

Abstract
Cardiac fibrosis in response to injury is a physiological response to wound healing. Efforts have been made to study and target fibroblast subtypes that mitigate fibrosis. However, fibroblast research has been hindered due to the lack of universally acceptable fibroblast markers to identify quiescent as well as activated fibroblasts. Fibroblasts are a heterogenous cell population, making them difficult to isolate and characterize. The presented protocol describes three different methods to enrich fibroblasts and myofibroblasts from uninjured and injured mouse hearts. Using a standard and reliable protocol to isolate fibroblasts will enable the study of their roles in homeostasis as well as fibrosis modulation.
Keywords: Biology, Issue 157, Isolation, αSMA, fluorescence activated cell sorting, fibroblasts, myofibroblast, collagen gel, immunofluorescence, MEFSK4
Introduction
Cardiac fibroblasts, cells of mesenchymal origin, play a significant role in maintaining the electrical conduction and mechanical forces in the heart in addition to the maintenance of cardiac architecture during homeostasis1. Following injury, these cells are activated, expand, and produce extracellular matrix (ECM) proteins2. Many preclinical studies have revealed fibroblasts as critical cellular regulators that maintain the structural integrity of an injured heart3 as well as main effector cells responsible for unchecked production and deposition of ECM proteins, resulting in stiff scar formation and heart failure4. Fibroblasts are a heterogenous group of cells, making it challenging to dissect their reparative function from pro-fibrotic maladaptive properties. Recently, the functional heterogeneity of two distinct fibroblast subtypes following myocardial injury have been defined, indicating the possibility of isolating different fibroblast subtypes and studying their role in wound healing5.
Obtaining a pure fibroblast population is crucial in delineating their functional role in repair and fibrosis. However, the presence of multiple fibroblast markers that recognize other cell types make it challenging to isolate a substantially pure fibroblast population6. Several elegant studies have devised clever ways to isolate cardiac fibroblasts from uninjured and injured myocardium. The most popular and well-established method of enriching fibroblasts is through selective adhesion following enzymatic tissue digestion7.
Additionally, fluorescence-activated cell sorting (FACS) of fibroblasts based on cell surface antigens has been successfully described8. In the study, following enzymatic digestion, the mesenchymal cells were sorted as lineage-negative (Lin: Ter119−CD45−CD31−) and gp38-positive (gp38+) from mouse hearts. Gp38+ve cells were confirmed to be fibroblasts based on their co-expression of col1α1 and other mesenchymal markers. Although most tissue digestion is completed after dissecting out the ventricle in a Petri dish, a recent study has investigated the use of a direct needle enzyme perfusion of the left ventricle to isolate myocytes and non-myocytes which include fibroblasts9. Fibroblasts were then isolated by selective adhesion in this case.
This protocol describes the isolation and enrichment of fibroblasts using three methods. The first is an already established method involving selective adhesion of fibroblasts following enzymatic digestion. The second method is used to primarily isolate injury-induced alpha smooth muscle expressing myofibroblasts. The third method involves sequential, magnetic depletion of an enzyme-digested cardiac cell suspension of hematopoietic and endothelial cells. Following depletion, fibroblasts/myofibroblasts are isolated based on the presence of the antigen MEFSK4 using magnetic beads. Recently, MEFSK4 has been described as an antigen present on quiescent as well as activated fibroblasts, making it a suitable marker for fibroblast identification and isolation. Naturally, all the methods described here have unique limitations. It is therefore highly recommended to check the purity of the isolated cell population by flow analysis, immunostaining, and semi-quantitative real-time PCR. However, these methodologies can be expanded upon, and additional markers can be added in order to exclude other contaminating populations prior to utilizing the fibroblast and myofibroblast populations for crucial experiments.
Protocol
This study strictly upholds the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The Vanderbilt University Institutional Animal Care and Use Committee approved the protocol (protocol number: M1600076–01).
1. Heart Dissection
1. Solution preparation
- KHB buffer
-
Using a stir bar, slowly dissolve 9.4 g of Krebs-Henseleit buffer (KHB) powder in 900 mL of DDI water.NOTE: Buffer will precipitate if stirred too quickly or for too long. KHB buffer must be cold during fibroblast isolation.
- Add 2.9 mM CaCl2 and 24 mM NaHCO3. Adjust the pH to 7.2–7.3 and dilute to a volume of 1 L with dH2O.
- Using a sterile filter (0.22 µm), store at 4 °C for up to 4 weeks. Keep on ice or at 4 °C for duration of isolation.
-
- Collagenase digestion cocktail
- Prepare digestion cocktail the day of fibroblast isolation. Determine the appropriate volume of digestion cocktail according to the number of hearts; 5 mL of cocktail per 1 heart.
- Prepare collagenase blend (see Table of Materials) and DNase I according to the manufacturer’s instructions.
- For 20 mL of total digestion cocktail, add 17.5 µL of DNase I, 180 µL of 1 M HEPES, and 500 µL of collagenase to an empty 50 mL conical tube.
- Add a sufficient volume of Hank’s Balanced Salt solution (HBSS) with Ca2+ and Mg2+ to obtain a 20 mL total volume.
- Red blood cell (RBC) lysis buffer
- Determine the total volume needed based on number of hearts (5 mL/heart). Dilute the 10x RBC lysis stock buffer to 1x using dH2O.
- Fibroblast media: 10% FBS in DMEM F-12
- Add 10% FBS to DMEM-F12 with L-glutamine and HEPES. Add 10 U/mL penicillin/streptomycin, 2.5 µg/mL anti-fungal, and 2.5 µg/mL mycoplasma prophylactic (see Table of Materials). Store at 4 °C.
