

Abstract
Draft genome sequences of five Calonectria species [including Calonectria aciculata, C. crousiana, C. fujianensis, C. honghensis and C. pseudoturangicola], Celoporthe dispersa, Sporothrix phasma and Alectoria sarmentosa are presented. Species of Calonectria are the causal agents of Eucalyptus leaf blight disease, threatening the growth and sustainability of Eucalyptus plantations in China. Celoporthe dispersa is the causal agent of stem canker in native Syzygium cordatum and exotic Tibouchina granulosa in South Africa. Sporothrix phasma was first discovered in the infructescences of Protea laurifolia and Protea neriifolia in South Africa. Alectoria sarmentosa is fruticose lichen belongs to the alectorioid clade of the family Parmeliaceae. The availability of these genome sequences will facilitate future studies on the systematics, population genetics, and genomics of these fungi.
Keywords: Alectoria sarmentosa, Calonectria species, Celoporthe dispersa, Eucalyptus leaf disease, Fungal pathogens, Sporothrix phasma
IMA GENOME-F 12A
Draft genome sequences of five Calonectria species from Eucalyptus plantations in China.
Introduction
Species in the genus Calonectria have a global distribution, particularly in tropical and subtropical regions of the world (Lombard et al. 2010c). These fungi include numerous important plant pathogens causing diseases on shoots, leave and roots of agricultural and forestry crops, which have led to significant economic impacts worldwide (Lombard et al. 2010c). The disease symptoms include cutting rot, damping off, leaf spots, leaf blight, shoot blight, defoliation as well as stem cankers and fruit rot (Crous 2002).
In the past 10 years, due to the influence of the phylogenetic species concept, many novel species of Calonectria have been described (Lombard et al. 2010a; Lombard et al. 2010b; Chen et al. 2011b; Xu et al. 2012; Alfenas et al. 2013a; Alfenas et al. 2013b; Lombard et al. 2016; Li et al. 2017; Liu and Chen 2017; Pham et al. 2019). This genus currently includes 171 recognized species residing in 10 different species complexes. Among these, 34 species, belonging to C. candelabra, C. cohounii, C. cylindrospora, C. kyotensis and C. reteaudii species complexes, were first discovered and described from China (Lombard et al. 2016; Li et al. 2017; Liu and Chen 2017; Pham et al. 2019). In 2015, a survey conducted in a relatively small area in southern China led to the discovery of 18 novel species from soil and symptomatic Eucalyptus plant tissues (Chen et al. 2011b; Lombard et al. 2015). This has highlighted the rich species diversity of Calonectria in China.
Despite their economic importance for Eucalyptus plantation forestry in China and other parts of the world, little is known regarding the biology and genetic determinants of virulence in Calonectria species. In this study, we sequenced the genomes of five important Calonectria species described from China. The overall aim was to facilitate future research regarding these important fungi, especially relating to their taxonomy, population genetics, and pathogenicity.
Sequenced strains
Calonectria aciculata: China: YunNan: isol. leaves of an E. urophylla × E. grandis hybrid clone, 16 Nov. 2014, S.F. Chen & J.Q. Li (PREM 61941 – holotype; CMW 47645 = CERC 5342 = CBS 142883 – ex-type culture).
Calonectria crousiana: China: FuJian: isol. leaves of Eucalyptus grandis, Aug. 2007 M.J. Wingfield (PREM 60453 – holotype; CMW 27249 = CBS 127198 – ex-type culture).
Calonectria fujianensis: China: FuJian: isol. leaves of Eucalyptus grandis, Aug. 2007 M.J. Wingfield (PREM 60460 – holotype; CMW 27257 = CBS 127201 – ex-type culture).
Calonectria honghensis: China: YunNan: isol. soil collected in a Eucalyptus plantation, 14 Nov. 2014, S.F. Chen & J.Q. Li (PREM 61943 – holotype; CMW 47669 = CERC 5572 = CBS 142885 – ex-type culture).
Calonectria pseudoturangicola: China: FuJian: isol. soil collected in the campus of Fujian Agriculture and Forestry University (FAFU), 14 Dec. 2014, S.F. Chen (PREM 61948 – holotype; CMW 47496 = CERC 7126 = CBS 142890 – ex-type culture).
