

Abstract
For infectious-disease outbreaks, clinical solutions typically focus on efficient pathogen destruction. However, the COVID-19 pandemic provides a reminder that infectious diseases are complex, multisystem conditions, and a holistic understanding will be necessary to maximize survival. For COVID-19 and all other infectious diseases, metabolic processes are intimately connected to the mechanisms of disease pathogenesis and the resulting pathology and pathophysiology, as well as the host defence response to the infection. Here, I examine the relationship between metabolism and COVID-19. I discuss why preexisting metabolic abnormalities, such as type 2 diabetes and hypertension, may be important risk factors for severe and critical cases of infection, highlighting parallels between the pathophysiology of these metabolic abnormalities and the disease course of COVID-19. I also discuss how metabolism at the cellular, tissue and organ levels might be harnessed to promote defence against the infection, with a focus on disease-tolerance mechanisms, and speculate on the long-term metabolic consequences for survivors of COVID-19.
Subject terms: Infectious diseases, SARS-CoV-2, Metabolic diseases
The importance of metabolism in the course of SARS-Cov-2 infection is highlighted by the metabolic comorbidities of COVID-19. In this Perspective, Ayres provides insight into how current knowledge of immunometabolism and metabolic diseases can inform the understanding of COVID-19 pathology, and proposes potential metabolism-based clinical solutions.
Main
The COVID-19 outbreak, caused by the novel coronavirus (CoV) called severe acute respiratory syndrome (SARS) coronavirus 2 (SARS-CoV-2), was first detected in late December of 2019 in Wuhan, China. The first cases outside China were reported in January 2020, and in March, the World Health Organization (http://who.int/) declared the outbreak a pandemic. At the time of writing of this Perspective, more than 200 countries have reported COVID-19 cases, and nearly seven million cases and 400,000 deaths have been reported globally, according to the World Health Organization.
The immediate response to any infectious-disease outbreak is to approach it from the pathogen perspective, because disease severity is assumed to be a direct function of pathogen burden1. However, the complexities of SARS-CoV-2 infection serve as an important reminder that this perspective is not sufficient for understanding the survival of infectious diseases2. Like that of most infections, the virulence of SARS-CoV-2 exists on a continuum: most individuals who acquire the infection experience mild disease, whereas a subset of individuals progress to severe or critical disease3. These severe and critical cases are driven by the host response to the infection, thus resulting in multisystem dysfunction and pathology. The pathologies seen in patients with COVID-19 are not necessarily new among infectious diseases. For example, extreme clotting and multiorgan damage can be consequences of a diverse array of infectious conditions that progress to critical stages. Instead, this pandemic has highlighted the necessity to change our perspective towards infectious diseases so that we can understand how to survive infections2. For COVID-19 and all other infectious diseases, this perspective will require an understanding of (1) the pathogenesis, pathology and pathophysiology of the infection; (2) how the pathology and resulting pathophysiology of preexisting conditions (beyond changes in immune function) influence susceptibility to developing disease once infection occurs; (3) the intrinsic defence mechanisms of the body that protect against damage and the resulting pathophysiology; and (4) how to develop treatments that alleviate the pathology and pathophysiology and complement antiviral-based approaches. Because COVID-19 is a multisystem condition, understanding the recovery and rehabilitation process in infectious-disease survivors is also important; however, this aspect of infectious-disease treatment is highly neglected. Survivors of severe or critical COVID-19 will exhibit an array of morbidities, possibly years after the infection. We must understand how the damage resulting from the infection and any patient treatments influences the development of new conditions in the recovery phase. If we can step beyond our focus on the virus, we will learn how to survive the infection and enable full recovery of patients, to maximize their healthspan and quality of life.
Metabolism will emerge as a critical regulator of susceptibility to, recovery from and survival after COVID-19. Infectious diseases and host metabolic processes are intimately connected, and changes in host metabolism occur at all levels—cellular, tissue, organ and physiological—during infection4–6. For COVID-19, these changes are clearest at the cellular level, at which the virus hijacks the host cell machinery to support viral replication and promote pathogenesis. However, host metabolic responses at the tissue, organ and physiological levels also occur during viral infections, and some of these responses are likely to reflect the adaptive mechanisms of the host to defend against the infection4,5. Although most attention has been paid to understanding how metabolism influences the host resistance response that destroys pathogens, recent evidence from other infectious diseases has demonstrated that metabolic processes are important mediators of host defence mechanisms that protect against the physiological damage that occurs during infections and consequently enable survival5,7–10. Furthermore, early clinical data on COVID-19 have demonstrated that people with type 2 diabetes (T2D) and other metabolic conditions that compromise overall metabolic health have greater risk of developing a more severe infection course than people who are metabolically healthy before acquiring the infection11. Although this finding has been largely attributed to these individuals’ being more permissive to viral infection and replication, the physiological complications caused by metabolic syndrome and T2D also probably render individuals more susceptible to developing COVID-19-associated pathologies independently of viral burden. Finally, people who survive SARS infections and other critical illnesses are predisposed to developing metabolic complications during their recovery process, thus suggesting that these infections, and potentially their treatments, might cause long-lasting collateral damage to metabolic health. In this Perspective, I discuss the relationship between metabolism and COVID-19. I discuss why preexisting metabolic abnormalities that compromise metabolic health, such as T2D and hypertension, may be important risk factors for severe and critical cases of infection, highlighting parallels between the pathophysiology of these metabolic abnormalities and the disease course of COVID-19. I also discuss how metabolism at the cellular, tissue and organ levels might be harnessed to promote defence against the infection, with a focus on disease-tolerance mechanisms, and I speculate on the long-term metabolic consequences for survivors of COVID-19.
Metabolic health and COVID-19 disease course
The effects of SARS-CoV-2 infection on host health are complex, and substantial variation is observed in the severity of disease (Fig. 1). COVID-19 cases can be classified as either asymptomatic or symptomatic. Asymptomatic individuals account for 25–50% of infected individuals and comprise carriers who test positive for SARS-CoV-2 and show no symptoms, as well as those who test negative for the virus but show seroconversion indicating a prior infection without ever having shown symptoms12 (http://CDC.gov/). During infection, these people maintain their health over time, exhibiting a maintenance-of-health phenotype6. Symptomatic individuals include those who exhibit sickness after a presymptomatic phase with no symptoms. Approximately 80% of these people exhibit a ‘mild’ disease course, whereas the other 20% progress to severe and critical stages associated with pneumonia, acute respiratory distress syndrome (ARDS) and respiratory failure, septic shock and multiorgan failure3.
Fig. 1. The disease phases of patients with COVID-19.
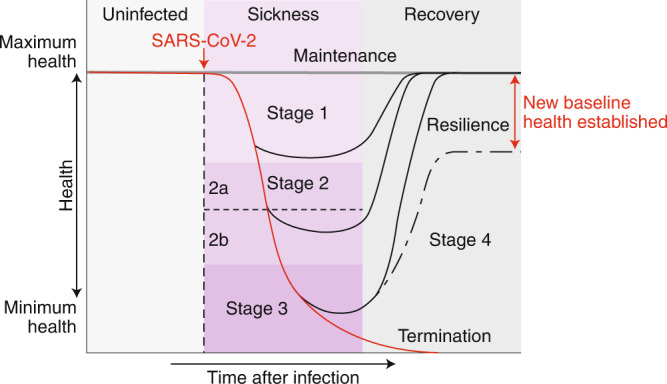
After infection, patients can remain healthy and show no signs of sickness (maintenance of health). For patients who become symptomatic, the disease course can be described by four stages. Stage 1 is mild, and patients exhibit fever, malaise and a dry cough. Stage 2 is characterized by a pneumonia phase without or with hypoxia (2a and 2b). Patients who progress further along the disease course develop acute respiratory distress syndrome, shock or multiorgan failure (stage 3 III). Patients who recover (stage 4) from the infection show a resilience phenotype. Some patients may never return to their original health state, thus establishing a new baseline for health. Patients who peak in stages 1 or 2 will bypass stage 2 or 3, respectively, and enter into their recovery phase.
The clinical course for symptomatic individuals can be divided into four stages (Fig. 1). Stage 1 begins when an individual becomes symptomatic3. Individuals typically develop a dry cough and fever, and they may lose their senses of taste and smell, and feel general malaise. For most people, with adequate self-care, the infection is limited to this stage3. Stage 2 describes the pulmonary phase of the infection3. People who enter this stage develop pulmonary inflammation and pneumonia, either without hypoxia (stage 2a) or with hypoxia (stage 2b)3. These people require hospitalization. Patients with prolonged hypoxia tend to require mechanical ventilation3. Patients can then progress to stage 3. These patients are in critical condition and can develop ARDS and extrapulmonary systemic hyperinflammation syndrome. In addition, they can develop shock, vasoplegia, respiratory failure, cardiopulmonary collapse, myocarditis, acute kidney injury and other extrapulmonary complications3. The prognosis for these patients is poor, and some continue to decline toward mortality, whereas the others enter stage 4, the recovery stage, and survive and exhibit a resilience-of-health phenotype4 (Fig. 1).
The metabolic health of an individual is represented by the proper functioning of organismal metabolic processes coordinated by multiple physiological systems. Disruption of these systems causes dysfunctional organismal metabolic processes and a decline in metabolic health (Fig. 2). The major risk factor for severe COVID-19 is poor metabolic health (Fig. 2). In past coronavirus outbreaks, T2D was one of the most common comorbidities in infected individuals13,14. In agreement with this finding, T2D, obesity and hypertension appear to be major comorbidities in people with COVID-19 and are associated with more severe and critical COVID-19 disease courses15–17. The reasons for this finding are likely to be multifactorial and are conventionally thought to involve immune dysfunction. For example, individuals with metabolic syndrome and T2D have impaired immune function in general18, and their antiviral response against SARS-CoV-2 may consequently be impaired. Moreover, the physiological complications of T2D and metabolic syndrome are also likely to have a synergistic role in SARS-CoV-2 pathogenesis, thus making patients more susceptible to developing severe pathology independently of viral burden. Similarly, among people with COVID-19 in the United States, obese younger individuals are more likely to require hospitalization and to develop more severe and critical disease19. This finding suggests that obesity may shift COVID-19 towards more severe and critical cases in younger adults. One possible explanation may be that obesity causes physical stress on ventilation by obstructing diaphragm excursion. Furthermore, diabetes increases the risk of pulmonary fibrosis, chronic obstructive pulmonary disorder and reduced respiratory function (Fig. 2). As patients with COVID-19 progress to the pulmonary stages of the infection and develop pneumonia and ARDS, these conditions further complicate patients’ breathing and hypoxic conditions, which lead to multiorgan damage.
Fig. 2. The parallels among metabolic syndrome, T2D and COVID-19.

