

SUMMARY
Inflammasome activation leads to pyroptotic cell death, thereby eliminating the replicative niche of virulent pathogens. Although inflammasome-associated cytokines IL-1β and IL-18 have an established role in T cell function, whether inflammasome activation in dendritic cells (DCs) is critical for T cell priming is not clear. Here, we find that conventional DCs (cDCs) suppress inflammasome activation to prevent pyroptotic cell death, thus preserving their ability to prime both CD4 and CD8 T cells. Transcription factors IRF8 and IRF4, in cDC1s and cDC2s, respectively, mediate suppression of inflammasome activation by limiting the expression of inflammasome-associated genes. Overexpression of IRF4 or IRF8 inhibits inflammasome activation in macrophages, while reduced expression of IRF8 leads to aberrant inflammasome activation in cDC1s and hampers their ability to prime CD8 T cells. Thus, activation of inflammasome in DCs is detrimental to adaptive immunity, and our results reveal that cDCs use IRF4 and IRF8 to suppress this response.
In Brief
The role of inflammasome activation in eliciting adaptive immune responses against pathogens is poorly understood. McDaniel et al. demonstrate that conventional dendritic cells use IRF4 and IRF8 to suppress the transcription of inflammasome-associated machinery. This resulting suppression of inflammasome activation allows DCs to prime T cell responses against virulent pathogens.
Graphical Abstract

INTRODUCTION
Myeloid cells play a central role in initiating both innate and adaptive immune responses. Macrophages and dendritic cells (DCs) sense their surroundings through the use of cell surface and cytosolic pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) and NOD-like receptors (NLRs). These PRRs recognize broadly conserved pathogen-associated molecular patterns (PAMPs) that can be produced by both virulent and non-virulent (commensal) microbes (Takeda et al., 2003). Microbial sensing by TLRs triggers a cascade that activates NF-κB signaling, resulting in the production of proinflammatory cytokines and chemokines that are necessary for acute protection of the host (West et al., 2006). Virulent pathogens that invade intracellularly or secrete tissue-injuring toxins are also sensed by cytosolic NLRs, leading to activation of the inflammasome (Meylan et al., 2006). Inflammasome activation is a highly regulated process consisting of two major steps (Martinon et al., 2002). Initial sensing of the pathogen by TLRs or other transmembrane PRRs mediates the first step, which results in the transcriptional upregulation of NLRs and other proteins involved in inflammasome activation, including pro-IL-1β. The second step requires sensing of various virulence factors, which causes oligomerization of the NLR with adaptor proteins and pro-caspase-1. Recruitment of pro-caspase-1 to these complexes results in its cleavage and activation, allowing further cleavage of caspase-1 targets including pro-IL-1β, pro-IL-18, and gasdermin-D (Thornberry et al., 1992; Shi et al., 2015). The active N terminus of gasdermin-D forms pores in the cellular membrane, which facilitates the secretion of mature IL-1β and IL-18 and subsequently commits the cell to an inflammatory cell death called pyroptosis (Fink and Cookson, 2006; Shi et al., 2015). Different inflammasome sensors respond to different virulence factors. For example, cytosolic flagellin activates the NLRC4 inflammasome, cytosolic DNA activates the AIM2 inflammasome, and a variety of ligands leading to potassium efflux and reactive oxygen species (ROS) production activate the NLRP3 inflammasome (Martinon et al., 2009).
Inflammasome activation is beneficial for early protection of the host from virulent pathogens, as pyroptosis eliminates intracellular pathogens’ replicative niche and exposes them to extracellular mediators that can kill them (Broz et al., 2012; Miao et al., 2010). Additionally, mature IL-1β and IL-18 released from the cell triggers a proinflammatory cascade, which leads to acute phase response and recruitment of neutrophils and monocytes to the site of infection (Martinon et al., 2009). Together, these events allow rapid protection from virulent pathogens, as inflammasome activation is known to occur within 30 min of initial pathogen sensing (von Moltke et al., 2013). Despite this innate response, long-term protection (as well as immunological memory for resistance to reinfection) also requires a robust antigen-specific adaptive immune response (Hess et al., 1996; Bhardwaj et al., 1998).
As professional antigen-presenting cells (APCs), conventional DCs (cDCs) act as a critical bridge between the innate and adaptive immune systems. Following pathogen detection, cDCs upregulate costimulatory molecules (such as CD80 and CD86), present pathogen-derived peptides on MHC-I or MHC-II, and secrete innate cytokines and chemokines (Larsen et al., 1992; Inaba et al., 2000). These three signals are necessary to activate and prime antigen-specific T cells, a process that can take several days to complete (Inaba et al., 2000; Jain and Pasare, 2017). On the basis of the initial PRRs engaged by a pathogen, the profile of secreted cytokines from the DCs is also altered to relay information about the nature of the pathogen to naive T cells (Gao et al., 2020; Huang et al., 2001). This pathogen-specific T cell response is critical for the long-term protection against many virulent pathogens (Lo et al., 1999; Bhardwaj et al., 1998; Hess et al., 1996).
Inflammasome-associated cytokines such as IL-1β and IL-18 are also known to be critical for T cell priming, clonal expansion, and effector differentiation (Ben-Sasson et al., 2009; Garlanda et al., 2013; Jain et al., 2018). Because of this association, it has been proposed that inflammasome activation is therefore important for protective T cell responses (Dostert et al., 2013). However, the direct role of inflammasome activation in the generation of antigen-specific T cell responses has never been fully investigated. Although inflammasome products such as the IL-1 family of cytokines are important for adaptive immunity (Ben-Sasson et al., 2009; Garlanda et al., 2013; Jain et al., 2018), release of these cytokines following inflammasome activation is generally concurrent with cell death via pyroptosis. Whether pyroptosis affects DC-mediated priming of T cell responses has neverbeen directly investigated. To better understand how APC-intrinsic inflammasome activation and T cell priming coexist during an infection, we rigorously investigated how myeloid cells respond to virulent pathogens. We found that activation of inflammasome in DCs is detrimental to T cell priming and that cDCs, unlike macrophages, do not undergo inflammasome activation in response to virulent pathogens. Furthermore, this lack of inflammasome activation was due primarily to the transcriptional suppression of inflammasome-associated machinery. Inhibition of inflammasome-dependent cell death preserved DCs’ ability to prime T cells in response to virulent pathogens. Mechanistically, we found the transcription factors IRF8 and IRF4 to play critical roles in regulating the expression of inflammasome sensors and adaptors in cDCs. Reduced expression of IRF8 in cDC1s led to cells becoming permissive to inflammasome stimuli and resulted in defective T cell priming. Conversely, overexpression of IRF8 or IRF4 in macrophages resulted in abrogation of inflammasome activation. Overall, our study demonstrates the necessity of inflammasome suppression by DCs in order to prime T cell responses and reveals a previously unappreciated role for IRF8 and IRF4 in the initiation of adaptive immunity.
RESULTS
Inflammasome Activation Is Detrimental to Adaptive Immunity
To elucidate the role of inflammasome in the initiation of adaptive immune responses, we used the virulent SL1344 strain of Salmonella (STm) that is known to infect both epithelial and myeloid cells. Intracellular infection with STm triggers inflammasome activation, as well as necrosis (Fink and Cookson, 2007). To test the importance of inflammasome activation for T cell priming, bone marrow-derived DCs (BMDCs) were pulsed with virulent STm and ovalbumin (OVA) for 1 h (Figure 1A). These DCs were then cultured with CFSE-labeled naive OT-II or OT-I T cells, which are specific to an OVA-derived peptide (Robertson et al., 2000) (Figure 1A). Interestingly, we found that BMDCs that sensed OVA in the presence of STm had a severe defect in their ability to prime naive T cells compared with BMDCs that sensed OVA in the presence of lipopolysaccharide (LPS) (Figure 1B). STm uses a type III secretion system encoded by Spi1 to deliver cytosolic virulence factors that are known to activate both NLRC4 and NLRP3 inflammasomes (Broz et al., 2010). Consistent with these reports, we found that following STm exposure, BMDCs underwent robust inflammasome activation as measured by caspase-1 cleavage, IL-1β secretion, and pyroptosis (Figures 1C–1E).
Figure 1. Inflammasome Activation Is Detrimental to Adaptive Immunity.
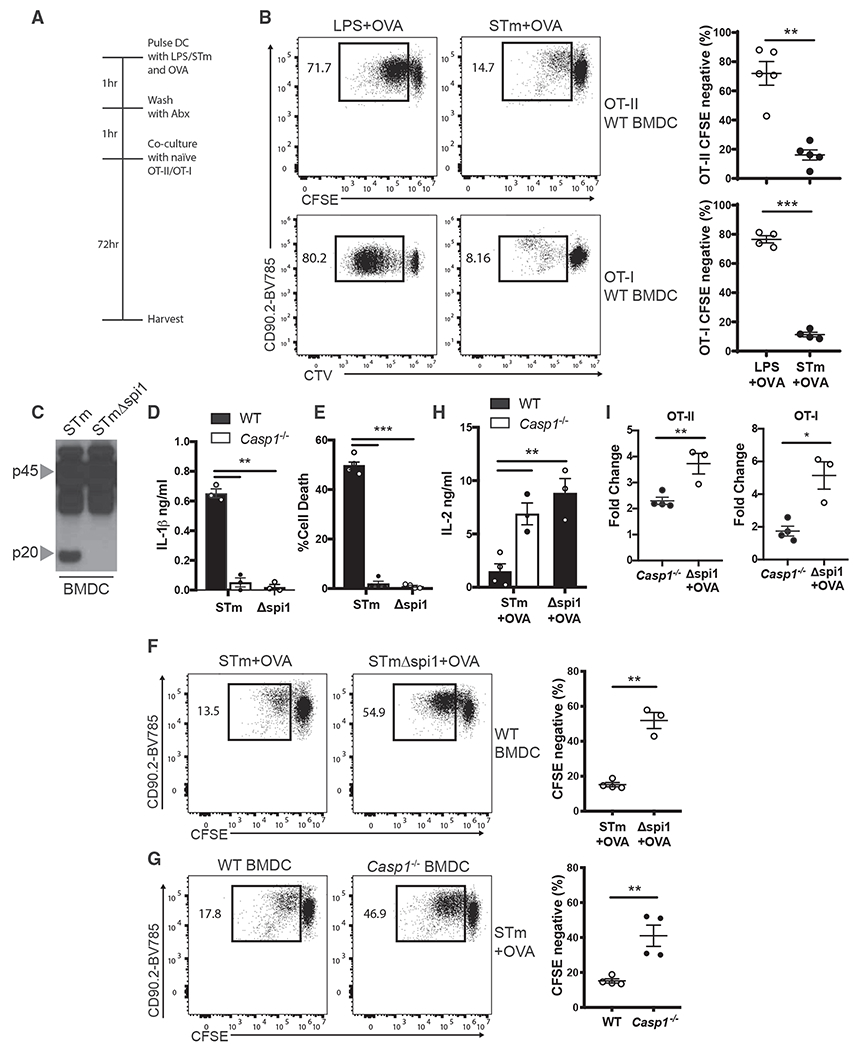
(A) Experimental schematic for culture of DCs and T cells.
(B) OT-II and OT-IT cell proliferation was quantified by flow cytometry following 72 h of culture with WTBMDCs that were pulsed with LPS or STm in the presence of OVA.
(C) Immunoblot analysis of pro-casp1 (p45) and cleaved casp1 (p20) in the lysates of WT BMDCs pulsed with STm or STmΔspi1 for 1 h.
(D and E) IL-1β was measured by ELISA (D), and cell death was measured by LDH release (E) in the supernatants of WT BMDCs from (C).
(F) OT-II T cell proliferation was quantified by flow cytometry following 72 h of culture with WT BMDCs that were pulsed with STm or STmΔspi1 and OVA.