Nucleotide sequence accession numbers
The draft genome data for the five Calonectria isolates have been deposited at DDBJ/EMBL/GenBank under BioProject PRJ562676. The accession numbers for each of the species are presented in Table 1.
Table 1.
Statistics of Calonectria genomes sequenced in this study
| Species | Isolate number | Accession number | Total Bases | Read Count | GC (%) | Scaffold number | Assembly size (Mb) | N50 (bp) | L50 | Coverage | Complete BUSCO (%) | Predicted gene models | Gene density (ORFs/Mb) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C. aciculata | CMW 47645 | VTGE01000000 | 2,442,674,772 | 9,731,772 | 47.721 | 221 | 61.6 | 675,696 | 25 | 39.7 | 98.7 | 15,556 | 252 |
| C. crousiana | CMW 27249 | VTGD01000000 | 6,593,014,992 | 26,266,992 | 48.718 | 358 | 58.1 | 419,924 | 46 | 113.5 | 98.5 | 14,967 | 257 |
| C. fujianensis | CMW 27257 | VTGC01000000 | 4,205,231,410 | 16,753,910 | 46.805 | 194 | 61.5 | 695,013 | 24 | 68.4 | 98.8 | 15,489 | 251 |
| C. honghensis | CMW 47669 | VTGB01000000 | 4,685,120,820 | 18,665,820 | 47.363 | 141 | 61.7 | 1,034,491 | 19 | 75.9 | 98.8 | 15,640 | 253 |
| C. pseudoturangicola | CMW 47496 | VTGA01000000 | 4,871,214,228 | 19,407,228 | 47.666 | 155 | 62.1 | 875,460 | 22 | 78.5 | 98.7 | 14,183 | 228 |
Materials and methods
Genomic DNA was extracted from single conidial cultures grown on malt yeast broth (2% malt extract, 0.5% yeast extract) using the method described by Duong et al. (2013). To verify the identification of all the sequenced isolates, PCR amplification and sequencing of the partial elongation factor gene (tef1) for the extracted DNA was carried out on each isolate. The tef1 sequences were then aligned against the sequences which developed in previous studies (Chen et al. 2011b; Li et al. 2017). After the identification, a phylogenetic tree reflecting the position of these five species in relation to other Calonectria species was subsequently produced based on the four gene regions (cmdA, his3, tef1 and but2). The sequences of representative isolates of the different species in this genus were obtained from GenBank, as reported by Liu and Chen (2017), and aligned using MAFFT version 7 (https://mafft.cbrc.jp/alignment/server/) (Katoh and Standley 2013). Phylogenetic analysis using maximum likelihood (ML), was conducted with PhyML v. 3.1 (Model = TIM2 + G; Guindon and Gascuel 2003). Confidence levels for the nodes were determined using 1000 bootstrap replicates. Final consensus trees were viewed and edited in MEGA 7.
The genomic DNA was submitted to Macrogen (South Korea), where one pair-end library with 550 bp median insert size was prepared using TruSeq DNA PCR-free protocol, and sequenced on Illumina Hiseq 2500 platform to get 250 bp pair-end reads. The quality of the data obtained was assessed using the software FastQC v. 0.11.5 (Afgan et al. 2016). Poor quality data and adapters were removed using the program Trimmomatic v. 0.36 (Bolger et al. 2014).
De novo assembly of the genome was carried out with SPAdes v. 3.9 (Bankevich et al. 2012) using trimmed pair-end data. Contigs that were smaller than 500 bp or with less than 20% of average K-mer coverage were removed from the assemblies. The filtered contigs were further placed into scaffolds with SSPACE-standard v. 3.0 (Boetzer et al. 2011) using the information from pair-end reads. Assembly gaps were filled or extended using GapFiller v. 1.10 (Boetzer and Pirovano 2012) with the paired-end data. Final assemblies were subjected to completeness assessment using the program Benchmarking Universal Single-Copy Orthologs (BUSCO) v. 2.0 (Simão et al. 2015) utilizing the dataset for Sordariomycetes. The program AUGUSTUS v. 3.2.2 was used to estimate the number of protein coding genes encoded by these genomes utilizing the species model for Magnaporthe grisea (Stanke et al. 2006).
