

Abstract
Brain astrocytes are implicated in estrogenic neuroprotection against bio-energetic insults, which may involve their glycogen energy reserve. Forebrain estrogen receptors (ER)-alpha (ERα) and -beta (ERβ) exert differential control of glycogen metabolic enzyme [glycogen synthase (GS); phosphorylase (GP)] expression in hypoglycemic male versus female rats. Studies were conducted using a rat hypothalamic astrocyte primary culture model along with selective ER agonists to investigate the premise that estradiol (E2) exerts sex-dimorphic control over astrocyte glycogen mass and metabolism. Female astrocyte GS and GP profiles are more sensitive to E2 stimulation than the male. E2 did not regulate expression of phospho-GS (inactive enzyme form) in either sex. Data also show that transmembrane G protein-coupled ER-1 (GPER) signaling is implicated in E2 control of GS profiles in each sex and alongside ERα, GP expression in females. E2 increases total 5’-AMK-activated protein kinase (AMPK) protein in female astrocytes, but stimulated pAMPK (active form) expression with equivalent potency via GPER in females and ERα in males. In female astrocytes, ERα protein was up-regulated at a lower E2 concentration and over a broader dosage range compared to males, whereas ERβ was increased after exposure to 1–10 nM versus 100 pM E2 levels in females and males, respectively. GPER profiles were stimulated by E2 in female, but not male astrocytes. E2 increased astrocyte glycogen content in female, but not male astrocytes; selective ERβ or ERα stimulation elevated glycogen levels in the female and male, respectively. Outcomes imply that dimorphic astrocyte ER and glycogen metabolic responses to E2 may reflect, in part, differential steroid induction of ER variant expression and/or regulation of post-receptor signaling in each sex.
Keywords: estrogen receptor-alpha, G protein-coupled estrogen receptor, glycogen synthase, glycogen phosphorylase, 5’-AMP-activated protein kinase, calcium/calmodulin-dependent protein kinase kinase-beta
Introduction:
The ovarian steroid hormone 17β-estradiol (E2) provides neuroprotection against injury or demise of vulnerable brain nerve cell populations during cerebral ischemia and other bio-energetic insults, and accounts in part for sex-dimorphic susceptibility to acute ischemic stress-associated tissue damage and behavioral deficits [Lebesque et al., 2009; Scott et al., 2012]. Estradiol activation of classical nuclear and membrane estrogen receptor signaling, including (estrogen receptor-alpha (ERα) [Dubal et al., 2001; Zhang et al., 2009; Spence et al., 2013], estrogen receptor-beta (ERβ) [Miller et al., 2005], and G-protein-coupled ER (GPER1/GPR30) [Gingerich et al., 2010], is implicated in hormone-mediated neuroprotection against bioenergetics brain injury. E2 acts directly on nerve cells [Elzer et al., 2010] to stabilize mitochondrial structure and function and to attenuate oxidative stress [Nilsen and Brinton, 2004; Simpkins and Dykens, 2008]. Estrogen-dependent neuroprotection is also achieved by indirect mechanisms involving neuroglia-neuron interaction [Azcoitia et al., 2010; Johann and Beyer, 2013; Spence et al., 2013; Acaz-Fonseca et al., 2014; Barreto, 2016; Ma et al., 2016].
Brain astrocytes promote optimal nerve cell function through glio-transmitter signaling, regulation of micro-circulatory function, and provision of metabolic substrate fuel [Stobart and Anderson, 2013]. Glucose, the primary energy source of the brain, is taken up from the circulation by astrocytes and either stored in glia as glycogen or converted to the oxidizable fuel L-lactate for trafficking to neurons [Laming et al., 2000]. Transfer of lactate between astrocyte and nerve cell compartments is accomplished by astrocyte-and neuron-specific monocarboxylate transporters (MCT1 and MCT2), respectively [Broer et al., 1997]. In the brain and periphery, glycogen metabolism is governed by opposing actions of the enzymes glycogen synthase (GS) and glycogen phosphorylase (GP), which respectively catalyze glycogen synthesis or breakdown. Brain glycogen undergoes dynamic turnover during normal brain activity and metabolic stasis, and is a vital metabolic fuel source during states of heightened activity or glucose deficiency, including seizure, sleep deprivation, and hypoglycemia [Gruetter, 2003; Brown, 2004]. Pharmacological augmentation of brain glycogen mass prolongs brain electrical activity during hypoglycemia and prevents nerve cell loss, indicating that this energy depot sustains neuronal function and survival during bioenergetic stress [Suh et al., 2007]. Reports that inhibition of GP activity [Alhamami et al., 2018] or monocarboxylate transporter function [Mahmood et al., 2019] augments metabolic neurotransmitter signaling of energy insufficiency in the ventromedial hypothalamic nucleus (VMN), a key brain gluco-regulatory structure, strongly suggests that astrocyte glycogen-derived fuel stream promotes neuro-metabolic stability.
E2 governs basal and hypoglycemic patterns of astrocyte GP expression in vivo in hypothalamic structures [VMN and lateral hypothalamic area (LHA)] involved in glucostasis [Tamrakar et al., 2015]. Our recent studies document sex differences in forebrain ER regulation of VMN GS and GP protein profiles during insulin-induced hypoglycemia [Mahmood et al., 2018]. Astrocytes exhibit sex-specific calcium flux responses to E2 in vitro [Kuo et al., 2010]. Current research utilized female and male hypothalamic astrocyte primary cultures as an experimental model to address the premise that E2 regulation of astrocyte glycogen metabolic enzyme protein expression and glycogen accumulation may vary according to sex. Hypothalamic astrocytes express multiple ER variants, e.g. ERα, ERβ, and GPER [Ibrahim et al., 2019; Mahmood et al., 2019]. In the present study, effects of selective ERα, ERβ, and GPER receptor agonists on astrocyte GS, GP, and glycogen content were evaluated in each sex. A corollary aim of this project was to assess E2 effects on ER receptor variant protein profiles to determine if sex-dimorphic regulation of glycogen metabolism by estradiol involves control of signal volume of one or more ER type(s). Subcellular localization of classical nuclear ER includes the cell cytoplasm and plasma membrane. E2 acts via astrocyte plasma membrane ERα to control intracellular calcium signaling [Micevych P et al., 2010; Micevych and Christensen, 2012]. The current study employed a membrane impermeable E2 analogue bovine serum albumin-conjugated E2 (E2/BSA) to investigate whether steroid control of astrocyte glycogen storage and mobilization is achieved in part by activation of cell surface ER.
The ultra-sensitive cellular energy gauge 5’AMP-activated protein kinase (AMPK) is activated via phosphorylation in response to cellular metabolic deficits. Phosphorylated AMPK (pAMPK) inhibits ATP-consuming biosynthetic pathways, while stimulating ATP-producing metabolic pathways such as fatty acid oxidation [Hardie, 2003; Kahn et al., 2005]. AMPK monitors both dynamic energy state, and energy reserves, signified by AMP/ATP ratio and glycogen mass, respectively [McBride et al., 2009; Jensen and Richter, 2015; Li et al., 2015]. Glucoprivic activation of AMPK is correlated with decreased GS activity and reductions in glycogen mass [Halse et al., 2003]. In vivo studies involving laser-catapult microdissection and Western blot analysis of hypothalamic astrocytes obtained from E2-replaced ovariectomized female rats demonstrate estrogen involvement in modulating glial AMPK reactivity to insulin-induced hypoglycemia [Tamrakar and Briski, 2015]. Additional studies were conducted here to examine whether E2 control of astrocyte glycogen metabolism correlates with altered activity of AMPK and its upstream stimulatory (calcium/calmodulin-dependent protein kinase kinase-β) and inhibitory (protein phosphatase-1) kinases.