(G) OT-II T cell proliferation was quantified by flow cytometry following 72 h of culture with WT or Casp1−/− BMDCs that were pulsed with STm and OVA.
(H) IL-2 was measured by ELISA in the supernatants of the cultures from (F) and (G).
(I) Fold change of rescued OT-II or OT-I proliferation was calculated from cell proliferation observed in (F) and (G) compared with WT BMDCs that were pulsed with STm and OVA.
Error bars indicate SEM; (B) and (D)–(I), paired t test; (C), data representative of three independent experiments. *p<0.05, **p<0.01, ***p<0.001.
We next asked if inflammasome activation and pyroptotic cell death in these BMDCs were responsible for their inability to prime naive CD4 and CD8 T cells. We specifically inhibited inflammasome activation in BMDCs and tested if this rescued T cell priming and differentiation. Wild-type (WT) BMDCs were pulsed with Spi1-deficient STm (Spi1ΔSTm), which failed to activate inflammasome (Figures 1C–1E). These BMDCs were then cultured with OT-II or OT-I T cells as described above. We found that adaptive immune responses to OVA, as determined by T cell proliferation and IL-2 production, were rescued in the absence of inflammasome activation (Figures 1F and 1H). In agreement with this, caspase-1-deficient BMDCs (Casp1−/−) pulsed with virulent STm also induced enhanced T cell proliferation and IL-2 production (Figures 1G and 1H). Both OT-II and OT-I T cells had a 2- to 6-fold increase in proliferation when BMDCs failed to undergo inflammasome activation (Figure 1I). These data provide compelling evidence to support the argument that inflammasome activation in APCs is detrimental to T cell priming.
Blocking Pyroptotic Cell Death of BMDCs Rescues T Cell Priming
To verify that this defect in priming capacity was due to elimination of the APCs via pyroptosis, we aimed to specifically block inflammasome-dependent cell death while leaving inflammasome activation otherwise intact. Gasdermin-D-deficient (Gasd−/−) macrophages have been reported to be protected from cell death following inflammasome activation (Shi et al., 2015). Although Gasd−/− BMDCs were protected from acute cell death (1 h), we found no difference in the rate of pyroptosis between WT and Gasd−/− BMDCs following 6 h of pathogen exposure (Figure 2A). This is presumably because other gasdermin family members, such as gasdermin-E, can mediate secondary pyroptosis (Tsuchiya et al., 2019). Extracellular glycine does not inhibit caspase-1 activation but has been shown to prevent pyroptosis mediated by gasdermin family members (Fink and Cookson, 2006). When WT BMDCs were cultured in the presence of glycine, we found a significant reduction in STm-induced pyroptosis for up to 12 h of pathogen exposure (Figure 2A). More important, the presence of extracellular glycine did not alter the levels of upregulated MHC-II or costimulatory molecules expressed by BMDCs following STm exposure (Figure 2B). WT BMDCs pulsed with STm in the presence of extracellular glycine led to significant rescue in both OT-II as well as OT-I T cell priming (Figures 2c and 2D). There was no increase in the ability of BMDCs to prime OT-II T cells in response to Spi1ΔSTm when cultured with extracellular glycine, suggesting that glycine has no direct effect on T cell priming (Figure 2E). These data provide direct and conclusive evidence that pyroptotic cell death, not altered DC maturation, following inflammasome activation prevents DCs from effectively priming T cells.
Figure 2. Preventing Pyroptotic Cell Death of BMDCs Rescues T Cell Priming.
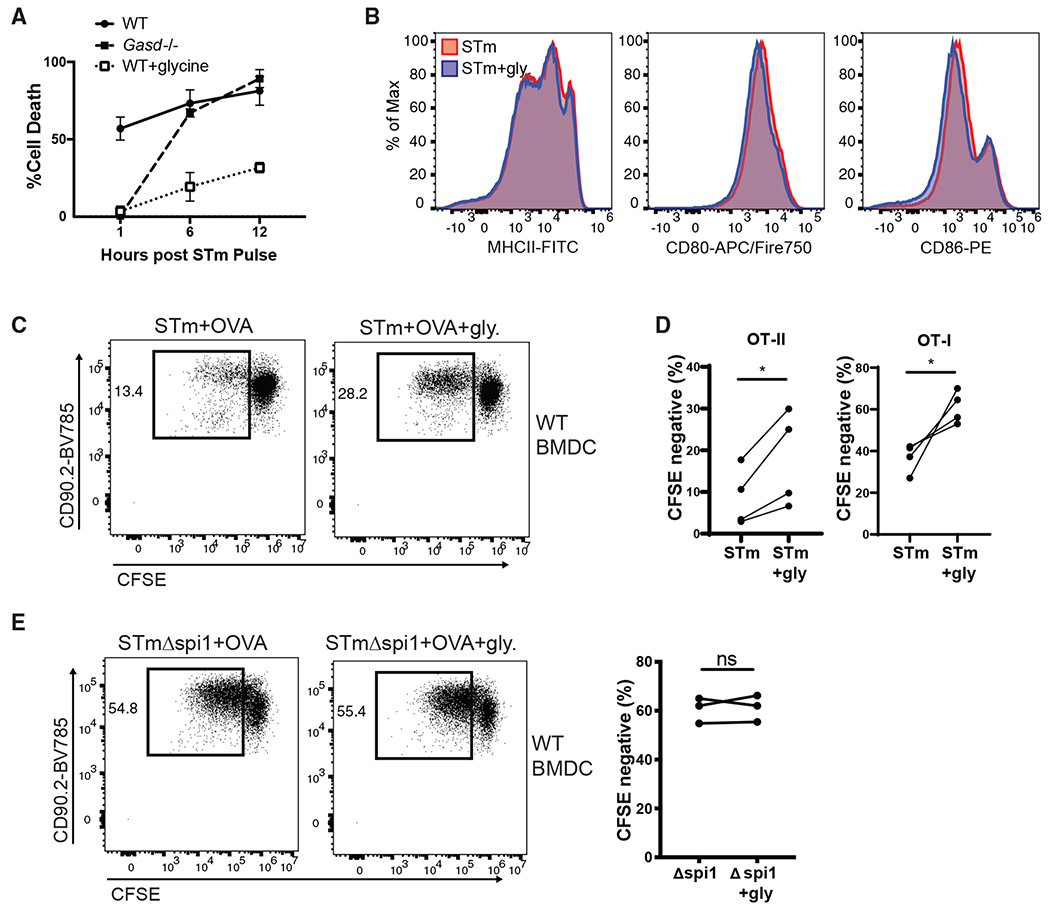
(A) Cell death was measured by LDH release in the supernatants of WT or Gasd−/− BMDCs following a 1 h STm pulse. Error bars indicate SEM.
(B) Expression of MHCII, CD80, and CD86 by WT BMDCs 6 h after a 1 h STm pulse in the presence or absence of 5 mM glycine was quantified by flow cytometry.
(C) Representative flow plot of OT-II T cell proliferation following 72 h of culture with WT BMDCs that were pulsed with STm and OVA in the presence or absence of 5 mM glycine.
(D) OT-II and OT-I T cell proliferation was quantified by flow cytometry following 72 h of culture with WT BMDCs that were pulsed with STm and OVA in the presence or absence of 5 mM glycine.
(E) OT-II T cell proliferation was quantified by flow cytometry following 72 h of culture with WT BMDCs that were pulsed with STmΔspi1 and OVA in the presence or absence of 5 mM glycine.
(D and E) Paired t test; (A and B) data representative of two independent experiments; (C) data representative of four independent experiments. *p<0.05; ns, not significant.
Splenic DCs Suppress Inflammasome Activation and Prime T Cells in Response to Virulent Pathogens
Long-term protection against pathogens requires robust induction of adaptive immunity (Wong and Pamer, 2003). More specifically, in vivo studies of Salmonella typhimurium have demonstrated a role for pathogen-specific CD4 and CD8 T cells in long-term clearance of the pathogen (Hess et al., 1996). Paradoxically, we have shown that BMDCs are defective in priming T cell responses against STm because of inflammasome activation. As GM-CSF-derived BMDCs are known to be a heterogeneous mixture of cells (Helft et al., 2015) that behave differently than in vivo differentiated DCs, we decided to investigate the responses of splenic DCs to virulent pathogens.
CD11c+ splenic DCs from WT mice were purified and pulsed with OVA in the presence of virulent STm or non-virulent Spi1ΔSTm and cultured with naive T cells, as described above. In contrast to BMDCs, the virulence of the pathogen did not compromise the ability of the splenic DCs to prime CD4 or CD8 T cells as measured by T cell proliferation and IL-2 production (Figures 3A and 3B). We next tested the ability of splenic DCs to undergo inflammasome activation, and surprisingly found minimal signs of inflammasome activation as measured by caspase-1 cleavage, IL-1β secretion, or pyroptosis (Figures 3C–3E). These data suggest that lack of inflammasome activation in splenic DCs allows them to prime T cells against virulent pathogens.
Figure 3. Splenic DCs Suppress Inflammasome Activation and Prime T Cells in Response to Virulent Pathogens.
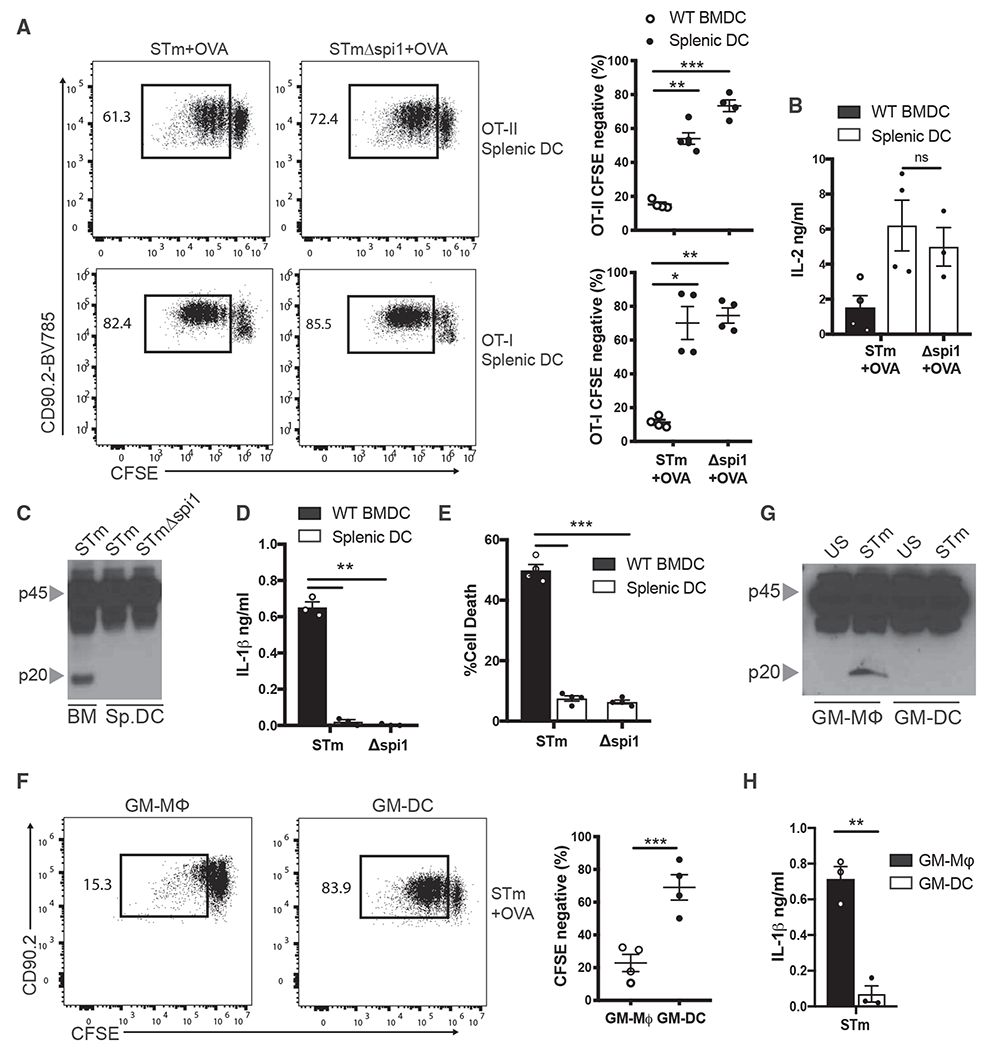
(A) OT-II and OT-I T cell proliferation was quantified by flow cytometry following 72 h of culture with WT CD11c+ splenic DCs that were pulsed with STm or STmΔspi1 in and OVA.