COVID-19, metabolic syndrome and T2D are multisystem diseases. The pathologies and pathophysiologies of metabolic syndrome and T2D affect the same systems that are damaged by COVID-19, thus probably predisposing patients to developing more severe pathology during the infection. Importantly, whereas we traditionally think about how metabolic syndrome and T2D make individuals more susceptible to infections because of diminished immune function, an understanding of the parallels between these conditions from a physiological perspective suggests that the greater susceptibility of these individuals to COVID-19 is likely to be partly because of an increased susceptibility to pathology and the resulting pathophysiology, rather than an inability to control the viral infection. COPD, chronic obstructive pulmonary disease.
Additional physiological changes caused by metabolic syndrome and T2D are likely to synergize with COVID-19, thereby further complicating the disease course. Metabolic syndrome and T2D are chronic inflammatory states (Fig. 2). Because the severe and critical phases of COVID-19 are driven by an excessive inflammatory response to the infection (cytokine storm), the heightened baseline inflammatory state in patients with preexisting compromised metabolic health may increase the likelihood that the inflammatory response will reach pathogenic levels, as well as the likelihood of physiological damage20 (Fig. 2). Similarly, diabetes and hypertension alone are risk factors for kidney disease. Diabetes leads to diabetic kidney disease, in which a decrease in kidney function21,22 leads to a build-up of toxic metabolites that can damage other organs (Fig. 2). The damage to the blood vessels caused by hypertension decreases blood flow to the kidneys and consequently results in kidney injury (Fig. 2). Because kidney injury is a common complication of COVID-19 (ref. 23), the kidney damage caused by these preexisting metabolic conditions may render the kidneys more susceptible to damage during SARS-CoV-2 infection.
Obesity, diabetes and hypertension increase the risk of stroke and cardiovascular complications, which are also observed in severe and critical cases of COVID-19 (refs. 24,25 and Fig. 2). Sustained hyperglycaemia and obesity damage the blood vessels and are risk factors for plaque build-up, which itself is sufficient to cause blood clots. People with COVID-19 exhibit an exacerbated coagulation response, which, in combination with preexisting damaged blood vessels with plaques, may increase the likelihood of stroke or pulmonary embolism. The effects of obesity and diabetes on the blood vessels can lead to hypertension, which in turn affects haemodynamics and alters the heart structure (Fig. 2). These changes can make the heart more susceptible to damage due to the host inflammatory response, hypoxia caused by ARDS, infection of the heart by the virus or other cardiac consequences in response to extrapulmonary multiorgan damage, thus potentially explaining why some people with COVID-19 exhibit cardiac damage and heart-attack-like signs of disease. Finally, in the brain, damage to blood vessels and nerves in people with diabetes causes vascular cognitive impairment, dementia and stroke. The dysregulated glycaemia in diabetics also causes seizures26. People with COVID-19 experience seizures, strokes and brain inflammation, and those with poor metabolic health may be more susceptible to developing these conditions because of the preexisting factors negatively affecting brain health (Fig. 2).
The prevalence of hypertension, cardiovascular disease, nephropathy and retinopathy in people with diabetes has implicated the renin–angiotensin system (RAS) in the pathogenesis of these disorders. Angiotensin II, the active RAS metabolite and a potent vasoconstrictor that increases peripheral vascular resistance, also causes insulin resistance and regulates sodium absorption in the kidneys. Treatment with RAS inhibitors in clinical trials has been found to decrease the vascular complications in people with diabetes27. Angiotensin-converting enzyme 2 (ACE2), the receptor that mediates entry of SARS-CoV-2 into host cells, antagonizes the effects of angiotensin II, inducing vasodilation. In people with T2D, ACE2 expression is elevated in multiple target tissues of SARS-CoV-2 (ref. 28), and this elevation has been proposed to be an adaptive mechanism that protects the body against diabetes-associated physiological dysfunction29. This increased expression may enhance SARS-CoV-2’s entry and subsequent replication, as well as its spread within the body in individuals with T2D. Thus, a physiological defence strategy used to support metabolic health in T2D may be maladaptive in the context of COVID-19.
Whereas chronic hyperglycaemia in diabetes leads to damage that can complicate the disease course in COVID-19, new evidence suggests that dysregulated glucose alone is detrimental in people with COVID-19 and T2D. A retrospective study of a cohort of more than 7,000 people with confirmed COVID-19 in China has suggested that controlled glycaemia is associated with improved outcomes in patients with COVID-19 and T2D30. Glycaemic control is likely to be necessary to regulate the host inflammatory response, to limit tissue susceptibility to inflammatory damage signals and to sustain physiological function during the severe and critical stages of infection. Thus, in addition to contributing to understanding of the pathophysiology of COVID-19 and how poor metabolic health predisposes individuals to a more severe disease course, manipulating organismal metabolic variables in patients may be sufficient to alter the disease course towards a more favourable one.
Long-lasting metabolic-health complications in survivors of severe or critical COVID-19
Patients with severe or critical COVID-19 who do survive may have a long road to recovery while experiencing the lasting effects of the infection and treatments31 (Fig. 1). One such lasting effect is likely to be the development of organismal metabolic abnormalities. Studies of patients who recovered from SARS-CoV or other critical illnesses support this idea. In one study from China, metabolic abnormalities were detected in people with SARS-CoV infection 12 years after recovery from the infection; these abnormalities included hyperlipidaemia and cardiovascular abnormalities as well as signs of abnormal glucose metabolism, such as hyperinsulinemia, insulin resistance, hyperglycaemia, type 1 diabetes or T2D32. Serum metabolomics showed that these individuals had disruptions in free fatty acids, phosphatidylinositol, lysophosphatidylcholine and lysophosphatidylinositol (LPI). LPIs have been suggested to regulate glucose homeostasis, including insulin release and activation of G-protein-coupled receptor 55 (GPR55), thus suggesting a possible mechanism for the abnormal glucose metabolism in the patients after recovery from SARS32. In another study of patients with SARS in China, 50% of individuals who had no prior history of T2D developed T2D during infection, and after 3 years, 5% of the patients still had diabetes33. Critically ill patients exhibiting similar pathologies to those of patients with COVID-19 generally experience long-lasting metabolic abnormalities34. For example, in one study, critically ill patients who experienced acute kidney injury had a higher incidence of new-onset diabetes than matched-control individuals during the recovery phase35.
The underlying cause of any long-lasting metabolic abnormality among survivors of critical or severe COVID-19 will probably be complex and multifactorial, involving mental, emotional and physiological factors that are consequences of both the infection and medical interventions (Fig. 3). Mentally, survivors of ARDS can exhibit long-term cognitive impairment that may be caused by the hypoxic and inflammatory states of the condition, as well as ventilation and sedation36. Individuals with poor episodic memory tend to experience uncontrolled eating because they have a diminished sensitivity to internal hunger states and satiety, thereby leading to weight gain and a high body mass index37. Emotionally, ARDS survivors have high rates of depression and post-traumatic stress disorder that tend to negatively influence metabolism and are driven by binge and compulsive eating, as well as a lessened motivation for physical activity38 (Fig. 3). Cognitive training and efforts to promote emotional health will be necessary for rehabilitation of survivors of severe or critical COVID-19 to maximize metabolic health and increase their likelihood of returning to their preinfection baseline health.
Fig. 3. Long-lasting effects of COVID-19 on metabolic health.

Damage caused by COVID-19 and related treatments can harm various systems in the body and may have long-lasting mental, emotional and physiological effects; these effects may result in metabolic abnormalities that hinder the recovery process.
Physiologically, damage to endocrine and metabolic organs including the pancreas, skeletal muscle, adipose tissue and liver may contribute to the development of new-onset metabolic syndrome in survivors of COVID-19 (Fig. 3). People with COVID-19 have been reported to develop pancreatic damage during the infection, as indicated by elevated levels of circulating pancreatic enzymes. The pancreas expresses ACE2, and binding of the SARS-CoV virus to its receptor damages islets of the pancreas, thus decreasing insulin release33, which in turn may contribute to the long-lasting diabetes observed in people who have recovered from SARS (Fig. 3).
People with ARDS and other critical conditions exhibit dramatic muscle wasting caused by metabolic dysregulation and the inflammatory response to the infection, as well as medical interventions including feeding tubes and immobilization (Fig. 3). Within the first week after admission to an intensive care unit, these patients can lose approximately 20% of their body mass39. In the United States, most survivors return to their prehospitalization weights within the first year after discharge; however, body-composition analysis has shown that the weight gain is due to an increase in adipose tissue without a gain in lean mass. ARDS survivors also experience myopathies that cause muscle weakness due to muscle-fibre dysfunction39. In survivors of sepsis, long-term muscle weakness and wasting are due to impaired mitochondrial and metabolic alterations in satellite cells that are necessary for muscle regeneration40. These changes may explain the overall decline in physical health associated with muscle weakness and changes in exercise capacity in survivors of critical illness41. The decreased exercise capacity and lean muscle mass can diminish insulin sensitivity in survivors. Although cachexia has not yet been reported in people with COVID-19, it may be likely in severe and critical cases. Rhabdomyolysis, a breakdown of skeletal muscle due to damage, has been reported in patients with COVID-19 (ref. 42) and is likely to contribute to long-term abnormalities in muscle health.
Stroke is a reported complication of COVID-19 (ref. 24). During recovery, patients with stroke are at risk of future vascular events43. Long-lasting changes in metabolism and energy balance appear to be important drivers of these future events. Weight loss and malnutrition are frequently observed in patients recovering from stroke43. This weight loss has traditionally been assumed to be due to cachexia; however, recent data suggest that sustained loss of adipose tissue may also contribute. In a stroke model, mice have been found to exhibit prolonged changes in body composition characterized by an initial loss of adipose tissue mass, which is subsequently sustained at lower levels43. This effect is associated with prolonged changes in the levels of plasma lipids including free fatty acids and triglycerides43. Sustained elevated levels of free fatty acids and triglycerides can lead to cardiovascular abnormalities in people who have recovered from SARS and potentially COVID-19 (Fig. 3).
Given the similarities among COVID-19, SARS-CoV and other critical illnesses, an association is likely to be found between the development of new-onset metabolic syndrome and a history of COVID-19 in the years to come. Because these potential long-term consequences are largely dependent on the types of medical interventions that a patient receives, as well as the societal variables that influence metabolic health, determining which consequences are specific to certain regions or countries and which consequences are more generalizable will be important. Thus, a focus on metabolic health must be integrated into patient rehabilitation programs using mental, emotional and physiological strategies to promote metabolic health and return patients to their baseline metabolic-health states.