Materials and Methods:
Primary Cell Culture:
Adult female Sprague-Dawley rats (3–4 months of age) were maintained under a 14 h light:10 h dark lighting schedule (lights on at 05.00 h), and allowed free access to standard laboratory rat chow (Harlan Teklad LM-485; Harlan Industries, Madison, WI) and tap water. Rats were acclimated to daily handling over a 7-day period prior to experimentation. All animal protocols were conducted in accordance with NIH guidelines for care and use of laboratory animals, and approved by the ULM Institutional Animal Care and Use Committee. The hypothalamus was dissected as a single block from each brain at the following boundaries: anterior: rostral border of optic chiasm; posterior: rostral margin of mammillary bodies; lateral: lateral border of tuber cinereum; dorsal: dorsum of third ventricle [Kuo et al., 2010]. Highly-purified primary astrocyte cultures were obtained using a modification of the method described by Schildge et al. [2013]. Briefly, tissue blocks were enzymatically digested in 2.5 mL 2.5% trypsin (prod. no. 15090–046; Thermo-Fisher Scientific, Waltham, MA) and 22.5 mL Hanks’ balanced salt solution (HBSS; prod. no. H2387, SigmaAldrich, St. Louis, MO), incubated (30 min.) in a 37°C water bath with shaking every 10 min., then centrifuged (5 min.) at 300 x g to obtain supernatant by careful decantation. Tissue was dissociated into a single-cell suspension by addition of 10 mL astrocyte plating media [DMEM high glucose media (prod. no. 12800–017; ThermoFisherSci.) supplemented with 10.0% heat-inactivated fetal bovine serum (FBS; GE Healthcare Bio-Sciences, Pittsburgh, PA) and 1.0% penicillin/streptomycin (prod. no. 15140–122, ThermoFisherSci.)] and vigorous pipetting (20–30 times) with a 10 mL pipette. Efficacy of cell isolation was confirmed by cell counting with a hemocytometer. Cells were suspended in an adjusted media volume of 20 mL astrocyte plating media, plated in poly-D-lysine (prod. no. A-003-E, MilliporeSigma, Burlington, MA) - coated T75 culture flasks at a concentration of 50 μg/mL, and incubated at 37 °C in a humidified environment in the presence of 5% CO2 with media changes at 48 and 120 hr post-plating. Approximately 7–8 days after plating, microglia were removed from cultures by aspiration after shaking at 180 rpm (30 min.). Cells were re-suspended in fresh media (20 mL) and shaken first on an orbital platform at 240 rpm (6–8 hr), then vigorously by hand (1 min.) before aspiration to discard oligodendrocyte precursor cells (OPC). Confluent astrocytes were washed twice with 10 mM phosphate-buffered saline, pH 7.4, then incubated at 37 °C (7–10 min.) with 5.0 mL trypsin-EDTA (prod. no. 25300–062, ThermoFisherSci.). Detached astrocytes were centrifuged at 180 x g (5 min.) and re-suspended in 40 mL fresh media. Cells derived from each confluent layer were plated in two T75 culture flasks, and incubated at 37 °C for 12–14 days with media changes every 2–3 days. Astrocytes were counted, plated, and grown for two weeks prior to experimentation. Cultures were routinely checked for purity by Western blotting using a primary antiserum against the astrocyte marker protein glial fibrillary acidic protein (GFAP) (prod. no. 3670S, Cell Signaling Technology, Danvers, MA). Cultures were determined to be > 95% pure astrocytes.
Western Blot Analysis:
Astrocytes were initially plated at the density of 1 × 106 cells/100 mm2 in poly-D-lysine – coated culture dishes, and maintained for 3-days in high glucose DMEM media until they reached approximately 70% confluency. Cells were then steroid-starved for 18 hr by incubation in high glucose media containing 5.0% charcoal-stripped FBS (prod. no. 12676029; ThermoFisherSci.). Cells were then incubated with HBSS media containing E2 (100 pM, 1 nM, 10 nM, 100 nM); estrogen receptor-alpha (ERα) agonist propylpyrazole triol (PPT); estrogen receptor-beta (ERβ) agonist diarylpropionitrile (DPN); transmembrane G protein-couple estrogen receptor (GPR30) agonist G1; or the membrane impermeable E2 analog E2/BSA, dissolved in dimethyl sulfoxide (DMSO), for a 4 hr period. Non-hormone and -drug – treated controls were incubated with media containing DMSO only. Astrocytes were detached from plates with 0.05% trypsin EDTA (prod. no. 25300–062; ThermoFisherSci.), washed in PBS, pelleted in lysis buffer [2.0% sodium dodecyl sulfate, 10.0% glycerol, 5% β-mercaptoethanol, 1 mM sodium orthovanadate (protease inhibitor), 60mM Tris-HCl, pH 6.8], heat-denatured (1 hr., 95°C), sonicated, centrifuged, and diluted with 4x Laemmli buffer (Bio-Rad, Hercules, CA). Sample protein concentrations were determined using protein assay kit reagents (Bio-Rad). Equal protein quantities per sample were separated by electrophoresis in Bio-Rad TGX 10–12% Stain-Free gels; gels were UV light-activated (1 min.) in a Bio-Rad ChemiDoc TM Touch Imaging System after separation. Proteins were transblotted overnight in Towbin buffer (30 V, 4 °C) to 0.45-μm PVDF membranes (ThermoFisherSci.). Membrane buffer washes and antibody incubations were carried out by Freedom Rocker™ Blotlbot® automation (Next Advance, Inc., Troy NY). Membranes were blocked (2 hrs.) with Tris-buffer saline (TBS), pH 7.4, containing 0.1% Tween-20 and 2% bovine serum albumin prior to overnight (4 °C) incubation with primary antisera raised in rabbit against glycogen synthase (GS, 1:1,000, prod. no. 3893S; Cell Signal. Technol.); phospho-glycogen synthase (pGS; 1:1,000, prod. no. 3891S; Cell Signal. Technol.); AMPKα1/2 (1:1,000, prod. no. 2532L; Cell Signal. Technol.); pAMPKα1/2 (1:1,000, prod. no. 2535L; Cell Signal. Technol.); glycogen phosphorylase-brain variant (GPBB; 1:1,000, prod. no. NBP1– 32799; Novus Biologicals, LLC, Littleton, CO); ERα (1:1,000, prod. no. NB100–91756; Novus Biol.); ERβ (1:1,000, prod. no. NB120–3577; Novus Biol.); GPR30 (1:1,000, prod. no. NLS-4271; Novus Biol.); in goat against calcium/calmodulin-dependent protein kinase kinaseβ (CAMKKβ; 1:1,000, prod. no. sc-9629; Santa Cruz Biotechnology, Inc., Santa Cruz, CA)]; or in mouse against protein phosphatase 1 (PP1; 1:1,000, prod. no. sc-7482; Santa Cruz Biotechnol.). Membranes were then incubated (1 hr.) with horseradish peroxidase-conjugated goat anti-rabbit (1:5,000; prod. no. NEF812001EA; PerkinElmer, Billerica, MA), donkey anti-goat (1:5,000; prod. no. sc-2020; Santa Cruz Biotechnol.), or goat anti-mouse (1:5,000; prod. no. NEF 822001EA, PerkinElmer) secondary antibodies, then exposed to Supersignal West Femto chemiluminescent substrate (prod. no. 34095; ThermoFisherSci.). Protein optical density (O.D.) signals were detected and quantified in the ChemiDoc MP System using Image Lab™ 6.0.0 software, build 25, 2017, and normalized to total in-lane protein. Precision plus protein molecular weight dual color standards (prod. no. 161–0374, Bio-Rad) were included in each Western blot analysis. Proteins analyses were carried out in triplicate experiments.
Glycogen UHPLC/Electrospray Ionization-Mass Spectrometric Analysis:
Cell lysate supernatant aliquots (10 μL) were hydrolyzed by incubation (2 hr.) with 10 μL 0.5 mg/ml amyloglucosidase and 10 μL 0.1M sodium acetate (pH 5.0), followed by sequential heating (100°C, 5 min) and cooling to room temperature. Glycogen concentrations were determined in a ThermoFisherScientific Vanquish UHPLC+ System equipped with Thermo Scientific™ Dionex™ Chromeleon™ 7 Chromatography Data System software, as described [Bheemanapally et al., 2019]. Column and autosampler temperatures were 35°C and 15°C, respectively. The auto-sampler needle was washed with 10% (v/v) methanol (10 sec). Hydrolyzed and non-hydrolyzed samples were derivatized with 100 μL 0.5 M 1-phenyl-3-methyl-5-pyrazolone (PMP) reagent supplemented with 0.3 M NaOH. After acidification with 400 μL 0.75% formic acid, derivatized samples were extracted with chloroform to remove excess PMP, 400 μL of supernatant was vacuum concentrated to remove organic solvents, frozen at −80°C, and lyophilized. Lyophilization product was diluted to 1.0 mL with 10 mM ammonium acetate, bath-sonicated (30 sec.), and centrifuged. Supernatant aliquots (250 μL) were transferred to 350 μL inserts, which were placed into 2 mL Surestop vials in an autosampler tray. D-(+)-Glucose-PMP was resolved using the Shodex™ Asahipak™ NH2P-40 3E column with a mobile phase (75:25 v/v), acetonitrile:10mM ammonium acetate (0.2 ml/min). D-(+)-Glucose-PMP ion chromatograms were extracted from Total Ion Current (TIC) at m/z 510.2 to generate area-under-the-curve data. Parameters for UHPLC-ESI-MS includes sheath gas pressure (SGP; 25 psig), auxiliary gas pressure (AGP; 4.6 psig), sweep gas pressure (SWGP; 0.5 psig), vaporizer temperature (VT; 150°C), ion transfer tube temperature (ITT; 150°C), source voltage (−2000V), foreline pressure (1.76 Torr; auto-set by instrument- and variable), source gas (nitrogen; Genius NM32LA 110V, 10–6520; Peak Scientific, Inchinnan, Scotland), and mass peak area detection algorithm (ICIS/Genesis) were maintained at optimum.
