

Abstract
Discovery and development of 210 new molecular entities [NMEs, new drugs] approved by the US Food and Drug Administration 2010–2016 was facilitated by 3D structural information generated by structural biologists worldwide and distributed on an open access basis by the Protein Data Bank [PDB]. The molecular targets for 94% of these NMEs are known. The PDB archive contains 5,914 structures containing one of the known targets and/or a new drug, providing structural coverage for 88% of the recently approved NMEs across all therapeutic areas. More than half of the 5,914 structures were published and made available by the PDB at no charge with no restrictions on usage >10 years before drug approval. Citation analyses revealed that these 5,914 PDB structures significantly impacted the very large body of publicly-funded research reported in publications on the NME targets that motivated biopharmaceutical company investment in discovery and development programs that produced the NMEs.
eTOC Blurb
Discovery/development of 210 new molecular entities (NMEs, new drugs) approved by the US Food and Drug Administration 2010–2016 was facilitated by open access to 3D structures stored in the Protein Data Bank (PDB). Nearly 6,000 relevant PDB structures contributed to approval of 88% of these NMEs across all therapeutic areas.
Introduction
The Protein Data Bank [PDB; (Berman et al., 2003)] is an enormously valuable gold standard, reference data resource for education/training and research (both basic and applied) across the biological and biomedical sciences. It was established in 1971 as the first open-access, digital data resource in biology with just seven protein structures (Protein Data Bank, 1971). Forty-seven years later, the PDB continues to serve as the single global repository for 3D structural data, making >146,000 experimentally determined structures [of proteins, DNA, RNA, and their complexes with drugs and/or other small molecules] freely available without restrictions on usage. Since 2003, the PDB has been managed jointly by the Worldwide Protein Data Bank [wwPDB] partnership (Berman et al., 2003) [including US Research Collaboratory for Structural Bioinformatics Protein Data Bank or RCSB PDB (Berman et al., 2000), Protein Data Bank in Europe (Velankar et al., 2010), Protein Data Bank Japan (Kinjo et al., 2017), and BioMagResBank (Ulrich et al., 2008)]. The wwPDB OneDep global system for deposition-validation-biocuration of incoming structures supports all PDB data depositors, helping to ensure that every structure archived in the PDB is well validated and expertly biocurated (Young et al., 2017; Gore et al., 2017; Young et al., 2018). wwPDB partners are committed to ensuring adherence to the FAIR Principles of indability-Accessibility-Interoperability-Reusability(Wilkinson et al., 2016). Publication of new macromolecular structures in most scientific journals is contingent on mandatory deposition to the PDB of the 3D atomic coordinates comprising the structure, together with experimental data and metadata. Many governmental/non-governmental research funders also require PDB deposition of macromolecular structural data.
Over the past two decades, structural biology and structure-guided drug discovery have become well established within the biopharmaceutical industry (Blundell, 2017; Klebe, 2013). 3D structures frequently provide information about how individual small-molecule weight ligands bind to their target proteins [e.g., imatinib in chronic myeloid leukemia (Capdeville et al., 2002; Nagar et al., 2002)]. Structural data have also proven useful in overcoming some of the myriad challenges inherent in turning biochemically-active compounds into potent drug-like molecules suitable for safety and efficacy testing in animals and humans (Stoll et al., 2011). In the realm of biologics [~20% of approved drugs over the past decade (Mullard, 2016)], 3D structural information is now routinely being used to drive engineering of monoclonal antibodies (Gilliland et al., 2012).
Previously published case studies [e.g., (Hu et al., 2018)] and largely anecdotal reports presented at scientific meetings leave no doubt as to the importance of individual contributions to drug discovery made by macromolecular crystallographers working in industry, but they do not address the impact on the biopharmaceutical industry by publicsector structural biologists and the data they contribute to the PDB archive. We, therefore, undertook a quantitative assessment of the impact of structural biologists and the PDB on the discovery and development of 210 New Medical Entities [NMEs or drugs] approved by the US Food and Drug Administration [FDA] between 2010 and 2016.
Results and Discussion
Overview:
We analyzed PDB archival holdings and identified 3D structures that include the 210 NMEs and/or their 150 known molecular targets [Table 1]. The 210 NMEs break down into three classes: small chemicals of molecular weight <1000 [Low Molecular Weight or LMW-NMEs: 81.4%, 171/210], proteins or peptide hormones with molecular weight ≥1000 [Biologic-NMEs: 17.1%, 36/210], and nucleic acid drugs [Antisense Oligonucleotide or ASO-NMEs: 1.5%, 3/210]. Known molecular targets of the 210 NMEs include proteins [147/150], a lipid [1/150], a small-molecule drug [1/150], and a glycolipid substrate of an enzyme [1/150]. 13 of the 210 NMEs have no known molecular target. Methods used in generating the findings reported in this Perspective are described in the Supplementary Materials. Primary data assembled from the PDB archive, US FDA documents, and the scientific literature are provided in Table S1.
Table 1.