Results and discussion
The tef1 gene from the PCR products confirmed the five Calonectria species, and a phylogenetic tree based on the four gene regions (cmdA, his3, tef1 and but2) reflecting the position of these five species in relation to other Calonectria species was produced (Fig. 1). The genomes of C. aciculata, C. crousiana, C. fujianensis, C. honghensis and C. pseudoturangicola were subsequently sequenced and assembled. Paired-end sequences of the libraries for the five isolates yielded from 9.7 to 26.2 million reads per library. These draft assemblies had scaffolds ranging from 141 to 358 in number. The assembled genome sizes were 58.1 Mb to 62.1 Mb in size. The N50 of the assemblies ranged from 419.9 Kb to 1034.5 Kb. The assemblies had BUSCO completeness scores ranging from 98.5 to 98.8%. The number of gene models ranged from 14,183 to 15,640. Statistics for all assembled genomes are presented in Table 1.
Fig. 1.

Maximum likelihood tree based on partial gene sequences of cmdA, his3, tef1 and but2 gene sequences (Chen et al. 2011b; Li et al. 2017; Liu and Chen 2017; Pham et al. 2019). Bootstrap values ≥65% are shown. The isolates used in this study are indicated in blue and bold
The statistics for the five Calonectria genomes were comparable to that of the draft genome sequence for C. pseudoreteaudii strain YA51, published by Ye et al. (2018), which had 507 scaffolds, 14,355 gene models, a total assembly size of 63.57 Mb, and a N50 of 1320 Kb. The availability of the genome sequences for the five Calonectria species presented in this study will enable comparative genomics studies to be conducted for these and various other species in the genus. They will also facilitate future investigations regarding the factors involved in pathogenicity, mating behaviour, and evolution of these important plant pathogens.
Authors: F.F. Liu, S.F. Chen * , B.D. Wingfield, M.J. Wingfield, T.A. Duong.
*Contact: shuaifei.chen@gmail.com
IMA GENOME-F 12B
Draft genome sequences of Celoporthe dispersa , a causal agent of canker diseases.
Introduction
The Cryphonectriaceae family includes several phytopathogens, and many of these pathogens cause severe damage to commercial plantations, especially Eucalyptus spp. (Chen et al. 2011a; Soares et al. 2018; Wang et al. 2018a). Celoporthe dispersa was first reported causing stem canker in native Syzygium cordatum and exotic Tibouchina granulosa in South Africa (Nakabonge et al. 2006). Pathogenicity trials conducted on E. grandis and T. granulosa showed that C. dispersa is pathogenic on both hosts, highlighting that C. dispersa could threaten commercial grown Eucalyptus trees in South Africa (Nakabonge et al. 2006). Within the Cryphonectriacea family, sequenced genomes are available for four species, including Chrysoporthe cubensis, Chr. deuterocubensis, Chr. austroaficana (Wingfield et al. 2015a; Wingfield et al. 2015b), and Cryphonectria parasitica (http://genome.jgi.doe.gov/Crypa2/Crypa2.info.html). The sequencing of this additional C. dispersa genome will be valuable for comparative genome studies within the Cryphonectriaceae family, and for improving disease management strategies, as well as preventing the threat to commercial plantations.
Sequenced strains
Celoporthe dispersa: South Africa, Limpopo Province, Tzaneen, Syzygium cordatum, 2003, M. Gryzenhout (CMW 9976 = CBS 118782, PREM 58897 – dried culture).
Nucleotide sequence accession numbers
The genome sequence of Celoporthe dispersa (isolate number CMW 9976) has been deposited in DDBJ/EMBL/GenBank databases under the accession number WAID00000000. The version described in this paper is WAID00000000.
Material and methods
Genomic DNA was extracted from freeze-dried mycelium of isolate CMW 9976 grown in malt yeast broth (2% Malt extract, 0.5% yeast extract; Biolab, Midrand, South Africa) using the Qiagen® Genomic-tip DNA extraction protocol for plants and fungi. Nanopore sequencing was conducted using the MinION sequencing device. The sequencing library was prepared using the Genomic DNA by Ligation (SQK-LSK109) protocol. The library was loaded on a MinION flowcell (R9.5.1) and sequencing was run for 48 h. Base calling was conducted using ONT Guppy basecalling software v 2.3.7.