Statistics:
Mean normalized protein O.D. and glycogen measures were evaluated between treatment groups within each sex by one-way analysis of variance and Student Newman Keuls post-hoc test. Differences of p < 0.05 were considered significant.
Results:
Effects of 17β-estradiol (E2) or selective ER agonists on expression of glycogen synthase (GS), the active enzyme form, in female versus male hypothalamic astrocytes are shown in Figure 1. In the female (Panel 1A), astrocytes treated with graded, e.g. 100 pM – 100 nM doses of E2 exhibited significant, equivalent up-regulation of GS content relative to non-E2 – treated controls [F(4,10) = 15.39; p < 0.0001] (Table 1).. Meanwhile, male astrocyte GS levels were significantly increased in response to 1–100 nM E2 dosages (Panel 1B) [F(4,10) = 7.741; p < 0.0001]. As depicted in Panel 1C, female rat astrocytes treated with the selective ERα agonist PPT, the selective ERβ agonist DPN, the membrane impermeable E2 analogue E2/BSA, or the GPER agonist G1 exhibited diminished GS expression compared to E2 [F(4,10) = 13.56; p < 0.0001] (Table 1). In this sex, GS levels were higher following incubation with G1 versus PPT or DPN. In male rat astrocytes, GS content did not differ between E2 versus PPT, DPN, or E2/BSA treatment groups, while GS expression was elevated in G1 versus E2 - treated cells (Panel 1D) [F(4,10) = 13.06; p < 0.0001]. Data in Figure 2 illustrate effects of E2 versus ER agonist incubation on astrocyte phospho-glycogen synthase (pGS; inactive enzyme form) protein content. E2 did not modify pGS expression in female (Panel 2A) [F(4,10) = 0.56; p = 0.56] or male astrocytes (Panel 2B) [F (4,10) = 0.8357; p = 0.5106] over the current dosage range. Female rat astrocytes treated with PPT, DPN, E2/BSA, or G1 exhibited elevated pGS content compared to E2 (Panel 2C) [F(4,10) = 45.40; p < 0.0001]; this augmentation was significantly greater after exposure to PPT or DPN versus E2/BSA or G1. Male rat astrocyte pGS levels did not differ between all treatment groups (Panel 2D) [F(4,10) = 06064; p = 0.6641].
Figure 1:
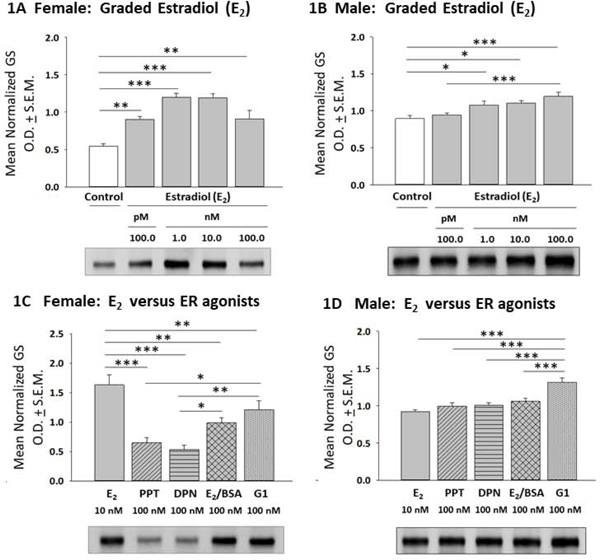
Effects of 17β-Estradiol (E2) or Selective Estrogen Receptor (ER) Agonists on Glycogen Synthase (GS) Expression in Female or Male Rat Hypothalamic Astrocyte Primary Cultures. After incubation of female [Panel 1A; p < 0.0001] or male [Panel 1B; p < 0.0001] rat hypothalamic astrocytes with varying (100 pM – 100 nM; gray bars) E2 concentrations or media lacking E2 (white bar), cell lysates were analyzed by Western blotting for GS protein expression. Data depict E2 effects on mean normalized GS protein optical density (O.D.) measures ± S.E.M. Panels 1C [female; p < 0.0001] and 1D [male; p < 0.0001] illustrate effects of E2 (solid gray bar) versus the selective ERα agonist PPT (diagonal-striped gray bar), the ERβ agonist DPN (horizontal-striped gray bar), membrane impermeable E2-BSA conjugate (cross-hatched gray bar), or the GPER agonist G1 (vertical-striped gray bar) on GS protein profiles. *p < 0.05; **p < 0.01; ***p < 0.001.
Table 1.
Summary of Effects of 17β-Estradiol (E2) versus Selective Estrogen Receptor Modulators on Hypothalamic Astrocyte Protein Expression
| Females | Males | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| E2 Dose response | PPTa | DPNb | E2/BSAc | G1d | E2 Dose response | PPT | DPN | E2/BSA | G1 | |
| 100 nM Compared to 10 nM β-E2 | 100 nM Compared to 10 nM β-E2 | |||||||||
| GSe |  |
 |
 |
 |
 |
 |
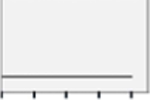
NSo | NS | NS |  |
| pGSf |  |
 |
 |
 |
 |
 |
NS | NS | NS | NS |
| GPg | 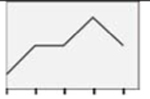 |
NS |  |
 |
NS | 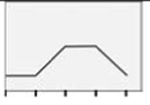 |
 |
 |
 |
 |
| ERαj |  |
NS | NS | NS |  |
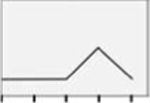 |
NS | NS | NS | NS |
| ERβk | 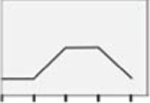 |
 |
 |
NS | NS | 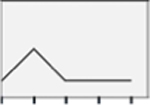 |
 |
 |
 |
NS |
| GPERl | 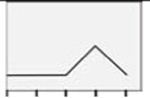 |
NS | NS | NS | NS |  |
NS | NS |  |
 |
| AMPKh | 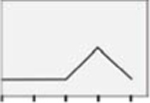 |
NS | NS | NS | NS | 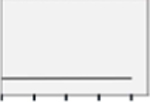 |
NS | NS | NS | NS |
| pAMPKi | 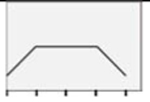 |
 |
 |
 |
 |
 |
 |
NS |  |
NS |
| CAMKKβm |  |
NS | NS | NS | NS | 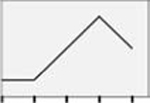 |
 |
NS |  |
 |
| PP1n | 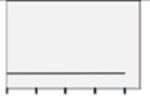 |
 |
 |
 |
NS |  |
NS |  |
NS | NS |
estrogen receptor-alpha (ERα) agonist propylpyrazole triol
estrogen receptor-beta (ERβ) agonist diarylpropionitrile (DPN)
estradiol-bovine serum albumin conjugate
transmembrane G protein-couple estrogen receptor (GPR30) agonist
glycogen synthase
phospho-GS
glycogen phosphorylase
5’-AMP-activated protein kinase
phospho-AMPK
estrogen receptor-alpha
estrogen receptor-beta
G protein-coupled estrogen receptor 1
calcium/calmodulin-dependent protein kinase kinase-beta
protein phosphatase-1
not significant
Figure 2:
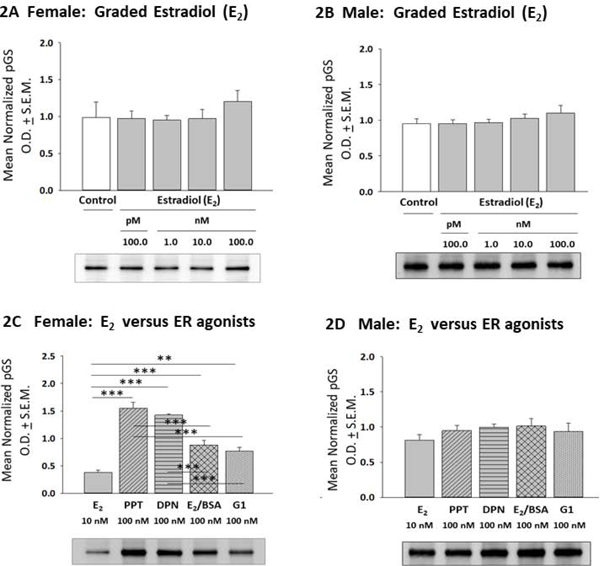
E2 versus ER Agonist Regulation of Phospho-GS (pGS) Expression in Female or Male Hypothalamic Astrocytes. Data depict mean normalized pGS protein O.D. measures ± S.E.M. for female [Panel 2A; p = 0.56] or male [Panel 2B; p = 0.5106] astrocytes exposed to graded E2 dosages. Panels 2C and 2D correspondingly show E2 versus ER agonist effects on female [p < 0.0001] or male [p = 0.6641] astrocyte pGS content. **p < 0.01; ***p < 0.001.
