm mitochondrial or nuclear localization of the sensors, prior to microscopy, 1×107 exponential phase cells were incubated with 5.5 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen) in SC-LEU medium for 40–60 min to stain nuclear or mitochondrial DNA (Hanna et al., 2016). The concentration or duration of DAPI exposure can be used to selectively stain nuclei versus mitochondria (Shadel and Seidel-Rogol, 2007; Williamson and Fennell, 1979). In our hands, a 40 min exposure preferentially stained the mitochondrial network, while >60 min exposures stained the nucleus. Laser scanning confocal microscopy was accomplished on a Zeiss ELYRA LSM 780 super-resolution microscope equipped with a 63×, 1.4 numerical aperture oil objective. DAPI was excited at 405 nm and emission was collected using 410–483 nm bandpass filters. eGFP was excited with the 488 nm line of an argon ion laser, while mKATE2 was excited using the 594 nm of a HeNe laser line. The 491–588 nm and 599–690 nm bandpass filters were used to filter emission for eGFP and mKATE, respectively. Images were collected using Zeiss software and analyzed with ImageJ 1.48v (Rasband, W.S., ImageJ, NIH, Bethesda, MD, http://rsb.info.nih.gov/ij/, 1997-2007). Auto-fluorescence of unlabeled cells was subtracted from the DAPI, eGFP, and mKATE2 channels to produce the images in Fig. 1B.
Confirmation of mitochondrial morphology in fission and fusion
To visualize the mitochondrial network, 1×107 exponential phase cells were incubated with 500 μM MitoTracker Red CM-H2Xros (Thermo Fisher Scientific) in SC-LEU medium for 45–60 min. Confocal microscopy was done on a Zeiss ELYRA LSM 780 super-resolution microscope equipped with a 63×, 1.4 numerical aperture oil objective. Mito Tracker Red was excited using the 594 nm of a HeNe laser line and emission was collected using a 415–735 nm bandpass filter. Images were collected using Zeiss software and analyzed with ImageJ 1.48v. The various mitochondrial network morphology types observed are outlined in Fig. S5A and ∼50 cells from each strain were analyzed to confirm the expected mitochondrial morphology (Fig. S5B). A representative image of the mitochondrial network from each strain is depicted in Fig. S5C. Fission and fusion mutants had characteristic elongated or punctate mitochondrial networks, respectively.
Mitochondrial fragmentation assay
Mitochondrial morphology was assessed in wild-type yeast (W303, MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1) transformed with the pYX142-Su9-RFP plasmid (Frederick et al., 2008). The pYX142-Su9-RFP plasmid expresses a mitochondria-targeted red fluorescent protein (RFP), allowing for visualization of mitochondria using a fluorescence microscope. Cells were grown overnight in SC medium with 2% glucose as the sole carbon source. The cells were then subcultured into fresh pre-warmed medium at an OD600 of 0.2 for 3 h, and then treated with SA at a final concentration of 500 μM and cultured for 4, 5, or 6 h post-SA treatment. Imaging was performed on an Olympus IX81-FV5000 confocal laser-scanning microscope with a 543 nm laser line, using a 100× oil objective. Images were acquired and processed with Fluoview 500 software (Olympus America). Mitochondrial fragmentation was quantified based on the ratio of the cells displaying punctated or dotted-like morphology versus normal ribbon-like mitochondrial networks. At least 300 cells were analyzed for each sample per replicate.
Mitochondrial membrane potential assay
The mitochondrial membrane potential was assayed on cells cultured in a similar manner to that described above for the assessment of heme-dependent changes in mitochondrial fragmentation, using a BD FACSCanto™ II system (Becton Dickinson) as previously described (Pan and Shadel, 2009). After culturing for the indicated times, cells were first washed with phosphate-buffered saline (PBS), and then stained with 200 nM 3,3′-Dihexyloxacarbocyanine iodide (DiOC6) (Molecular Probes) for 30 min at 30°C. Finally, the cells were washed twice with PBS and analyzed by flow cytometry using the FL3 channel without compensation. The data were collected, analyzed and plotted using BD FACSDiva software v6.1.1 (Becton Dickinson).