Materials
| Name | Company | Catalog Number | Comments |
|---|---|---|---|
| Reagents | |||
| Acetone | |||
| Anti-fungal (Amphotericin B- solubilized; Fungizone) | Sigma Aldrich | A9528 | |
| Bovine Serium Albumin (BSA) | Sigma | 9048–46-8 | |
| Calcium chloride | |||
| Citrate Buffer | |||
| Collagenase blend (Liberase Blendzyme 3 TH) | Roche Applied Science | ||
| DAPI | |||
| DDI water | |||
| DI water | |||
| DMEM-F12 with L-Glutamine and HEPES | Life technologies | 11330057 | |
| Dnase I(20U/mL) | BioRad | 7326828 | |
| Dulbecco's Phosphate-Buffered Saline (dPBS) without Ca2+ and Mg2+ | Gibco | 13190–144 | |
| 70% Ethanol | |||
| FC Blocker (Purified anti-mouse CD16/CD32) | Tonbo Biosciences | 70–0161 | |
| Fetal Bovine Serum (FBS) | Life technologies | 16000044 | |
| 10% goat serum | |||
| Hank's Balanced Salt Solution (HBSS) with Ca2+ and Mg2+ | Corning | 21–023-CV | |
| 1M HEPES | Corning | 25–060-Ci | |
| Krebs-Henseleit Buffer powder | Sigma | K3753 | |
| Mycoplasma prophylactic (Plasmocin) | Invivogen | ant-mpp | |
| Penicillin/Streptomycin | Thermo Fisher Scientific | 15140122 | |
| 1x Phosphate-Buffered Saline (PBS) | |||
| 10x Red Blood Cell Lysis Buffer | Miltenyi | 130–094-183 | |
| Slow-fade Mounting Media | |||
| Sodium azide | |||
| Sodium bicarbonate | |||
| TGFβ | |||
| Trypan Blue Stain (0.4%) | Gibco | 15250–061 | |
| Type 1 Rat Collagen | |||
| Antibodies | |||
| 7AAD (stock: 1 mg/mL solution in DMSO) | Molecular Probes | A1310 | dilution = 1:1000; RRID = |
| CD45-APC | BD Bioscience | 559864 | dilution = 1:200; RRID = AB_398672 |
| CD31-PE | BD Bioscience | 553373 | dilution = 1:200; RRID = AB_394819 |
| CD31 | BD Biosciences | 553370 | dilution = 1:250; RRID = AB_394816 |
| CD45 | BD Biosciences | 553076 | dilution = 1:250; RRID = AB_394606 |
| COL 1α1 | MD Bioproducts | 203002 | dilution = 1:1000; RRID = |
| Ghost dye violet 510 (Formulation: 1 uL/test in DMSO) | Tonbo Biosciences | 13–0870 | dilution = 1:1000; RRID = |
| Goat anti-mouse Alexa Fluor 488 | Molecular Probes | A11029 | dilution = 1:200; RRID = AB_138404 |
| Goat anti-rabbit-Cy3 | Southern Biotech | 4050–02 | dilution = 1:200; RRID = AB_2795952 |
| Goat anti-rabbit-FITC | Jackson Immunoresearch Laboratories | 711–165-152 | dilution = 1:200; RRID = AB_2307443 |
| Goat anti-rat Alexa Fluor 488 | Molecular Probes | A11006 | dilution = 1:200; RRID = AB_2534074 |
| Goat anti-rat Alexa Fluor 647 | Thermo-Fisher | A21247 | dilution = 1:200; RRID = AB_141778 |
| Periostin | Santa Cruz | SC67233 | dilution = 1:100; RRID = AB_2166650 |
| Vimentin | Sigma Aldrich | V2258 | dilution = 1:200; RRID = AB_261856 |
| α-smooth muscle actin (αSMA) | Sigma Aldrich | A2547 | dilution = 1:1000; RRID = AB_476701 |
| Fibroblast specific protein 1 (FSP1) | Millipore 07–2274 | 07–2274 | dilution = 1:100; RRID = AB_10807552 |
| CD45 Magnetic Beads | Miltenyi Biotec | 130–052-301 | |
| CD31 Magnetic Beads | Miltenyi Biotec | 130–087-418 | |
| Anti-feeder cells-APC (MEFSK4) | Miltenyi Biotec | 130–102-900 | dilution = 1:100; RRID = AB_2660619 |
| anti-APC Beads | Miltenyi Biotec | 130–090-855 | |
| Rat IgG-APC | Miltenyi Biotec | 130–103-034 | dilution = 1:100; RRID = AB_2661598 |
| Donkey anti-rat Alexa Fluor405 | Abcam | ab175670 | dilution = 1:100 |
| anti-AN2/NG2 | Miltenyi Biotec | 130–097-455 | dilution = 1:11; RRID = AB_2651235 |
| Other Materials | |||
| 0.22 µm Filter | Thermo Scientific | 723–2520 | |
| 10 cm2 Cell Culture Dish | Corning | 430167 | |
| 10 mL Pipet | Fisherbrand | 13–678-11E | |
| 40 µm Cell Strainer | Fisherbrand | 22363547 | |
| 5 mL Pipet | Fisherbrand | 13–678-11D | |
| 50 mL Conical Tube | Falcon | 352070 | |
| 6-well Plate | Corning | 3506 | |
| Flow Cytometry Tubes | Falcon | 352058 | |
| Forceps | |||
| Rocker | |||
| Single Edge Blade | PAL | 62–0177 | |
| Surgical Scissors | |||
| GFP-αSMA Reporter Mice | |||
| MACS Separator Magnetic Field | |||
| MACS Separation Column | |||
| Coverslips | |||
| Qaigen Rneasy Mini Kit | Qaigen | 74104 | |
| Ambion RNAqueous Micro Total Isolation Kit | Ambion | AM1931 | |
| BioRad iScript cDNA Syntehsis Kit | BioRad | 1708891 | |
| 48-well Plate | |||
| 30G Needle | |||
| 3 Laser Flow Cytometry Machine (BD LSRFortessa) | BD Biosciences | ||
| 4 Laser Flow Cytometry Machine (BD FACSAria III) | BD Biosciences | ||
| Flow Data Acquiring Software (BD FACSDiva Software v8.0a) | BD Biosciences | ||
| Flow Data Analysis Software (FlowJo Software) | BD Biosciences |
2. Heart dissection
Prepare a 6 well plate on ice with 2 mL of cold KHB per well to store hearts in during dissection. Utilize autoclaved surgical scissors and forceps.
Euthanize mice at 12 weeks of age or older by isoflurane overdose, and follow with cervical dislocation.
Alternatively, for activated fibroblast isolation, induce myocardial infarction in 12 week-old mice by coronary artery ligation10. Euthanize the mice 8–10 days following injury.
Spray body with 70% ethanol and orient so the ventral side is facing the experimenter. Pin or restrain appendages to prevent interference.