Breakdown of US FDA approved NMEs between 2010–2016 by drug type and therapeutic area.
| Therapeutic Area | NMEs US FDA Approved Between 2010–2016 | Low Molecular Weight NMEs | Biologic NMEs | Antisense Oligonucleotide NMEs | NMEs with Known Protein Targets | NMEs with Know NonProtein Target | NMEs with No Known Target |
|---|---|---|---|---|---|---|---|
| All | 210 | 171 | 36 | 3 | 194 | 3 | 13 |
| Anti-infective | 31 | 29 | 2 | 0 | 29 | 0 | 2 |
| Anti-neoplastic | 59 | 45 | 14 | 0 | 54 | 1 | 4 |
| Cardiovascular | 21 | 17 | 3 | 1 | 18 | 1 | 2 |
| Central Nervous System | 21 | 18 | 1 | 2 | 20 | 0 | 1 |
| Endocrine | 7 | 6 | 1 | 0 | 7 | 0 | 0 |
| Gastrointestinal | 6 | 5 | 1 | 0 | 5 | 0 | 1 |
| Immunologic | 21 | 13 | 8 | 0 | 21 | 0 | 0 |
| Metabolic | 27 | 22 | 5 | 0 | 24 | 1 | 2 |
| Respiratory | 13 | 12 | 1 | 0 | 12 | 0 | 1 |
| Miscellaneous | 4 | 4 | 0 | 0 | 4 | 0 | 0 |
In aggregate, we identified 5,914 unique PDB structures that contain a known protein target of an NME or one of the NMEs [hereafter referred to as “Relevant Structures”]. Our definition encompasses PDB structures containing the following: (i) a reference or a mutant/variant form of a human protein NME target; (ii) a reference or a mutant/variant form of a bacterial or a viral protein NME target; (iii) a LMW-NME bound to a reference or mutant/variant form of its target protein; (iv) a LMW-NME bound to a possible off-target protein; (v) a LMW-NME bound to a potential alternative target protein; (vi) a Biologic-NME; (vii) a Biologic-NME bound to a reference or mutant/variant form of its target protein; or (viii) a Biologic-NME bound to its small-molecule target.
These 5,914 Relevant Structures cover 86% [126/147] of the known protein targets of the NMEs and 88% [184/210] of NMEs [Table 2]. For most of the known protein targets of a given NME, the PDB archive contains more than one Relevant Structure [80%; 117/147]. In some cases, there are 10s to more than 100 Relevant Structures identified for a particular NME, reflecting frequent reliance on structural data by drug discovery teams operating in biopharmaceutical companies [Figure 1]. Finally, most drug targets do not function as macromolecules in isolation. They frequently engage in protein-protein interactions that underpin normal biological function and/or pathogenesis. It is, therefore, significant that a considerable fraction of the Relevant Structures [37%; 2,193/5,914] include both the NME target protein and one or more partner proteins, thereby providing broader structural insights into drug target biology.
Table 2.
PDB coverage of US FDA Approved NMEs between 2010–2016 with therapeutic area breakdown.
| Therapeutic Area | NMEs US FDA Approved Between 2010–2016 | NMEs with Relevant Structures in PDB1 | % PDB Coverage of NMEs with Known Targets | Low Molecular Weight NMEs in PDB Structures | Biologic NMEs in PDB Structures |
|---|---|---|---|---|---|
| All | 210 | 184 | 93% | 50 | 16 |
| Anti-infective | 31 | 27 | 93% | 7 | 1 |
| Anti-neoplastic | 59 | 54 | 98% | 26 | 6 |
| Cardiovascular | 21 | 16 | 89% | 5 | 1 |
| Central Nervous System | 21 | 18 | 90% | 1 | 1 |
| Endocrine | 7 | 7 | 100% | 1 | 1 |
| Gastrointestinal | 6 | 5 | 100% | 0 | 1 |
| Immunologic | 21 | 19 | 90% | 4 | 2 |
| Metabolic | 27 | 23 | 96% | 5 | 3 |