Harnessing metabolism for defence against COVID-19
The disease pathogenesis of COVID-19 is dependent on the direct effects of the virus on the host and the collateral damage of the host response to the infection1. An examination of the relationship between the clinical disease course and the virus dynamics as well as the host response to COVID-19 can help to conceptualize the different defence strategies that will be most effective for promoting patient survival in different stages of infection (Figs. 4 and 5). Systematic studies of the viral dynamics in patients with COVID-19 have shown that the viral load peaks within the first week of symptom onset and is followed by a steady decline over the second week44–47. There is no association between disease severity and viral load. As viral loads decline, the host-driven hyperinflammatory phase is initiated, thus driving severe and critical stages of infection. Resistance strategies, including the host immune response and antiviral therapies that destroy the virus, will be most effective for COVID-19 treatment if they are implemented when patients are asymptomatic or in stage 1 when viral loads peak. As patients progress to stage 2, when viral burdens decline, addressing the collateral damage of the host response to the infection through disease-tolerance and antivirulence strategies is more important, to sustain patients’ physiological functions and therefore their lives (Figs. 4 and 5). Clinical data for patients with influenza have demonstrated the importance of targeting the host rather than the virus in patients in severe or critical condition. Approximately 25% of critically ill patients who receive optimal antiviral therapy still die48. This finding suggests that the host response to the virus is a major factor determining the outcome of influenza, and this conclusion is also likely to be true for COVID-19. Furthermore, in mouse studies on SARS, promoting cooperation with the host and the virus has been found to confer 100% survival despite compromised antiviral immunity49. Clinical parameters and biomarkers will be important for determining a patient’s phase of disease and the most appropriate method for intervention6.
Fig. 4. Relationship between disease stage and therapy for patients with COVID-19.

After SARS-CoV2 infection, the virus replicates and reaches peak levels during stage 1, after which the levels steadily decline44–47. As viral levels decline, the host inflammatory response increases during the hyperinflammatory phase. Eventually this response decreases, and the recovery phase of the disease begins. An examination of the relationships among these parameters and the clinical course of the infection dictates which defence strategies will be most effective. Before infection, avoidance is the most clear defence strategy. After infection, patients will be in a presymptomatic (presymp.) phase of the infection, which is followed by stage 1 with fever, malaise and other mild symptoms. The viral levels peak and continue to decline as patients exit stage I, independently of whether patients will recover or progress to a severe or critical stage of infection. Antivirals are most effective for asymptomatic individuals and patients in stage 1. By stage 2, the host inflammatory response drives the disease, which continues into stage 3. Disease-tolerance and antivirulence strategies are most effective for patients in stages 2 and 3. Should patients survive, the host inflammatory response subsides, and the resolution phase begins; patients proceed toward recovery in stage 4. Disease-tolerance drugs are most effective for patients in stage 4. Patients who peak in disease severity in stages 1 or 2 can bypass stage 2 or 3, respectively, and enter into the recovery phase.
Fig. 5. A framework for defence strategies against COVID-19.
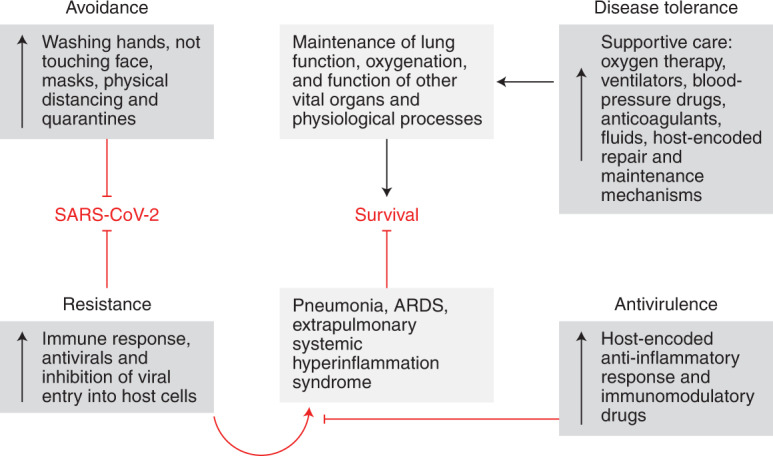
The ability of an infection to cause disease is largely dependent on the host response to the infection. Defensive health mechanisms evolved to promote maintenance or resilience in people challenged with infections6. These inducible mechanisms operate by enabling organisms to antagonize or withstand the pathogen. Antagonizing the pathogen is mediated by avoidance and resistance strategies. Avoidance mechanisms are innate and learned behavioural mechanisms that are largely triggered by sensory cues and prevent a host from becoming infected with a pathogen6. Resistance mechanisms are encoded by the immune system and destroy the pathogen after it has infected the host1. Withstanding the pathogen is mediated by disease-tolerance and antivirulence strategies—physiological defences that alleviate the fitness costs of the infection by limiting physiological damage and promoting health in the presence of the pathogen1. Antivirulence mechanisms are a neutralization strategy based on changes in host physiology that limit pathogenic signals during infection without affecting the pathogen’s ability to infect or replicate in the host6,9,10. Disease-tolerance mechanisms limit damage during infection by minimizing tissue susceptibility to damage cues, thus supporting maintenance of physiological function and promoting repair1,6. A critical distinction is the ways in which these strategies affect the health trajectory with respect to pathogen fitness: avoidance and resistance mechanisms promote maintenance or resilience by avoiding or eradicating the pathogen, whereas disease-tolerance and antivirulence mechanisms promote health by allowing the presence of the pathogen to be withstood6. This same framework is important for understanding public-health and medical interventions for COVID-19 that will influence patient disease course. For COVID-19, progression into severe and critical stages of the disease is driven by the hyperinflammatory response resulting from host resistance defences against the infection. Antivirulence strategies will neutralize these pathogenic signals to minimize damage. Disease-tolerance strategies will provide physiological defence in the face of these signals. Quarantines, physical-distancing and hygienic measures serve as avoidance strategies.
The current candidate strategies for COVID-19 therapeutics involve traditional antiviral-based strategies as well as immunomodulatory strategies to neutralize the host inflammatory response2. Recent evidence from studies on respiratory pathogens and other insults has provided strong support for targeting host metabolic processes for the effective treatment of patients with COVID-19. Below, I discuss how manipulation of host metabolism at the cellular, tissue and organ levels can be targeted at each stage of COVID-19 to help patients fight, survive and recover from the infection. I also discuss the consequences of defensive strategies for the metabolic health of the host. Although I largely focus on harnessing metabolism in the pulmonary environment, I speculate on how metabolism can be targeted to protect against the extrapulmonary damage that can occur in COVID-19.
Avoidance defences prevent COVID-19
Self- and government-initiated avoidance mechanisms have been essential for combating the COVID-19 pandemic. These include hand washing, avoiding touching the face, moving away from people who are sick, and wearing masks and other personal protective equipment. For COVID-19, avoidance mechanisms including quarantines and physical distancing have also been essential public-health measures to prevent the infection of naive individuals and in the movement to ‘flatten the curve’, aiming to decrease the amount of infection and to distribute the occurrence of infection over a greater time period to prevent the healthcare system from being overwhelmed (Figs. 4 and 5). Although avoidance defences are likely not to be driven by metabolic processes, some avoidance mechanisms may come at a substantial cost to the metabolic health of an individual (Fig. 6). For example, with physical distancing, there is substantial collateral damage to the organismal metabolism of an individual. In animal and humans studies, social isolation has been associated with the development of weight gain or obesity, increased adiposity, and a loss of lean mass and insulin resistance; socially isolated humans are also more likely to have newly diagnosed T2D50–53 (Fig. 6).
Fig. 6. Targeting host metabolism to defend against COVID-19.

Individuals can use four defence strategies against COVID-19. Avoidance mechanisms prevent an individual from acquiring the infection. Although these mechanisms are likely not to be driven by metabolic processes, the collateral damage from using some avoidance mechanisms can detrimentally affect metabolic health. Resistance strategies protect the host by destroying the infection. Various aspects of host metabolism can potentially be targeted to inhibit viral replication in the host cell and to boost the immune response of the host to destroy the pathogen. Targeting host metabolism may also be a viable strategy to promote both antivirulence defences to protect against pathogenic signals induced during the infection and disease-tolerance defences that limit tissue susceptibility to damage signals and enable them to function despite potentially experiencing damage, as well as to recover from the damage. T2AECs, type II alveolar epithelial cells; ROS, reactive oxygen species.
The underlying mechanisms leading to the development of the metabolic consequences for physical distancing are complex, involving physiological, mental and behavioural factors. The transition from an active to a more sedentary lifestyle caused by the inability to go outside or by depressive states negatively affects organismal metabolism52,53. An acute sedentary lifestyle causes loss of muscle mass, and greater glucose levels, insulin resistance and adiposity in humans52,53. Social isolation can limit individuals’ ability to consume a healthy diet; this limitation can be driven by emotional states of fear, such as the belief that leaving the home will result in contraction of a viral infection. Social isolation in experimental animals and in humans triggers hyperphagic and binge-feeding behaviour, as well as comfort feeding triggered by anxiety, fear or depression50,52,53. This response is likely to be due to the induction of various neurotransmitters involved in feeding and satiety, including serotonin, neuropeptide Y, α-melanocyte-stimulating hormone and cocaine- and amphetamine-regulated transcript peptide; these pathways may constitute the molecular mechanism underlying social-isolation-induced hyperphagia50. Indeed, central administration of cocaine- and amphetamine-regulated transcript (CART) during social isolation has been found to protect rats against hyperphagia, and blockade of CART prevents the effects of resocialization on feeding behaviour50. Finally, the stress response induced by social isolation causes behavioural and physiological changes that lead to unhealthful metabolic responses. For example, cortisol can intensify emotions and motivation for binge feeding; trigger insulin resistance and muscle atrophy; and directly alter lipid, glucose and amino acid metabolism50,54. Therefore, I propose that after the COVID-19 pandemic ends, our avoidance tactics will cause more people to develop metabolic syndrome, thus exacerbating the already existing pandemic of metabolic diseases. An additional important concern is that, because people with COVID-19 with preexisting metabolic diseases are more susceptible to developing severe or clinical disease courses11, if a second wave of the COVID-19 pandemic occurs, might a higher fraction of patients experience a more severe disease course because of our avoidance defences in the first wave of the pandemic? Therefore, people practicing these necessary avoidance behaviours must maintain a routine of exercise and healthful eating to promote metabolic health.
Avoidance defences practiced by individuals infected with COVID-19 may also have complications in patient disease course. Social isolation and quarantines will contribute to differences in metabolic health during the infection. Although whether these metabolic changes caused by social isolation will affect the disease course is unknown, the metabolism of a host is well established to dictate infection outcome6,9,10, and social interactions are known to be important for defence against infection55. Whether the effects of social isolation on infection defence are due to metabolic perturbations remains to be determined. Therefore, a potential conundrum exists between avoidance behaviours, such as quarantining and physical distancing, and infection defence. We must consider facilitating social interactions for patients while containing the infection. We also must understand the metabolic consequences of public-health and medical interventions for COVID-19 disease outcomes on people who become infected.
Resistance fights the fight against COVID-19
Avoidance mechanisms are not 100% effective, even in the most successful cases, and so other defensive strategies must be implemented. Resistance strategies protect the host by destroying the virus and are effective early in the course of COVID-19, when the viral levels peak (Figs. 4 and 5). Three strategies can be used to promote resistance against SARS-CoV-2 at this early phase: (1) preventing viral entry into host cells, (2) targeting the infected epithelial cells that the virus has hijacked to prevent replication and (3) targeting the host immune response to destroy the virus. No effective antiviral agents for COVID-19 treatment are currently available, and developing such therapies has been the priority in the therapeutic discovery response to the virus. The potential antiviral COVID-19 treatments currently being tested involve the traditional logic for combating viral infections56. However, evidence suggests that targeting host metabolism at both the host cell and immune cell levels may be effective antiviral strategies for treating COVID-19.