Cut the abdominal skin and muscle open but avoid piercing the liver. Cut vertically towards the sternum, and carefully open the thorax while avoiding piercing of the heart. Continue to cut through the ribcage to expose the heart.
Using forceps, gently lift the heart out of the chest, cutting away any lung or excess tissue attached to the outside of the heart. Remove the ventricle and place in one well of 6 well plate with cold KHB. Continue to dissect hearts in this manner until all samples have been isolated.
3. Enzymatic dissociation of heart
Using forceps, repeatedly squeeze and agitate the heart in KHB to remove excess blood. Transfer the heart to a clean sterile 10 cm2 plate. Using a single edge blade, quickly mince the heart into small pieces.
-
Add 1 mL of collagenase digestion cocktail and continue mincing until pieces are small enough to transfer with a 1 mL micropipette. Transfer pieces to a 50 mL conical tube with a 1 mL micropipette. Wash plate 2x with 2 mL of collagenase digestion cocktail.
NOTE: Cutting off a portion of the pipette tip can help collect larger pieces of heart that may otherwise become stuck in the pipette tip.
Incubate conical tube at 37 °C for 30 min with rocking or agitation. Secure tube as needed. Resuspend 10x with a 5 mL pipet until the contents are homogenous, and incubate the conical tube at 37 °C for 15 min with rotation or agitation.
-
Resuspend 10x with a 10 mL pipet until homogenous. Prime a 40 µm cell strainer by wetting the filter with 1–2 mL of KHB buffer on top of a new 50 mL conical tube. Add 25 mL of KHB buffer to digestion suspension, resuspend, and filter through a primed 40 µm cell strainer. Change filter as needed.
NOTE: As filtration slows, lightly tapping tube or using a 1 mL micropipette to draw suspension from underside of filter can help the cell suspension pass through a partially blocked 40 µm cell strainer.
Centrifuge at 400 × g and 4 °C for 10 min. Remove the supernatant and resuspend the pellet in 1x RBC lysis buffer (5 mL/heart). Incubate for 2 min at room temperature (RT).
Centrifuge at 400 × g and RT for 10 min. Remove supernatant then wash by resuspending pellet in 1 mL of KHB buffer.
Add 9 mL of KHB buffer and filter through primed 40 µm cell strainer into new 50 mL conical tube. Centrifuge at 400 × g and RT for 10 min.
Remove the supernatant, resuspend in 1 mL of fibroblast media or PBS, and determine the cell number. Fibroblasts can be isolated by the three different methods described below.
2. Isolation of Fibroblasts from Single Cell Suspension
1. Fibroblast isolation: differential plating (Figure 1)
Figure 1: Schematic of fibroblast isolation using three different approaches.
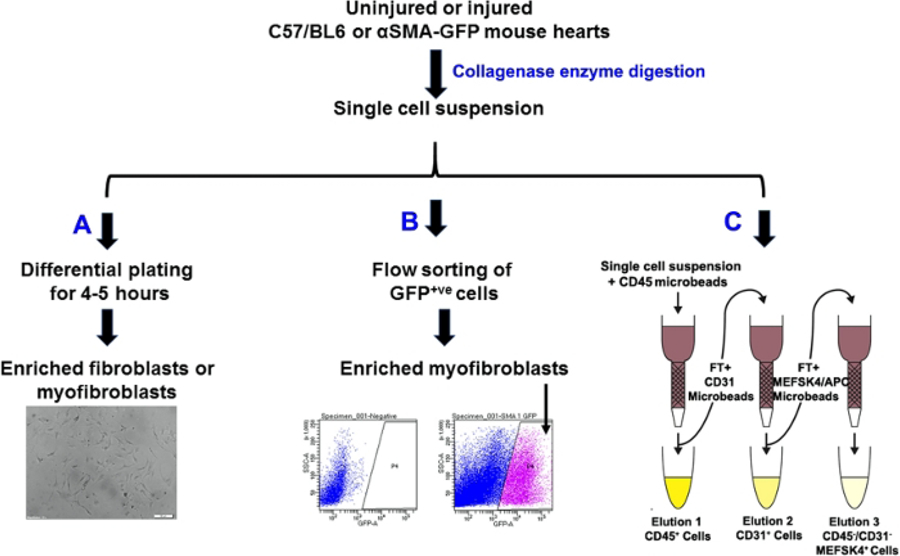
(A) differential plating, (B) GFP+ cell sorting of αSMA positive cells, and (C) magnetic bead based isolation of fibroblasts. Representative bright field of the cells in culture following differential plating. Scale bar = 50 µM.
Prepare a 6-well plate by adding 2 mL of fibroblast media per well and swirling the plate to cover the well bottom.
Resuspend cells in 1 mL of media per heart. Plate 1 mL of cell suspension per well such that one heart (theoretically) is being plated per well. For example, six hearts would be resuspended in 6 mL of media, then plated into six wells with 1 mL of cell suspension per well.
Swirl plate to evenly distribute cells. Add an additional 1–2 mL media per well for a total volume of up to 5 mL per well, and incubate at 37 °C for 4 h.
Fibroblasts will be separated by selective adhesion after 4 h. Remove unattached and dead cells by removing media. Wash attached cells with 2 mL of PBS and add 2–4 mL sterile fibroblast media per well. Incubate at 37 °C until confluent. Change media every 2–4 days.
2. Fibroblast Isolation: FACS using a GFP reporter mouse model (Figure 1)
- FACS buffer
- Prepare 15 mL of 5% fetal bovine serum (FBS) in dPBS without Ca2+ and Mg2+. Store on ice or at 4°C.
- FC blocker solution
- Prepare 0.5 µL FC blocker (purified anti-mouse CD16/CD32) in 25 µL of FACS (1:50 dilution). 25 µL of FC blocker solution is required per sample. Store on ice or at 4 °C.
-
Sample preparation and antibody staining
NOTE: Antibody dilutions can be prepared in advance, but this may risk light or temperature exposure.- Prepare 7AAD or Ghost dye violet 510 that is 2x the required dilution in FACS buffer. See Table 1 for antibodies and dilutions.
- Resuspend 500,000 freshly isolated cells in 25 µL of FC blocker solution to prevent nonspecific binding of the FC antibody region to an FC receptor. Incubate for 5 min at RT.
- Add 7AAD or Ghost dye violet 510 antibody to the final volume of 25 µL of cell suspension. Incubate for 30–60 min on ice in the dark.