| Respiratory | 13 | 11 | 92% | 0 | 0 |
| Miscellaneous | 4 | 3 | 75% | 1 | 0 |
Totals include all NME protein targets with ≥95% sequence identity to the UniProt (uniprot.org) reference sequence for the target protein, with the exception of 3 NME protein targets where ≥50% sequence identity was used.
Figure 1.
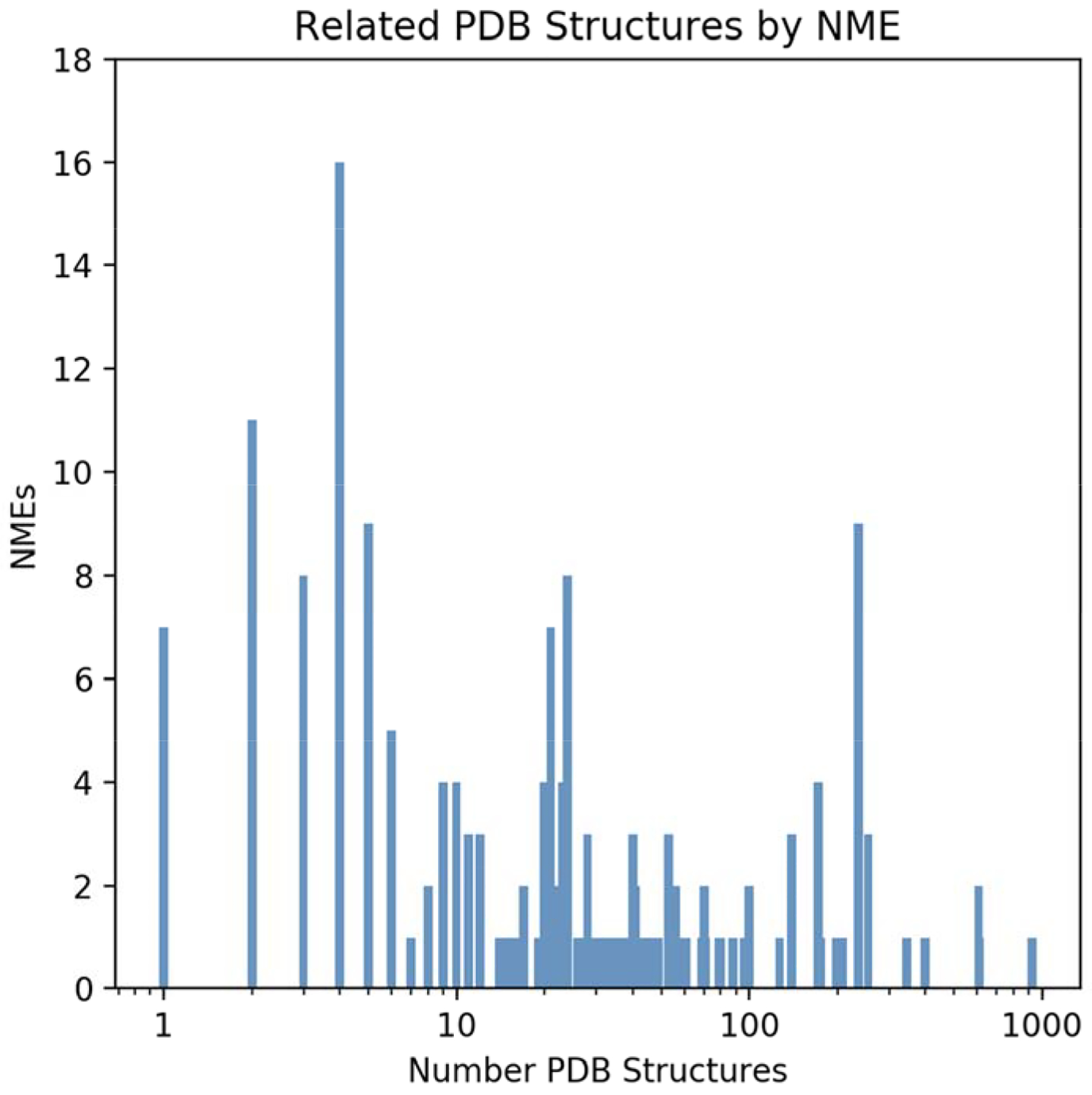
Distribution of the number of Relevant Structures/NME.
Experimental Methods used to Determine the Relevant Structures:
More than 95% of Relevant Structures [median resolution limit 2.08Å] were determined using macromolecular crystallography [MX], the dominant experimental method in structural biology today. The remainder were determined by either nuclear magnetic resonance spectroscopy [NMR; ~4%] or 3D electron microscopy [3DEM; ~1%]. For the ~145,000 structures currently housed in the PDB archive, the relative breakdown by experimental method used for structure determination is as follows: MX: 89.5%; NMR: 8.5%; 3DEM: 1.6%; Other: 0.4%.
Types of Institutions Depositing Relevant Structures:
In most cases, the earliest instance of a Relevant Structure for a given NME came from research groups in academe [73%], with the remainder coming from industry [21%] or from combined academic-industrial research teams [6%]. Institution type information for the earliest Relevant Structure for each NME was derived manually from the author affiliations listed in the publication reporting the structure determination. Other 3D structures of protein-ligand complexes contributed to the PDB by industrial researchers are impacting to drug discovery by providing data useful for methods development. For example, a large number of “post-competitive” co-crystal structures have been generously donated by biopharmaceutical companies to the NIGMS-NIH funded Drug Design Data Resource [D3R; drugdesigndata.org] for use in blind challenges of computational docking/scoring of ligands (Gathiaka et al., 2016).
Open-access Availability and Publication of Relevant Structures:
More than half of the 5,914 Relevant Structures were deposited to the PDB well before the NME was approved by the US FDA for clinical use. The median time between deposition and approval exceeded 10 years [Figure 2]. Virtually all of the 5,914 Relevant Structures [92%, 5,441/5,914] were reported in a PubMed-indexed publication around the time of PDB deposition. In aggregate, the PDB archive has records of 2,541 primary publications describing one or more of the Relevant Structures. These papers had garnered 204,660 literature citations as of August 2018, giving an average of >80 citations/primary publication. For reference, the average number of literature citations/primary publication across the entire PDB archive is ~50 (Burley et al., 2018). Galkina Cleary and coworkers (Galkina Cleary et al., 2018) recently published a study of these same 210 NMEs and their molecular targets. They documented that research studies on the 150 known molecular targets were reported in more than 2 million PubMed-indexed publications. Although we cannot draw definitive conclusions from these data, it appears that ~10% of worldwide research on the known molecular targets of the 210 NMEs was influenced by 3D structural data made freely accessible from the PDB.
Figure 2.