Recent studies on RNA viruses have demonstrated the potential for targeting host cell metabolism as an antiviral strategy to treat infections. CoV pathogenesis involves entry of the viral RNA genome into host cells, in a process driven by interactions between the spike protein of the virus and ACE2 on the host cell, followed by proteolytic cleavage of the spike protein and endocytosis-mediated viral entry into the host cell. The RNA genome is deposited into the host cell and translated, and the replication and transcription complexes necessary for translation and replication then form. Subsequently, virions are packaged, assembled and released, and subsequently infect new cells57. In epithelial cells infected with human CoV, lipidomic analyses have shown an accumulation of saturated and unsaturated fatty acids and phospholipids, thus suggesting that these changes in the lipid profiles of infected cells may be important for viral replication58,59. In agreement with this idea, inhibition of proteolytic processing of the transcription factor SREBP through treatment with the compound AM580 in epithelial cells in vitro, and in infected mice in vivo, has been found to inhibit viral replication by acting on multiple steps including decreasing intracellular lipid-droplet formation, double-membrane-vesicle formation and palmitoylation of viral proteins59,60. Furthermore, AM580 decreases palmitolyation of the surface glycoprotein haemagglutinin of the RNA virus HIN1, thereby impairing replication of influenza59,60. This finding suggests that targeting lipid metabolism in infected epithelial host cells may be a potential antiviral strategy for COVID-19 treatment (Fig. 6).
Children diagnosed with respiratory viral infections have elevated glucose uptake in their lungs61. In agreement with this finding, analyses of influenza-infected human bronchial epithelial cells from paediatric donors have indicated increased c-Myc, glycolysis and glutaminolysis, which are required for viral replication. Treatment with the drug BEZ235, an inhibitor of the PI3K–mTOR signalling pathway, decreases viral titres and infection-associated pathologies, owing to the ablation of c-Myc induction and the restoration of PI3K–mTOR pathway homeostasis61. In Caco-2 cells, infection with SARS-CoV-2 has been found to upregulate carbon metabolism. Treatment with the glycolysis inhibitor 2-deoxy-d-glucose (2DG) prevents replication of SARS-CoV-2 in Caco-2 cells, thus suggesting that targeting glucose metabolism of the host may be a viable antiviral strategy for treating COVID-19 (ref. 62 and Fig. 6).
With SARS and MERS, there is a delayed antiviral response with respect to viral load49. Similarly, with SARS-CoV-2, a reduced antiviral response is observed with respect to viral load63. Induction of type I interferon (IFN), an important component of the antiviral response of the host, is triggered in response to recognition of pathogen-associated molecular patterns by the innate immune system. In BALB/c mice, SARS-CoV infection reaches nearly maximal viral load by 16 hours after infection, with a delayed type I IFN response. Intranasal administration of the type I IFN IFN-β 6 hours after infection promotes resistance, decreasing viral load early in the infection and increasing survival in mice49. This finding suggests that controlling viral load early is necessary for preventing disease progression and that promoting the IFN-I response early in the infection course may be an effective antiviral-based strategy in patients with COVID-19 (Fig. 6). A recent report has shown how the antiviral response to RNA viruses can be metabolically targeted to promote an IFN-I response64. The retinoic-acid-inducible gene I–like receptor (RLR) family detects cytosolic viral RNA and, after activation, leads to the induction of the mitochondrial antiviral signalling protein (MAVS), which is required for activation of interferon-regulatory transcription factor (IRF3) and IFN-I. MAVS localizes to the mitochondria and interacts with hexokinase-2 (HK-2). RLR activation abolishes this interaction, thus decreasing the levels of glycolytic intermediates downstream of HK-2. Growth of cells in low-glucose conditions or with a hexokinase inhibitor enhances the type I IFN response, whereas lactate produced by glycolysis suppresses the IFN-I response by binding directly to MAVS. In mice, fasting-induced hypoglycaemia increases IFN-I. Treatment of mice with a lactate dehydrogenase A inhibitor results in a more robust IFN-I response and greater resistance to RNA viruses64.
The studies above provide several demonstrations of how manipulating host metabolism to promote resistance defences by targeting the immune response or viral replication within the host cell may be an effective treatment strategy for patients with COVID-19. Several excellent reviews describe immunometabolism and antiviral defence, and suggest additional ways to potentially manipulate host metabolism to promote resistance defences65–67. These treatments may be effective for COVID-19 if they are implemented early in the infection to minimize disease severity.
Physiological defences promote survival and recovery
Although antiviral agents will be effective for asymptomatic and stage 1 patients with mild clinical symptoms, promoting the ability of patients to withstand the virus will be necessary after patients enter stages 2 and 3 (Figs. 4 and 5). As patients enter these phases, the infection transitions from a viral-driven disease to one driven by the host response. The inflammation caused by pneumonia in stage 2 leads to a hyperinflammatory phase characterized by the release of cytokines from alveolar macrophages and recruitment of neutrophils to the pulmonary environment. These immune cells release platelet-activating factor and proteases that damage the pulmonary environment by acting on the alveolar epithelial–capillary endothelial barrier. Disruption of this barrier causes fluid and debris accumulation in the alveoli, decreased surfactant secretion and alveolar collapse, thus decreasing gas exchange and leading to hypoxia, dyspnoea, confusion, cyanosis and tachypnoea. At later stages, fibrosis in the lung can occur because of dysregulated repair responses. The systemic inflammation and hypoxia cause extrapulmonary pathologies in vital organs including the brain, heart and kidneys.
Physiological defences enable patients to withstand the infection by (1) blocking the pathogenic responses that cause damage with antivirulence-based strategies and (2) limiting tissue, organ or physiological susceptibility to damage and maintaining physiological function in the face of damage with disease-tolerance strategies1,6 (Fig. 5). The host response during the hyperinflammation phase that drives pathology is well understood. This understanding has informed the current pursuit of candidate treatments that act as antivirulence mechanisms by blocking the cytokine storm that leads to ARDS and extrapulmonary pathologies such as anti-IL-6 therapy68. Less is known about the mechanisms of disease tolerance, and treatment therefore has been limited to supportive care to improve disease tolerance by promoting lung function to meet oxygen demands, as well as to maintain blood pressure and prevent clotting2. However, on the basis of knowledge of other lung infections and lung injury conditions, as well as normal lung physiology, predictions can be made as to how disease tolerance to COVID-19 might be induced. In both cases, recent studies have demonstrated that disease tolerance and antivirulence defences are largely driven by metabolic processes7–10. Below, I discuss ways in which host metabolism can be targeted to promote the physiological defences of the host, focusing largely on antivirulence and disease-tolerance methods to limit susceptibility to damage and promote repair in the pulmonary environment. I also address how metabolism can be leveraged to defend against extrapulmonary damage in people with COVID-19.
Antivirulence metabolic strategies to block pathogenic signals
Metabolic programming is essential for regulating inflammatory responses, and targeting host cellular metabolism is a clear way to promote antivirulence in patients with severe or critical COVID-19. In a mouse model of lipopolysaccharide (LPS)-induced lung injury, inhibition of glutamine metabolism with the glutamine analogue 6-diazo-5-oxo-l-norleucine (DON) decreases lung inflammation, as characterized by diminished neutrophil infiltration, and lower levels of interstitial macrophages and proinflammatory cytokines and chemokines. These effects are associated with earlier expression of the growth factor amphiregulin and faster recovery from lung damage69. In agreement with this finding, in mice with LPS-induced lung injury, glutamine treatment exacerbates cytokine production and neutrophil recruitment to the pulmonary environment. However, in other models with various methods of lung injury induction and routes of glutamine administration, glutamine has been found to be protective against ARDS. The differences in effects on disease pathogenesis are likely to be explained by the differences in the route of glutamine administration, timing and specific insult triggering ARDS70 (Fig. 6).
Targeting mitochondria may serve an important antivirulence mechanism for surviving ARDS. Reactive oxygen species regulate the activation of the transcription factor NF-κB and the inflammatory response. Treatment of LPS-exposed neutrophils with either rotenone, an inhibitor of complex I of the mitochondria, or metformin results in increased intracellular superoxide and hydrogen peroxide. These effects are associated with the inhibition of LPS-induced NF-κB activation and proinflammatory-cytokine production. In a mouse model of LPS-induced lung injury, treatment with rotenone or metformin results in inhibition of complex I in the lung and protection against lung injury, including decreased neutrophil infiltration, proinflammatory cytokines and lung oedema, thus suggesting that targeting mitochondrial function in neutrophils may promote antivirulence defence during COVID-19. Metformin also induces autophagy, and CoVs may use autophagy for replication71; consequently, the timing of metformin treatment in patients with COVID-19 may be tricky in severe and critical cases, because the treatment may promote viral replication if it is administered too early in the infection course (Fig. 6).
The cytokine storm seen in patients with SARS has been suggested to be driven by a delayed IFN-I response72. A robust anorexic response has been reported patients with COVID-19 (ref. 73). Because fasting-induced hypoglycaemia affects the cellular antiviral response, activating MAVS and an IFN-I response during viral infections, a potential cellular metabolic target to suppress IFN-I during these late stages may involve activation of HK-2, for example by feeding or glucose administration, or by treatment with lactate. Indeed, in a mouse model of influenza infection, overriding the anorexic response or oral administration of glucose has been found to be sufficient to protect mice against infection7. The relationship between glycolysis and IFN-I in the lungs demonstrates the complexities of promoting antivirulence defences. Inhibiting glycolysis would theoretically be beneficial early in the course of COVID-19 because it might induce the IFN-I response, thus promoting resistance defences; however, later during the host-response phase of the disease, IFN-I should be downregulated by promoting glycolysis, in an antivirulence mechanism to inhibit the cytokine storm (Fig. 6).
The differential effects of glycolysis on the induction of different proinflammatory cytokines will further complicate the candidacy of glycolysis as an antivirulence target for COVID-19. IL-1β is a proinflammatory cytokine that is elevated in people with COVID-19 (ref. 74). Treatment of mice with the glycolysis inhibitor 2DG has been found to inhibit IL-1β maturation during LPS treatment75. Furthermore, treatment of LPS-treated mice with 2DG protects against LPS-induced death7. Glycolysis supports IL-1β maturation in vivo through generation of 3-phosphoglycerate, a precursor for de novo serine synthesis76. Treatment of LPS-challenged mice with PH-739-005N, a small-molecule inhibitor of phosphoglycerate dehydrogenase (a substrate in de novo serine synthesis) is sufficient to protect mice against LPS-induced lethality and maturation of IL-1β (ref. 76 and Fig. 6).