- Wash by resuspending in 1 mL of FACS buffer. Centrifuge at 500 × g for 5 min at 4°C.
- Resuspend pellet in recommended flow cytometry sample volume of FACS buffer (300 µL) and transfer to flow cytometry tube.
-
Fluorescence-activated cell sorting (FACS)
NOTE: GFP-αSMA reporter mice were a contribution from Dr. Ivo Kalajzic11. This mouse expresses GFP under the alpha smooth muscle cell (αSMA) promoter. αSMA has been used as a marker of activated fibroblasts (myofibroblasts)12.- For activated fibroblast (αSMA expressing myofibroblast) isolation, induce myocardial infarction in 12 week-old GFP-αSMA mice by coronary artery ligation. Sacrifice the mice 8–10 days following injury.
- Following single cell suspension from the injured mice hearts, sort αSMA+ve fibroblasts for green fluorescent protein (GFP). Use unstained uninjured fibroblasts to set the background signal in the GFP channel post-compensation.
- Gate for live GFP+ve cells by gating for 7AAD−ve/GFP+ve cells or Ghost dye violet 510−ve/GFP+ve cells and sort GFP expressing αSMA+ve myofibroblasts. Collect the cells in fibroblast media.
Table 1:
FACS dyes and antibodies.
| Antibody | Cell Target | Dilution |
|---|---|---|
| 7AAD | Dead cells | 1:1000 |
| Ghost dye violet 510 | Dead cells | 1:1000 |
| APC-CD45 | Hematopoietic cells | 1:200 |
| PE-CD31 | Endothelial cells | 1:200 |
| anti-AN2/NG2 | Pericytes | 1:11 |
| Donkey anti-rat alexa fluor 405 | Secondary Antibody | 1:100 |
| Anti-feeder cells-APC (MEFSK4) | Fibroblasts | 1:100 |
| Rat IgG-APC | Isotype Control | 1:100 |
3. Fibroblast isolation: magnetic bead-based isolation of fibroblasts (Figure 1)
-
Equilibration buffer
NOTE: Always prepare fresh buffer for isolations.-
Prepare 0.5% BSA and 2 mM EDTA in PBS. Degas the buffer by stirring the solution while attaching a vacuum to the lid of the container.NOTE: Stirring removes excess gas from the solution that is then removed through the vacuum such that bubbles will not clog the separation column upon usage.
-
- Magnetic labeling: CD45+ hematopoietic cells
- Centrifuge isolated cells from mice hearts at 500 × g for 5 min, then remove the supernatant.
- Resuspend the cell pellet in 1 mL of equilibration buffer. Count the cells using a hemocytometer.
- Centrifuge the cell suspension as above and resuspend cell pellet in 90 μL equilibration buffer per 1 × 107 total cells. Add 10 μL of CD45+ magnetic beads per 1 × 107 total cells. Mix well and incubate for at least 15 min at 4°C.
- Wash cells by adding 2 mL equilibration buffer per 1 × 107 total cells, then centrifuge at 500 × g for 10 min at 4°C.
- Remove supernatant, count the cells using a hemocytometer and resuspend up to 1 × 107 total cells in 2 mL of equilibration buffer. If more than 107 cells are present, scale the buffer linearly. Pass cells through a 40 μm filter to prevent cell aggregations from clogging the separation column matrix.
- Magnetic separation: CD45+ hematopoietic cells
- Place separation column in the magnetic field of a suitable separator and equilibrate column with at least 3 mL of PBS.
- Collect unlabeled cells in the flowthrough (FT), and wash column 3x with 3 mL of equilibration buffer. Collect washes with FT. Remove the column from the separator. Place the column on a 15 mL conical tube.
- Flush out magnetically labeled CD45+ cells by pipetting 5 mL of equilibration buffer onto the column and firmly plunging the cells with a plunger supplied with the column.
- Centrifuge eluent as well as FT/wash fractions at 500 × g. Count the cells using a hemocytometer.
- Magnetic labeling and separation: CD31+ endothelial cells
- Repeat protocol for CD45+ magnetic labeling and separation (sections 2.3.2–2.3.3), except use CD31+ magnetic beads to incubate with the FT and wash portions from the CD45+ isolation.
- Magnetic labeling and separation: MEFSK4+ fibroblasts
- Repeat protocol for CD45+ magnetic labeling using the MEFSK4 anti-feeder-APC antibody instead of magnetic beads. Centrifuge the FT and Wash portions from the CD31+ isolation at 500 × g for 5 min, then remove supernatant. Resuspend the cell pellet in 1 mL of equilibration buffer, and count the cells using a hemocytometer.
- Add 10 µL of MEFSK4 anti-feeder-APC antibody per 1 × 107 cells. Incubate for at least 15 min at 4 °C.
- Wash MEFSK4 antibody bound cells by adding 5 mL of equilibration buffer per 1 × 107 total cells, then centrifuge at 500 × g for 5 min.
- Remove supernatant and resuspend in anti-APC beads, using the same volume of MEFSK4 anti-feeder-APC antibody used. Incubate on ice for 15 min at 4 °C.
- Centrifuge at 500 × g for 10 min. Remove supernatant, resuspend in 2 mL of equilibration buffer per 1 × 107 total cells, and proceed with magnetic separation as previously described (section 2.3.3). Magnetically separated MEFSK4+ve/ CD45−ve/CD31−ve fibroblasts can be used for purity analyses and other downstream applications.
3. Purity and Functionality Analysis of Isolated Fibroblast Population
1. FACS population purity analysis: αSMA-GFP cell analysis (Figure 2)
Figure 2: FACS analysis of single cells isolated from αSMA-GFP mice hearts following MI.