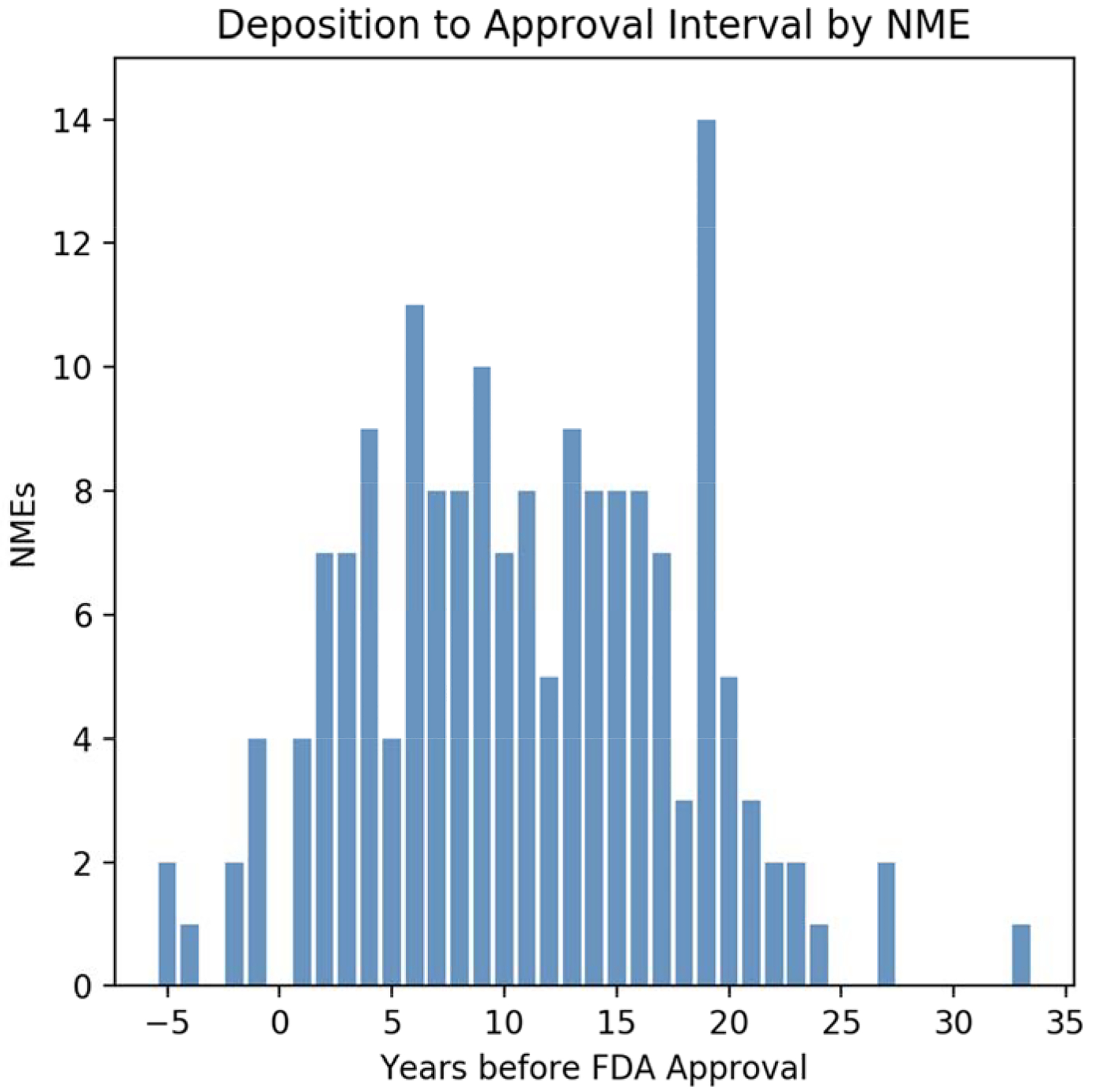
Distribution of the lead time between earliest PDB deposition of a Relevant Structure for a particular NME and subsequent US FDA approval of that NME.
Low Molecular Weight-NMEs:
171 of the 210 NMEs are small molecules with molecular weight <1000. Approximately 92% of these LMW-NMEs [158/171] have known protein targets. We identified 5,364 Relevant Structures containing a known protein target of a LMW-NME [Table 2]. In addition, we identified 277 Relevant Structures containing one of the LMW-NMEs [Table 2]. In a significant minority of cases [18%, 30/171], the number of Relevant Structures for a given LMW-NME exceeded 100 [Figure 1], reflecting the importance of structure-guided drug discovery for some drug targets [e.g., thrombin (Nar, 2012); HIV-protease inhibitors (Wlodawer and Vondrasek, 1998)].
US FDA approvals of anti-neoplastic agents benefited very considerably from PDB structural data. 55 of the 59 anti-cancer NMEs have Relevant Structures for their known protein targets. 26 of the 59 anti-cancer NMEs have Relevant Structures containing one of the NMEs. Some of these new drugs have a wealth of Relevant Structures represented in the PDB archive. For example, bosutinib has a total of 179 Relevant Structures, including those of its protein target [human Src] alone, mutant forms of Src, phosphorylated forms of Src, bosutinib bound to Src, other small-molecule inhibitors bound to Src, and bosutinib bound to proteins other than Src. Beginning in 1994 with publication and PDB deposition of the first human Src structure [PDB ID: 1shd; (Gilmer et al., 1994)] these 179 structures provided detailed insights into the biology of Src as a non-receptor tyrosine kinase, its role as a driver of tumor growth, inhibition with a variety of small-molecules, and eventual targeting with a US FDA approved drug, the LWM-NME bosutinib.
For some of the LMW-NMEs, structural data for drug-protein complexes were only deposited to the PDB and published after the for-profit company had completed its proprietary structure-guided drug discovery campaign [e.g., vemurafenib targeting the Val600→Glu or V600E mutant form of B-Raf protein kinase found in ~50% of late stage metastatic melanomas, PDB ID: 3og7; (Bollag et al., 2010)]. For some Central Nervous System NME targets [e.g., ion channels and G-protein coupled receptors], some of the structure determinations dependent on 3DEM (Renaud et al., 2018) were only deposited to the PDB and published around the time of NME approval. Open access to these and other PDB structural data forthcoming from 3DEM will almost certainly influence future US FDA NME approvals in this challenging yet important therapeutic area.