(B) IL-2 was measured by ELISA in the supernatants of the cultures from (A).
(C) Immunoblot analysis of pro-casp1 (p45) and cleaved casp1 (p20) in the lysates of WT BMDCs and CD11c+ splenic DCs pulsed with STm or STmΔspi1 for 1 h.
(D and E) IL-1β was measured by ELISA (D), and (E) cell death was measured by LDH release in the supernatants of WT BMDCs and CD11c+ splenic DCs from (C).
(F) OT-II T cell proliferation was quantified by flow cytometry following 72 h of culture with FACS-sorted GM-Macs (CD11c+MHCIIintCD115+) and GM-DCs (CD11c+MHCII+CD115−) that were pulsed with STm and OVA.
(G) Immunoblot analysis of pro-casp1 (p45) and cleaved casp1 (p20) in the lysates of FACS-sorted GM-Macs and GM-DCs pulsed with STm for 1 h.
(H) IL-1β was measured by ELISA in the supernatants of GM-Macs and GM-DCs from (G).
See also Figures S1 and S2. Error bars indicate SEM; (A, B, D–F, and H) paired t test; (C and G) data representative of three independent experiments. *p<0.05, **p<0.01, ***p<0.001; ns, not significant.
GM-CSF-derived BMDCs constitute a small subpopulation that resembles cDCs (GM-DCs, CD11c+MHCII+CD115−), as well as other subpopulations that more closely resemble macrophages (GM-Macs, CD11c+MHCIIintCD115+) (Helft et al., 2015). We asked if this GM-DC population behaved similarly to splenic DCs in their ability to prime T cells in response to a virulent pathogen. Indeed, following a pulse with virulent STm, sorted GM-DCs (Figure S1) were significantly better at priming OT-II T cells than were GM-Macs (Figure 3F). We tested if the capacity of GM-DCs to prime T cells correlated with their inability to undergo inflammasome activation. Consistent with their ability to prime T cells, GM-DCs did not undergo caspase-1 cleavage or secrete IL-1β in response to virulent STm (Figures 3G and 3H). The marginal T cell proliferation seen in total BMDCs pulsed with virulent STm was likely due to the presence of GM-DCs in the culture. These results show that cDCs, irrespective of whether they are derived from bone marrow precursors or are resident in the spleen, are resistant to STm-induced inflammasome activation. Furthermore, these data underscore the importance of APCs to resist inflammasome activation in response to virulent pathogens in order to prime T cells.
Splenic DCs and GM-DCs Do Not Undergo Inflammasome Activation in Response to Canonical Ligands
We next examined if splenic DCs were resistant specifically to STm-induced inflammasome activation or if they had evolved to be resistant to all inflammasome-activating ligands. We pretreated splenic DCs with LPS or Pam3, then stimulated with inflammasome-activating ligands ATP (NLRP3) (Figures S2A and S2B), FlaTox (NLRC4) (Figures S2B and S2C), or intracellular pol-y(dA:dT) (AIM2) (Figure S2D). Although previous work has suggested that splenic DCs can undergo inflammasome activation in response to these ligands (Ghiringhelli et al., 2009; Kang et al., 2013), we found minimal caspase-1 cleavage, IL-1β secretion, or pyroptosis following stimulation (Figures S2A–S2F). These results are in agreement with a recent publication that also showed that splenic DCs are broadly resistant to inflammasome activation in response to a variety of ligands (Erlich et al., 2019). Previous work has demonstrated that rapid inflammasome activation occurs when both TLR and inflammasome ligands are sensed simultaneously (Lin et al., 2014). In our experimental system, we found that splenic DCs were also unable to cleave caspase-1 in response to simultaneous TLR and NLRP3 activation (Figure S2G). We also investigated the ability of GM-Macs and GM-DCs to undergo canonical inflammasome activation. Similar to their response against STm, GM-Macs exhibited robust caspase-1 cleavage and IL-1β secretion in response to NLRP3-activating ligands, while GM-DCs were resistant to NLRP3 inflammasome-activating ligands (Figures S2H and S2I). These data show that both bona fide DCs generated in vitro or resident in the spleen share a common feature of lacking the ability to activate inflammasome in response to virulent bacteria or canonical ligands.
Splenic DCs and GM-DCs Express Lower Levels of Inflammasome Machinery
In myeloid cells, sensing of PAMPs through TLRs leads to the upregulation of NLRs and adaptor proteins necessary for inflammasome activation (Guarda et al., 2011). This prepares cells to respond rapidly to a subsequent inflammasome signal, such as intracellular flagellin or bacterial toxins (Latz et al., 2013). Although it is possible that cDCs express a dominant-negative regulator of inflammasome activation, we first tested if cDCs were producing the machinery necessary for and associated with inflammasome activation such as NLRP3, NLRC4, ASC, caspase-1, and pro-IL-1β. Although splenic DCs produce the same amount of pro-caspase-1 protein as BMDCs, we found that following LPS stimulation, splenic DCs and GM-DCs have reduced transcriptional levels of Nlrp3, Nlrc4, Pycard, and Il1b compared with BMDCs or GM-Macs, respectively (Figures S2J and S2K). This deficiency in production of inflammasome-associated machinery was also seen at the protein level (Figures S2L and S2M). Although Nlrc4 transcripts were slightly decreased in splenic DCs, the NLRC4 protein was nearly undetectable, suggesting the existence of additional mechanisms of post-transcriptional and post-translational regulation. These results suggest that there is a broad transcriptional downregulation of inflammasome-associated machinery in splenic DCs and GM-DCs compared with total BMDCs or purified GM-Macs, respectively.
IRF8 and IRF4 Expressed by cDC1s and cDC2s Bind to and Suppress Expression of Inflammasome-Associated Genes
Transcriptional programs that govern the differentiation of cDC subsets are well studied, but the distinct roles of these lineage-defining transcription factors in DC function, following their development and differentiation, is only recently being investigated (Naik et al., 2007; Tamura et al., 2005). Conventional lymphoid-organ DCs develop from a common DC progenitor in the bone marrow and further differentiate into cDC1s (CD11c+CD8α+XCR1+) through transcription factors BATF3 and IRF8, or cDC2s (CD11c+CD4+SIRPa+) through IRF4 (Naik et al., 2007; Tamura et al., 2005; Guilliams et al., 2014). Many differences between cDC1s and cDC2s have been described, including their roles in priming CD8 or CD4 T cells, respectively (Schnorrer et al., 2006; Dudziak et al., 2007). Although expression of IRF8 or IRF4 is vital to the development of cDC1 or cDC2 cells, respectively, whether these transcription factors continue to regulate the functions of fully developed DCs is not completely clear. In previously published studies, IRF8 expression by cDC1s has been correlated with maintaining peripheral tolerance (Qiu et al., 2009; Idoyaga et al., 2013; Orabona et al., 2006). Further work has suggested that IRF4 can functionally promote a tolerogenic DC phenotype by negatively regulating the transcription of proinflammatory cytokines and inducing the expression of PD-L2 and RALDH (Vander Lugt et al., 2017). These results suggest a post-developmental role for IRF8 and IRF4 in promoting tolerogenic and anti-inflammatory DC responses. Therefore, we asked if these transcription factors were also important for preventing inflammasome activation in splenic DCs. Because total splenic DCs, composed of both cDC1s and cDC2s, failed to undergo inflammasome activation, we specifically examined the role of cDC1 intrinsic IRF8 and cDC2 intrinsic IRF4 in suppressing inflammasome activation.
Using fluorescence-activated cell sorting (FACS), we sorted splenic cDC1 and cDC2 cells from naive mice and found these populations to have significantly higher expression of IRF8 or IRF4, respectively, compared with splenic macrophages (Figure 4A). Furthermore, this higher expression of IRF8 or IRF4 was inversely correlated with the cells’ ability to produce inflammasome-dependent IL-1β (Figure S3A), as well as the expression of multiple genes associated with inflammasome activation (Figure 4B). Because of this correlation and the aforementioned reports (Qiu et al., 2009; Idoyaga et al., 2013; Orabona et al., 2006; Vander Lugt et al., 2017), we hypothesized that IRF8 in cDC1s or IRF4 in cDC2s could function as a negative regulator of inflammasome activation through transcriptional suppression of inflammasome-associated genes. To test this idea, we analyzed previously reported chromatin immunoprecipitation sequencing (ChIP-seq) datasets (Kc et al., 2014; Glasmacher et al., 2012). In agreement with our hypothesis, both IRF8 and IRF4 binding peaks were enriched at the promoter regions of Nlrp3, Il1b, and Aim2, suggesting that their transcription could be regulated by binding of these transcription factors (Figure 4C). We also identified IRF8 and IRF4 binding sites in the intronic regions of Pycard and Nlrc4 genes (Figure S3B). Binding of introns by transcription factors has been reported to negatively regulate gene transcription (Kikuchi et al., 2000; Tokuhiro et al., 2003). The presence of Ets-IRF elements (EICEs), which are known to recruit IRF4 or IRF8 via Ets transcription factors PU.1 and Spi-B, were verified for each region (Figures S3C and S3D; Lambert et al., 2018; Grant et al., 2011; Glasmacher et al., 2012; Vander Lugt et al., 2014). No IRF4 or IRF8 binding sites were found in the promoter or intronic regions of Nlrx1 (Figure S3B), a known negative regulator of inflammation and inflammasome activation (Li et al., 2016), suggesting that IRF8 and IRF4 may be regulating only a specific set of inflammasome-associated genes.
Figure 4. Expression of IRF8 and IRF4 by cDC1s and cDC2s Negatively Correlates with the Expression of Inflammasome-Associated Genes.