In addition to undergoing glycolysis, glucose flows through the hexosamine-biosynthesis pathway. The end product of this pathway is uridine diphosphate N-acetylglucosamine (UDP-GlcNAc). O-linked β-N-acetylglucosamine transferase (OGT) mediates the transfer of UDP-GlcNAc to serine or threonine residues on target proteins. During influenza A virus infection, OGT is induced; OGT then binds interferon regulatory factor-5 (IRF5), and IRF5 is subsequently O-GlcNacylated. This process is necessary for K63-linked ubiquitination of IRF5 and subsequent cytokine production, thereby leading to the cytokine storm induced by influenza infection. Genetic analyses in mice have confirmed that OGT mediates the influenza-induced cytokine storm via IRF5 in vivo77. Thus, inhibition of the hexosamine-biosynthetic pathway may be a feasible antivirulence strategy for respiratory infections including SARS-CoV-2 (Fig. 6).
Peroxisome proliferator-activate receptor γ (PPARγ), is a nuclear receptor that regulates various aspects of metabolism and inflammation, including polarization of M2 macrophages and restriction of excessive production of inflammatory cytokines. During influenza A infection in mice, PPARγ is downregulated in alveolar macrophages in an IFN-I-dependent manner78. Mice deficient in PPARγ in macrophages and other Lyz2-positive cells exhibit a greater inflammatory response during influenza A infection than wild-type mice, as well as increased death78. In agreement with these findings, mice treated with prophylactic or therapeutic agonists of PPARγ, exhibit decreased morbidity and mortality when challenged with influenza infection79. Interestingly, in obesity, PPARγ is dysregulated, thus suggesting that the increased susceptibility to SARS-CoV-2 in people with metabolic diseases may be partly due to a dysregulated inflammatory response mediated by defects in PPARγ activity, and that targeting PPARγ in these patients may be a therapeutic option (Fig. 6).
The studies above provide some examples of how manipulating host cellular metabolism may be an effective strategy for promoting antivirulence defences against COVID-19. However, because the antivirulence strategies are directly linked to resistance strategies, antivirulence strategies have inherent complexities and might potentially compromise resistance defences and trigger more severe disease. Several recent reviews on immunometabolism and metabolic reprogramming in immune cells provide additional examples of how to target metabolism to downregulate the inflammatory response for antivirulence purposes65–67 (Fig. 6).
Disease tolerance limits susceptibility to damage
There are two potential concerns regarding antivirulence-based approaches. First, these approaches may increase patient susceptibility to the virus or secondary bacterial infections, because they block immune responses2,80. Second, there is an important temporal aspect to the infection that must be considered. To be effective, antivirulence strategies must be implemented early and before damage has occurred. Thus, focusing on promoting disease tolerance seems reasonable to limit tissue susceptibility to damage, sustain physiological function in the presence of this damage and initiate a recovery response2. Timing will certainly be important, and combinatorial strategies are likely to be most effective in patients with COVID-19.
One way to potentially limit susceptibility to the inflammatory damage during COVID-19 is to limit epithelial cell sensitivity by adapting and surviving in the presence of pathogenic signals6. In a mouse model of acute pulmonary endotoxemia–induced damage, alveolar epithelial cells show decreased expression of genes involved in mitochondrial biogenesis and fatty acid oxidation, including Ppargc1a and Cpt1a, as well as impaired fatty acid oxidation. The deletion of Ppargc1a, the gene encoding PGC-1A, from alveolar epithelial cells renders mice more susceptible to pulmonary damage caused by intranasal treatment with LPS, an effect associated with increased caspase-3 cleavage in epithelial cells, thus suggesting that the absence of fatty acid oxidation in the alveolar epithelial cells is necessary for the survival of these cells in response to acute lung injury81. PGC-1A cooperates with PPARα in the transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes, thereby inducing fatty acid oxidation82. The treatment of mice with a PPARα agonist is sufficient to protect against LPS-induced lung injury; consequently, PGC-1A may promote disease tolerance in response to acute lung injury by promoting fatty acid oxidation in alveolar epithelial cells and consequently protect against cell death81. Therefore, promoting fatty acid oxidation in alveolar epithelial cells may be useful for increasing survival in patients with COVID-19 by maintaining the epithelial barrier. Similarly, repair of the lung epithelium and restoration of lung epithelial function is necessary for the repair of pulmonary damage caused by ARDS. This process involves proliferation of type II alveolar epithelial cells and their differentiation into type I alveolar epithelial cells. Promoting the survival of type II epithelial cells through fatty acid oxidation may be necessary for the repair process by sustaining the number of cells that can differentiate into type I cells and therefore may be important for the recovery phase of COVID-19 (Fig. 6).
PGC-1A performs many additional functions in cellular metabolism, including mitochondrial biogenesis, oxidative phosphorylation, gluconeogenesis and glycogenolysis; these functions may be important for promoting disease tolerance in acute lung injury. The glucose catabolism associated with these functions may contribute to the protective effects of PGC-1A. In a mouse model of mechanical-stretching injury of the lung, stretching of pulmonary epithelial cells has been found to result in inhibition of succinate dehydrogenase, thus leading to increased levels of succinate83. Succinate activates hypoxia-inducible factor (HIF) 1α in a normoxic manner in alveolar epithelial cells83 and consequently facilitates the adaptation of these epithelial cells to mechanical stress by increasing the glycolytic capacity of the cells, tricarboxylic acid flux and mitochondrial respiration, thus increasing in the amount of ATP produced by alveolar epithelial cells. Treatment of mice with dimethyl-oxaloglycine, a pharmacological stabilizer of HIF1α, protects against lung inflammation and pulmonary oedema, and improves gas exchange, thereby increasing survival83. These data suggest that targeting glycolysis in alveolar epithelial cells in COVID-19 during the ARDS stage may be a useful strategy to promote disease tolerance during infection. Furthermore, this strategy may be useful to protect against the collateral lung damage caused by the use of mechanical ventilators, which can contribute to lung injury. Although these data suggest that glycolysis may be beneficial for tolerance during the ARDS phase of COVID-19, as discussed earlier, targeting glycolysis in patients with COVID-19 will be complicated by the differential effects that glycolysis appears to have on multiple aspects of host defence, including resistance, antivirulence and disease tolerance. Therefore, there are important temporal and cellular target considerations for drugging this process for COVID-19 treatment (Fig. 6).
Beyond maintaining the alveolar epithelial barrier, maintaining the integrity of the capillary endothelial barrier is also essential for preventing lung vascular permeability. Dysfunction of the endothelial cell barrier in ARDS results in accumulation of fluid and macromolecules in the interstitium and alveolar space, thus leading to pulmonary oedema. The kinase AMPK is a critical regulator of normal endothelial cell function and maintenance of endothelial barrier integrity. AMPK is also a critical regulator of cellular metabolism. In a mouse model of LPS-induced acute lung injury, AMPK activity is inhibited in the lung. AMPKα-knockout mice, deficient in AMPK, are more susceptible than wild-type mice to LPS-induced acute lung injury and to increased barrier dysfunction, thereby resulting in inflammation, oedema and congestion. Treatment of mice with the drug AICAR, which activates AMPK, increases barrier integrity, and decreases pulmonary vascular permeability and overall morbidity in response to acute lung injury84. Thus, to promote survival of patients with COVID-19 via improving disease tolerance by maintaining barrier integrity in response to the hyperinflammatory state, targeting metabolism in pulmonary endothelial cells in addition to alveolar epithelial cells may be an important strategy (Fig. 6).
Pulmonary surfactant is a lipoprotein complex, composed of 90% lipids and 10% proteins, that is secreted by epithelial cells and coats the alveolar surface of the lung. It promotes ventilation by decreasing the surface tension between air and fluid, thus facilitating gas exchange and preventing alveolar collapse. Alveolar surface tension increases during ARDS, and collapse can occur and subsequently contribute to poor gas exchange and hypoxia. This response is due to changes in the amount of surfactant secreted into the pulmonary environment, and in phospholipid, fatty acid, neutral lipid and surfactant-apoprotein composition85. In patients with ARDS, profiling of the phospholipid composition of the bronchoalveolar lavage fluid has shown that the composition changes, with decreased phosphatidylglycerol levels and a compensatory increase in the amounts of minor components86. Levels of palmitate, the most abundant fatty acid in surfactant, are also diminished in people with ARDS86. Work in animal models of acute lung injury suggests that changes in surfactant metabolism may be responsible for these alterations during ARDS87. These findings suggest that manipulating surfactant metabolism in pulmonary epithelial cells to maintain proper composition and secretion may be beneficial for promoting disease tolerance in COVID-19 by maintaining alveolar integrity and structure (Fig. 6).
Surfactant physiology appears to also be regulated metabolically at the organismal level. Mice deficient in leptin have diminished lung expression of parathyroid-hormone-related protein, a critical regulator of surfactant production by alveolar epithelial cells88. However, whether leptin is protective in the context of ARDS is questionable. In a mouse model of bleomycin-induced fibrosis, leptin-receptor-deficient (db/db) mice are protected against developing lung fibrosis. This protection is correlated with elevated levels of PPARγ, which inhibits the transcriptional response to TGF-β1, a cytokine that contributes to the pathogenesis of ARDS fibrosis89. Interestingly, obese animal models, compared with control animals with healthful weight, have altered pulmonary lipid deposition, surfactant levels and composition90. Furthermore, obese rats show an increase in lung size, thus causing a surfactant deficiency relative to the lung surface area, along with changes in respiratory function90. These findings suggest that leptin may be protective during the survival phase of ARDS but maladaptive in the recovery phase of ARDS. Moreover, alterations in surfactant physiology may contribute to the increased risk associated with metabolic syndrome and COVID-19 (Fig. 6).
Alveolar-fluid accumulation occurs in both pneumonia and ARDS, and people with COVID-19 that progresses to ARDS have pulmonary oedema, which contributes to hypoxia. Type II alveolar epithelial cells substantially contribute to the resolution of oedema by mediating fluid reabsorption. This process is dependent on the active transport of Na+ across the alveolar epithelium, in a process primarily mediated by apical sodium channels and basolateral Na/K-ATPase. Transport of Na+ and K+ across the cell maintains the ionic gradients and consumes ~40% of the cellular ATP. The hypoxic environment in the alveoli during ARDS and pneumonia decreases the amounts of these transporters at the plasma membrane, thus impairing fluid reabsorption. The hypoxic environment leads to mitochondrial reactive-oxygen-species-mediated activation of the CRAC channel, thereby leading to Ca2+/CaMKKb-dependent AMPK activation and subsequent endocytosis of Na/K-ATPase transporters and impaired alveolar fluid clearance91. Therefore, maintaining the levels of Na/K-ATPase transporters at the epithelial cell plasma membrane may be an important disease-tolerance mechanism to promote fluid reabsorption. This maintenance may be achieved via HIF. Endocytosis of Na/K-ATPase is mediated by PKCζ. During hypoxia, HIF upregulates the ubiquitin ligase HOIL-1L, which targets the kinase PKCζ for degradation. Consequently, the amount of Na/K-ATPase transporters at the plasma membrane is stabilized, thus resulting in protection against hypoxia-induced lung injury92 (Fig. 6).