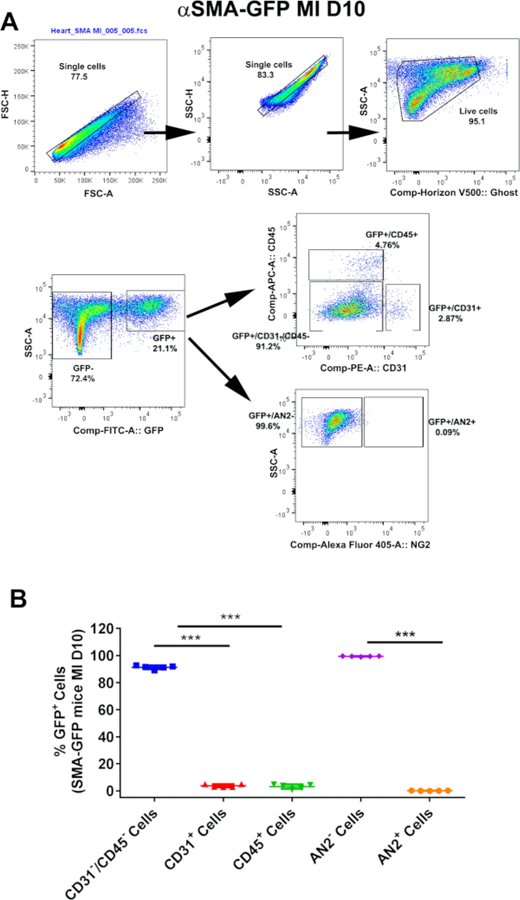
(A) Representative FACS gating scheme demonstrating GFP+ cells co-expressing CD31, CD45, or AN2 from αSMA-GFP mice injured hearts 10 days after myocardial infarction (MI). (B) Graphical quantification of the presented FACS data for post-MI hearts; n = 5 experiments were performed independently (***p < 0.0001 as calculated using one-way ANOVA with Tukey’s multiple comparisons test). This figure is adapted from Saraswati et al.10.
Resuspend freshly isolated cells in 25 µL of Fc blocker solution to prevent the nonspecific binding of antibodies. Incubate for 5 min at RT.
Optional: add 25 µL of 7AAD or Ghost dye violet 510 to cell suspension. Incubate for 30–60 min on ice in the dark.
Wash by resuspending in 1 mL of FACS buffer. Centrifuge at 500 × g for 5 min.
Resuspend pellet in 50 µL of FACS buffer. Add CD31-PE, CD45-APC, and AN2/NG2 antibodies directly to cell suspension. Incubate for 15 min on ice (Table 1).
Wash by resuspending in 1 mL FACS buffer. Centrifuge at 400 × g for 5 min.
Add donkey anti-rat AlexaFluor 405 secondary antibody to the cells labeled with unconjugated primary antibody. Incubate for 30 min on ice.
Wash by resuspending in 1 mL of FACS buffer. Centrifuge at 400 × g for 5 min.
-
Resuspend pellet in recommended flow cytometry sample volume of FACS buffer (300 µL) and transfer to a labeled flow cytometry tube for flow analysis.
NOTE: FACS analysis to characterize the expression of MEFSK4 antigen on isolated fibroblasts is described in section 3.3.
2. Fibroblast purity analysis: immunofluorescence (Figure 3A)
Figure 3: Purity analyses of fibroblasts isolated from uninjured and injured mice hearts.
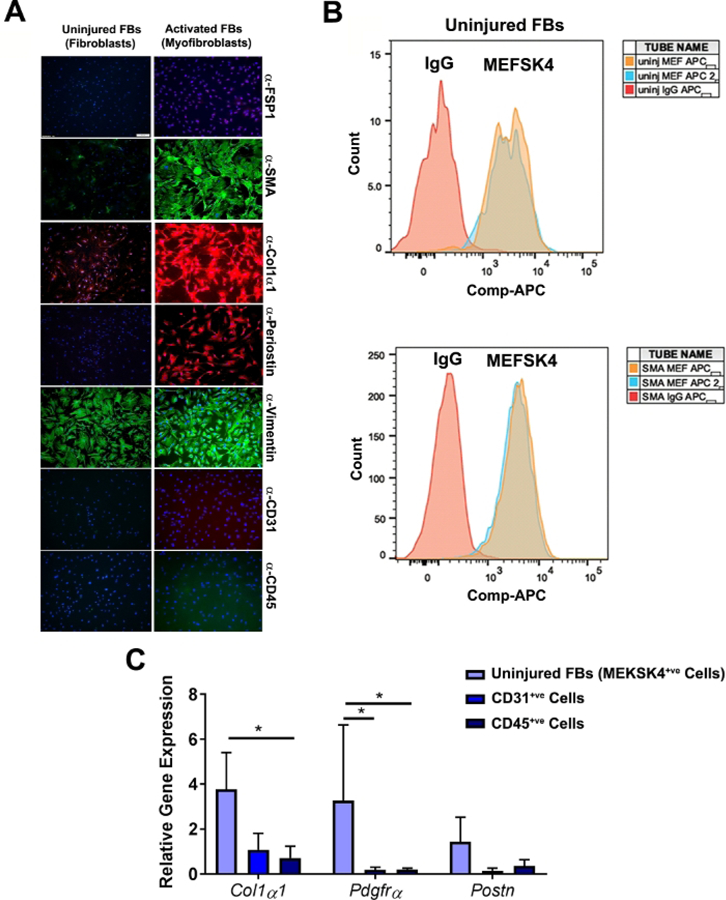
(A) Immunofluorescence staining of cell populations (P0) from heart of uninjured, or injured αSMA-GFP mice sorted by FACS. Both uninjured and activated cells express fibroblast (FB) markers, such as COL1α1, and vimentin, but not hematopoietic marker CD45 or the endothelial marker CD31. Cells isolated from injured αSMA-GFP mice heart expressed activated fibroblast markers, αSMA, and periostin, which were not present in the cells isolated from uninjured mice hearts. Nuclei were stained with DAPI, n = 3 experiments were performed independently. Scale bar = 100 µm. (B) Representative FACS overlay histogram of uninjured and activated fibroblasts (P3–P5) showing the expression of the fibroblast marker MEF-SK4. For a negative control, rat IgG was used, n = 2 experiments were performed independently. (C) Relative fold change of Col1α1, Pdgfrα, and Postn transcripts in uninjured MEKSK4+ve fibroblasts, n = 3 experiments were performed independently (*p < 0.05 as calculated using two-way ANOVA with Tukey’s multiple comparisons test). (A) and (B) are adapted from Saraswati et al.10.
Seed 30,000 primary fibroblasts (P0–P1) per well on coverslips placed in a 24 well plate and culture until 80% confluent. Or concentrate sorted CD45+ and CD31+ cells (30,000 cells per well) on a coverslip by cytospin at 400 x× g (a method used to deposit cells directly and evenly onto a coverslip in a 24 well plate).