Biologics-NMEs:
36 of the 210 NMEs are biologics [i.e., proteins or peptide hormones with molecular weight ≥1000]. More than 90% of these Biologics-NMEs [33/36] have known protein targets. The number of unique protein targets of all Biologics-NMEs equals 25. Not surprisingly, monoclonal antibodies are most numerous [24/33], followed by peptide hormones [6/33], antibody-drug conjugates [2/33], and one enzyme [Clostridium histolyticum collagenase; approved for treating Dupuytren’s contractures]. The remaining three Biologics-NMEs that do not have protein targets include an antilipid antibody [dinutuximab; approved for treating high-risk pediatric neuroblastomas], an anti-drug antibody [idarucizumab; approved for reversal of the effects of the small molecule thrombin inhibitor dabigatran], and a beta glucocerebrosidase enzyme [taliglucerase alfa; approved for treating Gaucher’s lipid storage disease]. Structures of both idarucizumab [PDB IDs: 4jn1, 4jn2; (Schiele et al., 2013)] and taliglucerase alfa [earliest of 24 PDB IDs: 1ogs; (Dvir et al., 2003)] are represented in the PDB. There are no Biologics-NMEs with unknown molecular targets.
Relevant Structures for 94% [31/33] of the Biologic-NMEs with known protein targets were detected. In aggregate, we found 474 Relevant Structures containing a known protein target of a Biologic-NME [Table 2]. We also identified 41 Relevant Structures containing a Biologic-NME [Table 2]. Approximately one third [13/36] of the Biologic-NMEs are represented in the PDB with structures of the drug itself [e.g., metreleptin, PDB ID: 1ax8; (Zhang et al., 1997)]. Finally, 10 Biologic-NMEs are represented in the PDB as one or more co-crystal structures of the drug bound to its known target protein, including parathyroid hormone bound to the extracellular portion of its G protein-coupled receptor (Pioszak and Xu, 2008)]. Given the complexity of designing a Biologic-NME and determining optimal usage in clinical settings, it is not surprising that PDB structures play many different roles in biologics drug discovery and development (Hu et al., 2018).
Structural information can also assist in improving properties of proteins developed as Biologic-NMEs. A powerful example is provided by recombinant human insulin and its analogs, which in aggregate were prescribed nearly 58 million times in the US during calendar year 2016 (Kane, 2018). There are more than 100 insulin structures in the PDB. The first insulin structure was deposited in 1980 [PDB ID 1ins; (Blundell et al., 1971)]. Knowledge of the 3D structure of insulin decisively influenced the molecular designs of recently approved analogs with pharmacokinetic and pharmacodynamic properties markedly different from those of earlier formulations of wild-type bovine, porcine, and human insulins [e.g., degludec, an ultralong-acting insulin, PDB IDs: 4ajx, 4ajz, 4ak0, 4akj; (Steensgaard et al., 2013)].
Eight of the monoclonal antibody Biologic-NMEs and one of the antibody-drug conjugate Biologic-NMEs are represented in the PDB as co-crystal structures of the Fab fragment bound to its known target protein. Such co-crystal structures provide important insights into how these drugs work. For example, structural studies of anti-her2 antibodies determined that they bind to distinct antigenic epitopes, explaining the molecular underpinnings of effective combination therapy for breast cancer [reviewed in (Olson, 2012)] with pertuzumab [PDB ID: 1s78; (Franklin et al., 2004)] and trastuzumab [PDB ID: 1n8z; (Cho et al., 2003)].
Structural biologists have also helped to explain how immune check-point inhibitors prevent down regulation of T-cells by tumors displaying the cell surface proteins PD-L1and PD-L2. The PDB archive contains multiple structures of PD-1 [earliest PDB ID: 1npu; (Zhang et al., 2004)], PD-L1[earliest PDB ID: 3bis; (Lin et al., 2008)], PD-L2 [earliest PDB ID: 3bov; (Lazar-Molnar et al., 2008)], PD-1/PD-L1 complexes [earliest PDB ID: 3bik; (Lin et al., 2008)], and PD-1/PD-L2 complexes [earliest PDB ID: 3bp5; (Lazar-Molnar et al., 2008)]. Two of the recently approved monoclonal antibody Biologic-NMEs target PD-1. Structures of both nivolumab [earliest PDB ID: 5ggq; (Lee et al., 2016)] and pembrolizumab [earliest PDB ID: 5dk3; (Scapin et al., 2015)] are available from the PDB . The PDB archive also contains multiple structures of nivolumab/PD-1 complexes [earliest PDB ID: 5ggr; (Lee et al., 2016)] and pembrolizumab/PD-1 complexes [earliest PDB ID: 5jxe; (Na et al., 2017)]. Biopharmaceutical company recognition that PD-1 was a bona fide target for anti-neoplastic immunotherapy came in no small part from ~750 PDB structures that together revealed the molecular mechanisms underpinning antigen presentation to T-cell receptors and T-cell regulation, beginning with the crystal structure of the major histocompatibility complex published and deposited to the PDB in 1987 [PDB ID: 1hla; (Bjorkman et al., 1987)].
Antisense Oligonucleotide Drugs:
Three of the 210 recently approved NMEs are antisense oligonucleotides (ASO-NMEs) that target mRNAs and not proteins, including Eteplirsen [targeting dystrophin], mipomersen [targeting apo-lipoprotein B 100], and nusinersen [targeting survival motor neuron or SMN Protein]. There are, however, Relevant Structures of the proteins encoded by the target mRNAs in the PDB for both eteplirsen/dystrophin [earliest PDB ID: 1dxx; (Norwood et al., 2000)] and nusinersen/SMN protein [earliest PDB ID: 4a4f; (Tripsianes et al., 2011)]. While these Relevant Structures did not influence the design of the ASO-NMEs per se, they did contribute to the very significant body of “pre-competitive” data pertaining to the biology of the proteins translated from each of the mRNAs and their respective roles in human disease.