Supplementary Material
Acknowledgements
We acknowledge the core facilities at the Petit Institute for Bioengineering and Biosciences at the Georgia Institute of Technology for use of the shared equipment and services.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: O.M.-G., A.E.M., O.K., A.R.R.; Methodology: O.M.-G., A.R.R.; Validation: A.R.R.; Formal analysis: O.M.-G., M.M.W., I.B., A.E.M., O.K., A.R.R.; Investigation: O.M.-G., M.M.W., A.S., J.V.D., I.B., A.R.R.; Resources: O.M.-G., M.M.W., A.S., J.V.D., I.B., A.R.R.; Writing - original draft: O.M.-G., A.R.R.; Writing - review & editing: O.M.-G., M.M.W., J.V.D., I.B., A.E.M., O.K., A.R.R.; Supervision: O.K., A.R.R.; Project administration: A.R.R.; Funding acquisition: A.E.M., O.K., A.R.R.
Funding
This work was supported by the National Institutes of Health (ES025661 to A.R.R., GM108975 to O.K., DK111653 to A.E.M.), the National Science Foundation (MCB-1552791 to A.R.R.), the Blanchard Professorship (to A.R.R.), Georgia Institute of Technology (to A.R.R.), the U.S. Department of Education GAANN Program (Grant P200A120081 to O.M.-G.), and the University of Nebraska-Lincoln Molecular Mechanisms of Disease graduate program (to J.V.D.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.237917.supplemental
References
- Abrams A. J., Hufnagel R. B., Rebelo A., Zanna C., Patel N., Gonzalez M. A., Campeanu I. J., Griffin L. B., Groenewald S., Strickland A. V. et al. (2015). Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat. Genet. 47, 926-932. 10.1038/ng.3354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arosio P., Knowles T. P. J. and Linse S. (2015). On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys. 17, 7606-7618. 10.1039/C4CP05563B [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atamna H. and Frey W. H. II (2004). A role for heme in Alzheimer's disease: heme binds amyloid beta and has altered metabolism. Proc. Natl. Acad. Sci. USA 101, 11153-11158. 10.1073/pnas.0404349101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atamna H., Killilea D. W., Killilea A. N. and Ames B. N. (2002). Heme deficiency may be a factor in the mitochondrial and neuronal decay of aging. Proc. Natl. Acad. Sci. USA 99, 14807-14812. 10.1073/pnas.192585799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldogh I. R., Nowakowski D. W., Yang H.-C., Chung H., Karmon S., Royes P. and Pon L. A. (2003). A protein complex containing Mdm10p, Mdm12p, and Mmm1p links mitochondrial membranes and DNA to the cytoskeleton-based segregation machinery. Mol. Biol. Cell 14, 4618-4627. 10.1091/mbc.e03-04-0225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess S. M., Delannoy M. and Jensen R. E. (1994). MMM1 encodes a mitochondrial outer membrane protein essential for establishing and maintaining the structure of yeast mitochondria. J. Cell Biol. 126, 1375-1391. 10.1083/jcb.126.6.1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschlen S., Amillet J.-M., Guiard B., Fournier A., Marcireau C. and Bolotin-Fukuhara M. (2003). The S. Cerevisiae HAP complex, a key regulator of mitochondrial function, coordinates nuclear and mitochondrial gene expression. Comp. Funct. Genomics 4, 37-46. 10.1002/cfg.254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D. C. (2012). Fusion and fission: interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 46, 265-287. 10.1146/annurev-genet-110410-132529 [DOI] [PubMed] [Google Scholar]