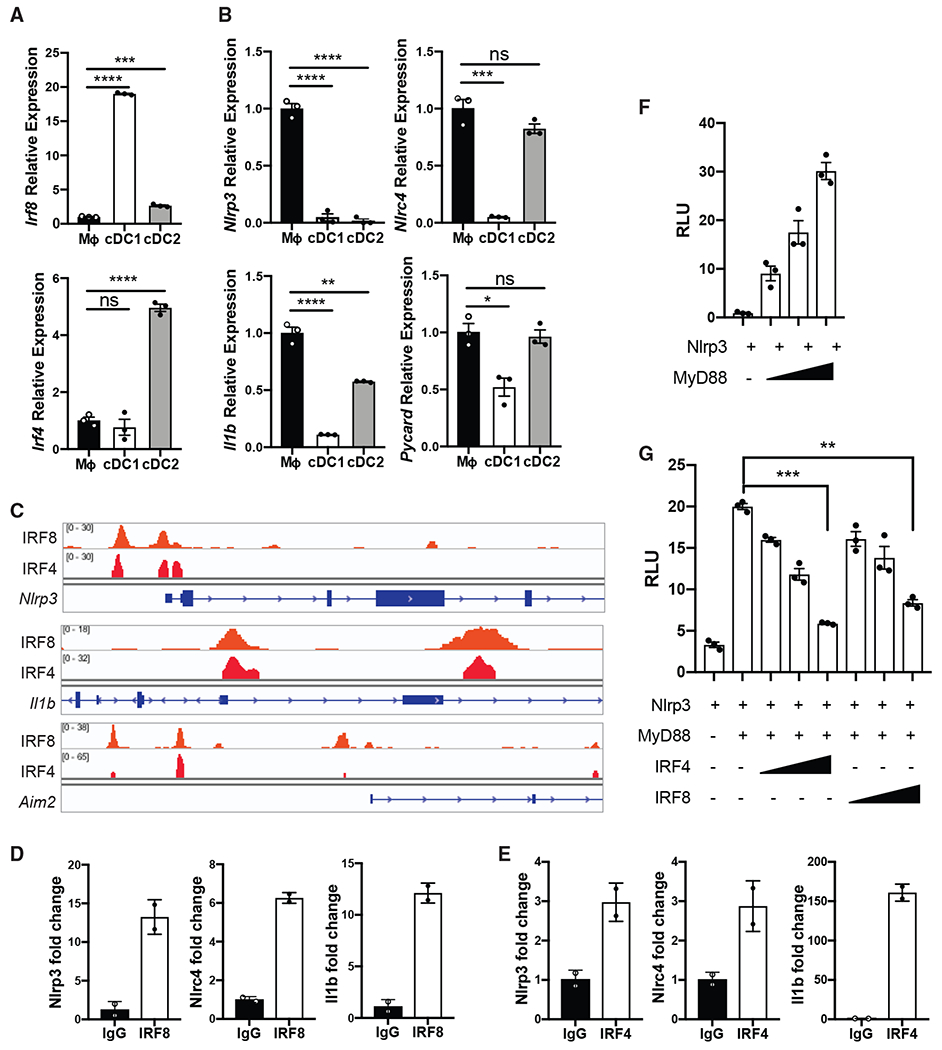
(A) qPCR of Irf8 and Irf4 mRNA in the lysates of FACS-sorted WT CD11b+ splenic macrophages, cDC1s (CD11c+CD8α+XCR1+), and cDC2s (CD11c+CD4+SIRPa+).
(B) qPCR of designated mRNA in the lysates of FACS-sorted splenic macrophages, cDC1s, and cDC2s following 4 h of LPS stimulation (100 ng/mL).
(C) ChIP-seq data from GSE53311 (Kc et al., 2014) and GSE40727 (Glasmacher et al., 2012) were analyzed using IGV.
(D and E) qPCR of designated promoter and intronic regions was performed on chromatin immunoprecipitated with IRF8 or (E) IRF4.
(F) Relative luciferase units (RLU) from 293T cells following 18 h of transfection with NLRP3 promoter reporter construct and increasing doses of MyD88.
(G) RLU from 293T cells following 18 h of transfection with NLRP3 promoter reporter construct, MyD88, and increasing doses of IRF4 or IRF8.
See also Figure S3. Error bars indicate SEM; (A, B, and G) paired t test; (D and E) data representative of two independent experiments. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; ns, not significant.
To confirm the direct binding of IRF8 and IRF4 to these inflammasome-associated transcripts, we performed ChIP-qPCR on splenic DCs. Indeed, we found significant enrichment of IRF8 and IRF4 binding at Nlrp3, Nlrc4, Il1b, and Pycard promoter or intronic regions (Figures 4D, 4E, and S3E). There was no binding of either transcription factor to a negative control region (Figure S3E). To verify that binding of IRF8 or IRF4 to these promoter regions was directly repressing gene expression, we constructed a luciferase expressing plasmid under the direct control of a putative NLRP3 promoter. We found that overexpression of MyD88, which leads to NF-κB activation, leads to activation of this NLRP3 promoter reporter (Figure 4F). When we co-transfected plasmids that express IRF8 or IRF4 into these cells, it led to dose-dependent suppression of MyD88-driven NLRP3 promoter activity (Figure 4G). In addition to demonstrating direct suppression of an inflammasome sensor by both IRF4 and IRF8, these results suggest that IRF8 in cDC1s and IRF4 in cDC2s are likely mediating broader transcriptional repression of inflammasome-associated gene.
Forced Expression of IRF8 and IRF4 Suppresses Inflammasome Activation in BMDCs and Macrophages
If expression of IRF8 or IRF4 is sufficient to prevent inflammasome activation in cDCs, we posited that overexpression of these transcription factors in a cell that is permissive to inflammasome activation should now hinder this response. We observed that the inflammasome-activating subpopulation of BMDCs, GM-Macs, had lower expression of IRF8 and IRF4 than the inflammasome resistant GM-DCs (Figure S4A). We therefore asked if overexpression of IRF8 or IRF4 in GM-Macs would suppress inflammasome activation in response to canonical ligands. BMDCs were retrovirally transduced with IRF8 or IRF4 (Figure S4B) and stimulated with NLRP3-activating ligands. BMDCs transduced with either IRF8 or IRF4 exhibited reduced active caspase-1, as detected intracellularly using FLICA reagent, compared with control transduced cells (Figure 5A). More important, purified GM-Macs that were transduced with IRF8 or IRF4 also showed significantly less IL-1β secretion in response to inflammasome activation than control transduced GM-Macs (Figure 5B).
Figure 5. IRF8 Activity Suppresses Inflammasome Activation in BMDCs and Macrophages.
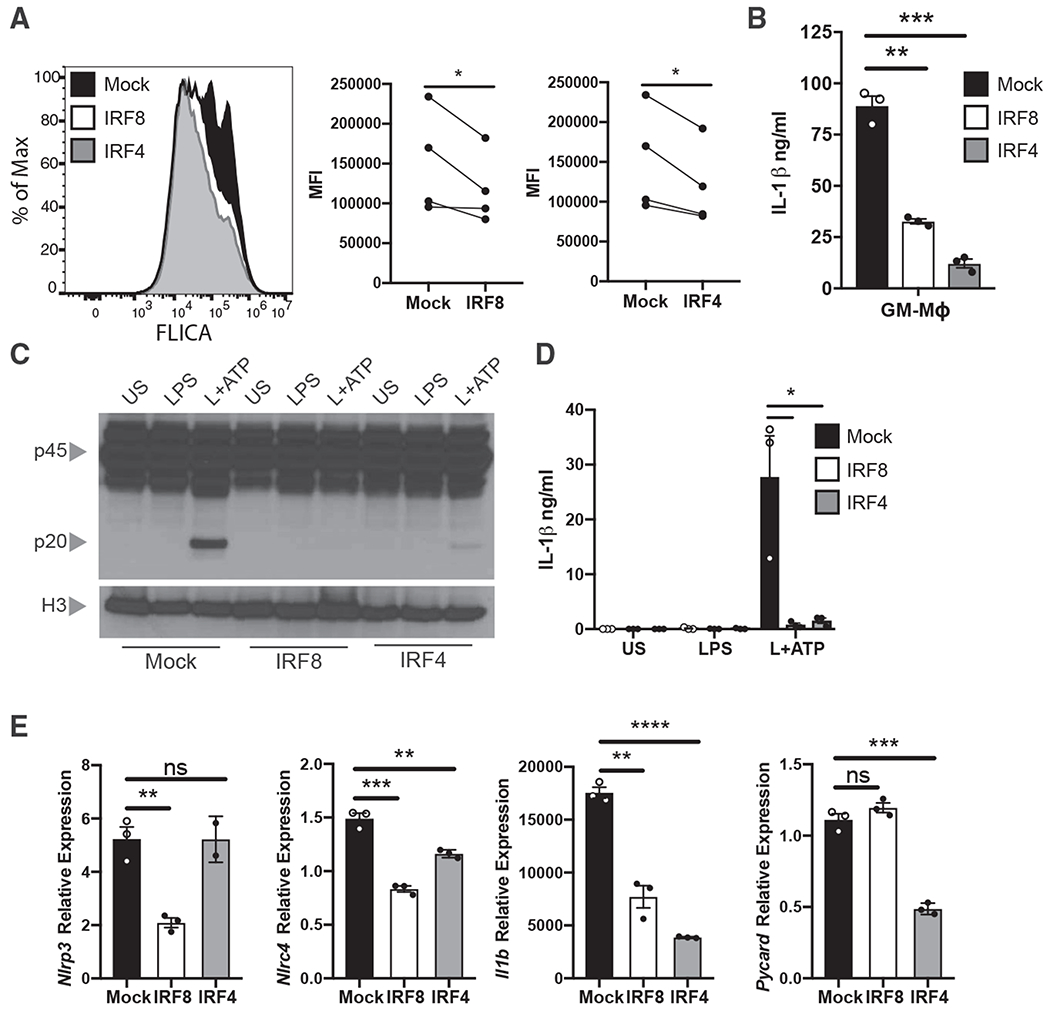
(A) Caspase-1 cleavage (hCD4+FLICA+) was quantified by flow cytometry following NLRP3 inflammasome activation (LPS+ATP) in BMDCs that were transduced with retrovirus expressing IRF8, IRF4, or control plasmids. Mean fluorescence intensity (MFI) from four independent experiments is quantified.
(B) IL-1β was measured by ELISA in the supernatants of FACS-sorted hCD4+ GM-Macs following NLRP3 inflammasome activation. See also Figure S1.
(C) Immunoblot analysis of pro-casp1 (p45) and cleaved casp1 (p20) in the lysates of iBMDMs transduced with lentivirus expressing IRF8, IRF4, or control plasmids and stimulated with NLRP3 inflammasome ligands.
(D) IL-1β was measured by ELISA in the supernatants of iBMDMs from (C).
(E) qPCR of designated mRNA in the lysates of mock, IRF8, or IRF4 transduced iBMDMs following 4 h of LPS stimulation.
See also Figure S4. Error bars indicate SEM; (A, B, D, and E) paired t test; (C) data representative of three to four independent experiments. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; ns, not significant.
To establish a direct link between transcription factor expression and the capacity of a cell to undergo inflammasome activation, we overexpressed IRF8 or IRF4 in immortalized BMDMs (iBMDMs) (Figures S4C–S4E). Macrophages are known to undergo robust inflammasome activation in response to inflammasome ligands and have low steady-state expression of IRF8 and IRF4 (Figure S4F). iBMDMs transduced with retrovirus expressing IRF8 or IRF4 had reduced ability to undergo caspase-1 cleavage and IL-1β secretion in response to NLRP3 inflammasome ligands (Figures 5C and 5D). Furthermore, IRF8 and IRF4 transduced iBMDMs had reduced transcriptional expression of various inflammasome-associated genes (Figure 5E). There was incomplete suppression of Nlrp3 by IRF4 and Pycard by IRF8, which suggests that complete transcriptional suppression may require the presence of a cofactor not endogenous to iBMDMs. Overall, these data provide further evidence that IRF8 and IRF4 act as negative regulators of inflammasome activation through transcriptional downregulation of inflammasome-associated machinery.
Irf8 Haploinsufficiency Permits cDC1 Inflammasome Activation and Reduces Their Ability to Prime CD8 T Cells in Response to Virulent Pathogen
We next asked if removing the regulatory effects of IRF8 from fully developed cDC1s would allow inflammasome activation in these cells. As IRF8 activity is critical for cDC1 differentiation, we used Irf8 heterozygous cDC1s which have reduced expression of IRF8 (Figure 6A). Haploinsufficiency of Irf8 reduces the proportion of cDC1s that are present in the spleen (Sichien et al., 2016; Grajales-Reyes et al., 2015), but injection of Flt3L-expressing melanoma cells allowed ~20%–40% of total splenic DCs to be of cDC1 lineage. Although WT Flt3L-induced splenic DCs failed to undergo inflammasome activation, pulsing of Irf8+/− splenic DCs with virulent STm, but not Spi1ΔSTm, resulted in caspase-1 cleavage and IL-1β secretion (Figures 6B and 6C). Additionally, LPS-stimulated Irf8+/− cDC1 cells exhibited increased expression of various genes necessary for inflammasome activation compared with WT cDC1s (Figure 6D). Expression of Nlrp3 was not affected, therefore suggesting that even limited expression of IRF8 is sufficient to suppress its transcription in cDC1s. The increased expression of other inflamma-some-associated genes, however, appears to be sufficient to drive inflammasome activation in response to STm. Although IRF8 is an important transcription factor that regulates cDC1 development, our results ascribe a critical post-developmental role for IRF8 in suppressing inflammasome activation in mature cDC1 cells.