Patients who survive the inflammatory phase of ARDS enter the repair phase of the condition. Repair of pulmonary tissue is essential for restoring lung function and is mediated by disease-tolerance mechanisms; however, dysregulation of this process can cause pulmonary fibrosis—an ‘out-of-control’ wound with a scar that forms around the alveoli, which can lead to lung malfunction and stiffness, thereby causing a disruption in gas exchange and potentially death from respiratory failure. This phase is often called the late fibrotic stage. Approximately 20–30% of patients with COVID-19 have decline in lung function after surviving severe or critical stages of the infection, thus suggesting that they may be experiencing pulmonary fibrosis93. There is no cure for fibrosis, but if it is identified early, therapies including anti-inflammatory agents and anti-growth factors may be effective for decreasing fibrosis development. Drugs that target host metabolism may also protect against pulmonary fibrosis. Mitochondrial dysfunction and altered lipid metabolism in alveolar type II epithelial cells are associated with the pathogenesis of pulmonary fibrosis. In a mouse model of bleomycin-induced pulmonary fibrosis, the levels of elongation of long-chain fatty acid family member 6 (Elovl6), the rate-limiting enzyme catalysing the elongation of fatty acids, is decreased in fibrotic lungs. ElovI6 deficiency increases susceptibility of lung fibrosis associated with alveolar-wall thickening94. In the fibrotic lung, expression of the mitochondrial lipid-remodelling enzyme lysocardiolipin acyltransferase, is decreased. Overexpression of this enzyme protects against lung fibrosis95. In fibrotic lungs, epithelial cells accumulate large aggregates of misfolded proteins, which cause endoplasmic reticulum stress. Similarly, inhibition of stearoyl-CoA desaturase, an enzyme involved in the desaturation of fatty acids, causes endoplasmic reticulum stress and pulmonary fibrosis in mice96, thus suggesting that epithelial cell metabolism may control the endoplasmic reticulum stress in fibrotic lungs. Chu et al. have reported that fibrotic patients have higher palmitate levels in their lungs than control individuals. In mice fed a palmitate-rich Western-type diet, mice have a more severe response to bleomycin-induced lung injury, and also show elevated expression of endoplasmic reticulum stress markers and lung fibrosis. In vitro palmitate is directly toxic to lung epithelial cells by causing endoplasmic reticulum stress. Deletion of the CD36 lipid receptor abolishes these toxic effects, thus demonstrating that sensing of lipids by pulmonary epithelial cells can trigger endoplasmic reticulum stress, which in turn may facilitate the fibrotic response in lungs during lung injury. However, deletion of the CD36 lipid receptor abolishes these toxic effects97. Thus, targeting pulmonary metabolism may be an additional therapeutic option in patients with COVID-19, to promote their recovery and increase their likelihood of returning to their original health state before the infection (Fig. 6).
Targeting pulmonary physiology in patients with COVID-19 will be essential for the treatment of severe or critically ill patients, and although disease-tolerance strategies will probably be most useful, antivirulence strategies may provide some benefit in a narrow therapeutic window. The effectiveness of disease-tolerance strategies is not limited to patients who reach severe or critical points in their disease course. For example, drugs that target the alveolar epithelial cells and limit their sensitivity to inflammatory damage could be administered early to limit damage to the lung. This is an underappreciated aspect of infectious-disease defence and therapy in general, and is likely to be the strategy most amenable to metabolic targeting strategies.
Extrapulmonary events in patients with COVID-19
Although this discussion has focused on the pulmonary effects of the hyperinflammatory phase of COVID-19, extrapulmonary effects also occur in patients, including damage to the heart and other components of the cardiovascular system, skeletal muscle, brain and kidney, as well as gastrointestinal effects (Fig. 2). The causes of these extrapulmonary events are not clear, but many non-exclusive explanations exist for these events. First, many of these tissues express ACE2, and therefore the organ damage in these patients may be the direct consequence of SARS-CoV-2 infecting these tissues. Second, the damage can be a consequence of the direct effects of the host inflammatory response. Third, the damage can be a consequence of the organ dysfunction and the resulting decline in the physiological function of other organs. For example, the respiratory failure causes hypoxia, which can damage other physiological systems. Similarly, cardiovascular damage can limit the delivery of oxygenated blood to other systems. Acute kidney injury can lead to the accumulation of metabolites in the blood that are toxic to other systems. Regardless of the cause, the most logical defence strategy for patients presenting with these complications is disease tolerance to (1) limit the damage; (2) enable the organ to perform its physiological function despite experiencing damage; and (3) promote repair of the organ6. Targeting metabolism is a viable option to mediate these goals6.
In COVID-19, studies from Wuhan have indicated that 20–45% of patients experience heart damage from the infection98. Similar percentages of patients have been reported to have abnormal blood clotting during the infection99. Cardiac damage can decrease the capacity of the heart to provide oxygenated blood to the rest of the body. This effect, combined with blood clotting, can contribute to the extrapulmonary hypoxia that leads to organ failure. Because the heart and cardiovascular system express ACE2, this effect can be due to direct infection by the virus or to the collateral damage of the host response during the infection (Fig. 2). The heart has an excellent capacity to adapt to pathogenic signals and injury by modulating cardiac metabolism. Fatty acid oxidation is the primary energy substrate for the heart under homeostatic conditions. During heart failure, cardiomyocyte energetics is altered, with a shift from fatty acids to glucose as the preferred substrate. During ischaemic injury, the heart remodels and exhibits hypertrophy to adapt to the injury, and this response is associated with an upregulation of glycolysis in cardiomyocytes100. This important disease-tolerance mechanism enables the heart to perform its physiological functions in the presence of pathogenic signals despite being damaged6. Shifting to glucose metabolism promotes myocardial contractile efficiency, thus optimizing the ratio of ATP production to oxygen consumption. In a porcine model, increasing glucose oxidation and decreasing fatty acid metabolism improves cardiac efficiency101 and may serve as a method to promote disease tolerance in patients with COVID-19 by facilitating the adaptation of the heart to the diseased state (Fig. 6).
Patients with COVID-19 have been reported to exhibit symptoms in the brain and central nervous system, including stroke and seizures102 (Fig. 2). Cells in specific regions of the brain express ACE2, but whether the virus actually infects the brain is unclear. Certainly, stroke and seizures can be a consequence of the collateral damage of the host response, including respiratory failure and damage to the cardiovascular system. In a mouse model of poly(I:C) to mimic viral inflammation, glucose metabolism by the brain is necessary to facilitate neuronal adaptation to the unfolded-protein response occurring during infection7. Decreased glucose availability to the brain due to fasting results in lethal seizures7. This finding is consistent with observations in people with epilepsy, whose seizures are associated with impairments in oxidative glucose metabolism103. Therefore, targeting glucose metabolism in the brain may be a viable disease-tolerance strategy for a broad range of diseases that cause central symptoms, including COVID-19 (Fig. 6).
Like other organs, the kidneys exhibit mitochondrial dysfunction and decreased levels of intracellular ATP during severe or critical illness caused by acute inury104,105. In a sepsis mouse model involving caecal ligation and puncture, renal phosphorylation of AMPK is elevated. Activation of AMPK with the AMP analog AICAR protects mice against acute kidney injury induced by caecal ligation and puncture, and this effect is associated with decreased inflammation and endothelial cell activation and injury106. In this case, AMPK appears to act as an antivirulence mechanism, because AMPK activity downregulates the inflammatory and endothelial cell response that serves as the damage signal to the kidney. Because AMPK is also a critical regulator of energy, part of the renal protection mediated by AMPK activity in response to acute kidney injury might involve regulating energy balance and promoting disease tolerance of the organ, thus enabling the kidneys to function during the disease phase (Fig. 6).
Skeletal muscle atrophy is a complication of hypercapnia that results from poor gas exchange in the lung. Rhabdomyolysis, a form of atrophy due to the breakdown of damaged muscle, has been reported in patients with late stages of COVID-19 (ref. 42 and Fig. 2). This condition is life threatening and may be related to the acute renal injury observed in people with COVID-19. In a mouse model of acute lung injury caused by bacterial pneumonia, induction of IGF-1 in the white adipose tissue promotes disease tolerance to bacterial pneumonia via signalling to the skeletal muscle, thus maintaining skeletal muscle size8 (Fig. 6). These examples demonstrate how metabolism in organs can be regulated to promote an individual’s physiological defences during an infection. Elucidation of the full spectrum of physiological defence mechanisms and how metabolism can be targeted to exploit these mechanisms during COVID-19 and other diseases will be important in further investigations6.
Immediate clinical consequences
Data from the COVID-19 pandemic, past SARS outbreaks and other viral and critical illness settings demonstrate that host metabolism must be considered. Metabolic abnormalities are a risk factor for severe and critical conditions, thus demonstrating that metabolic parameters must be managed to control the COVID-19 disease course. Glycaemic management is associated with better outcomes in COVID-19. Management through measures such as insulin administration or GLP1 for hyperglycaemic cases and glucose for hypoglycaemic cases may be beneficial for patient care. Metabolic strategies that promote patient disease tolerance may also have therapeutic potential, and strategies that can have beneficial effects in multiple systems would be ideal. Because the regulation of fatty acid oxidation appears to be important for promoting disease tolerance in various organ and physiological systems, one such potential candidate would be administration of PPAR agonists. PPAR agonists have been shown to have tissue-protective roles in the heart during ischaemic challenge, in the kidneys in acute-injury settings and against pulmonary damage in ARDS models107,108. Other drugs that modulate fatty acid oxidation, such as trimetazidine or perhexiline, also have protective effects in the heart, kidneys and lungs107,109, and may be viable avenues to pursue. Because COVID-19 is a new infection, the most effective metabolic targets to treat the infection are not yet known. Although this pathogen is new, the pathologies are old and generalizable for several severe and critical conditions. Although the upstream pathways activated by SARS-CoV-2 will be distinct from those in other diseases, they are likely to ultimately converge on the same pathways that lead to pathology and death6, thus suggesting that some commonalities are likely between COVID-19 and other diseases that cause similar pathologies.
Conclusions
The relevant way to think about infectious-disease pandemics is in the context of when the next pandemic will occur rather than whether there will be another pandemic. With the next outbreak, it is unlikely that effective vaccine- or antimicrobial-based strategies will be available to fight the infection; the ‘enemy’ will be unknown until it is upon us. In each pandemic, the first line of defence has been supportive care to promote patient disease tolerance and to gain time for patients to recover from the infection. An examination of the pathologies of infectious-disease outbreaks over the past century demonstrates that although the pathogens are different, the pathologies are the same. The COVID-19 pandemic has served as an important reminder that we must broaden our focus in how we think about infectious diseases. We must approach infectious diseases from a more holistic viewpoint to understand how pathology occurs; how physiological function changes as a consequence of the damage; what methods we can use to defend against the pathology and physiological dysfunction and the immune response; and how to destroy the pathogen. We must begin developing disease-tolerance-based strategies so that when the next pandemic occurs, we will be better equipped to treat infected people before effective vaccines are developed. We must understand how the pathophysiologies of preexisting conditions influence the disease course of COVID-19 and other infections, and how these conditions may render patients more susceptible to infection-induced damage. We also must understand the recovery process of patients with infectious diseases and how the infection sequelae can make patients susceptible to developing other conditions. Evidence from COVID-19, past SARS outbreaks and critical infectious conditions in general highlight the importance of understanding the relationship between metabolic processes and health for infection susceptibility, treatments and recovery. Targeting host metabolism at the cellular, tissue, organ and physiological levels is a viable approach to promoting defence against COVID-19 at various stages along the disease course. Once we think beyond the traditional view of infectious diseases, we will be better able to treat them.