Fix cells with cold acetone for 15 min. Wash 3x times with PBS.
Block slides in 10% goat serum. Incubate slides with primary antibodies overnight (Table 2).
Wash slides 3x in PBS. Incubate secondary antibodies for 2 h (Table 2).
Counterstain slides, and mount with one drop of DAPI in slow-fade mounting media.
Table 2:
Immunofluorescence primary and secondary antibodies.
| Primary Antibody | Dilution |
|---|---|
| α-smooth muscle actin (αSMA) | 1:1000 |
| Fibroblast specific protein 1 (FSP1)1:250 | 1:100 |
| COL 1α1 | 1:1000 |
| Periostin | 1:100 |
| Vimentin | 1:200 |
| CD31 | 1:250 |
| CD45 | 1:250 |
| Secondary Antibody | Dilution |
| Goat anti-mouse Alexa Fluor 488 | 1:200 |
| Goat anti-rabbit-FITC | 1:200 |
| Goat anti-rabbit-Cy3 | 1:200 |
| Goat anti-rat Alexa Fluor 488 | 1:200 |
| Goat anti-rat Alexa Fluor 647 | 1:200 |
3. FACS population purity analysis: MEFSK4 probing (Figure 3B)
Resuspend freshly isolated cells in 25 µL of FC blocker solution to prevent the nonspecific binding of antibodies. Incubate for 5 min at RT.
Optional: add 25 µL of 7AAD or Ghost dye violet 510 to cell suspension. Incubate for 30–60 min on ice in the dark.
Wash by resuspending in 1 mL of FACS buffer. Centrifuge at 500 × g for 5 min.
Resuspend pellet in 50 µL of FACS buffer. Add MEFSK4 antibody directly to cell suspension. Incubate for 15 min on ice (Table 1).
Wash by resuspending in 1 mL of FACS buffer. Centrifuge at 400 × g for 5 min.
Add rat IgG-APC to the cells (Table 1). Incubate for 30 min on ice.
Wash by resuspending in 1 mL of FACS buffer. Centrifuge at 400 × g for 5 min.
Resuspend pellet in recommended flow cytometry sample volume of FACS buffer (300 µL) and transfer to flow cytometry tube for flow analysis.
4. Fibroblast purity analysis: semi-quantitative real-time rtPCR (Figure 3C)
RNA isolation and semiquantitative real-time PCR
Following the enrichment of fibroblasts, isolate RNA using an RNA isolation kit (see Table of Materials). Follow the manufacturer’s instructions.
Complete first strand DNA synthesis using a cDNA synthesis kit (see Table of Materials), following the manufacturer’s instructions.
Perform semiquantitative real-time PCR5.
5. Fibroblast functionality analysis: collagen gel contractility assay (Figure 4)
Figure 4: Functional characterization of fibroblasts isolated from uninjured and injured mouse hearts.
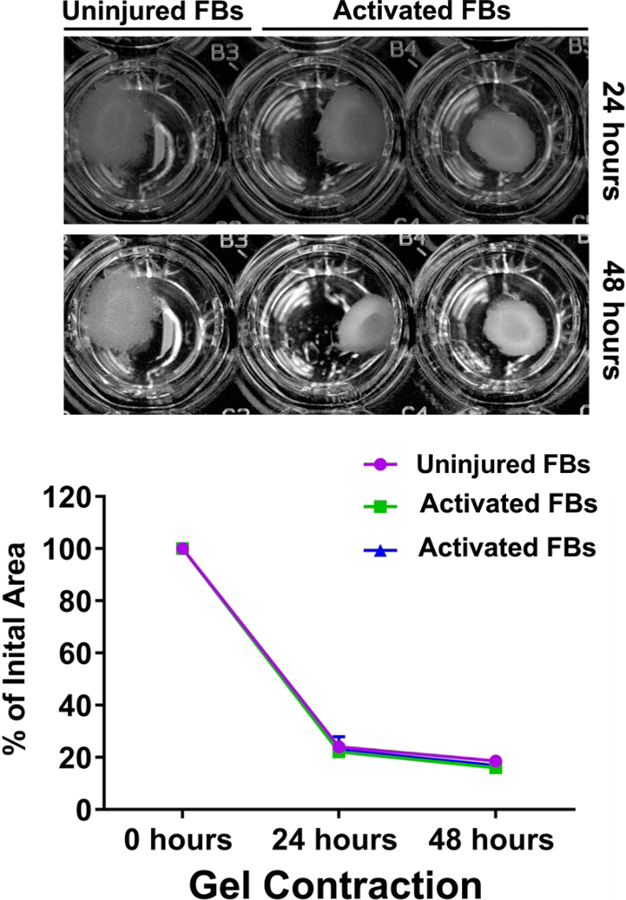
Representative figure of collagen gel contraction in the presence of uninjured and injured activated αSMA+ fibroblasts (P3–P5). The graph represents the percentage change in the initial gel area after 24 h and 48 h of contraction when incubated with uninjured and injured activated αSMA+ fibroblasts, n = 2 experiments were performed independently. This figure is adapted from Saraswati et al.10.
- Collagen solution
- Prepare 20 mM HEPES and 44 mM NaHCO3 in DMEM. Add 1.67 mg of type 1 rat collagen per 1 mL of DMEM with HEPES and NaHCO3.
- TGFβ-supplemented DMEM
- Prepare 10% FBS in DMEM supplemented with antibiotics and anti-fungal. Add TGFβ to a final concentration of 1 ng/mL.
- Cell/collagen mixture and plating
- Prepare cell suspension (P3–P5) and determine required volume to obtain 3.3 × 105 cells.
- Add suspension volume with 3.3 × 105 cells to enough collagen solution to obtain 1 mL total volume. NOTE: Rat collagen type 1 concentration should now be 1.5 mg/mL.
- In a 48 well plate, seed 300 µL of cell-collagen mix per well (~1 × 105 cells/well). Incubate at 37 °C for 15–20 min until gelled.