Drugs with No Known Molecular Targets:
Only 6% [13/210] of the NMEs approved between 2010–2016 have no known molecular target. These new drugs came primarily from phenotypic screening, which remains an important means of identifying lead compounds for drug discovery (Moffat et al., 2017).
Cost of Relevant Structures Deposited to the PDB:
The global cost of all publicly-funded research focused on the known protein targets of the 210 NMEs is significant. Identification of 5,914 Relevant Structures allowed us to estimate the total cost of the structure determinations themselves at ~$600 million [using an average cost/structure of ~$100,000 in today’s currency extrapolated from metrics reported during the NIH NIGMS-funded Protein Structure Initiative] (Burley et al., 2008). Galkina Cleary et al. (2018) estimated that in excess of $100 billion in NIH funding [~20% of the NIH budget between 2000 and 2016] helped support research on the known molecular targets of the 210 NMEs approved between 2010 and 2016. N.B.: Most of NIH-funded research on the 150 known molecular targets analyzed by Galkina Cleary et al. (2018) was deemed to have been basic and non-proprietary (or pre-competitive).
Neither of these cost estimates include the enormous sums of money expended by various biopharmaceutical companies to discover, develop, and secure US FDA approval of the 210 NMEs. The Tufts University Center for the Study of Drug Development recently reported the average cost per new drug approval at ~$2.87 billion [in 2013 US dollars] (DiMasi et al., 2016). Combining this industry estimate with our findings and those of Galkina Cleary et al. (2018) suggests that aggregate public-and private-sector expenditures that ultimately yielded the 210 new drug approvals exceeded $700 billion.
Impact of the PDB on Drugs Approved Outside the US:
Not captured in our analyses is the impact of PDB structures on contemporaneous approvals of NMEs outside the US [i.e., Europe, Japan]. For example, tafamidis, a small-molecule transthyretin stabilizer, was approved in 2011 by the European Medicines Agency to delay peripheral nerve impairment in adults with transthyretin-related hereditary amyloidosis. The first 3D structure of transthyretin a.k.a. prealbumin, the protein target of tafamidis, was deposited to the PDB in 1977 [PDB ID: 2pab; (Blake et al., 1978)]. Subsequently, a co-crystal structure of the transthyretin-tafamidis complex was both published (Penchala et al., 2013) and deposited to the PDB [PDB ID: 4his]. Tafamidis is currently undergoing clinical trials in the US (clinicaltrials.gov).
Drug Discovery and Development Eco-system:
Documentation of the impact of PDB data on recent drug approvals in the US and elsewhere is consistent with our previously published work that reported mentions of the PDB in >50,000 issued patents and patent applications in process worldwide during 2016 (Burley et al., 2018). The findings reported herein are also consistent with the holistic picture of the drug discovery and development eco-system painted by Galkina Cleary et al. (2018). We identified a wealth of contributions by hundreds if not thousands of structural biologists to an extensive body of basic research that underpinned discovery and development of new drugs. For the avoidance of doubt, we acknowledge that many other biomedical research disciplines (e.g., genome sequencing, high-throughput screening, medicinal and computational chemistry) are also of tremendous importance in drug discovery and development efforts. Finally, our analyses of the impact of 3D structure provide further support for the view that life-changing biomedical innovations (e.g., NME approvals) depend critically on open sharing of data within scientific ecosystems and generation of knowledge therefrom (Beierlein et al., 2017).
Postscript
The PDB is now in its 48th year of continuous operation, reflecting an unwavering community commitment to open access sharing of 3D structural data for biological macromolecules. Our analyses document that structural biologists in both academe and the biopharmaceutical industry have contributed substantially to both pre-competitive and proprietary research that led to US FDA approval of 184 NMEs between 2010 and 2016. Deposition of 3D structural data to the PDB at the time of structure publication ensures that this valuable information is immediately shared with the global biomedical research community to further basic and applied research. Open access to 3D structures of presumed drug targets directly facilitated structure-guided discovery of many of the 184 NMEs, thereby increasing efficiency and lowering costs of drug discovery campaigns in biopharmaceutical companies. Our work underscores the importance of adherence to the FAIR Principles (Wilkinson et al., 2016), which help ensure the broadest possible use of biomedical research data generated with government/private philanthropic funding as public goods that enable basic and applied research and education across the sciences.
Supplementary Material
Highlights.
Open access to PDB structure data facilitates discovery and development of new drugs
210 new molecular entities (drugs) were approved by the US Food and Drug Administration 2010–2016
Molecular targets are known for 94% of these 210 NMEs
PDB contains 5,914 structures relevant to 88% of these new drugs across all therapeuticareas
Acknowledgements
We thank the more than 30,000 structural biologists who have deposited structures to the PDB since 2000. We also thank Drs. Eddy Arnold, Helen M. Berman, Kenneth J. Breslauer, R. Andrew Byrd, Kirk L. Clark, S. David Kimball, Christopher J. Molloy, Cathy Peishoff, Jonathan H. Watanabe, and Cynthia Wolberger for their insightful comments, and gratefully acknowledge contributions from all members of the Research Collaboratory for Structural Bioinformatics PDB and our Worldwide Protein Data Bank partners. The RCSB PDB is jointly funded by the National Science Foundation, the National Institute of General Medical Sciences, the National Cancer Institute, and the Department of Energy (NSF-DBI 1338415).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Information is provided describing the assembly and analysis of the data set reported in this article.