- Chiabrando D., Marro S., Mercurio S., Giorgi C., Petrillo S., Vinchi F., Fiorito V., Fagoonee S., Camporeale A., Turco E. et al. (2012). The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J. Clin. Invest. 122, 4569-4579. 10.1172/JCI62422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S., Martins de Brito O., Dal Zilio B. and Scorrano L. (2004). OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 101, 15927-15932. 10.1073/pnas.0407043101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVay R. M., Dominguez-Ramirez L., Lackner L. L., Hoppins S., Stahlberg H. and Nunnari J. (2009). Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J. Cell Biol. 186, 793-803. 10.1083/jcb.200906098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert P. S., Hess R. A., Frykholm B. C. and Tschudy D. P. (1979). Succinylacetone, a potent inhibitor of heme biosynthesis: effect on cell growth, heme content and δ-aminolevulinic acid dehydratase activity of malignant murine erythroleukemia cells. Biochem. Biophys. Res. Commun. 88, 1382-1390. 10.1016/0006-291X(79)91133-1 [DOI] [PubMed] [Google Scholar]
- Elbaz-Alon Y., Rosenfeld-Gur E., Shinder V., Futerman A. H., Geiger T. and Schuldiner M. (2014). A dynamic interface between vacuoles and mitochondria in yeast. Dev. Cell 30, 95-102. 10.1016/j.devcel.2014.06.007 [DOI] [PubMed] [Google Scholar]
- Frederick R. L., Okamoto K. and Shaw J. M. (2008). Multiple pathways influence mitochondrial inheritance in budding yeast. Genetics 178, 825-837. 10.1534/genetics.107.083055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J. R., Lackner L. L., West M., DiBenedetto J. R., Nunnari J. and Voeltz G. K. (2011). ER tubules mark sites of mitochondrial division. Science 334, 358-362. 10.1126/science.1207385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz R. D. and Schiestl R. H. (1991). Applications of high efficiency lithium acetate transformation of intact yeast cells using single-stranded nucleic acids as carrier. Yeast 7, 253-263. 10.1002/yea.320070307 [DOI] [PubMed] [Google Scholar]
- Gray L. T., Puig Lombardi E., Verga D., Nicolas A., Teulade-Fichou M.-P., Londoño-Vallejo A. and Maizels N. (2019). G-quadruplexes sequester free heme in living cells. Cell Chem Biol 26, 1681-1691.e5. 10.1016/j.chembiol.2019.10.003 [DOI] [PubMed] [Google Scholar]
- Hanna D. A., Harvey R. M., Martinez-Guzman O., Yuan X., Chandrasekharan B., Raju G., Outten F. W., Hamza I. and Reddi A. R. (2016). Heme dynamics and trafficking factors revealed by genetically encoded fluorescent heme sensors. Proc. Natl. Acad. Sci. USA 113, 7539-7544. 10.1073/pnas.1523802113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna D. A., Martinez-Guzman O. and Reddi A. R. (2017). Heme gazing: illuminating eukaryotic heme trafficking, dynamics, and signaling with fluorescent heme sensors. Biochemistry 56, 1815-1823. 10.1021/acs.biochem.7b00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna D. A., Hu R., Kim H., Martinez-Guzman O., Torres M. P. and Reddi A. R. (2018). Heme bioavailability and signaling in response to stress in yeast cells. J. Biol. Chem. 293, 12378-12393. 10.1074/jbc.RA118.002125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessenberger M., Zerbes R. M., Rampelt H., Kunz S., Xavier A. H., Purfürst B., Lilie H., Pfanner N., van der Laan M. and Daumke O. (2017). Regulated membrane remodeling by Mic60 controls formation of mitochondrial crista junctions. Nat. Commun. 8, 15258 10.1038/ncomms15258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon T., Lee H. C., Hu Z., Iyer V. R. and Zhang L. (2005). The heme activator protein Hap1 represses transcription by a heme-independent mechanism in Saccharomyces cerevisiae. Genetics 169, 1343-1352. 10.1534/genetics.104.037143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hönscher C., Mari M., Auffarth K., Bohnert M., Griffith J., Geerts W., van der Laan M., Cabrera M., Reggiori F. and Ungermann C. (2014). Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev. Cell 30, 86-94. 10.1016/j.devcel.2014.06.006 [DOI] [PubMed] [Google Scholar]