-
Use a 30 G needle to help separate gel from the well walls. Add 600 µL of TGFβ and FBS supplemented DMEM to each well. Image plates on a reflective scanner at 24 h and 48 h.NOTE: All Immunofluorescence experiments were performed on a flow cytometry machine equipped with three lasers (405 nm, 488 nm, and 640 nm). Data were acquired using a flow data acquiring software (Table of Materials). Further data analysis was performed using flow data analysis software. AlexaFluor 405 and Ghost Dye Violet 510 were excited with the 405 nm laser and collected using a 450/50 BP and 525/50 BP filter, respectively. GFP and PE were excited by the 488 nm laser and collected using the 530/30 BP and 575/26 BP filters, respectively. Either APC or AlexaFluor 647 was excited by the 640 nm laser and collected using a 670/14 BP filter. All cell sorting experiments were performed on a flow cytometry machine (Table of Materials) equipped with four lasers (405 nm, 488 nm, 561 nm and 640 nm). 7-AAD was excited using the 561 nm laser and collected with a 670/14 BP filter. GFP and Ghost Dye Violet 510 were collected using the same laser/filter combinations as described above. All sorting experiments utilized a 100 um nozzle with a 17 psi pressure configuration for increased downstream viability of the target cells.
Representative Results
Flow gating scheme demonstrating myofibroblast isolation using αSMA-GFP reporter mice
Uninjured hearts showed no detectable GFP+ cells in αSMA-GFP reporter mouse model; hence, they were used to establish a gate for the background signal of the GFP channel post-compensation (Figure 2). αSMA+ cells were sorted based on the presence of GFP expression from the injured left ventricle 10 days following MI. A small percentage of endothelial (GFP+/CD31+ cells; SD = 3.8% ± 0.0164; n = 5) and hematopoietic (GFP+/CD45+ cells; SD = 3.18% ± 0.0112; n = 5) cells also expressed GFP in the injured αSMA-GFP mouse hearts (Figure 2A). However, GFP+/CD31-/CD45- cells did not express AN2, a pericyte marker.
Uninjured (quiescent) and injured (activated, αSMA+GFP+) cells expressed fibroblast markers
GFP+ cells isolated from αSMA-GFP mice expressed αSMA, collagen type 1 alpha-1 chain (COL1α1), vimentin, and periostin when analyzed by IF analysis. Uninjured fibroblasts isolated by selective adhesion expressed vimentin but did not demonstrate expression of the activated fibroblast markers: αSMA, periostin, and COL1α1 (Figure 3A). Both uninjured and activated fibroblasts expressed the MEFSK4 antigen when analyzed by flow analysis (Figure 3B). Magnetically isolated MEFSK4+ve cells from uninjured mice hearts expressed markers of fibroblasts: Col1a1, pdgfrα, and periostin. In contrast, magnetically isolated CD45 and CD31 positive cells had negligible expression of fibroblast markers.
Fibroblasts and myofibroblasts demonstrated the ability to contract collagen
In cell culture on stiff plastic, fibroblasts have been shown to contract collagen gels in the presence of TGFβ, demonstrating their functional capability of contraction13,14. This in vitro characteristic of fibroblasts is very similar to the connective tissue contraction that happens during tissue repair as well as other biological processes. Both uninjured fibroblasts, isolated by selective adhesion, and myofibroblasts, isolated and sorted from αSMA-GFP mice, demonstrated an ability to contract collagen (Figure 4).
Discussion
Fibroblasts are a heterogenous group of cells, identified by diverse set of markers. The protein markers that have been used to identify fibroblasts are discoidin domain receptor 2 (DDR2), fibronectin, vimentin, collagen I and III, and Thy115,16,17,18,19,20. Whereas vimentin has been used to identify uninjured quiescent cardiac fibroblasts, fibroblast specific protein 1, αSMA, and periostin have been shown to identify injury-induced activated fibroblasts, with αSMA being the most common marker to detect activated fibroblasts6,12,21. Additionally, Tcf21 and MEFSK4 proteins have gained recent recognition in recognizing both quiescent fibroblasts found in uninjured cardiac tissue as well as activated fibroblasts including myofibroblasts found in injured mouse hearts21,22.
This protocol utilizes three different approaches to isolate and enrich fibroblasts and activated fibroblasts including myofibroblasts. The fibroblast’s ability to preferentially adhere to plastic is used in the first approach for isolation. Following enzymatic digestion with liberase, the single cell suspension of cells is seeded on a plastic dish to preferentially adhere. The inability of many non-fibroblast cells to adhere to the polystyrene surface of Petri dishes allows us to remove all media from the dish and remain with a relatively pure population of fibroblasts. A caveat of using this technique is although fibroblasts will preferentially adhere to the polystyrene dish, some contaminating non-fibroblast cells may also attach, leaving a non-homogenous population of cells.
The second isolation technique utilizes FACS to separate αSMA expressing myofibroblasts from other cells. In the transgenic mouse model employed here, GFP is exclusively expressed along with αSMA, so myofibroblasts containing αSMA can be detected by a FACS machine through the fluorescent capabilities of GFP. This isolation procedure enables us to obtain a population of cells that is approximately 99% myofibroblast. The purity analyses of these cells have been extensively described by Saraswati et al.10.
The third isolation technique is an efficient way to isolate both uninjured and activated fibroblasts by magnetic bead-based separation of MEFSK4 expressing cell. By allowing a single cell suspension to bind to anti-CD45 and anti-CD31 magnetic beads and become immobilized in a matrix due to magnetic field effects, this allowed the separation of any hematopoietic as well as endothelial cells that may have contaminated the fibroblast isolation. As MEFSK4 has been recently used as a reliable marker to identify fibroblasts, an antibody that will bind to MEFSK4 expressing cells is able to be applied. After binding a magnetic bead to the antibody, creating a complex that allows isolation of fibroblasts, the magnetic bead-cell complex is passed through a matrix in a magnetic field, and a highly enriched fibroblast population is obtained. The purity of the isolated fibroblast population should be assessed by immunostaining, RTPCR, and flow cytometry analyses.
As with any other technique, there are limitations with the techniques described in this manuscript. The limitation of the selective adhesion protocol and magnetic bead-based isolation is that these methods do not differentiate between quiescent and activated fibroblasts. In order to enrich activated fibroblasts, the isolation should be performed 8–10 days following myocardial infarction. Additionally, it is important to check the purity of the isolation with other fibroblast markers. MEFSK4-positive fibroblast purity has been demonstrated only by RTPCR, it is recommended to test (by immunostaining and flow cytometry analysis) with other fibroblast markers and markers that recognize contaminating cell types including hematopoietic (CD45), endothelial (CD31), and pericytes (AN2). If possible, other fibroblast specific markers could be used to further sort or magnetically isolate the fibroblast population.