Author Information The authors declare no competing financial interests.
References
- Beierlein JM, McNamee LM, Walsh MJ, Kaitin KI, DiMasi JA, and Ledley FD (2017). Landscape of Innovation for Cardiovascular Pharmaceuticals: From Basic Science to New Molecular Entities. Clin. Ther 39, 1409–1425 e1420. [DOI] [PubMed] [Google Scholar]
- Berman HM, Henrick K, and Nakamura H (2003). Announcing the worldwide Protein Data Bank. Nat. Struct. Biol 10, 980. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, and Bourne PE (2000). The Protein Data Bank. Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, and Wiley DC (1987). Structure of the human class I histocompatibility antigen, HLA-A2. Nature 329, 506. [DOI] [PubMed] [Google Scholar]
- Blake CC, Geisow MJ, Oatley SJ, Rerat B, and Rerat C (1978). Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J. Mol. Biol 121, 339–356. [DOI] [PubMed] [Google Scholar]
- Blundell TL (2017). Protein crystallography and drug discovery: recollections of knowledge exchange between academia and industry. IUCr J. 4, 308–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell TL, Cutfield JF, Cutfield SM, Dodson EJ, Dodson GG, Hodgkin DC, Mercola DA, and Vijayan M (1971). Atomic positions in rhombohedral 2-zinc insulin crystals. Nature 231, 506–511. [DOI] [PubMed] [Google Scholar]
- Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, et al. (2010). Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467, 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley SK, Berman HM, Christie C, Duarte J, Feng Z, Westbrook J, Young J, and Zardecki C (2018). RCSB Protein Data Bank: Sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci. 27, 316–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley SK, Joachimiak A, Montelione GT, and Wilson IA (2008). Contributions to the NIH-NIGMS Protein Structure Initiative from the PSI Production Centers. Structure 16, 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdeville R, Buchdunger E, Zimmermann J, and Matter A (2002). Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discov 1, 493–502. [DOI] [PubMed] [Google Scholar]
- Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW Jr., and Leahy DJ (2003). Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421, 756–760. [DOI] [PubMed] [Google Scholar]
- DiMasi JA, Grabowski HG, and Hansen RW (2016). Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 47, 20–33. [DOI] [PubMed] [Google Scholar]
- Dvir H, Harel M, McCarthy AA, Toker L, Silman I, Futerman AH, and Sussman JL (2003). X-ray structure of human acid-beta-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 4, 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, and Sliwkowski MX (2004). Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 5, 317–328. [DOI] [PubMed] [Google Scholar]
- Galkina Cleary E, Beierlein JM, Khanuja NS, McNamee LM, and Ledley FD (2018). Contribution of NIH funding to new drug approvals 2010–2016. Proc. Natl. Acad. Sci. USA 115, 2329–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gathiaka S, Liu S, Chiu M, Yang H, Stuckey JA, Kang YN, Delproposto J, Kubish G, Dunbar JB Jr., Carlson HA, et al. (2016). D3R grand challenge 2015: Evaluation of protein-ligand pose and affinity predictions. J. Comput. Aided Mol. Des 30, 651–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliland GL, Luo J, Vafa O, and Almagro JC (2012). Leveraging SBDD in protein therapeutic development: antibody engineering. Methods Mol. Biol 841, 321–349. [DOI] [PubMed] [Google Scholar]
- Gilmer T, Rodriguez M, Jordan S, Crosby R, Alligood K, Green M, Kimery M, Wagner C, Kinder D, Charifson P, et al. (1994). Peptide inhibitors of src SH3-SH2-phosphoprotein interactions. J. Biol. Chem 269, 31711–31719. [PubMed] [Google Scholar]
- Gore S, Sanz Garcia E, Hendrickx PMS, Gutmanas A, Westbrook JD, Yang H, Feng Z, Baskaran K, Berrisford JM, Hudson BP, et al. (2017). Validation of the Structures in the Protein Data Bank. Structure 25, 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T, Sprague ER, Fodor M, Stams T, Clark KL, and Cowan-Jacob SW (2018). The impact of structural biology in medicine illustrated with four case studies. J. Mol. Med. (Berlin) 96, 9–19. [DOI] [PubMed] [Google Scholar]
- Kane SP (2018). ClinCalc DrugStats Database version 19.0. (ClinCalc LLC; ). [Google Scholar]
- Kinjo AR, Bekker GJ, Suzuki H, Tsuchiya Y, Kawabata T, Ikegawa Y, and Nakamura H (2017). Protein Data Bank Japan (PDBj): updated user interfaces, resource description framework, analysis tools for large structures. Nucleic Acids Res. 45, D282–D288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebe G (2013). Drug Design - Methodology, Concepts, and Mode-of-Action. (Berlin: Springer: ). [Google Scholar]
- Lazar-Molnar E, Yan Q, Cao E, Ramagopal U, Nathenson SG, and Almo SC (2008). Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc. Natl. Acad. Sci. USA 105, 10483–10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Lee HT, Shin W, Chae J, Choi J, Kim SH, Lim H, Won Heo T, Park KY, Lee YJ, et al. (2016). Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nature Comm. 7, 13354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DY, Tanaka Y, Iwasaki M, Gittis AG, Su HP, Mikami B, Okazaki T, Honjo T, Minato N, and Garboczi DN (2008). The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc. Natl. Acad. Sci. USA 105, 3011–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat JG, Vincent F, Lee JA, Eder J, and Prunotto M (2017). Opportunities and challenges in phenotypic drug discovery: an industry perspective. Nat. Rev. Drug Discov 16, 531–543. [DOI] [PubMed] [Google Scholar]