Figure 6. Irf8 Haploinsufficiency Restores Inflammasome Activation in cDC1s and Reduces Their Ability to Prime CD8 T Cells in Response to a Virulent Pathogen.
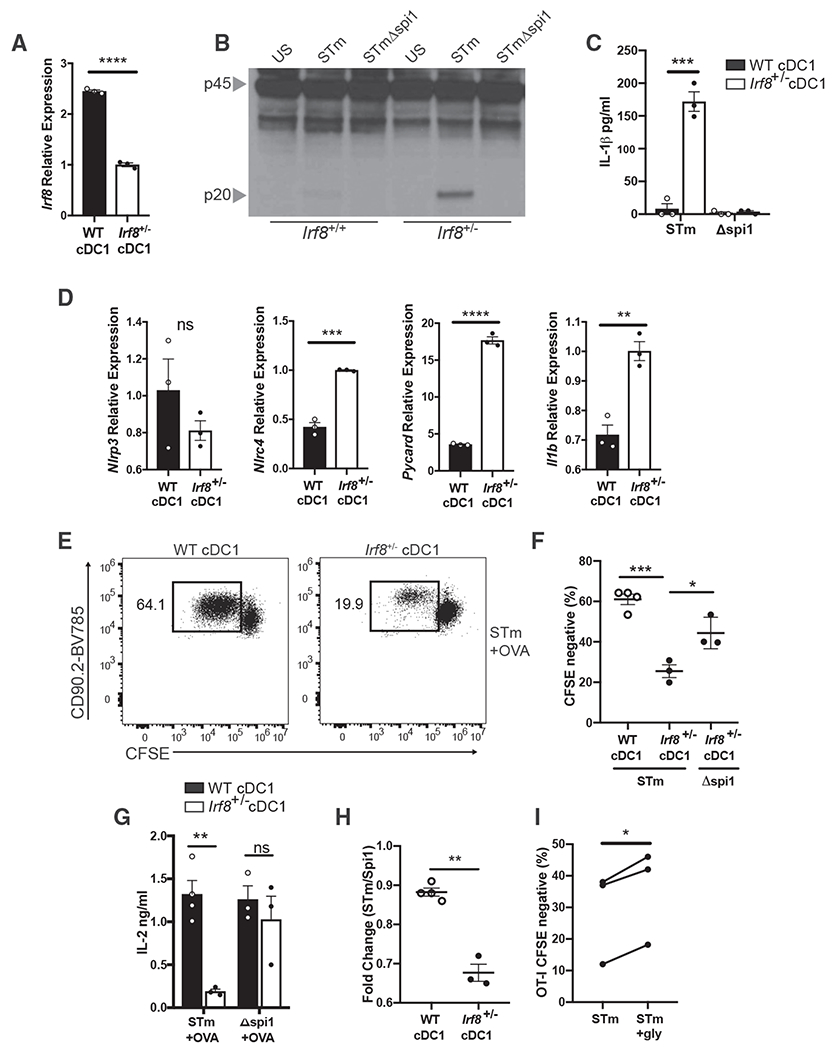
(A) qPCR of Irf8 mRNA in the lysates of FACS-sorted WT and Irf8+/− splenic cDC1s.
(B) Immunoblot analysis of pro-casp1 (p45) and cleaved casp1 (p20) in the lysates of FACS-sorted WT and Irf8+/− splenic DCs following pulse with STm or STmΔspi1 for 1 h.
(C) IL-1β was measured by ELISA in the supernatants of WT and Irf8+/− cDC1s from (B).
(D) qPCR of designated mRNA in the lysates of WT and Irf8+/− cDC1s following 4 h of LPS stimulation.
(E and F) (E) Representative data for flow cytometry of OT-I T cell proliferation and (F) quantification following 72 h of culture with FACS-sorted WT or Irf8+/− cDC1s that were pulsed with STm or STmΔspi1 and OVA.
(G) IL-2 was measured by ELISA in the supernatants of the cultures from (E).
(H) Fold change of OT-I proliferation was calculated from cDC1s of the same genotype pulsed with STm compared with STmΔspi1.
(I) OT-I T cell proliferation was quantified by flow cytometry following 72 h of culture with Irf8+/− cDC1s that were pulsed with STm and OVA in the presence or absence of 5 mM glycine.
Error bars indicate SEM; (A, C, D, and F–I) paired t test; (B and E) data representative of two independent experiments. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; ns, not significant.
We next asked if this reduced expression of IRF8 leading to inflammasome sensitivity would hamper the ability of cDC1s to prime CD8 T cells in response to virulent pathogens. We isolated cDC1 cells from WT or Irf8+/− mice and pulsed them with STm and OVA as described previously (Figure 1A). Irf8+/− cDC1s led to significantly less OT-I T cell proliferation and IL-2 production in response to virulent STm than WT cDC1s (Figures 6E–6G). Furthermore, although WT cDC1s had a similar ability to prime T cells in response to virulent or non-virulent pathogens, there was a significant decrease in the ability of Irf8+/− cDC1s to prime T cells in response to virulent STm compared with non-virulent Spi1ΔSTm (Figures 6F–6H). These data agree with our previous findings that Irf8+/− cDC1s fail to suppress inflammasome activation in response to virulent pathogens. To conclusively show that this defect in T cell priming was due to pyroptosis of DCs, we blocked cell death while leaving inflammasome activation otherwise intact by culturing the cDC1s in the presence of glycine. Irf8+/− cDC1s that were pulsed with STm in the presence of glycine led to significantly more OT-I T cell proliferation than in the absence of glycine (Figure 6I). Overall, these results conclusively demonstrate that inflammasome activation in APCs is detrimental to T cell priming and that cDCs have evolved to use specific transcription factors to mediate suppression of inflammasome activation in response to virulent pathogens.
DISCUSSION
Innate immune cells are exposed to a diverse array of microbial insults during an infection, and it is important that these cells respond to them appropriately in order to mount protective immune responses. Our study has revealed that cDCs suppress inflammasome activation and pyroptosis in response to virulent STm. We find that inflammasome activation in both BMDCs and cDCs is detrimental to the induction of adaptive immunity, as rapid pyroptosis prevents these cells from priming T cells. Blocking pyroptotic cell death did not alter MFIC and costimulatory molecule expression but rescued the ability of DCs to prime T cells. It seems that cDCs have naturally evolved to suppress inflammasome activation to protect themselves from pyroptotic cell death, thus preserving their ability to prime T cells against virulent pathogens. Further investigation found that IRF8 or IRF4 expressed by cDC1s or cDC2s, respectively, mediate suppression of inflammasome activation by negatively regulating the transcription of inflammasome-associated genes. Loss of IRF8 in cDC1s permitted these cells to undergo inflammasome activation, while overexpression of these transcription factors in macrophages suppressed inflammasome activation.
Differences between DCs and macrophages have been extensively investigated in the past. As professional APCs, DCs are specialized in their ability to carry antigen from the peripheral tissues to secondary lymphoid organs, where they upregulate MHC and costimulatory molecules to facilitate naive T cell priming (Dieu et al., 1998; Wilson and Villadangos, 2005). They also have a limited capacity for protein degradation, which allows the preservation of T cell epitopes and aids in priming a more robust T cell response (Delamarre et al., 2005). Macrophages, on the other hand, are not as efficient at priming T cells, because of their lower surface expression of MHC and differential emphasis on complete digestion of phagocytosed material (Mellman et al., 1998). Here we show that another stark difference between these two cells types is their ability to express sensors and effectors of inflammasome. This difference, in turn, has major implications for how these cell types respond to virulent infections.
Although we focus here on the role of inflammasome activation in the early stages of adaptive immune responses (T cell priming), it is important to note that DC-intrinsic inflammasome activation may have different roles in later stages of infection. Following pathogenic insult, monocyte-derived DCs are recruited to sites of infection and can aid in further T cell responses and pathogen clearance (León et al., 2007). Monocyte-derived DCs are phenotypically and functionally different than steady-state cDCs and have been shown to have the capacity for inflammasome activation (Erlich et al., 2019). Overall, our data demonstrate that cDCs, whether they are differentiated in vivo or in vitro, do not undergo inflammasome activation in response to virulent pathogens and suggest the possibility that cDCs acquired such properties to facilitate priming of productive T cell responses irrespective of the nature of the invading pathogen.
IRF8 is known to be required for the differentiation of multiple cell types, including microglia, basophils, plasmacytoid DCs (pDCs), and cDC1s, while IRF4 has a well-defined role in the differentiation of cDC2s and germinal center B cells (Tamura et al., 2000; Wang and Morse, 2009). pDCs express high levels of IRF8 and have been previously shown to be resistant to inflammasome activation (Erlich et al., 2019). Interestingly, this deficiency in inflammasome activation correlates with reduced expression of NLRP3 and pro-IL-1β. Although IRF8 deficiency in pDCs has been reported to alter their function, it remains to be investigated if their ability to activate inflammasome is restored in this setting (Sichien et al., 2016). Post-developmental functions of IRF4 and IRF8 in cell types other than DCs have also been reported. Following CD4T cell commitment to the Th17 lineage, IRF8 suppresses RORγt and inhibits further Th17 differentiation, thereby protecting the host from excessive tissue damage (Ouyang et al., 2011). In macrophages, IRF8 was shown to be important for the upregulation of an inflammatory gene program following stimulation with IFNγ (Langlais et al., 2016). IRF4 has an established post-developmental role in T and B cell function, as it is required for optimal cell proliferation and cytokine production (Mittrücker et al., 1997). Furthermore, IRF4 and IRF8 are known to negatively regulate each other, and their expression can often dictate differential cell states. B cells, for example, use these transcription facts in a redundant manner during pre-B cell differentiation, and a unique manner during activation (Xu et al., 2015).
At the molecular level, the ability of IRF8 and IRF4 to function as activators or repressors is directly influenced by the regulatory region that they target as well as the presence or absence of cofactors (Langlais et al., 2016; Biswas et al., 2010a). Further studies have shown that the phosphorylation status of IRF8 and IRF4 may also determine if it activates or represses gene transcription (Fragale et al., 2011; Biswas et al., 2010b). Although IRF4 and IRF8 play critical roles in DC development and maintenance of the tolerogenic status, whether they play an active role in regulating DC function following pathogen sensing has never been examined. Our data clearly demonstrate that IRF8 and IRF4 play fundamental roles in suppressing the expression of inflammasome-associated genes in cDC1s and cDC2s, respectively. Although the gene expression of Nlrc4 in cDC2s appears to be the same as splenic macrophages, the protein expression of NLRC4 appears to be drastically reduced in total splenic DCs, suggesting that there may be additional post-transcriptional and post-translational events that regulate its expression. This would be consistent with a recent study that reported a post-transcriptional regulatory mechanism for NLRP3 in colonic macrophages (Filardy et al., 2016). Furthermore, it remains to be investigated which specific cofactors expressed by cDC1s and cDC2s may be mediating the inhibitory effects of IRF8 and IRF4 binding to inflammasome-associated genes. The incomplete regulation of Nlrp3 by IRF4 suggests that there could be multiple layers of regulation present for these genes. For example, we find that overexpression of IRF4 in macrophages does not alter Nlrp3 levels but significantly reduces Pycard expression. In contrast, overexpression of IRF8 in macrophages downregulates the expression of Nlrp3 without altering levels of Pycard. The limiting levels of either the sensor (Nlrp3) or the adaptor (Pycard) results in the net outcome of NLRP3 inflammasome inhibition. A previous report has suggested that IRF8 positively regulates Nlrc4 gene transcription in macrophages (Karki et al., 2018), however, we find that overexpression of IRF8 in macrophages downregulates the expression of both Nlrp3 and Nlrc4. Overall our results support the idea that IRF8 and IRF4 negatively regulate a broad set of inflammasome-associated genes in cDCs, thereby impeding their ability to cleave caspase-1 and preserving their capacity to prime adaptive immune responses.