Nanopore reads were error-corrected using Canu v 1.8 (Koren et al. 2017). The genome was assembled using smartdenovo (Istace et al. 2017), with corrected reads from Canu as input. The assembly was polished using base level signal from the ONT raw reads using the program Nanopolish (Jain et al. 2018). The program AUGUSTUS (Stanke and Morgenstern 2005) was used for prediction of protein coding genes present in C. dispersa genome. The Fusarium graminearum augustus species model was used as this is the most closely related species to C. dispersa available. The assembled genome completeness was evaluated using the Benchmarking Universal Single-Copy Orthologs tool, BUSCO (Simão et al. 2015). BUSCO was done on all contigs bigger than 1 Kb, using the fungal lineage dataset.
Results and discussion
Phylogenetic analysis using partial gene sequence of translation elongation factor of the sequenced genome confirmed the taxonomic identity as C. dispersa (Fig. 2). The assembly of C. dispersa consisted of 19 scaffolds, with the N50 of 1,993,378 bp. The calculated genome size was around 40 Mb and with a CG content of 52.9%. This assembly was also predicted to have 12,078 ORFs based on the gene models for Fusarium graminearum. Based on BUSCO analysis, this draft genome assembly had 94% completeness confirming the presence of these core eukaryotic genes. Out of this, 93% were present as single-copy genes. In our analysis, 1% of the BUSCO orthologs were found to be duplicated and 3.4% of the genes were missing. Only 39 BUSCO orthologs were classified as missing or fragmented out of the possible 1315 groups searched.
Fig. 2.

Neighbor Joining (NJ) tree of the partial gene sequences of translation elongation factor 1-α of selected reference sequences of different Celoporthe species. Holocryphia capensis was used as an outgroup in our analysis. MAFFT version 7 (Katoh and Standley 2013) was used for making alignments. The phylogenetic analysis was conducted in MEGA 7 (Tamura et al. 2013). The optimal tree with the sum of branch length = 26.62500000 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. The Celoporthe dispersa CMW 9976 isolate used in this study is shown in blue and bold
The estimated genome size and gene number for C. dispersa is similar to that of other species in Cryphonectriaceae, such as Chrysoporthe austroafricana (44.6 Mb, 13,484) (Wingfield et al. 2015a), Chr. cubensis (42.6 Mb, 13,121) (Wingfield et al. 2015b), Chr. deuterocubensis (43.9 Mb, 13,772) (Wingfield et al. 2015b) and Cryphonectria parasitica (43.9 Mb, 11,184) (http://genome.jgi.doe.gov/Crypa2/Crypa2.home.html). The availability of the C. dispersa genome from this study will enable various genomic comparisons between species of Cryphonectriaceae to be conducted. Additionally, the genome can be used to study pathogenicity, mating type and other important factors in phytopathology. The availability of the C. dispersa genome will enable us to include this genome within the Cryphonectriaceae during genomic comparison studies.
Authors: B.D. Wingfield * , T.A. Duong, M. Sayari, M. A. Ferreira, A. M. Kanzi.
*Contact: brenda.wingfield@fabi.up.ac.za
IMA GENOME-F 12C
Draft genome sequence of Sporothrix phasma.
Introduction
The Ophiostomatales, defined by the single family Ophiostomatacea, accommodates 11 well-defined genera including Sporothrix (De Beer et al. 2016a; De Beer et al. 2016b). Sporothrix was recently separated from Ophiostoma based on analyses of sequence data for multiple gene regions (De Beer et al. 2016a). This genus now accommodates more than 50 species that reside in six species complexes and five additional groups (De Beer et al. 2016a).