- Kawano S., Tamura Y., Kojima R., Bala S., Asai E., Michel A. H., Kornmann B., Riezman I., Riezman H., Sakae Y. et al. (2018). Structure-function insights into direct lipid transfer between membranes by Mmm1-Mdm12 of ERMES. J. Cell Biol. 217, 959-974. 10.1083/jcb.201704119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keng T. and Guarente L. (1987). Constitutive expression of the yeast HEM1 gene is actually a composite of activation and repression. Proc. Natl. Acad. Sci. USA 84, 9113-9117. 10.1073/pnas.84.24.9113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knorre D. A., Sokolov S. S., Zyrina A. N. and Severin F. F. (2016). How do yeast sense mitochondrial dysfunction? Microb. Cell 3, 532-539. 10.15698/mic2016.11.537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B., Currie E., Collins S. R., Schuldiner M., Nunnari J., Weissman J. S. and Walter P. (2009). An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325, 477-481. 10.1126/science.1175088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B., Osman C. and Walter P. (2011). The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proc. Natl. Acad. Sci. USA 108, 14151-14156. 10.1073/pnas.1111314108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba T., Holman H. A., Kubara K., Yasukawa K., Kawabata S., Okamoto K., MacFarlane J. and Shaw J. M. (2011). Structure-function analysis of the yeast mitochondrial Rho GTPase, Gem1p: implications for mitochondrial inheritance. J. Biol. Chem. 286, 354-362. 10.1074/jbc.M110.180034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y., Nomura K., Katoh Y., Yamashita R., Kaneko K. and Furuyama K. (2016). Novel mechanisms for heme-dependent degradation of ALAS1 protein as a component of negative feedback regulation of Heme biosynthesis. J. Biol. Chem. 291, 20516-20529. 10.1074/jbc.M116.719161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S. and Bandyopadhyay U. (2005). Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 157, 175-188. 10.1016/j.toxlet.2005.03.004 [DOI] [PubMed] [Google Scholar]
- Lackner L. L. (2019). The expanding and unexpected functions of mitochondria contact sites. Trends Cell Biol. 29, 580-590. 10.1016/j.tcb.2019.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S., Chao J. T., Tavassoli S., Wong A. K. O., Choudhary V., Young B. P., Loewen C. J. R. and Prinz W. A. (2014). A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria. PLoS Biol. 12, e1001969 10.1371/journal.pbio.1001969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald P. J., Stepanyants N., Mehrotra N., Mears J. A., Qi X., Sesaki H. and Ramachandran R. (2014). A dimeric equilibrium intermediate nucleates Drp1 reassembly on mitochondrial membranes for fission. Mol. Biol. Cell 25, 1905-1915. 10.1091/mbc.e14-02-0728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacVicar T. and Langer T. (2016). OPA1 processing in cell death and disease - the long and short of it. J. Cell Sci. 129, 2297-2306. 10.1242/jcs.159186 [DOI] [PubMed] [Google Scholar]
- Medlock A. E., Shiferaw M. T., Marcero J. R., Vashisht A. A., Wohlschlegel J. A., Phillips J. D. and Dailey H. A. (2015). Identification of the mitochondrial Heme metabolism complex. PLoS ONE 10, e0135896 10.1371/journal.pone.0135896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeusen S., DeVay R., Block J., Cassidy-Stone A., Wayson S., McCaffery J. M. and Nunnari J. (2006). Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell 127, 383-395. 10.1016/j.cell.2006.09.021 [DOI] [PubMed] [Google Scholar]
- Michener J. K., Nielsen J. and Smolke C. D. (2012). Identification and treatment of heme depletion attributed to overexpression of a lineage of evolved P450 monooxygenases. Proc. Natl. Acad. Sci. USA 109, 19504-19509. 10.1073/pnas.1212287109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murley A. and Nunnari J. (2016). The emerging network of mitochondria-organelle contacts. Mol. Cell 61, 648-653. 10.1016/j.molcel.2016.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murley A., Lackner L. L., Osman C., West M., Voeltz G. K., Walter P. and Nunnari J. (2013). ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. eLife 2, e00422 10.7554/eLife.00422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness F., Achstetter T., Duport C., Karst F., Spagnoli R. and Degryse E. (1998). Sterol uptake in Saccharomyces cerevisiae heme auxotrophic mutants is affected by ergosterol and oleate but not by palmitoleate or by sterol esterification. J. Bacteriol. 