Astrocyte glycogen phosphorylase (GP) protein responses to E2 or ER agonist treatment are presented in Figure 3. In female astrocytes (Panel 3A), E2 incubation elevated GP expression at each dose level; GP content was significantly higher after treatment with 10 nM versus 100 pM or 100 nM β-E2 [F(4,10) = 10.65; p < 0.0001]. On the other hand, male astrocyte GP profiles were augmented by 1 or 10 nM E2 (Panel 3B) [F(4,10) = 7.140; p = 0.003]. Incubation with female astrocytes with E2, PPT, or G1 resulted in equivalent GP levels, whereas DPN and E2/BSA treatment had a lesser stimulatory effect compared to E2 (Panel 3C) [F(4,10) = 4.62; p = 0.02]. In male astrocytes, (Panel 3D), E2 had a greater stimulatory effect on GP expression relative to individual ER agonists [F(4,10) = 4.865; p = 0.005] (Figure 1F).
Figure 3:
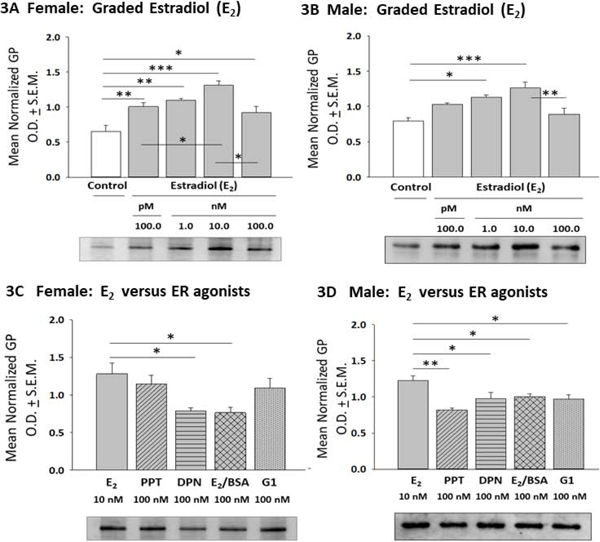
Effects of E2 versus ER Agonist Treatment on Female or Male Hypothalamic Astrocyte Glycogen Phosphorylase (GP) Expression. Data depict mean normalized astrocyte GP O.D. values ± S.E.M. following incubation of female [Panel 3A; p < 0.0001] or male [Panel 3B; p = 0.003] astrocytes with graded E2 concentrations. Panels 3C and 3D show effects of E2 versus selective ER agonist exposure on female [p = 0.02] and male astrocyte [p = 0.005] GP content. *p < 0.05; **p < 0.01; ***p < 0.001.
Data in Figure 4 depict regulation of female versus male astrocyte ERα protein expression by E2 and selective ER agonists. As illustrated in Panel 4A, E2 doses of 1 or 10 nM stimulated female astrocyte ERα content [F(4,10) = 9.19; p =0.0002]. Panel 4B shows that male rat astrocyte ERα expression was elevated only in response to the latter, e.g. 10 nM E2 dose (Panel 4B) [F(4,10) = 2.496; p = 0.05]. In the female, incubation with E2, PPT, DPN, or E2/BSA had equivalent effects on ERα content, whereas G1 had a comparatively diminished stimulatory effect (Panel 4C) [F(4,10) = 29.88; p < 0.0001]. As shown in Panel 4D, male astrocytes incubated with E2, PPT, DPN, E2/BSA or G1 exhibited comparable levels of ERα expression [F(4,10) = 1.22; p = 0.3383]
Figure 4:

Effects of E2 versus ER Agonist Treatment on Female or Male Hypothalamic Astrocyte ER-Alpha (ERα) Protein Expression. Data depict mean normalized female [Panel 4A; p = 0.0002] or male [Panel 4B: p = 0.05] astrocyte ERα O.D. values ± S.E.M. after graded E2 exposure. Panels 4C and 4D show effects of E2 versus ER agonist exposure on female [p < 0.0001] or male [p = 0.3383] astrocyte ERα protein profiles. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 5 depicts effects of E2 versus selective ER agonists on hypothalamic astrocyte ERβ profiles. In female astrocytes (Panel 5A), E2 dosing at 1 or 10 nM increased ERβ protein expression [F(4,10) = 7.41; p = 0.001]. In male astrocytes (Panel 5B), exposure to only the minimum, e.g. 100 pM E2 dose stimulated ERβ expression [F(4,10) = 8.52; p <0.0001]. As shown in Panel 5C, female astrocytes incubated with PPT or DPN showed elevated ERβ content relative to E2, E2/BSA, or G1 treatment [F(4,10) = 5.13; p = 0.017]. Data in Panel 5D indicate that PPT, DPN and E2/BSA had greater potency in stimulating male astrocyte ERβ protein levels than E2, whereas G1 caused less stimulation than E2 [F(4,10) = 20.15; p < 0.0001].
Figure 5:
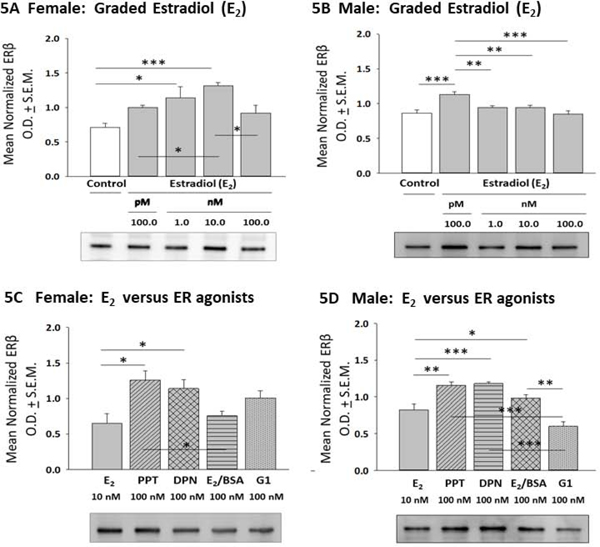
Effects of E2 versus ER Agonist Incubation on Female or Male Hypothalamic Astrocyte ER-Beta (ERβ) Protein Expression. Data depict mean normalized female [Panel 5A; p = 0.001] or male [Panel 5B; p < 0.0001] astrocyte ERβ O.D. measures ± S.E.M. after treatment with graded E2 dosages. Panels 5C and 5D illustrate effects of E2 versus selective ER agonists on female [p = 0.017] or male [p < 0.0001] astrocyte ERβ protein expression. *p < 0.05; **p < 0.01; ***p < 0.001.
As shown in Figure 6, female rat astrocytes exhibited increased GPER protein content after exposure to 10 nM (but not other) E2 dose levels (Panel 6A) [F(4,10) = 4.14; p =0.02], while male astrocyte GPER content was resistant to E2 over the dosage range used here (Panel 6B) [F(4,10) = 0.6238; p =0.6498]. E2, PPT, DPN, E2/BSA, and G1 all had equivalent stimulatory effects on female astrocyte GPER expression [F(4,10) = 2.06; p = 0.14] (Panel 6C). Male astrocyte GPER content (Panel 6D) was increased to a significantly greater extent after treatment with E2/BSA or G1 compared to E2, PPT, or DPN [F(4,10) = 13.61; p < 0.0001].
Figure 6:
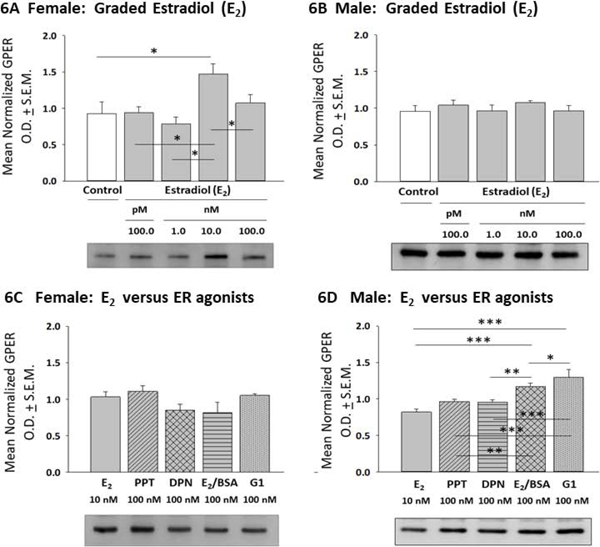
E2 versus Selective ER Agonist Regulation of Transmembrane G Protein-Coupled ER (GPER) Expression in Female or Male Hypothalamic Astrocytes. Data show mean normalized GPER O.D. measures ± S.E.M. for female [Panel 6A; p =0.02] or male [Panel 6B; p =0.6498] astrocytes incubated with graded concentrations of E2. Panel versus selective ER agonists on female hypothalamic astrocytes [p = 0.14], and 6D: Effect of E2 versus selective ER agonists on male hypothalamic astrocytes [p < 0.0001]. *p < 0.05; **p < 0.01; ***p < 0.001.