Our study finds a critical and overlapping post-developmental role for IRF8 and IRF4 in cDC function as they suppress the inflammasome response, thus maintaining DC viability during virulent pathogenic infection. This active suppression of inflammasome has an important benefit for adaptive immunity, as it allows DCs to prime CD4 and CD8 T cells irrespective of the nature of the invading pathogen. Interestingly, because IL-1β and IL-18 play a critical role in priming, differentiation, and functioning of T cells, these data also suggest that DCs potentially use inflammasome-independent pathways to produce these cytokines (Jain et al., 2020). Overall, our data establish that DCs have acquired the ability of being impartial to the virulence status of the invading microbe, thereby allowing them to suppress inflammasome activation and prime CD4 and CD8 T cell responses.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Chandrashekhar Pasare (chandrashekhar.pasare@cchmc.org). All unique/stable reagents generated in this study are available from the lead contact with a completed Material Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Mouse tissues including spleen and bone marrow were harvested as per protocols approved by IACUC at UT Southwestern Medical Center and Cincinnati Children’s Hospital Medical Center. C57BL/6J (JAX:000664), B6.OTII (JAX:004194), B6.OTI (JAX:003831), and IRF8−/− (JAX:018298) mice were purchased from Jackson Laboratory. Casp1Δ10 mice were a gift from Drs. Russell Vance and Isabella Rauch University of California, Berkeley. Mice were bred and maintained in the specific pathogen free facility at UTSW and CCHMC, provided with sterilized food and water ad libitum. Age- and sex-matched mice between 6 and 12 weeks of age were used for all experiments.
Primary cell cultures
Complete RPMI media (RPMI1640 (Hyclone), L-glutamine, penicillin-streptomycin, sodium pyruvate, β-mercaptoethanol (Sigma)) was used throughout the experiments. Mouse cell progenitors were isolated from bone marrow (femurs and tibias). Following RBC lysis, cells were plated at 0.75x10^6/ml in BMDC media (5% FCS containing complete RPMI + rGMCSF (Biolegend, 20ng/ml)). Media was replaced on day 2 and day 4, then cells were harvested for experiments on day 5 by gently flushing each well.
Cell lines
293T cells were cultured in complete DMEM media (DMEM (Hyclone), L-glutamine, sodium pyruvate, β-mercaptoethanol (Sigma), 10% FCS). iBMDM cells and B16-Flt3L were cultured in complete RPMI media (RPMI1640 (Hyclone), L-glutamine, penicillin-streptomycin, sodium pyruvate, β-mercaptoethanol (Sigma), 10% FCS).
Bacterial Strains
Salmonella typhimurium (SL1344) and STmΔspi1 (SPI1::Cm, [Lawley et al., 2006]) was streaked on a Luria-Bertani (LB) agar plate with streptomycin antibiotics. A single colony was picked and expanded in LB broth containing antibiotics overnight. Bacteria was grown to log phase (OD600 = 0.5x10^6 cfu) on the day of infection, extensively washed, and resuspended in complete RPMI media containing 1% FCS.
METHOD DETAILS
Isolation of splenic DCs
Spleens were harvested from WT mice and CD11c+ cells were magnetically sorted using positive selection on AutoMacs (Miltenyi). Approximately 95% of cells were CD11c+MHCII+. Flt3L expressing B16 melanoma cells were subcutaneously injected into the mouse flank and allowed to grow for 10-15 days before spleens were harvested (Mach et al., 2000) for cDC isolation.
Isolation of lymphocytes
Spleen and lymph nodes were harvested from 6-10 weeks old OT-II or OT-I mice. Naive CD4 or CD8 T cells were isolated according to the MojoSort kit protocol (Biolegend). The purity of naive OT-II and OT-I T cells was consistently monitored and maintained at > 95%. Cells were washed extensively with PBS, then labeled with CFSE (Biolegend) or CellTrace Violet (Thermo Fisher), then washed extensively with complete RPMI. For all experiments DC donor mice and naive T cell donor mice were age- and sex-matched.
Bacterial pulse and culture of DCs
DCs were infected at MOI:1 for 1hr, then incubated with gentamycin (200μg/ml) for 1 hr. Cells were washed twice with complete media, then cultured with labeled naive T cells at a ratio of 1:5 DCs to T cells for 72hrs.
Viral production and BMDC transduction
IRF4-hCD4, IRF8-hCD4, and empty-hCD4 retrovirus was produced by transfection of 293T with respective expression plasmids and packaging plasmid pCL-Eco. Viral supernatant was collected after 60hrs and filtered priorto use. IRF4-GFP, IRF8-GFP, and empty-GFP lentivirus was produced by transfection of 293T with respective expression plasmids, VSV-G, and PAX2. Viral supernatant was collected after 60hrs and filtered prior to use.
For retroviral transduction of BMDC precursors, following RBC lysis cells were plated at 10x10^6/ml in 2ml of retroviral supernatant containing 8μl/ml polybrene. Cells were spinfected at 2500 rpm, 32°C, for 90min, then 3ml of BMDC media was added to each well and cells were incubated overnight. The next morning, ~70% of the media was removed from the wells and spinfection was repeated with fresh retroviral supernatant. On day 5 cells were harvested for experiments.
Enzyme-linked immunosorbent assay (ELISA)
Supernatants were stored at −80°C upon harvest. Capture antibodies were diluted and used to coat ELISA plate overnight at 4°C. Plates were blocked with PBS containing 10% FCS. Samples were diluted in PBS containing 10% FCS, loaded in duplicate, and incubated overnight at 4°C. Capture and detection antibodies were used according to manufacturer’s instruction (IL-1β, R&D Systems; IL-2, BD). Protein concentration was quantified using OPD colorimetric assay.
Immunoblot analysis
Cells were washed with PBS and lysed using RIPA buffer containing protease inhibitors. Protein concentration was measured by a detergent resistant Bradford assay, and equal amounts of protein were boiled with SDS-containing Laemmli sample buffer. Western blotting was performed following standard procedure. Primary antibodies used were anti-caspase-1 (Genentech), anti-NLRP3 (Adipogen), anti-NLRC4 (LSBio), anti-ASC (SCBT), anti-IL1b (R&D), anti-IRF8 (Biolegend), anti-IRF4 (Biolegend), anti-histone H3 (CST), or anti-β-actin (SCBT). HRP (Jackson Lab) conjugated secondary antibodies were used. Proteins were visualized by using Chemi-luminescent SuperSignal (Thermo Fisher) and ECL signal was recorded on X-ray films using a developer (Kodak).
Quantitative real-time PCR
Cells were washed with PBS, lysed in Trizol, and stored at −80°C. Total RNA was isolated with chloroform and isopropanol, then treated with DNase1 (QIAGEN) and cleaned up with RNeasy mini kit (QIAGEN). cDNA synthesis was performed with M-MLV reverse transcriptase (Invitrogen) in the presence of RNase inhibitor (Promega). Quantitative RT-PCR was performed using SYBR green 2x Mastermix (Applied Biosystems) and measured using the QuantStudio 7 Flex Real-Time PCR System (Thermo Fisher). Data was normalized to Hprt. The primers used in this study are as follows: mouse Nlrp3 forward: 5′-GACACGAGTCCTGGTGACTTT-3′ reverse: 5′-GTGTCATTCCACTCTGGCTGG-3′; mouse Pycard forward: 5′-GTCTTAGGGGCGGAAACCAA-3′ reverse: 5′-TGGTCCACAAAGTGTCCTGT-3′; mouse Casp1 forward: 5′-AAGAATTTGCTGCCTGCCCA-3′ reverse: 5′-CCTTGTTTCTCTCCACGGCAT-3′; mouse Il1b forward: 5′-TGTGCTCTGCTTGTGAGGTGCTG-3′ reverse: 5′-CCCTGCAGCTGGAGAGTGTGGA-3′; mouse Nlrc4 forward: 5′-TCACCACGGATGACGAACAG-3′ reverse: 5′-GTCAATCAGACCACCTGGCA-3′; mouse Irf4 forward: 5′-AGATTCCAGGTGACTCTGTG-3′ reverse: 5′-TGCCCTGTCAGAGTATTTC-3′; mouse Irf8 forward: 5′-CCTATGACACACACCATTCAGC-3′ reverse: 5′-AGAGACGGCAGCCTTCAA-3′; mouse Hprt forward: 5′-CAGTCCCAGCGTCGTGATTA-3′ reverse: 5′-TGGCCTCCCATCTCCTTCAT-3′.
Pyroptosis assay
Cells were plated and cultured in 1% FCS containing complete RPMI. Supernatants were collected from cultured cells after the specified time. LDH reagent was reconstituted and added to samples according to manufacturer’s instruction (Takara).
Inflammasome activation
DCs were harvested then plated at 1x10^6/ml in 1% or 10% FCS containing complete RPMI depending on downstream analyses. Cells were stimulated with 100ng/ml LPS or PAM3 for 4hrs, then NLRP3 (5mM ATP), NLRC4 (1μg/ml PA + 1μg/ml LFn-FlaA) (von Moltke et al., 2012), or AIM2 (LF2k + 2μg/ml poly(dA:dT)) ligands for 30min. Cells were immediately harvested on ice. Supernatants were collected and cells were treated based on downstream application.
Glycine was dissolved in PBS and filter-sterilized prior to use. A final concentration of 5mM was added to cell culture to prevent inflammasome dependent pyroptosis.
Flow cytometry and cell sorting
For surface marker staining, cells were blocked with Fc Shield (anti-mouse CD16/CD32, Tonbo) for 10min then incubated with antibodies of interest for 30 min. Samples were washed extensively with sterile FACS buffer (PBS, 2% FCS, 2mM EDTA) then analyzed using Novocyte 3001 (ACEA Biosciences). Cells were gated on singlets and dead cells were excluded using Zombie Yellow live/dead staining (Biolegend). Data were analyzed using FlowJo software (BD).
For cell sorting, samples were stained, washed, then sorted by Moflo XDP (Beckman Coulter) directly into 10% FCS containing complete RPMI media. Cells were washed once with complete RPMI media before culturing.
FLICA staining
BMDCs were retrovirally transduced with mock, IRF8, or IRF4 expressing virus. Non-adherent cells were harvested, stained for transduction marker hCD4, and subjected to inflammasome activation. FLICA was added to a final concentration of 1X at the same time ATP was added to the culture and incubated for 30min at 37°C. Cells were washed in accordance with manufacturer’s instruction (ImmunoChemistry) and PI was added to samples prior to analysis on a flow cytometer.
Lentiviral transduction of iBMDMs
iBMDMs (1x10^6) were plated in 2 mL of lentiviral supernatant containing 8 μl/ml polybrene. Cells were spinfected at 2500 rpm, 32°C, for 90min, then 3ml of 10% FCS containing complete RPMI media was added to each well and cells were incubated overnight. 48hrs later, cells were collected and GFP+ cells were sorted as single cells into 96 well plates. Clones were expanded and characterized based on their expression of the desired construct.
ChIP-qPCR
WT BMDCs and splenic DCs were crosslinked with 1% formaldehyde for 10 minutes at room temperature followed by glycine quenching. Samples were washed with ice cold PBS and pelleted followed by lysis with SDS buffer and sonication using a Covaris sonicator. Equal amounts of DNA were used for IgG (SCBT), IRF8 (SCBT), and IRF4 (CST) immunoprecipitation. Immunoprecipitated DNA was eluted and amplified with the following primers for the promoter or intronic regions: chip-Nlrp3 forward: 5′-CCATCCAGATGAGTAACTGCCA-3′ reverse: 5′-ACTCACCCAGATAGCAGCCT-3′; chip-Pycard forward: 5′-ACTGCCATGCAAAGCATCCA-3′ reverse: 5′-TCAGGGTAGCAATAGCCTCTTC-3′; chip-Il1b forward: 5′-GTCCAACTTGTTTTCCCTCCC-3′ reverse: 5′-CCCTGCAGCTGGAGAGTGTGGA-3′; chip-Nlrc4 forward: 5′-GAGAAGCAGGTTAGGTAGGGC-3′ reverse: 5′-CAGGACACCAGCAGGAACTC-3′; chip-negative forward: 5′-TCTGTGTTCACAGGTTGGGT-3′ reverse: 5′-CCACGCCTACCAGGAAATCT-3′.