- Mullard A (2016). 2015 FDA drug approvals. Nat. Rev. Drug Discov 15, 73–76. [DOI] [PubMed] [Google Scholar]
- Na Z, Yeo SP, Bharath SR, Bowler MW, Balikci E, Wang CI, and Song H (2017). Structural basis for blocking PD-1-mediated immune suppression by therapeutic antibody pembrolizumab. Cell Res. 27, 147–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar B, Bornmann WG, Pellicena P, Schindler T, Veach DR, Miller WT, Clarkson B, and Kuriyan J (2002). Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 62, 4236–4243. [PubMed] [Google Scholar]
- Nar H (2012). The role of structural information in the discovery of direct thrombin and factor Xa inhibitors. Trends Pharm. Sci 33, 279–288. [DOI] [PubMed] [Google Scholar]
- Norwood FL, Sutherland-Smith AJ, Keep NH, and Kendrick-Jones J (2000). The structure of the N-terminal actin-binding domain of human dystrophin and how mutations in this domain may cause Duchenne or Becker muscular dystrophy. Structure 8, 481–491. [DOI] [PubMed] [Google Scholar]
- Olson EM (2012). Maximizing human epidermal growth factor receptor 2 inhibition: a new oncologic paradigm in the era of targeted therapy. J. Clin. Oncol 30, 1712–1714. [DOI] [PubMed] [Google Scholar]
- Penchala SC, Connelly S, Wang Y, Park MS, Zhao L, Baranczak A, Rappley I, Vogel H, Liedtke M, Witteles RM, et al. (2013). AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc. Natl. Acad. Sci. USA 110, 9992–9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioszak AA, and Xu HE (2008). Molecular recognition of parathyroid hormone by its G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 105, 5034–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protein Data Bank. (1971). Crystallography: Protein Data Bank. Nature New Biol. 233, 223–223.20480989 [Google Scholar]
- Renaud J-P, Chari A, Ciferri C, Liu W. t., Rémigy H-W, Stark H, and Wiesmann C (2018). Cryo-EM in drug discovery: achievements, limitations and prospects. Nat. Rev. Drug Discov 17, 471. [DOI] [PubMed] [Google Scholar]
- Scapin G, Yang X, Prosise WW, McCoy M, Reichert P, Johnston JM, Kashi RS, and Strickland C (2015). Structure of full-length human anti-PD1 therapeutic IgG4 antibody pembrolizumab. Nat. Struct. Mol. Biol 22, 953–958. [DOI] [PubMed] [Google Scholar]
- Schiele F, van Ryn J, Canada K, Newsome C, Sepulveda E, Park J, Nar H, and Litzenburger T (2013). A specific antidote for dabigatran: functional and structural characterization. Blood 121, 3554–3562. [DOI] [PubMed] [Google Scholar]
- Steensgaard DB, Schluckebier G, Strauss HM, Norrman M, Thomsen JK, Friderichsen AV, Havelund S, and Jonassen I (2013). Ligand-controlled assembly of hexamers, dihexamers, and linear multihexamer structures by the engineered acylated insulin degludec. Biochemistry 52, 295–309. [DOI] [PubMed] [Google Scholar]
- Stoll F, Goller AH, and Hillisch A (2011). Utility of protein structures in overcoming ADMET-related issues of drug-like compounds. Drug Discov. Today 16, 530–538. [DOI] [PubMed] [Google Scholar]
- Tripsianes K, Madl T, Machyna M, Fessas D, Englbrecht C, Fischer U, Neugebauer KM, and Sattler M (2011). Structural basis for dimethylarginine recognition by the Tudor domains of human SMN and SPF30 proteins. Nat. Struct. Mol. Biol 18, 1414–1420. [DOI] [PubMed] [Google Scholar]
- Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Z, et al. (2008). BioMagResBank. Nucleic Acids Res. 36, D402–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velankar S, Best C, Beuth B, Boutselakis CH, Cobley N, Sousa Da Silva AW, Dimitropoulos D, Golovin A, Hirshberg M, John M, et al. (2010). PDBe: Protein Data Bank in Europe. Nucleic Acids Res. 38, D308–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson MD, Dumontier M, Aalbersberg IJ, Appleton G, Axton M, Baak A, Blomberg N, Boiten JW, da Silva Santos LB, Bourne PE, et al. (2016). The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 3, 160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodawer A, and Vondrasek J (1998). Inhibitors of HIV-1 protease: a major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct 27, 249–284. [DOI] [PubMed] [Google Scholar]
- Young JY, Westbrook JD, Feng Z, Peisach E, Persikova I, Sala R, Sen S, Berrisford JM, Swaminathan GJ, Oldfield TJ, et al. (2018). Worldwide Protein Data Bank biocuration supporting open access to high-quality 3D structural biology data. Database (Oxford) 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JY, Westbrook JD, Feng Z, Sala R, Peisach E, Oldfield TJ, Sen S, Gutmanas A, Armstrong DR, Berrisford JM, et al. (2017). OneDep: Unified wwPDB System for Deposition, Biocuration, and Validation of Macromolecular Structures in the PDB Archive. Structure 25, 536–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Basinski MB, Beals JM, Briggs SL, Churgay LM, Clawson DK, DiMarchi RD, Furman TC, Hale JE, Hsiung HM, et al. (1997). Crystal structure of the obese protein leptin-E100. Nature 387, 206–209. [DOI] [PubMed] [Google Scholar]
- Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Lorenz M, Cammer M, Chen L, Zhang ZY, Edidin MA, et al. (2004). Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity 20, 337–347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.