Acknowledgements
J.S.A. is supported by the NOMIS Foundation, The Keck Foundation, an NIH Pioneer award and NIH grant R01 AI114929.
Competing interests
The author declares no competing interests.
Footnotes
Peer review information Primary Handling Editor: George Caputa.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Schneider DS, Ayres JS. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat. Rev. Immunol. 2008;8:889–895. doi: 10.1038/nri2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ayres JS. Surviving COVID-19: a disease tolerance perspective. Sci. Adv. 2020;6:eabc1518. doi: 10.1126/sciadv.abc1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siddiqi HK, Mehra MR. COVID-19 illness in native and immunosuppressed states: a clinical–therapeutic staging proposal. J. Heart Lung Transplant. 2020;39:405–407. doi: 10.1016/j.healun.2020.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ayres JS. Immunometabolism of infections. Nat. Rev. Immunol. 2020;20:79–80. doi: 10.1038/s41577-019-0266-9. [DOI] [PubMed] [Google Scholar]
- 5.Troha K, Ayres JS. Metabolic adaptations to infections at the organismal level. Trends Immunol. 2020;41:113–125. doi: 10.1016/j.it.2019.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ayres JS. The biology of physiological health. Cell. 2020;181:250–269. doi: 10.1016/j.cell.2020.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang A, et al. Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell. 2016;166:1512–1525.e1512. doi: 10.1016/j.cell.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schieber AM, et al. Disease tolerance mediated by microbiome E. coli involves inflammasome and IGF-1 signaling. Science. 2015;350:558–563. doi: 10.1126/science.aac6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rao S, et al. Pathogen-mediated inhibition of anorexia promotes host survival and transmission. Cell. 2017;168:503–516.e512. doi: 10.1016/j.cell.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanchez KK, et al. Cooperative metabolic adaptations in the host can favor asymptomatic infection and select for attenuated virulence in an enteric pathogen. Cell. 2018;175:146–158.e115. doi: 10.1016/j.cell.2018.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bornstein SR, Dalan R, Hopkins D, Mingrone G, Boehm BO. Endocrine and metabolic link to coronavirus infection. Nat. Rev. Endocrinol. 2020;16:297–298. doi: 10.1038/s41574-020-0353-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu, X. & Yang, R. COVID-19 transmission through asymptomatic carriers is a challenge to containment. Influenza Other Respir. Viruses10.1111/irv.12743 (2020). [DOI] [PMC free article] [PubMed]
- 13.Booth CM, et al. Clinical features and short-term outcomes of 144 patients with SARS in the greater Toronto area. JAMA. 2003;289:2801–2809. doi: 10.1001/jama.289.21.JOC30885. [DOI] [PubMed] [Google Scholar]
- 14.Yang JK, et al. Plasma glucose levels and diabetes are independent predictors for mortality and morbidity in patients with SARS. Diabet. Med. 2006;23:623–628. doi: 10.1111/j.1464-5491.2006.01861.x. [DOI] [PubMed] [Google Scholar]
- 15.Deng SQ, Peng HJ. Characteristics of and public health responses to the coronavirus disease 2019 outbreak in China. J. Clin. Med. 2020;9:E575. doi: 10.3390/jcm9020575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang D, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. 2020;323:1061–1069. doi: 10.1001/jama.2020.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang, J. J. et al. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy10.1111/all.14238 (2020). [DOI] [PubMed]
- 18.Casqueiro J, Casqueiro J, Alves C. Infections in patients with diabetes mellitus: a review of pathogenesis. Indian J. Endocrinol. Metab. 2012;16(Suppl. 1):S27–S36. doi: 10.4103/2230-8210.94253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kass DA, Duggal P, Cingolani O. Obesity could shift severe COVID-19 disease to younger ages. Lancet. 2020;395:1544–1545. doi: 10.1016/S0140-6736(20)31024-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ayres JS, Schneider DS. Tolerance of infections. Annu. Rev. Immunol. 2012;30:271–294. doi: 10.1146/annurev-immunol-020711-075030. [DOI] [PubMed] [Google Scholar]
- 21.Tziomalos K, Athyros VG. Diabetic nephropathy: new risk factors and improvements in diagnosis. Rev. Diabet. Stud. 2015;12:110–118. doi: 10.1900/RDS.2015.12.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tedla FM, Brar A, Browne R, Brown C. Hypertension in chronic kidney disease: navigating the evidence. Int. J. Hypertens. 2011;2011:132405. doi: 10.4061/2011/132405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henry BM, Lippi G. Chronic kidney disease is associated with severe coronavirus disease 2019 (COVID-19) infection. Int. Urol. Nephrol. 2020;52:1193–1194. doi: 10.1007/s11255-020-02451-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Avula A, et al. COVID-19 presenting as stroke. Brain Behav. Immun. 2020;87:115–119. doi: 10.1016/j.bbi.2020.04.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akhmerov A, Marbán E. COVID-19 and the heart. Circ. Res. 2020;126:1443–1455. doi: 10.1161/CIRCRESAHA.120.317055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seaquist ER. The final frontier: how does diabetes affect the brain? Diabetes. 2010;59:4–5. doi: 10.2337/db09-1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruilope LM, Solini A. RAS blockade for every diabetic patient: pro and con. Diabetes Care. 2011;34(Suppl. 2):S320–S324. doi: 10.2337/dc11-s248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Batlle D, Jose Soler M, Ye M. ACE2 and diabetes: ACE of ACEs? Diabetes. 2010;59:2994–2996. doi: 10.2337/db10-1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bindom SM, Lazartigues E. The sweeter side of ACE2: physiological evidence for a role in diabetes. Mol. Cell. Endocrinol. 2009;302:193–202. doi: 10.1016/j.mce.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu L, et al. Association of blood glucose control and outcomes in patients with COVID-19 and pre-existing type 2 diabetes. Cell Metab. 2020;31:1068–1077.e3. doi: 10.1016/j.cmet.2020.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan JC. Recovery pathway of post-SARS patients. Thorax. 2005;60:361–362. doi: 10.1136/thx.2004.035972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Q, et al. Altered lipid metabolism in recovered SARS patients twelve years after infection. Sci. Rep. 2017;7:9110. doi: 10.1038/s41598-017-09536-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang JK, Lin SS, Ji XJ, Guo LM. Binding of SARS coronavirus to its receptor damages islets and causes acute diabetes. Acta Diabetol. 2010;47:193–199. doi: 10.1007/s00592-009-0109-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jivanji CJ, Asrani VM, Windsor JA, Petrov MS. New-onset diabetes after acute and critical illness: a systematic review. Mayo Clin. Proc. 2017;92:762–773. doi: 10.1016/j.mayocp.2016.12.020. [DOI] [PubMed] [Google Scholar]
- 35.Lin YF, et al. New-onset diabetes after acute kidney injury requiring dialysis. Diabetes Care. 2018;41:2105–2110. doi: 10.2337/dc17-2409. [DOI] [PubMed] [Google Scholar]
- 36.Sasannejad C, Ely EW, Lahiri S. Long-term cognitive impairment after acute respiratory distress syndrome: a review of clinical impact and pathophysiological mechanisms. Crit. Care. 2019;23:352. doi: 10.1186/s13054-019-2626-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Higgs S, Spetter MS. Cognitive control of eating: the role of memory in appetite and weight gain. Curr. Obes. Rep. 2018;7:50–59. doi: 10.1007/s13679-018-0296-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang M, et al. Psychiatric symptoms in acute respiratory distress syndrome survivors: a 1-year national multicenter study. Crit. Care Med. 2016;44:954–965. doi: 10.1097/CCM.0000000000001621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan KS, et al. Evaluating muscle mass in survivors of acute respiratory distress syndrome: a 1-year multicenter longitudinal study. Crit. Care Med. 2018;46:1238–1246. doi: 10.1097/CCM.0000000000003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rocheteau P, et al. Sepsis induces long-term metabolic and mitochondrial muscle stem cell dysfunction amenable by mesenchymal stem cell therapy. Nat. Commun. 2015;6:10145. doi: 10.1038/ncomms10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pfoh ER, et al. Physical declines occurring after hospital discharge in ARDS survivors: a 5-year longitudinal study. Intensive Care Med. 2016;42:1557–1566. doi: 10.1007/s00134-016-4530-1. [DOI] [PubMed] [Google Scholar]
- 42.Jin M, Tong Q. Rhabdomyolysis as potential late complication associated with COVID-19. Emerg. Infect. Dis. 2020;26:1618–1620. doi: 10.3201/eid2607.200445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haley, M. J. et al. Stroke induces prolonged changes in lipid metabolism, the liver and body composition in mice. Transl. Stroke Res.10.1007/s12975-019-00763-2 (2019). [DOI] [PMC free article] [PubMed]
- 44.To KK, et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. Lancet Infect. Dis. 2020;20:565–574. doi: 10.1016/S1473-3099(20)30196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He X, et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 2020;26:672–675. doi: 10.1038/s41591-020-0869-5. [DOI] [PubMed] [Google Scholar]
- 46.Zou L, et al. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N. Engl. J. Med. 2020;382:1177–1179. doi: 10.1056/NEJMc2001737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wölfel R, et al. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020;581:465–469. doi: 10.1038/s41586-020-2196-x. [DOI] [PubMed] [Google Scholar]
- 48.Louie JK, et al. Treatment with neuraminidase inhibitors for critically ill patients with influenza A (H1N1)pdm09. Clin. Infect. Dis. 2012;55:1198–1204. doi: 10.1093/cid/cis636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Channappanavar R, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. 2016;19:181–193. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun M, et al. Metabolic effects of social isolation in adult C57BL/6 mice. Int. Sch. Res. Notices. 2014;2014:690950. doi: 10.1155/2014/690950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brinkhues S, et al. Socially isolated individuals are more prone to have newly diagnosed and prevalent type 2 diabetes mellitus: the Maastricht study. BMC Public Health. 2017;17:955. doi: 10.1186/s12889-017-4948-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Altenburg TM, Rotteveel J, Serné EH, Chinapaw MJ. Effects of multiple sedentary days on metabolic risk factors in free-living conditions: lessons learned and future recommendations. Front. Physiol. 2016;7:616. doi: 10.3389/fphys.2016.00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Panahi S, Tremblay A. Sedentariness and health: is sedentary behavior more than just physical inactivity? Front. Public Health. 2018;6:258. doi: 10.3389/fpubh.2018.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jeong IK. The role of cortisol in the pathogenesis of the metabolic syndrome. Diabetes Metab. J. 2012;36:207–210. doi: 10.4093/dmj.2012.36.3.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kappeler PM, Cremer S, Nunn CL. Sociality and health: impacts of sociality on disease susceptibility and transmission in animal and human societies. Phil. Trans. R. Soc. Lond. B. 2015;370:20140116. doi: 10.1098/rstb.2014.0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kupferschmidt K, Cohen J. Race to find COVID-19 treatments accelerates. Science. 2020;367:1412–1413. doi: 10.1126/science.367.6485.1412. [DOI] [PubMed] [Google Scholar]