E2 versus ER agonist regulation of hypothalamic astrocyte AMPK expression is illustrated in Figure 7. E2 significantly enhanced AMPK expression in female rat astrocytes at a discrete (10 nM) dosage (Panel 7A) [F(4,10) = 3.25; p = 0.03], whereas male rat astrocytes were resistant to steroid control of AMPK (Panel 7B) [F(4,10) = 0.7027; p = 0.61]. As shown in Panel 7C, incubation with E2, PPT, DPN, E2/BSA, or G1 had similar effects on female astrocyte AMPK content [F(4,10) = 0.84; p =0.53]. Male astrocyte AMPK levels did not differ after exposure to E2, PPT, DPN, E2/BSA, or G1 (Panel 7D) [F(4,10) = 2.422; p =0.08]. Figure 8 shows effects of E2 or selective ER agonist treatment on hypothalamic astrocyte phospho-AMPK (pAMPK) expression. As indicated in Panel 8A, female astrocyte pAMPK levels were increased to a comparable degree after exposure to 100 pM - 10 nM E2 [F(4,10) = 9.19; p = 0.0002]. Uniform stimulation of male astrocyte pAMPK content occurred over the same E2 dosage range (Panel 8B). [F(4,10) = 11.5; p =0.0002]. Female astrocytes expressed higher levels of pAMPK protein after incubation with PPT, DPN, E2/BSA, or G1 versus E2, and G1 showed the highest activity in increasing pAMPK (Panel 8C) [F(4,10) = 44.94; p < 0.0001]. Male astrocytes treated with PPT or E2/BSA expressed higher or lower levels of pAMPK, respectively, relative to E2 (Panel 8D) [F(4,10) = 44.42; p < 0.0001].
Figure 7:
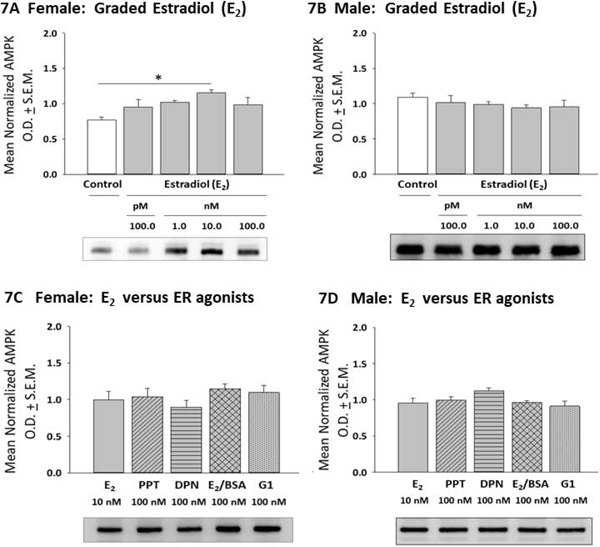
Effects of E2 versus Selective ER Agonist Treatment on 5’-AMP-Activated Protein Kinase (AMPK) Expression in Female or Male Hypothalamic Astrocytes. Data depict mean normalized AMPK O.D. measures ± S.E.M. for female [Panel 7A; p = 0.03] or male [Panel 7B; p = 0.61] astrocytes incubated with varying E2 doses. Panels 7C and 7D show effects of E2 versus selective ER agonists on female [p = 0.53] or male [p = 0.08] hypothalamic astrocyte AMPK content. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 8:
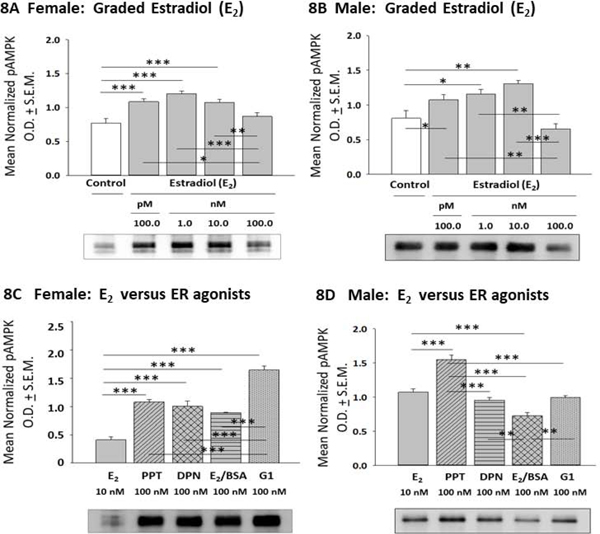
Effects of E2 or ER Agonist Treatment on Female or Male Astrocyte Phospho-AMPK (pAMPK) Expression. Data depict mean normalized pAMPK O.D. measures ± S.E.M. for female [Panel 8A; p = 0.0002] or male [Panel 8B; p = 0.0002] astrocytes incubated with graded E2 concentrations. Panels 8C and 8D illustrate effects of E2 versus selective ER agonists on female [p < 0.0001] or male [p < 0.0001] astrocyte pAMPK protein expression. *p < 0.05; **p < 0.01; ***p < 0.001.
Effects of E2 or ER agonist treatment on astrocyte calcium/calmodulin-dependent protein kinase kinase-beta (CAMKKβ) protein profiles are illustrated in Figure 9. Data show that the current E2 dosing paradigm did not affect female astrocyte CAMKKβ content (Panel 9A) [F(4,10) = 2.37; p =0.10]. In contrast, male astrocyte CAMKKβ levels were significantly increased in response to 1, 10 and 100 nM E2 (Panel 9B) [F(4,10) = 28.22; p <0.0001]. As shown in Panel 9C, female astrocytes exhibited comparable levels of CAMKKβ expression in response to E2, PPT, DPN, E2/BSA, or G1 [F(4,10) = 0.93; p =0.92]. In contrast, male astrocyte showed lesser reactivity to PPT, E2/BSA and G1 compared to E2 (Panel 9D [F(4,10) = 7.538; p = 0.0013]. E2 or ER agonist effects on astrocyte protein phosphatase-1 (PP1) protein expression are shown in Figure 10. Exposure to E2 did not affect female (Panel 10A) [F(4,10) = 0.62; p =0.65] or male (Panel 10B) [F(4,10) = 1.092; p =0.3960] PP1 levels. Female astrocytes incubated with PPT, DPN, or E2/BSA exhibited higher PP1 expression versus the E2 treatment group (Panel 10C) [F(4,10) = 7.87; p =0.003]. Male astrocytes showed elevated PP1 profiles after exposure to DPN versus E2 (Panel 10D) [F(4,10) = 4.249; p =0.0066].
Figure 9:
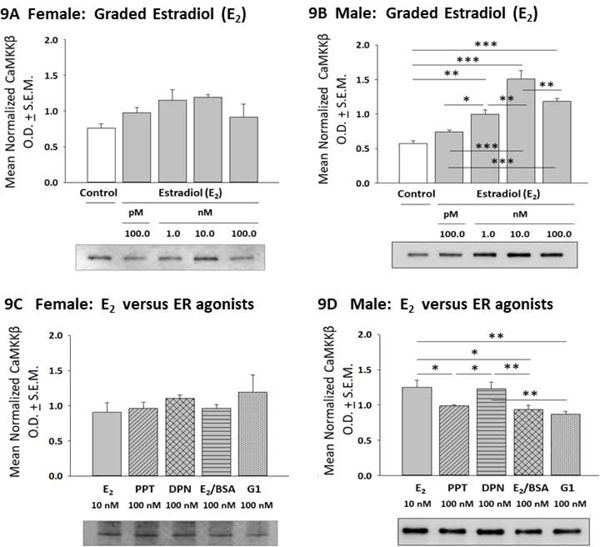
Effects of E2 versus ER agonist Treatment on Calcium/Calmodulin-Dependent Protein Kinase Kinase-Beta (CAMKKβ) Expression in Female or Male Hypothalamic Astrocytes. Data depict mean normalized CAMKKβ O.D. values ± S.E.M. for female [Panel 9A; p = 0.10] or male [Panel 9B; p < 0.0001] astrocytes incubated with graded doses of E2. Panels 9C and 9D illustrate effects of E2 versus selective ER agonists on female [p = 0.92] or male [p = 0.0013] astrocyte CAMKKβ content. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 10:
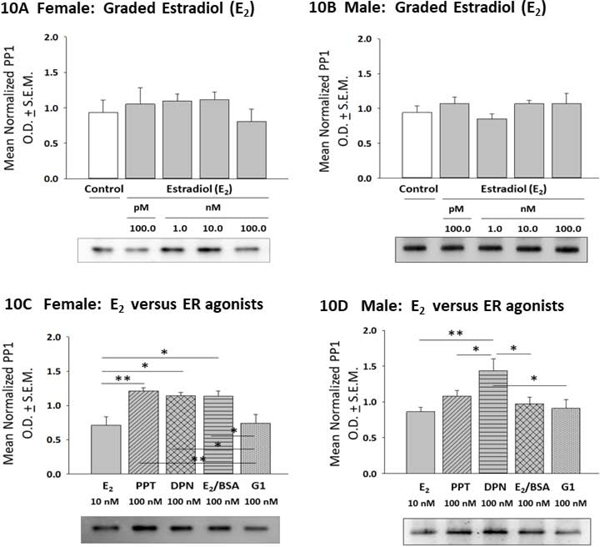
Effects of E2 versus Selective ER Agonists on Protein Phosphatase-1 (PP1) Profiles in Female or Male Hypothalamic Astrocytes. Data depict mean normalized PP1 O.D. measures ± S.E.M. for female [Panel 10A; p = 0.65] or male [Panel 10B; p = 0.3960] astrocytes treated with graded E2 dosages. Panels 10C and 10D show effects of E2 versus ER agonist incubation on female [p = 0.003] or male [p =0.0066] PP1 protein profiles. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 11 depicts effects of E2 or selective ER agonists on female versus male hypothalamic astrocyte glycogen content. Data in Panel 11A show that in female astrocytes, E2 and DPN caused significant glycogen up-regulation, with β-E2 exhibiting greater potency than DPN [F(6,14) = 4.249; p =0.0066]. Male astrocyte glycogen levels were refractory to E2, but were increased in response to PPT [F(4,10) = 4.249; p = 0.0066].