Luciferase reporter assay
The mouse NLRP3 promoter (−4000 bp to 0 bp upstream of the transcription start site) was cloned from cDNA into pGL4.21.293T cells were transfected with luciferase reporter plasmid, pRL-TK, and expression plasmids (100ng MyD88, titrating IRF4/IRF8 25ng-500ng) using Lipofectamine 2000 (Invitrogen). Cells were incubated for 18hrs and collected using passive lysis buffer (Promega). Luciferase activity was determined by Dual-Glo Luciferase Assay (Promega).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed in Prism (Graphpad) using unpaired or paired Student’s t test as indicated in the figure legends. Data are presented as mean ± SEM. Significance was considered at *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns = not significant.
DATA AND CODE AVAILABILITY
This study did not generate any unique datasets or code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-mouse IL-2 | BD | Cat# 554424; RRID: AB_395383 |
| Biotin rat anti-mouse IL-2 | BD | Cat# 554426; RRID: AB_395384 |
| Rat anti-mouse caspase-1 | Genentech | 4B4 |
| anti-mouse NLRP3 mAb | Adipogen | Cat# AG-20B-0014; RRID: AB_2490202 |
| Rabbit anti-mouse NLRC4 | LSBio | LS-C407885 |
| Rabbit anti-mouse ASC (N-15) | SCBT | 22514 |
| Goat anti-mouse IL1b | RnD | Cat# AF-401-NA; RRID: AB_416684 |
| Rat anti-mouse IRF8 | Biolegend | Cat# 656501; RRID: AB_2562396 |
| Rat anti-mouse IRF4 | Biolegend | Cat# 646402; RRID: AB_2280462 |
| Histone H3 Rabbit Ab | CST | Cat# 9715; RRID: AB_331563 |
| Beta-tubulin Rabbitt Ab | CST | Cat# 2146; RRID: AB_2210545 |
| Anti-mouse CD16/CD32 | Tonbo | Cat# 70-0161; RRID: AB_2621487 |
| Anti-mouse CD90.2-BV785 | Biolegend | Cat# 105331; RRID: AB_2562900 |
| Anti-mouse CD11c-APC | Invitrogen | Cat# 17-0114-82; RRID: AB_469346 |
| Anti-mouse MHCII-FITC | Biolegend | Cat# 107606; RRID: AB_313321 |
| Anti-mouse CD80-APC/Fire750 | Biolegend | Cat# 104740; RRID: AB_2687095 |
| Anti-mouse CD86-PE | BD | Cat# 553692; RRID: AB_394994 |
| Normal goat IgG | SCBT | Cat# sc-2028; RRID: AB_737167 |
| ICSBP (C-19) | SCBT | Cat# sc-6058; RRID: AB_649510 |
| IRF4 D9P5H | CST | Cat# 15106; RRID: AB_2798709 |
| Bacterial and Virus Strains | ||
| Salmonella typhimurium, SL1344 | Laboratory of Denise Monack, Lawley et al. (2006) | N/A |
| Salmonella typhimurium, SPI1::Cm | Laboratory of Denise Monack, Lawley et al. (2006) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| LPS | Sigma | L2880 |
| Pam3 | Invivogen | tlrl-pms |
| ATP | Sigma | A1852 |
| FlaTox | von Moltke et al. (2012) | N/A |
| Poly(dA:dT) | Invitrogen | tlrl-patn |
| Glycine | Fisher | BP381 |
| EndoFit Ovalbumin | Invivogen | Vac-pova |
| Gentamycin Reagent | GIBCO | 15750-060 |
| OPD | Sigma | P1526 |
| CellTrace Violet | Thermo Fisher | C34571 |
| CFSE | Biolegend | 423801 |
| GM-CSF | Biolegend | 576306 |
| Lipofectamine 2000 | Invitrogen | 116680 |
| Polybrene | Sigma | TR-1003 |
| Critical Commercial Assays | ||
| LDH Assay | Takara | MK401 |
| FAM-FLICA | ImmunoChemistry | 97 |
| Dual-Glo Luciferase Assay | Promega | E2920 |
| Mouse IL-1b DuoSet ELISA | RnD | DY401 |
| Naive CD4 Mojosort | Biolegend | 480039 |
| Naive CD8 Mojosort | Biolegend | 480043 |
| Zombie Yellow Fixable Viability Kit | Biolegend | 423103 |
| Experimental Models: Cell Lines | ||
| 293T | Laboratory of Ruslan Medzhitov | N/A |
| iBMDM | Laboratory of Kate Fitzgerald | N/A |
| B16-Flt3L | Mach et al. (2000) | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | 000664 |
| Mouse: OTII | Jackson Laboratory | 004194 |
| Mouse: OTI | Jackson Laboratory | 003831 |
| Mouse: IRF8−/− | Jackson Laboratory | 018298 |
| Mouse: Casp1d10 | Jackson Laboratory | 032662 |
| Oligonucleotides | ||
| Primers, see STAR Methods, Quantitative real-time PCR section | This paper | N/A |
| Recombinant DNA | ||
| pCL-ECO | Naviaux et al., 1996 | RRID: Addgene_12371 |
| VSV.G | Reya et al., 2003 | RRID: Addgene_14888 |
| psPAX2 | Laboratory of Didier Trono | RRID: Addgene_12260 |
| pGL4.21 | Promega | E6761 |
| pGL4.21-Nlrp3-4kb | This paper | N/A |
| pRL-TK | Promega | E2241 |
| MSCV-IRES-CD2-MYD88 | Laboratory of C. Pasare | N/A |
| MSCV-IRF4-hCD4 | Laboratory of Harinder Singh | N/A |
| MSCV-IRF8-hCD4 | Laboratory of Harinder Singh | N/A |
| pHIV-IRF4-GFP | This paper | N/A |
| pHIV-IRF8-GFP | This paper | N/A |
| Software and Algorithms | ||
| Graphpad Prism 7 | Graphpad Software | N/A |
| FlowJo | FlowJo, LLC | N/A |
| BioRender | BioRender | N/A |
Highlights.
Virulent pathogens fail to activate inflammasomes in conventional dendritic cells
Inflammasome activation in DCs is detrimental to the induction of adaptive immunity
Suppression of pyroptosis in DCs is necessary for priming of naive T cells
cDCs use IRF4 and IRF8 to suppress transcription of inflammasome-associated genes
ACKNOWLEDGMENTS
We thank all the members of the Pasare lab for their insight, helpful discussions, and critical reading of this manuscript. Special thanks to Lisa Waggoner for maintaining the mouse colony and the proper functioning of the lab. We thank Denise Monack (Stanford) for sharing WT STm and Spi1ΔSTm, Russell Vance (Berkeley) for sharing FlaTox, Jon Kagan (Harvard) for sharing Gasd−/− bones, and Omer Donmez (Cincinnati Children’s Hospital Medical Center [CCHMC]) for ChIP-qPCR expertise. This work was supported by grants from the National Institutes of Health (AI113125, AI123176, and GM120196) to C.P. and by the National Science Foundation Graduate Research Fellowship under grant 2017220107 to M.M.M.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107604.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, Dinarello CA, and Paul WE (2009). IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc. Natl. Acad. Sci. U S A 106, 7119–7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj V, Kanagawa O, Swanson PE, and Unanue ER (1998). Chronic Listeria infection in SCID mice: requirements for the carrier state and the dual role of T cells in transferring protection or suppression. J. Immunol 160, 376–384. [PubMed] [Google Scholar]
- Biswas PS, Bhagat G, and Pernis AB (2010a). IRF4 and its regulators: evolving insights into the pathogenesis of inflammatory arthritis? Immunol. Rev 233, 79–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas PS, Gupta S, Chang E, Song L, Stirzaker RA, Liao JK, Bhagat G, and Pernis AB (2010b). Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J. Clin. Invest 120, 3280–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, and Monack DM (2010). Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J. Exp. Med 207, 1745–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, and Monack DM (2012). Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490, 288–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delamarre L, Pack M, Chang H, Mellman I, and Trombetta ES (2005). Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science 307, 1630–1634. [DOI] [PubMed] [Google Scholar]
- Dieu MC, Vanbervliet B, Vicari A, Bridon JM, Oldham E, Aït-Yahia S, Brière F, Zlotnik A, Lebecque S, and Caux C (1998). Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J. Exp. Med 188, 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostert C, Ludigs K, and Guarda G (2013). Innate and adaptive effects of inflammasomes on T cell responses. Curr. Opin. Immunol 25, 359–365. [DOI] [PubMed] [Google Scholar]
- Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee HW, Park CG, et al. (2007). Differential antigen processing by dendritic cell subsets in vivo. Science 315, 107–111. [DOI] [PubMed] [Google Scholar]
- Erlich Z, Shlomovitz I, Edry-Botzer L, Cohen H, Frank D, Wang H, Lew AM, Lawlor KE, Zhan Y, Vince JE, and Gerlic M (2019). Macrophages, rather than DCs, are responsible for inflammasome activity in the GM-CSF BMDC model. Nat. Immunol 20, 397–406. [DOI] [PubMed] [Google Scholar]
- Filardy AA, He J, Bennink J, Yewdell J, and Kelsall BL (2016). Posttranscriptional control of NLRP3 inflammasome activation in colonic macrophages. Mucosal Immunol. 9, 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink SL, and Cookson BT (2006). Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol 8, 1812–1825. [DOI] [PubMed] [Google Scholar]
- Fink SL, and Cookson BT (2007). Pyroptosis and host cell death responses during Salmonella infection. Cell. Microbiol 9, 2562–2570. [DOI] [PubMed] [Google Scholar]
- Fragale A, Stellacci E, Ilari R, Remoli AL, Lanciotti A, Perrotti E, Shytaj I , Orsatti R, Lawrence HR, Lawrence NJ, et al. (2011). Critical role of IRF8 in negative regulation of TLR3 expression by Src homology 2 domain-containing protein tyrosine phosphatase-2 activity in human myeloid dendritic cells. J. Immunol 186, 1951–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Deason K, Jain A, Irizarry-Caro RA, Dozmorov I, Coughlin LA, Rauch I, Evers BM, Koh AY, Wakeland EK, and Pasare C (2020).Transcriptional profiling identifies caspase-1 as a T cell–intrinsic regulator of Th17 differentiation. J. Exp. Med 217, e20190476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlanda C, Dinarello CA, and Mantovani A (2013). The interleukin-1 family: back to the future. Immunity 39, 1003–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, et al. (2009). Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med 15, 1170–1178. [DOI] [PubMed] [Google Scholar]
- Glasmacher E, Agrawal S, Chang AB, Murphy TL, Zeng W, Vander Lugt B, Khan AA, Ciofani M, Spooner CJ, Rutz S, et al. (2012). A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science 338, 975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grajales-Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, Kc W, Kretzer NM, Briseño CG, Durai V, Bagadia P, et al. (2015). Batf3 maintains autoactivation of Irf8 for commitment of a CD8α(+) conventional DC clonogenic progenitor. Nat. Immunol 16, 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarda G, Zenger M, Yazdi AS, Schroder K, Ferrero I, Menu P, Tardivel A, Mattmann C, and Tschopp J (2011). Differential expression of NLRP3 among hematopoietic cells. J. Immunol 186, 2529–2534. [DOI] [PubMed] [Google Scholar]
- Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E,Tussiwand R, and Yona S (2014). Dendriticcells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat. Rev. Immunol 14, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, Goubau D, and Reis e Sousa C (2015). GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendriticcells. Immunity 42, 1197–1211. [DOI] [PubMed] [Google Scholar]
- Hess J, Ladel C, Miko D, and Kaufmann SH (1996). Salmonella typhimurium aroA- infection in gene-targeted immunodeficient mice: major role of CD4+ TCR-alpha beta cells and IFN-gamma in bacterial clearance independent of intracellular location. J. Immunol 156, 3321–3326. [PubMed] [Google Scholar]
- Huang Q, Liu D, Majewski P, Schulte LC, Korn JM, Young RA, Lander ES, and Hacohen N (2001). The plasticity of dendritic cell responses to pathogens and their components. Science 294, 870–875. [DOI] [PubMed] [Google Scholar]