Using αSMA-GFP mice to isolate and sort myofibroblasts is a reliable technique to obtain an activated fibroblast population. However, a negligible percentage of hematopoietic and endothelial cells has been observed in the flow analysis. To improve upon this technique, CD45+ve/CD31+ve and AN2+ve cells should be excluded from the GFP+ve/αSMA+ve cell sorting. Since αSMA is a widely accepted marker of myofibroblasts, the αSMA-GFP reporter mouse model is a valuable tool that should be exploited to study myofibroblasts in the context of myocardial injuries.
There are several crucial troubleshooting steps that must be taken into account. Digestion time can be decreased if cell viability and yield is affected. A stir bar should not be used to stir the digestion mixture, as this affects cell viability. The tube should be secured on a rocker or in a shaking incubator to agitate the digestion mixture gently. Resuspending the digested tissue 10x with a 5 mL or 10 mL pipette is crucial for proper dissociation of cells.
Proper red blood cell lysis of the single cell suspension must be utilized if cells are going to be sorted or analyzed by flow cytometry. For magnetic bead isolation, degassing of the buffer is essential to prevent the introduction of any air bubbles in the column. The column used for magnetic bead cell isolation should not be reused between different magnetic bead-conjugated cells. For example, a new column should be used to separate CD45+ cells, which should be discarded after elution of the CD45+ cells, then another new one for the CD31+ cell isolation.
In our hands, we have not seen contamination of pericytes in isolated/sorted fibroblasts. However, MEFSK4 has been shown to recognize pericytes22. It is therefore recommended to use an additional step to sort out/magnetically deplete pericytes (AN2) from the single cells. Although this protocol is validated in 12 week-old mice, the technique may be used for younger or older mice.
Acknowledgments
The authors want to thank Dr. Ivo Kalajzic for the αSMA-GFP mice. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health (NIH) under Award Number R01GM118300 (S.S.), National Institute of Biomedical Imaging and Bioengineering of the NIH under Award Number R21EB019509 (P.P.Y.), and Scientist Development Grant of the American Heart Association under Award Number 17SDG33630187 (S.S.). Flow cytometry analyses were performed at the VUMC Flow Cytometry Shared Resource which is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404).
Footnotes
Video Link
The video component of this article can be found at https://www.jove.com/video/60909/
Disclosures
The authors have nothing to disclose.
References
- 1.Ivey MJ, Tallquist MD Defining the Cardiac Fibroblast. Circulation Journal. 80 (11), 2269–2276 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prabhu SD, Frangogiannis NG The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circulatory Research. 119 (1), 91–112 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duan J et al. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO Journal. 31 (2), 429–442 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shinde AV, Frangogiannis NG Fibroblasts in myocardial infarction: a role in inflammation and repair. Journal of Molecular and Cellular Cardiology. 70, 74–82 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saraswati S, Marrow SMW, Watch LA, Young PP Identification of a pro-angiogenic functional role for FSP1-positive fibroblast subtype in wound healing. Nature Communications. 10 (1), 3027 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kong P, Christia P, Saxena A, Su Y, Frangogiannis NG Lack of specificity of fibroblast-specific protein 1 in cardiac remodeling and fibrosis. American Journal of Physiology-Heart and Circulatory Physiology. 305 (9), H1363–1372 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furtado MB, Nim HT, Boyd SE, Rosenthal NA View from the heart: cardiac fibroblasts in development, scarring and regeneration. Development. 143 (3), 387–397 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Stellato M, Czepiel M, Distler O, Blyszczuk P, Kania G Identification and Isolation of Cardiac Fibroblasts From the Adult Mouse Heart Using Two-Color Flow Cytometry. Frontiers in Cardiovascular Medicine. 6, 105 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ackers-Johnson M et al. A Simplified, Langendorff-Free Method for Concomitant Isolation of Viable Cardiac Myocytes and Nonmyocytes From the Adult Mouse Heart. Circulatory Research. 119 (8), 909–920 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saraswati S, Marrow SMW, Watch LA, Young PP Identification of a pro-angiogenic functional role for FSP1-positive fibroblast subtype in wound healing. Nature Communications. 10 (1), 3027 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalajzic Z et al. Use of an alpha-smooth muscle actin GFP reporter to identify an osteoprogenitor population. Bone. 43 (3), 501–510 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC Cardiac Fibrosis: The Fibroblast Awakens. Circulatory Research. 118 (6), 1021–1040 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bell E, Ivarsson B, Merrill C Production of a tissue-like structure by contraction of collagen lattices by human fibroblasts of different proliferative potential in vitro. Proceedings of the National Academy of Sciences USA. 76 (3), 1274–1278 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Montesano R, Orci L Transforming growth factor beta stimulates collagen-matrix contraction by fibroblasts: implications for wound healing. Proceedings of the National Academy of Sciences USA. 85 (13), 4894–4897 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldsmith EC et al. Organization of fibroblasts in the heart. Developmental Dynamics. 230 (4), 787–794 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Bagchi RA, Lin J, Wang R, Czubryt MP Regulation of fibronectin gene expression in cardiac fibroblasts by scleraxis. Cell Tissue Research. 366 (2), 381–391 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Goodpaster T et al. An immunohistochemical method for identifying fibroblasts in formalin-fixed, paraffin-embedded tissue. Journal of Histochemistry and Cytochemistry. 56 (4), 347–358 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chapman D, Weber KT, Eghbali M Regulation of fibrillar collagen types I and III and basement membrane type IV collagen gene expression in pressure overloaded rat myocardium. Circulatory Research. 67 (4), 787–794 (1990). [DOI] [PubMed] [Google Scholar]
- 19.Vasquez C, Benamer N, Morley GE The cardiac fibroblast: functional and electrophysiological considerations in healthy and diseased hearts. Journal of Cardiovascular Pharmacology. 57 (4), 380–388 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hudon-David F, Bouzeghrane F, Couture P, Thibault G Thy-1 expression by cardiac fibroblasts: lack of association with myofibroblast contractile markers. Journal of Molecular and Cellular Cardiology. 42 (5), 991–1000 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Kanisicak O et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nature Communications. 7, 12260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinto AR et al. Revisiting Cardiac Cellular Composition. Circulatory Research. 118 (3), 400–409 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]