Figure 11:
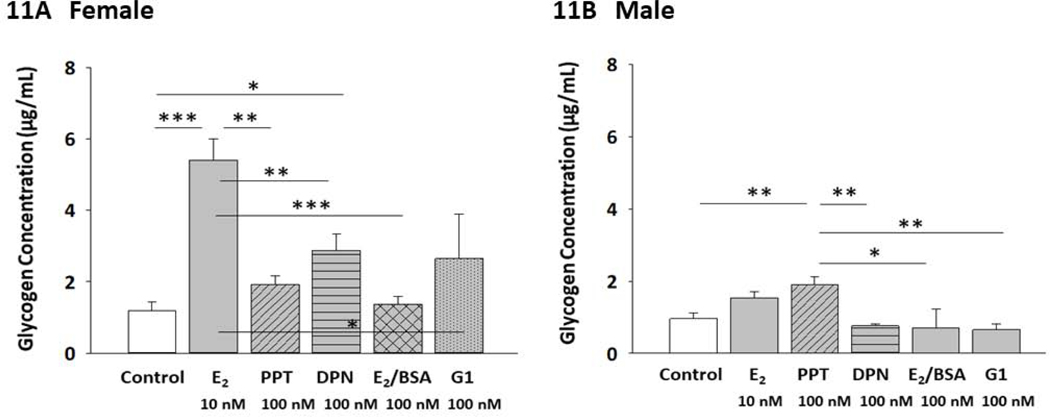
Effects of E2 versus Selective ER Agonists on Female or Male Astrocyte Glycogen Content. Panels 11A and 11 B depict treatment effects on mean cellular glycogen levels in female [p = 0.0006] or male [p = 0.006] astrocytes, respectively. *p < 0.05; **p < 0.01; ***p < 0.001.
Discussion:
Glycogen is a complex carbohydrate that provides the brain with an alternative fuel source to blood glucose that becomes depleted during energy deficiency or heightened neurological activity. Astrocytes convert glucose to L-lactate for trafficking to neurons as a substrate for mitochondrial oxidative respiration. Estradiol (E2) can act through multiple mechanisms such as modulation of neuro-glial metabolic coupling, to protect the brain against bio-energetic insults. Previously, in vivo studies showed that ERs impose sex-specific effects on hypothalamic glycogen metabolic enzyme protein expression during insulin-induced hypoglycemia [Mahmood et al., 2018]. Current research utilized a primary astrocyte culture model to address the hypothesis that E2 may exert differential effects on female versus male hypothalamic astrocyte GS (active and inactive forms) and/or GP profiles and glycogen accumulation, and that sex-specific regulation of glycogen metabolism may involve distinctive ER variant signaling. Results document greater GS and GP sensitivity to E2 stimulation in female versus male astrocytes, but lack of E2 regulation of GS phosphorylation status in either sex. GPER had highest efficacy among ER variants in both sexes in increasing GS levels, and was equal to ERα in stimulating GP profiles in female astrocytes. E2 increased astrocyte glycogen content in female, but not male astrocytes, indicating sex-dimorphic estrogenic regulation of hypothalamic glycogen accumulation under conditions of energy sufficiency. These results may mirror sex-specific E2 – induced ER variant expression, namely up-regulation of ERα in males and females with (females) or without (males) concurrent augmentation of ERβ, and/or regulation of post-receptor signaling. Further studies are needed to determine if and how E2 may promote dissimilar glycogen amassment in the female versus male hypothalamus in vivo, as well as examine what effects these sex differences may play in glycogen maintenance and turnover, and neuron function under conditions of metabolic homeostasis and energy imbalance.
The present studies document differential glycogen metabolic GS and GP protein sensitivity to E2 in female versus male hypothalamic astrocytes, as the minimum steroid dose (100 pM) stimulated these protein profiles in the former, but not the latter sex. In each sex, both proteins were up-regulated over a range (1 – 100 nM) of higher E2 dosages. Interestingly, E2 treatment did not alter expression of pGS, the inactive enzyme form. It is possible that E2 - associated increases in total GS protein content may result in corresponding augmentation of GS enzyme activity. However, it should be noted that current work did not seek to generate experimental confirmation of direct correlations between net changes in expression profiles versus activity of GS or other enzymes investigated here. Levels of astrocyte total GP protein content provide less clear insight on potential E2 regulation on GP-mediated glycogen breakdown as phosphorylated GP (pGP) is the active enzyme form, and experimental tools for analysis of pGP activity are not currently available. Our working assumption that E2 may promote an ‘as-yet-to-be-determined’ proportionate increase in active GP in one or both sexes remains to be verified. An unpredicted, yet intriguing outcome of this research is evidence for unambiguous sex-specific E2 regulation of hypothalamic astrocyte glycogen accumulation in vitro, including an absence of hormonal effect on glycogen reserves in the male. Since E2 up-regulates both GS and GP protein profiles in male astrocytes, a plausible explanation is that this hormone may elicit equivalent, matched adjustments in the rate of glucose monomer incorporation into versus liberation from glycogen in males, resulting in elevated glycogen turnover in the absence of overall change in storage mass. Meanwhile, estrogenic augmentation of astrocyte glycogen content is likely a clear indicator of preferential E2 stimulation of glucose assimilation versus release from glycogen, which could reflect, in part, steroidal induction of opposite changes in GS (up-regulated) versus GP (down-regulated) activity. It would be informative to learn if E2 increases glycogen accumulation and turnover in the female. Present findings support the need for further research to investigate whether hypothalamic astrocyte glycogen amassment varies by sex in vivo as a consequence of E2 regulation. Since hypoglycemia causes sex-dimorphic adjustments in VMN glycogen content in male versus female rats [Ali et al., 2019; Napit et al., 2019]; it would be informative to learn if E2 is involved in these dissimilar responses.
In female astrocytes, E2 stimulation of GS expression was most closely mimicked by the GPER agonist G1, while steroidal augmentation of GP profiles was emulated by the ERα agonist PPT and G1. Interestingly, PPT and the ERβ agonist DPN were significantly less potent than E2 in increasing GS, while DPN had less stimulatory effect on GP content versus E2. These findings raise the prospect that in the female, ERα signaling via ERα and ERβ may possibly mitigate simultaneous GPER induction of GS expression, whereas ERβ may negatively modulate ERα and GPER regulation of GP profiles. In male astrocytes, G1 more effectively enhanced astrocyte GS levels compared to E2 and other agonists, suggesting that E2 stimulation of GS mediated by GPER may be offset by concurrent hormone interaction with other ER variants. Evidence that male astrocyte GP levels were more potently stimulated by E2 relative to selective ER agonists suggests that steroid up-regulation of this protein may involve synergistic/additive effects of multiple ER variants. Although E2 did not alter astrocyte pGS expression in either sex, female astrocytes incubated with PPT or DPN exhibited dramatic up-regulation of pGS compared to E2, whereas G1 exposure has a lesser, but still significant stimulatory effect on this protein. These data infer that ERα and ERβ signals to limit total GP expression and to enhance pGS levels are likely counter-balanced by input from other ERs. Data show that G1 and E2/BSA exhibited similar stimulatory potencies toward GS protein expression in females and toward GP profiles in the male, suggesting that GPER is a principal membrane site of E2 action on these proteins. Nonetheless, E2/BSA was less efficacious than E2 in up-regulating GS or GP in the female or GP in the male, inferring that E2 control likely involves non-membrane as well as membrane ER.