- Idoyaga J, Fiorese C, Zbytnuik L, Lubkin A, Miller J, Malissen B, Mucida D, Merad M, and Steinman RM (2013). Specialized role of migratory dendritic cells in peripheral tolerance induction. J. Clin. Invest 123, 844–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K, Turley S, Iyoda T, Yamaide F, Shimoyama S, Reis e Sousa C, Germain RN, Mellman I, and Steinman RM (2000). The formation of immunogenic major histocompatibility complex class II-peptide ligands in lysosomal compartments of dendritic cells is regulated by inflammatory stimuli. J. Exp. Med 191, 927–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Irizarry-Caro RA, McDaniel MM, Chawla AS, Carroll KR, Overcast GR, Philip NH, Chervonsky AV, Oberst A, Katz JD, and Pasare C (2020). T cells instruct myeloid cells to produce inflammasome-independent IL-1β and cause autoimmunity. Nat. Immunol 21, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, and Pasare C (2017). Innate control of adaptive immunity: beyond the three-signal paradigm. J. Immunol 198, 3791–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Song R, Wakeland EK, and Pasare C (2018). T cell-intrinsic IL-1R signaling licenses effector cytokine production by memory CD4 T cells. Nat. Commun 9, 3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TB, Yang SH, Toth B, Kovalenko A, and Wallach D (2013). Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38, 27–40. [DOI] [PubMed] [Google Scholar]
- Karki R, Lee E, Place D, Samir P, Mavuluri J, Sharma BR, Balakrishnan A, Malireddi RKS, Geiger R, Zhu Q, et al. (2018). IRF8 regulates transcription of Naips for NLRC4 inflammasome activation. Cell 173, 920–933.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kc W, Satpathy AT, Rapaport AS, Briseño CG, Wu X, Albring JC, Russler-Germain EV, Kretzer NM, Durai V, Persaud SP, et al. (2014). L-Myc expression by dendritic cells is required for optimal T-cell priming. Nature 507, 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi M, Miki T, Kumagai T, Fukuda T, Kamiyama R, Miyasaka N, and Hirosawa S (2000). Identification of negative regulatory regions within the first exon and intron of the BCL6 gene. Oncogene 19, 4941–4945. [DOI] [PubMed] [Google Scholar]
- Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, and Weirauch MT (2018). The human transcription factors. Cell 175, 598–599. [DOI] [PubMed] [Google Scholar]
- Langlais D, Barreiro LB, and Gros P (2016). The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J. Exp. Med 213, 585–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen CP, Ritchie SC, Pearson TC, Linsley PS, and Lowry RP (1992). Functional expression of the costimulatory molecule, B7/BB1, on murine dendritic cell populations. J. Exp. Med 176, 1215–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz E, Xiao TS, and Stutz A (2013). Activation and regulation of the inflammasomes. Nat. Rev. Immunol 13, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley TD, Chan K, Thompson LJ, Kim CC, Govoni GR, and Monack DM (2006). Genome-wide screen for Salmonella genes required for long-term systemic infection of the mouse. PLoS Pathog. 2, e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- León B, López-Bravo M, and Ardavín C (2007). Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity 26, 519–531. [DOI] [PubMed] [Google Scholar]
- Li H, Zhang S, Li F, and Qin L (2016). NLRX1 attenuates apoptosis and inflammatory responses in myocardial ischemia by inhibiting MAVS-dependent NLRP3 inflammasome activation. Mol. Immunol 76, 90–97. [DOI] [PubMed] [Google Scholar]
- Lin KM, Hu W, Troutman TD, Jennings M, Brewer T, Li X, Nanda S, Cohen P, Thomas JA, and Pasare C (2014). IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc. Natl. Acad. Sci. U S A 111,775–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WF, Ong H, Metcalf ES, and Soloski MJ (1999). T cell responses to Gram-negative intracellular bacterial pathogens: a role for CD8+ T cells in immunity to Salmonella infection and the involvement of MHC class Ib molecules. J. Immunol 162, 5398–5406. [PubMed] [Google Scholar]
- Mach N, Gillessen S, Wilson SB, Sheehan C, Mihm M, and Dranoff G (2000). Differences in dendritic cells stimulated in vivo by tumors engineered to secrete granulocyte-macrophage colony-stimulating factor or Flt3-ligand. Cancer Res. 60, 3239–3246. [PubMed] [Google Scholar]
- Martinon F, Burns K, and Tschopp J (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426. [DOI] [PubMed] [Google Scholar]
- Martinon F, Mayor A, and Tschopp J (2009). The inflammasomes: guardians of the body. Annu. Rev. Immunol 27, 229–265. [DOI] [PubMed] [Google Scholar]
- Mellman I, Turley SJ, and Steinman RM (1998). Antigen processing for amateurs and professionals. Trends Cell Biol. 8, 231–237. [DOI] [PubMed] [Google Scholar]
- Meylan E, Tschopp J, and Karin M (2006). Intracellular pattern recognition receptors in the host response. Nature 442, 39–44. [DOI] [PubMed] [Google Scholar]
- Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, and Aderem A (2010). Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol 11, 1136–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittrücker HW, Matsuyama T, Grossman A, Kündig TM, Potter J, Shahinian A, Wakeham A, Patterson B, Ohashi PS, and Mak TW (1997). Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science 275, 540–543. [DOI] [PubMed] [Google Scholar]
- Naik SH, Sathe P, Park HY, Metcalf D, Proietto AI, Dakic A, Carotta S, O’Keeffe M, Bahlo M, Papenfuss A, et al. (2007). Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat. Immunol 8, 1217–1226. [DOI] [PubMed] [Google Scholar]
- Naviaux RK, Costanzi E, Haas M, and Verma IM (1996). The pCL Vector System: Rapid Production of Helper-Free, High-Titer, Recombinant Retroviruses. J. Virol 70, 5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orabona C, Puccetti P, Vacca C, Bicciato S, Luchini A, Fallarino F, Bianchi R, Velardi E, Perruccio K, Velardi A, et al. (2006). Toward the identification of a tolerogenic signature in IDO-competent dendritic cells. Blood 107, 2846–2854. [DOI] [PubMed] [Google Scholar]
- Ouyang X, Zhang R, Yang J, Li Q, Qin L, Zhu C, Liu J, Ning H, Shin MS, Gupta M, et al. (2011). Transcription factor IRF8 directs a silencing programme for TH17 cell differentiation. Nat. Commun 2, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu CH, Miyake Y, Kaise H, Kitamura H, Ohara O, and Tanaka M (2009). Novel subset of CD8alpha+ dendritic cells localized in the marginal zone is responsible for tolerance to cell-associated antigens. J. Immunol 182,4127–4136. [DOI] [PubMed] [Google Scholar]
- Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz l. , Nusse R, and Weissman IL (2003). A Role for Wnt Signalling in Self-Renewal of Haematopoietic Stem Cells. Nature 423, 409–414. [DOI] [PubMed] [Google Scholar]
- Robertson JM, Jensen PE, and Evavold BD (2000). DO11.10 and OT-II T cells recognize a C-terminal ovalbumin 323-339 epitope. J. Immunol 164, 4706–4712. [DOI] [PubMed] [Google Scholar]
- Schnorrer P, Behrens GM, Wilson NS, Pooley JL, Smith CM, El-Sukkari D, Davey G, Kupresanin F, Li M, Maraskovsky E, et al. (2006). The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture. Proc. Natl. Acad. Sci. U S A 103, 10729–10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Sichien D, Scott CL, Martens L, Vanderkerken M, Van Gassen S, Plantinga M, Joeris T, De Prijck S, Vanhoutte L, Vanheerswynghels M, et al. (2016). IRF8 transcription factor controls survival and function of terminally differentiated conventional and plasmacytoid dendritic cells, respectively. Immunity 45, 626–640. [DOI] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, and Akira S (2003). Toll-like receptors. Annu. Rev. Immunol 21, 335–376. [DOI] [PubMed] [Google Scholar]
- Tamura T, Nagamura-Inoue T, Shmeltzer Z, Kuwata T, and Ozato K (2000). ICSBP directs bipotential myeloid progenitor cells to differentiate into mature macrophages. Immunity 13, 155–165. [DOI] [PubMed] [Google Scholar]
- Tamura T, Tailor P, Yamaoka K, Kong HJ, Tsujimura H, O’Shea JJ, Singh H, and Ozato K (2005). IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversity. J. Immunol 174, 2573–2581. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, et al. (1992). A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356, 768–774. [DOI] [PubMed] [Google Scholar]
- Tokuhiro S, Yamada R, Chang X, Suzuki A, Kochi Y, Sawada T, Suzuki M , Nagasaki M, Ohtsuki M, Ono M, et al. (2003). An intronic SNP in a RUNX1 binding site of SLC22A4, encoding an organic cation transporter, is associated with rheumatoid arthritis. Nat. Genet 35, 341–348. [DOI] [PubMed] [Google Scholar]
- Tsuchiya K, Nakajima S, Hosojima S, Thi Nguyen D, Hattori T, Manh Le T, Hori O, Mahib MR, Yamaguchi Y, Miura M, et al. (2019). Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun 10, 2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Lugt B, Khan AA, Hackney JA, Agrawal S, Lesch J, Zhou M, Lee WP, Park S, Xu M, DeVoss J, et al. (2014). Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat. Immunol 15, 161–167. [DOI] [PubMed] [Google Scholar]
- Vander Lugt B, Riddell J, Khan AA, Hackney JA, Lesch J, DeVoss J, Weirauch MT, Singh H, and Mellman I (2017). Transcriptional determinants of tolerogenic and immunogenic states during dendritic cell maturation. J. Cell Biol. 216, 779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke J,Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, Brown CR, Krantz BA, Leppla SH, Gronert K, and Vance RE (2012). Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 490, 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke J, Ayres JS, Kofoed EM, Chavarría-Smith J, and Vance RE (2013). Recognition of bacteria by inflammasomes. Annu. Rev. Immunol 31, 73–106. [DOI] [PubMed] [Google Scholar]
- Wang H, and Morse HC 3rd. (2009). IRF8 regulates myeloid and B lymphoid lineage diversification. Immunol. Res 43, 109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Koblansky AA, and Ghosh S (2006). Recognition and signaling by toll-like receptors. Annu. Rev. Cell Dev. Biol. 22, 409–437. [DOI] [PubMed] [Google Scholar]
- Wilson NS, and Villadangos JA (2005). Regulation of antigen presentation and cross-presentation in the dendritic cell network: facts, hypothesis, and immunological implications. Adv. Immunol 86, 241–305. [DOI] [PubMed] [Google Scholar]
- Wong P, and Pamer EG (2003). CD8 T cell responses to infectious pathogens. Annu. Rev. Immunol 21, 29–70. [DOI] [PubMed] [Google Scholar]
- Xu H, Chaudhri VK, Wu Z, Biliouris K, Dienger-Stambaugh K, Rochman Y, and Singh H (2015). Regulation of bifurcating B cell trajectories by mutual antagonism between transcription factors IRF4 and IRF8. Nat. Immunol 16, 1274–1281. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.