- 57.Oberfeld B, et al. SnapShot: COVID-19. Cell. 2020;181:954–954.e1. doi: 10.1016/j.cell.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yan B, et al. Characterization of the lipidomic profile of human coronavirus-infected cells: implications for lipid metabolism remodeling upon coronavirus replication. Viruses. 2019;11:E73. doi: 10.3390/v11010073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mayer KA, Stöckl J, Zlabinger GJ, Gualdoni GA. Hijacking the supplies: metabolism as a novel facet of virus-host interaction. Front. Immunol. 2019;10:1533. doi: 10.3389/fimmu.2019.01533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yuan S, et al. SREBP-dependent lipidomic reprogramming as a broad-spectrum antiviral target. Nat. Commun. 2019;10:120. doi: 10.1038/s41467-018-08015-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smallwood HS, et al. Targeting metabolic reprogramming by influenza infection for therapeutic intervention. Cell Rep. 2017;19:1640–1653. doi: 10.1016/j.celrep.2017.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bojkova, J. et al. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature10.1038/s41586-020-2332-7 (2020). [DOI] [PMC free article] [PubMed]
- 63.Blanco-Melo D, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045.e1039. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang W, et al. Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell. 2019;178:176–189.e115. doi: 10.1016/j.cell.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rao M, Dodoo E, Zumla A, Maeurer M. Immunometabolism and pulmonary infections: implications for protective immune responses and host-directed therapies. Front. Microbiol. 2019;10:962. doi: 10.3389/fmicb.2019.00962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Loftus RM, Finlay DK. Immunometabolism: cellular metabolism turns immune regulator. J. Biol. Chem. 2016;291:1–10. doi: 10.1074/jbc.R115.693903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell. 2017;169:570–586. doi: 10.1016/j.cell.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mehta P, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–1034. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vigeland CL, et al. Inhibition of glutamine metabolism accelerates resolution of acute lung injury. Physiol. Rep. 2019;7:e14019. doi: 10.14814/phy2.14019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oliveira GP, de Abreu MG, Pelosi P, Rocco PR. Exogenous glutamine in respiratory diseases: myth or reality? Nutrients. 2016;8:76. doi: 10.3390/nu8020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maier HJ, Britton P. Involvement of autophagy in coronavirus replication. Viruses. 2012;4:3440–3451. doi: 10.3390/v4123440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Acharya, D., Liu, G. & Gack, M. U. Dysregulation of type I interferon responses in COVID-19. Nat. Rev. Immunol.10.1038/s41577-020-0346-x (2020). [DOI] [PMC free article] [PubMed]
- 73.Lechien, J. R. et al. Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): a multicenter European study. Eur. Arch. Otorhinolaryngol.10.1007/s00405-020-05965-1 (2020). [DOI] [PMC free article] [PubMed]
- 74.Conti P, et al. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents. 2020;34:1. doi: 10.23812/CONTI-E. [DOI] [PubMed] [Google Scholar]
- 75.Tannahill GM, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rodriguez AE, et al. Serine metabolism supports macrophage IL-1β production. Cell Metab. 2019;29:1003–1011.e1004. doi: 10.1016/j.cmet.2019.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Q, et al. O-GlcNAc transferase promotes influenza A virus–induced cytokine storm by targeting interferon regulatory factor–5. Sci. Adv. 2020;6:eaaz7086. doi: 10.1126/sciadv.aaz7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hung CC, et al. The intestinal fatty acid propionate inhibits Salmonella invasion through the post-translational control of HilD. Mol. Microbiol. 2013;87:1045–1060. doi: 10.1111/mmi.12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Moseley CE, Webster RG, Aldridge JR. Peroxisome proliferator-activated receptor and AMP-activated protein kinase agonists protect against lethal influenza virus challenge in mice. Influenza Other Respir. Viruses. 2010;4:307–311. doi: 10.1111/j.1750-2659.2010.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kimmig, L. M. et al. IL6 inhibition in critically ill COVID-19 patients is associated with increased secondary infections. Preprint at medRxiv10.1101/2020.05.15.20103531 (2020). [DOI] [PMC free article] [PubMed]
- 81.Cui H, et al. Impairment of fatty acid oxidation in alveolar epithelial cells mediates acute lung injury. Am. J. Respir. Cell Mol. Biol. 2019;60:167–178. doi: 10.1165/rcmb.2018-0152OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Eckle T, et al. HIF1A reduces acute lung injury by optimizing carbohydrate metabolism in the alveolar epithelium. PLoS Biol. 2013;11:e1001665. doi: 10.1371/journal.pbio.1001665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xing J, et al. Inhibition of AMP-activated protein kinase accentuates lipopolysaccharide-induced lung endothelial barrier dysfunction and lung injury in vivo. Am. J. Pathol. 2013;182:1021–1030. doi: 10.1016/j.ajpath.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fessler MB, Summer RS. Surfactant lipids at the host-environment interface. Metabolic sensors, suppressors, and effectors of inflammatory lung disease. Am. J. Respir. Cell Mol. Biol. 2016;54:624–635. doi: 10.1165/rcmb.2016-0011PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Günther A, et al. Surfactant alteration and replacement in acute respiratory distress syndrome. Respir. Res. 2001;2:353–364. doi: 10.1186/rr86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bezerra FS, et al. Exogenous surfactant prevents hyperoxia-induced lung injury in adult mice. Intensive Care Med. Exp. 2019;7:19. doi: 10.1186/s40635-019-0233-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Oruqaj L, et al. Effect of high fat diet on pulmonary expression of parathyroid hormone-related protein and its downstream targets. Heliyon. 2016;2:e00182. doi: 10.1016/j.heliyon.2016.e00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jain M, et al. Leptin promotes fibroproliferative acute respiratory distress syndrome by inhibiting peroxisome proliferator-activated receptor-γ. Am. J. Respir. Crit. Care Med. 2011;183:1490–1498. doi: 10.1164/rccm.201009-1409OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Inselman LS, Chander A, Spitzer AR. Diminished lung compliance and elevated surfactant lipids and proteins in nutritionally obese young rats. Lung. 2004;182:101–117. doi: 10.1007/s00408-003-1048-4. [DOI] [PubMed] [Google Scholar]
- 91.Gusarova GA, et al. Hypoxia leads to Na,K-ATPase downregulation via Ca2+ release-activated Ca2+ channels and AMPK activation. Mol. Cell. Biol. 2011;31:3546–3556. doi: 10.1128/MCB.05114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Magnani ND, et al. HIF and HOIL-1L-mediated PKCζ degradation stabilizes plasma membrane Na,K-ATPase to protect against hypoxia-induced lung injury. Proc. Natl Acad. Sci. USA. 2017;114:e10178–e10186. doi: 10.1073/pnas.1713563114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shi H, et al. Radiological findings from 81 patients with COVID-19 pneumonia in Wuhan, China: a descriptive study. Lancet Infect. Dis. 2020;20:425–434. doi: 10.1016/S1473-3099(20)30086-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sunaga H, et al. Deranged fatty acid composition causes pulmonary fibrosis in Elovl6-deficient mice. Nat. Commun. 2013;4:2563. doi: 10.1038/ncomms3563. [DOI] [PubMed] [Google Scholar]
- 95.Huang LS, et al. The mitochondrial cardiolipin remodeling enzyme lysocardiolipin acyltransferase is a novel target in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014;189:1402–1415. doi: 10.1164/rccm.201310-1917OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Romero F, et al. Lipid synthesis is required to resolve endoplasmic reticulum stress and limit fibrotic responses in the lung. Am. J. Respir. Cell Mol. Biol. 2018;59:225–236. doi: 10.1165/rcmb.2017-0340OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chu SG, et al. Palmitic acid-rich high-fat diet exacerbates experimental pulmonary fibrosis by modulating endoplasmic reticulum stress. Am. J. Respir. Cell Mol. Biol. 2019;61:737–746. doi: 10.1165/rcmb.2018-0324OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shi, S. et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol.10.1001/jamacardio.2020.0950 (2020). [DOI] [PMC free article] [PubMed]
- 99.Klok FA, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020;191:145–147. doi: 10.1016/j.thromres.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ardehali H, et al. Targeting myocardial substrate metabolism in heart failure: potential for new therapies. Eur. J. Heart Fail. 2012;14:120–129. doi: 10.1093/eurjhf/hfr173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chavez PN, et al. Effect of hyperglycemia and fatty acid oxidation inhibition during aerobic conditions and demand-induced ischemia. Am. J. Physiol. Heart Circ. Physiol. 2003;284:H1521–H1527. doi: 10.1152/ajpheart.00974.2002. [DOI] [PubMed] [Google Scholar]
- 102.Karimi N, Sharifi Razavi A, Rouhani N. Frequent convulsive seizures in an adult patient with COVID-19: a case report. Iran. Red Crescent Med. J. 2020;22:e102828. [Google Scholar]
- 103.McDonald T, Puchowicz M, Borges K. Impairments in oxidative glucose metabolism in epilepsy and metabolic treatments thereof. Front. Cell. Neurosci. 2018;12:274. doi: 10.3389/fncel.2018.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hsiao HW, et al. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock. 2012;37:289–296. doi: 10.1097/SHK.0b013e318240b52a. [DOI] [PubMed] [Google Scholar]
- 105.Bartz RR, et al. Staphylococcus aureus sepsis induces early renal mitochondrial DNA repair and mitochondrial biogenesis in mice. PLoS ONE. 2014;9:e100912. doi: 10.1371/journal.pone.0100912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Escobar DA, et al. Adenosine monophosphate-activated protein kinase activation protects against sepsis-induced organ injury and inflammation. J. Surg. Res. 2015;194:262–272. doi: 10.1016/j.jss.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lionetti V, Stanley WC, Recchia FA. Modulating fatty acid oxidation in heart failure. Cardiovasc. Res. 2011;90:202–209. doi: 10.1093/cvr/cvr038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fogo AB. PPARγ and chronic kidney disease. Pediatr. Nephrol. 2011;26:347–351. doi: 10.1007/s00467-010-1602-2. [DOI] [PubMed] [Google Scholar]
- 109.Park JH, et al. Effects of post ischemia-reperfusion treatment with trimetazidine on renal injury in rats: insights on delayed renal fibrosis progression. Oxid. Med. Cell. Longev. 2018;2018:1072805. doi: 10.1155/2018/1072805. [DOI] [PMC free article] [PubMed] [Google Scholar]