Present data document distinctive sex-dimorphic effects of E2 on hypothalamic astrocyte ER protein expression. Female astrocyte ERα protein was up-regulated at a lower E2 concentration and over a broader dosage range compared to males, and astrocyte ERβ expression was increased at distinctive E2 dosage levels in females (1–10 nM) versus males (100 pM); and 3) E2 elevated GPER profiles in female, but not male astrocytes. These findings support the view that sex-specific E2 effects on astrocyte glycogen metabolic protein profiles and glycogen accumulation may ensue from differential hormonal regulation of ER variant expression and/or regulation of post-receptor signaling in the female versus the male. E2 and ER agonists had similar potency in stimulating ERα profiles in each sex, with the exception of lesser G1 efficacy in females. In both sexes, astrocyte ERβ expression was up-regulated to a greater extent by PPT and DPN versus E2, suggesting that ERα and ERβ signals that augment this ER variant may be offset by E2 action on other ER. E2 and ER agonists elevated GPER profiles to an equal extent in the female, while G1 had greater potency stimulate GPER in males compared to E2 or other ER agonists, suggesting that GPER self-upregulation may be curbed by other ER during E2 exposure.
In astrocytes of both sexes, E2/BSA had equivalent potency to E2, PPT, and DPN, and either greater or equal potency to G1 in females and males, respectively, to up-regulate ERα. These findings imply that in both sexes, E2 controls ERα expression via membrane ERα and ERβ action, in addition to GPER activity in the male. In female astrocytes, E2/BSA had equal ability to E2 and G1 to increase ERβ expression, suggesting that hormone regulation of this protein is mediated by GPER. In contrast, E2/BSA had similar potency to E2, but greater potency than G1 to elevate male astrocyte ERβ, suggesting that E2 action involves classical receptors associated with plasma membrane. Female astrocyte GPER profiles was augmented equally by E2/BSA and E2, pointing to the membrane as a principal site of regulatory action. Meanwhile, male astrocyte GPER content was increased to a greater extent by E2/BSA and G1 compared to E2, suggesting that a principal membrane site of estradiol stimulatory action that may be curtailed by ER.
Previous studies showed that E2 was required for hypoglycemia-associated activation of female hypothalamic astrocyte AMPK [Tamrakar and Briski, 2015]. Current research addressed the premise that E2 may impose sex-specific regulatory effects on energy sensor expression and/or activation state in hypothalamic astrocytes and that hormonal control of astrocyte glycogen metabolism may be correlated with E2 effects on AMPK activity and upstream stimulatory CaMKKβ and/or inhibitory PP1 kinase expression. Results suggest that during energy homeostasis, E2 may exert sex-dimorphic effects on hypothalamic astrocyte total AMPK content, yet cause comparable augmentation of pAMPK expression in females versus males. Outcomes show that up-regulated AMPK activity coincides with increased glycogen accumulation in females, but not males, suggesting that E2 may exert sex-specific control of downstream sensor targets. Further studies are needed to investigate possible AMPK involvement in observed E2 effects on glycogen mass and metabolic enzyme expression in the female. Individual ER agonists exhibited greater potency to increase pAMPK than E2 in female rats, suggesting that broad ER activation may result in synergistic inhibition of E2 action. In males, the ERα agonist was more potent than E2 in increasing pAMPK, inferring that ERα-mediated effects may be blunted by other ER activation. In female astrocytes, E2/BSA had greater potency than E2, equivalent efficacy to PPT and DPN, and lesser potency than G1 in stimulating pAMPK. These findings suggest that ERα and ERβ stimulation of sensor activity is initiated foremost at the plasma membrane, and may involve blunting of GPER signaling. In male astrocytes, E2/BSA was less effective than E2 and G1 in promoting pAMPK expression, which infers that membrane and non-membrane ERs are involved and that stimulatory G1 input is curtailed.
E2 up-regulated CaMKKβ protein expression in male but not, female astrocytes, which infers that relative contribution of phosphorylation versus allosteric mechanisms of AMPK activation in these glia may vary between the sexes. E2/BSA had lesser potency than E2, in stimulating male astrocyte CaMKKβ expression; thus, non-membrane as well as membrane ERs likely mediate steroid regulation of this protein profile. Equivalent potencies of E2/BSA and G1 infer that membrane E2 action primarily involves GPER.
In summary, present research investigated the premise that E2 regulation of astrocyte glycogen mass and metabolism is sex-dimorphic. Results reveal discriminative effects of E2 on astrocyte total GS and GP profiles in the two sexes [Figure 12], but clarify an absence of estrogenic control of GS phosphorylation state. Findings that E2/BSA has lesser potency than E2 to stimulate GS or GP in the female or GP in the male infer that hormonal regulation of these proteins involves non-membrane and membrane ER, specifically GPER. E2 elevated total AMPK content in female but not male astrocytes, but augmented pAMPK profiles in each sex with comparable potency to GPER agonist in females and ERα agonist in males; estrogen-dependent astrocyte downstream targets of AMPK, including glycogen, appears to vary between the two sexes. Evidence for sex-contingent E2 effects on astrocyte ERα, ERβ, and GPER protein expression support the view that differential regulation of astrocyte glycogen content and metabolic enzyme protein profiles by E2 is plausibly linked, in part, to characteristic hormonal induction of ER variant expression and/or regulation of post-receptor signaling in each sex.
Figure 12.
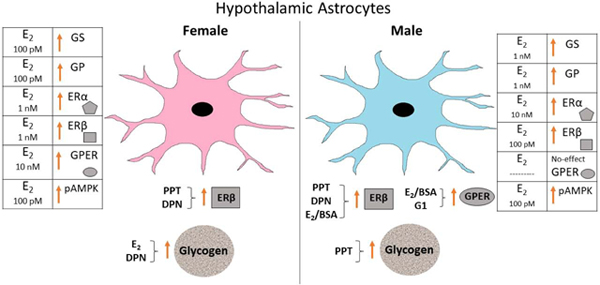
Estradiol (E2) and Estrogen Receptor (ER) Agonist Regulation of Female versus Male Hypothalamic Astrocyte Glycogen Metabolic Enzyme and ER Protein Expression. Tables on right (female) and left (male) sides denote minimum E2 dosages that significantly augmented astrocyte GS, GP, ERα, ERβ, GPER, and pAMPK protein expression in each sex. Sex dimorphic effects of E2 and individual ER agonists on astrocyte ER variant profiles and glycogen content are presented below illustrations of female versus male astrocytes.
Highlights:
Hypothalamic astrocytes from either sex were incubated with estradiol (E2) or selective ER agonists.
E2 more potently stimulated glycogen synthase and phosphorylase expression in females versus male
G protein-coupled ER-1 (GPER) mediates E2 effects on GS profiles in each sex and GP in females.
E2 increased astrocyte glycogen content in female, but not male astrocytes
Acknowledgments
Funding: This research was supported by NIH DK 109382.