180, 1913-1919. 10.1128/JB.180.7.1913-1919.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. T., Lewandowska A., Choi J.-Y., Markgraf D. F., Junker M., Bilgin M., Ejsing C. S., Voelker D. R., Rapoport T. A. and Shaw J. M. (2012). Gem1 and ERMES do not directly affect phosphatidylserine transport from ER to mitochondria or mitochondrial inheritance. Traffic 13, 880-890. 10.1111/j.1600-0854.2012.01352.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y. and Shadel G. S. (2009). Extension of chronological life span by reduced TOR signaling requires down-regulation of Sch9p and involves increased mitochondrial OXPHOS complex density. Aging (Albany NY) 1, 131-145. 10.18632/aging.100016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer K., Kim K.-S., Kogan S. and Guarente L. (1989). Functional dissection and sequence of yeast HAP1 activator. Cell 56, 291-301. 10.1016/0092-8674(89)90903-3 [DOI] [PubMed] [Google Scholar]
- Piel R. B. III, Shiferaw M. T., Vashisht A. A., Marcero J. R., Praissman J. L., Phillips J. D., Wohlschlegel J. A. and Medlock A. E. (2016). A novel role for Progesterone Receptor Membrane Component 1 (PGRMC1): a partner and regulator of ferrochelatase. Biochemistry 55, 5204-5217. 10.1021/acs.biochem.6b00756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel R. B. III, Dailey H. A. Jr. and Medlock A. E. (2019). The mitochondrial heme metabolon: insights into the complex(ity) of heme synthesis and distribution. Mol. Genet. Metab. 128, 198-203. 10.1016/j.ymgme.2019.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puy H., Gouya L. and Deybach J.-C. (2010). Porphyrias. Lancet 375, 924-937. 10.1016/S0140-6736(09)61925-5 [DOI] [PubMed] [Google Scholar]
- Reddi A. R. and Culotta V. C. (2013). SOD1 integrates signals from oxygen and glucose to repress respiration. Cell 152, 224-235. 10.1016/j.cell.2012.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddi A. R. and Hamza I. (2016). Heme mobilization in animals: a Metallolipid's journey. Acc. Chem. Res. 49, 1104-1110. 10.1021/acs.accounts.5b00553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachar M., Anderson K. E. and Ma X. (2016). Protoporphyrin IX: the Good, the Bad, and the Ugly. J. Pharmacol. Exp. Ther. 356, 267-275. 10.1124/jpet.115.228130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassa S. (2004). Why heme needs to be degraded to iron, biliverdin IXalpha, and carbon monoxide? Antioxid Redox Signal. 6, 819-824. 10.1089/1523086041798006 [DOI] [PubMed] [Google Scholar]
- Schipper H. M., Song W., Zukor H., Hascalovici J. R. and Zeligman D. (2009). Heme oxygenase-1 and neurodegeneration: expanding frontiers of engagement. J. Neurochem. 110, 469-485. 10.1111/j.1471-4159.2009.06160.x [DOI] [PubMed] [Google Scholar]
- Schrepfer E. and Scorrano L. (2016). Mitofusins, from Mitochondria to Metabolism. Mol. Cell 61, 683-694. 10.1016/j.molcel.2016.02.022 [DOI] [PubMed] [Google Scholar]
- Schuler M.-H., Di Bartolomeo F., Mårtensson C. U., Daum G. and Becker T. (2016). Phosphatidylcholine affects inner membrane protein translocases of mitochondria. J. Biol. Chem. 291, 18718-18729. 10.1074/jbc.M116.722694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadel G. S. and Seidel-Rogol B. L. (2007). Diagnostic assays for defects in mtDNA replication and transcription in yeast and humans. Methods Cell Biol. 80, 465-479. 10.1016/S0091-679X(06)80023-3 [DOI] [PubMed] [Google Scholar]