Abbreviations:
- E2
17β-estradiol
- AMPK
5’-AMP-activated protein kinase
- CaMKKβ
calcium/calmodulin-dependent protein kinase kinase-beta
- DPN
estrogen receptor-beta (ERβ) agonist diarylpropionitrile
- E2/BSA
estradiol-bovine serum albumin conjugate
- ERα
estrogen receptor-alpha
- ERβ
estrogen receptor-beta
- GP
glycogen phosphorylase
- GPER
G protein-coupled estrogen receptor-1
- GS
glycogen synthase
- pAMPK
phosphoAMPK
- pGS
phosphoGS
- PP1
protein phosphatase-1
- PPT
estrogen receptor-alpha (ERα) agonist propylpyrazole triol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Acaz-Fonseca E, Sanchez-Gonzalez R, Azcoitia I, Arevalo MA, Garcia-Segura LM. Role of astrocytes in the neuroprotective actions of 17β-estradiol and selective estrogen receptor modulators. Mol Cell Endocrinol. 2014; 389: 48–57. [DOI] [PubMed] [Google Scholar]
- Alhamami HN, Alshamrani A, Briski KP. Effects of the glycogen phosphorylase inhibitor 1,4-dideoxy-1,4-imino-D-arabinitol on ventromedial hypothalamic nucleus 5’-adenosine monophosphate-activated protein kinase activity and metabolic neurotransmitter biosynthetic enzyme protein expression in eu-versus hypoglycemic male rats. Physiol. Reports 2018; 103: 236–249. [Google Scholar]
- Ali MH, Napit PR, Mahmood ASMH, Bheemanapally K, Alhamami HN, Uddin MM, Mandal KS, Ibrahim MMH, Briski KP. Hindbrain estrogen receptor regulation of ventromedial hypothalamic glycogen metabolism and glucoregulatory transmitter expression in the hypoglycemic male rat. Neuroscience 2019; 409:253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azcoitia I, Santos-Galindo M, Arevalo MA, Garcia-Segura LM. Role of astroglia in the neuroplastic and neuroprotective actions of estradiol. Eur. J. Neurosci. 2010; 32: 1995–2002. [DOI] [PubMed] [Google Scholar]
- Barreto GE. Targeting astrocytes in brain injuries: A translational research approach. Prog. Neurobiol. 2016; 144: 1–4. [DOI] [PubMed] [Google Scholar]
- Bheemanapally K, Ibrahim MMH, Briski KP. Development of a combinatory high-resolution microdissection/high-sensitivity high performance liquid chromatographic-mass spectrometric technique for small tissue volume analysis of rat brain glycogen. J. Pharmaceut. Biomed. Anal. 2019; 10.1016/j.jpba.2019.112884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brann DW, Dhandapani K, Wakade C, Mahesh VB, Khan MM. Neurotrophic and neuroprotective actions of estrogen: basic mechanisms and clinical implications. Steroids 2007; 72: 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci U S A. 2001; 98: 1952–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elzer JG, Muhammad S, Wintermantel TM, Regnier-Vigouroux A, Ludwig J, Schütz G, Schwaninger M. Neuronal estrogen receptor-alpha mediates neuroprotection by 17beta-estradiol. J. Cereb. Blood Flow Metab. 2010;.30:.935–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Segura LM, Wozniak A, Azcoitia I, Rodriguez JR, Hutchison RE, Hutchison JB. Aromatase expression by astrocytes after brain injury: implications for local estrogen formation in brain repair. Neuroscience. 1999; 892: 567–578. [DOI] [PubMed] [Google Scholar]
- Gingerich S, Kim GL, Chalmers JA, Koletar MM, Wang X, Wang Y, Belsham DD. Estrogen receptor alpha and G-protein coupled receptor 30 mediate the neuroprotective effects of 17beta-estradiol in novel murine hippocampal cell models. Neuroscience. 2010;170:54–66. [DOI] [PubMed] [Google Scholar]
- Halse R, Fryer LG, McCormack JG, Carling D, Yeaman SJ. Regulation of glycogen synthase by glucose and glycogen: a possible role for AMP-activated protein kinase. Diabetes 2003; 52: 9–15. [DOI] [PubMed] [Google Scholar]
- Hardie DG. Minireview: The AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology 2003; 144: 5179–5183. [DOI] [PubMed] [Google Scholar]
- Ibrahim MMH, Alhamami HN, Briski KP. Norepinephrine regulation of ventromedial hypothalamic nucleus metabolic transmitter biomarker and astrocyte enzyme and receptor expression: role of 5’-AMP-activated protein kinase. Brain Research 2019; 1711: 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TE, Richter EA. Regulation of glucose and glycogen metabolism during and after exercise. J. Physiol. 2012; 590: 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johann S, Beyer C. Neuroprotection by gonadal steroid hormones in acute brain damage requires cooperation with astroglia and microglia. J. Steroid Biochem. Mol. Biol. 2013; 137: 71–81. [DOI] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metabol. 2005; 1: 15–25. [DOI] [PubMed] [Google Scholar]
- Kuo J, Hamid N, Bondar G, Dewing P, Clarkson J, Micevych P. Sex differences in hypothalamic astrocyte response to estradiol stimulation. Biol. Sex Differ. 2010; 1:7. doi: 10.1186/2042-6410-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo J, Hamid N, Bondar G, Prossnitz ER, Micevych P. Membrane estrogen receptors stimulate intracellular calcium release and progesterone synthesis in hypothalamic astrocytes. J. Neurosci. 2010; 30: 12950–12957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laming PR, Kimelberg H, Robinson S, Salm A, Hawrylak N, Muller C, Roots B, Ng K. Neuronal-glial interactions and behavior. Neurosci. Biobehav. Rev. 2000; 24: 295–340. [DOI] [PubMed] [Google Scholar]
- Lebesgue D, Chevaleyre V, Zukin RS, Etgen AM. Estradiol rescues neurons from global ischemia-induced cell death: multiple cellular pathways of neuroprotection. Steroids. 2009; 74: 555–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang L, Zhou EX, Ke J, de Waal PW, Gu X, Tan MHE, Wang D, Wu D, Xu EH, Melcher K. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Research 2015; 25: 50–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Guo H, Zhang L, Tao L, Yin A, Liu Z, Li Y, Dong H, Xiong L, Hou W. Estrogen replacement therapy-induced neuroprotection against brain ischemia-reperfusion injury involves the activation of astrocytes via estrogen receptor β. Sci. Rep. 2016; 6:21467. doi: 10.1038/srep21467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood ASMH Uddin MM, Mandal SK Ibrahim MMH, Alhamami HN Briski KP. Sex differences in forebrain estrogen receptor regulation of hypoglycemic patterns of counter-regulatory hormone secretion and ventromedial hypothalamic nucleus gluco-regulatory neurotransmitter and astrocyte glycogen metabolic enzyme expression. Neuropeptides 2018; 72:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood ASMH, Bheemanapally K, Mandal SK, Ibrahim MMH, Briski KP Norepinephrine control of ventromedial hypothalamic nucleus glucoregulatory neurotransmitter expression in the female rat: role monocarboxylate transporter function. Mol. Cell. Neurosci. 2019; 95:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride A, Ghilagaber S, Nikolaev A, Hardie DG. The Glycogen-Binding Domain on the AMPK β Subunit Allows the Kinase to Act as a Glycogen Sensor. Cell Metab. 2009; 9: 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchenthaler I, Dellovade TL, Shughrue PJ. Neuroprotection by estrogen in animal models of global and focal ischemia. Ann. N.Y. Acad. Sci. 2003; 1007: 89–100. [DOI] [PubMed] [Google Scholar]
- Micevych P, Bondar G, Kuo J. Estrogen actions on neuroendocrine glia. Neuroendocrinology 2010; 91: 211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micevych P, Christensen A. Membrane-initiated estradiol actions mediate structural plasticity and reproduction. Front Neuroendocrinol. 2012. October;33(4):331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller NR, Jover T, Cohen HW, Zukin RS, Etgen AM. Estrogen can act via estrogen receptor alpha and beta to protect hippocampal neurons against global ischemia-induced cell death. Endocrinology 2005; 146: 3070–3079. [DOI] [PubMed] [Google Scholar]
- Napit PR, Ali MH, Shakya M, Mandal SK, Bheemanapally K, Mahmood ASMH, Ibrahim MMH, Briski KP. Hindbrain estrogen receptor regulation of counter-regulatory hormone secretion and ventromedial hypothalamic nucleus glycogen content and glucoregulatory transmitter signaling in hypoglycemic female rats. Neuroscience 2019; 411: 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen J, Brinton RD. Mitochondria as therapeutic targets of estrogen action in the central nervous system.Curr Drug Targets CNS Neurol Disord. 2004. Aug;3(4):297–313. [DOI] [PubMed] [Google Scholar]
- Saldanha CJ, Duncan KA, Walters BJ. Neuroprotective actions of brain aromatase. Front Neuroendocrinol. 2009; 30: 106–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildge et al. , JOVE 2013; 10.3791/50079 [DOI]
- Scott E, Zhang QG, Wang R, Vadlamudi R, Brann D. Estrogen neuroprotection and the critical period hypothesis. Front. Neuroendocrinol. 2012; 33: 85–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins JW, Dykens JA. Mitochondrial mechanisms of estrogen neuroprotection. Brain Res Rev. 2008; 57: 421–430. [DOI] [PubMed] [Google Scholar]
- Spence RD, Wisdom AJ, Cao Y, Hill HM, Mongerson CR, Stapornkul B, Itoh N, Sofroniew MV, Voskuhl RR. Estrogen mediates neuroprotection and anti-inflammatory effects during EAE through ERα signaling on astrocytes but not through ERβ signaling on astrocytes or neurons. J. Neurosci. 2013; 33: 10924–10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh SW, Bergher JP, Anderson CM, Treadway JL, Fosgerau K, Swanson RA. Astrocyte glycogen sustains neuronal activity during hypoglycemia: studies with the glycogen phosphorylase inhibitor CP-316,819 ([R-R*,S*]-5-chloro-N-[2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmethyl)propyl]-1H-indole-2-carboxamide). J Pharmacol Exp Ther. 2007; 321: 45–50. [DOI] [PubMed] [Google Scholar]
- Tamrakar P, Briski KP. Estradiol regulation of hypothalamic astrocyte adenosine 5’-monophosphate-activated protein kinase activity: role of hindbrain catecholamine signaling. Brain Res. Bull. 2015; 110: 47–53. [DOI] [PubMed] [Google Scholar]
- Wender R, Brown AM, Fern R, Swanson RA, Farrell K, Ransom BR. Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J. Neurosci. 2000; 20:6804–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QG, Raz L, Wang R, Han D, De Sevilla L, Yang F, Vadlamudi RK, Brann DW. Estrogen attenuates ischemic oxidative damage via an estrogen receptor alpha-mediated inhibition of NADPH oxidase activation. J. Neurosci. 2009;29:13823–13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QG, Wang R, Tang H, Dong Y, Chan A, Sareddy GR, Vadlamudi RK, Brann DW. Brain-derived estrogen exerts anti-inflammatory and neuroprotective actions in the rat hippocampus. Mol. Cell. Endocrinol. 2014; 389: 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]