- Shen J., Sheng X., Chang Z. N., Wu Q., Wang S., Xuan Z., Li D., Wu Y., Shang Y., Kong X. et al. (2014). Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 7, 180-193. 10.1016/j.celrep.2014.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R. S. and Hieter P. (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces ceresiviae. Genetics 122, 19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogo L. F. and Yaffe M. P. (1994). Regulation of mitochondrial morphology and inheritance by Mdm10p, a protein of the mitochondrial outer membrane. J. Cell Biol. 126, 1361-1373. 10.1083/jcb.126.6.1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeny E. A., Singh A. B., Chakravarti R., Martinez-Guzman O., Saini A., Haque M. M., Garee G., Dans P. D., Hannibal L., Reddi A. R. et al. (2018). Glyceraldehyde-3-phosphate dehydrogenase is a chaperone that allocates labile heme in cells. J. Biol. Chem. 293, 14557-14568. 10.1074/jbc.RA118.004169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuta T., Scharwey M. and Langer T. (2014). Mitochondrial lipid trafficking. Trends Cell Biol. 24, 44-52. 10.1016/j.tcb.2013.07.011 [DOI] [PubMed] [Google Scholar]
- Wang X., Su B., Siedlak S. L., Moreira P. I., Fujioka H., Wang Y., Casadesus G. and Zhu X. (2008). Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 105, 19318-19323. 10.1073/pnas.0804871105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. (2008). Molecular machinery of mitochondrial fusion and fission. J. Biol. Chem. 283, 13501-13505. 10.1074/jbc.R800011200 [DOI] [PubMed] [Google Scholar]
- Williamson D. H. and Fennell D. J. (1979). Visualization of yeast mitochondrial DNA with the fluorescent stain “DAPI”. Methods Enzymol. 56, 728-733. 10.1016/0076-6879(79)56065-0 [DOI] [PubMed] [Google Scholar]
- Wu M. L., Ho Y. C., Lin C. Y. and Yet S. F. (2011). Heme oxygenase-1 in inflammation and cardiovascular disease. Am. J. Cardiovasc. Dis. 1, 150-158. [PMC free article] [PubMed] [Google Scholar]
- Xue Y., Schmollinger S., Attar N., Campos O. A., Vogelauer M., Carey M. F., Merchant S. S. and Kurdistani S. K. (2017). Endoplasmic reticulum-mitochondria junction is required for iron homeostasis. J. Biol. Chem. 292, 13197-13204. 10.1074/jbc.M117.784249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Keel S. B., Shimamura A., Liu L., Gerds A. T., Li H. Y., Wood B. L., Scott B. L. and Abkowitz J. L. (2016). Delayed globin synthesis leads to excess heme and the macrocytic anemia of Diamond Blackfan anemia and del(5q) myelodysplastic syndrome. Sci. Transl. Med. 8, 338ra67 10.1126/scitranslmed.aaf3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman M. J., Hobbs A. E. A., Burgess S. M., Srinivasan M. and Jensen R. E. (2004). Mmm2p, a mitochondrial outer membrane protein required for yeast mitochondrial shape and maintenance of mtDNA nucleoids. J. Cell Biol. 164, 677-688. 10.1083/jcb.200308012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. and Hach A. (1999). Molecular mechanism of heme signaling in yeast: the transcriptional activator Hap1 serves as the key mediator. Cell. Mol. Life Sci. 56, 415-426. 10.1007/s000180050442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Bermingham-McDonogh O., Turcotte B. and Guarente L. (1993). Antibody-promoted dimerization bypasses the regulation of DNA binding by the heme domain of the yeast transcriptional activator HAP1. Proc. Natl. Acad. Sci. USA 90, 2851-2855. 10.1073/pnas.90.7.2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Hach A. and Wang C. (1998). Molecular mechanism governing heme signaling in yeast: a higher-order complex mediates heme regulation of the transcriptional activator HAP1. Mol. Cell. Biol. 18, 3819-3828. 10.1128/MCB.18.7.3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Bu P., Zeng J. and Vancura A. (2017). Increased heme synthesis in yeast induces a metabolic switch from fermentation to respiration even under conditions of glucose repression. J. Biol. Chem. 292, 16942-16954. 10.1074/jbc.M117.790923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zick M., Duvezin-Caubet S., Schäfer A., Vogel F., Neupert W. and Reichert A. S. (2009). Distinct roles of the two isoforms of the dynamin-like GTPase Mgm1 in mitochondrial fusion. FEBS Lett. 583, 2237-2243. 10.1016/j.febslet.2009.05.053 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.