Species of Sporothrix are found in various habitats including on wood, in soil, and in association with arthropod vectors (Roets et al. 2007; Seifert et al. 2013; Lopes-Bezerra et al. 2018). Several Sporothrix species cause human and animal diseases, the best-known of which is S. shenckii (Teixeira et al. 2014), but most are considered saprotrophs. Some species also occupy the very unusual ecological habitat within the floral heads (infructescences) of Protea spp., which represent an important component of the Cape Floristic Region of South Africa (Cowling and Richardson 1995; Roets et al. 2006; Roets et al. 2009b). Twelve Sporothrix species have been collected and described in association with Protea spp. (Roets et al. 2006; Roets et al. 2008; Roets et al. 2010; Ngubane et al. 2018).
Sporothrix phasma was described by Roets et al. (2006) where it was first discovered in the infructescence of Protea laurifolia and Protea neriifolia. This species, together with others occupying the unusual Protea niche, were shown to be vectored by mites and Protea pollinating beetles (Roets et al. 2009a). However, this system is very complex and it has recently been shown that the S. phasma spore-carrying mites, are phoretic on larger mites, which in turn are phoretic on Protea-pollinating birds (Theron-De Bruin et al. 2018). In order to better understand the processes that have allowed Sporothrix species to adapt to different and diverse habitats, the genome of S. phasma was sequenced. The broader intention was that this sequence will contribute to the basal genomic data required to study the biology, ecology and, in some cases, pathogenicity of these fungi.
Sequenced strains
South Africa, isolated from Protea laurifolia, 2005, F. Roets, (culture CBS 119721 = CMW 20676 (ex- holotype); PREM 58941- dried culture).
Nucleotide sequence accession number
The genomic sequence of Sporothrix phasma (CMW 20676, CBS 119721) has been deposited at DDBJ/EMBL/GenBank under the accession number WJIH00000000. The version described in this paper is version WJIH01000000.
Materials and methods
Sporothrix phasma isolate CMW 20676 was obtained from the culture collection (CMW) of the Forestry and Agricultural Biotechnology Institute (FABI), the University of Pretoria, South Africa. Genomic DNA was extracted using the method described by Duong et al. (2013). Two pair-end libraries (350 bp and 550 bp average insert size) were prepared and sequenced using the Illumina HiSeq 2000 platform with 100 bp read length. Trimmomatic v. 0.38 (Bolger et al. 2014) was used for quality and adapter trimming. The program SPAdes v. 3.11.1 (Bankevich et al. 2012) was used to assemble the genome. SSPACE-standard v. 3.0 (Boetzer et al. 2011) was used for further scaffolding of SPAdes scaffolds. GapFiller v. 1–10 (Boetzer and Pirovano 2012) was used to fill or extend the assembly gaps. The Benchmarking Universal Single-Copy Orthologs (BUSCO v. 3.1.0) program (Simão et al. 2015) was used to assess the completeness of the assembly using the Sordariomyceta odb9 dataset. Maker v. 2.31.8 (Holt and Yandell 2011) was used to predict the number of protein coding genes present in the assembled genome.
The taxonomic placement of S. phasma in the genus Sporothrix was investigated by phylogenetic analysis of four combined gene regions, the Large Subunit (LSU) of the nuclear ribosomal RNA (rRNA) gene, the Internal Transcribed Spacer (ITS) regions, β tubulin (BT) gene and calmodulin (CAL) gene. The sequences of representative isolates in this genus were obtained from GenBank, as reported by De Beer et al. (2016a), and aligned online with MAFFT v. 7 (Katoh and Standley 2013). A maximum likelihood analysis was performed with the sequence data, using RaxML v. 8.2.4 (Stamatakis 2014) on the CIPRES Science Gateway v. 3.3 (Miller et al. 2010) and 1000 bootstrap replicates were performed to obtain branch support values. The genus Ophiostoma was used as outgroup.
Results and discussion
More than 6 million read pairs were obtained after the quality trimming. De-novo assembly using SPAdes resulted in 487 scaffolds which were larger than 500 bp. The number of final scaffolds was reduced to 279 after scaffolding with SSPACE and filling gaps with GapFiller. The current assembly has an N50 of 306 Kb and size of 30.2 Mb, with an overall GC content of 57.36%. The assembly included 96.8% complete, 1.2% fragmented, and 2.0% missing, BUSCOs. Maker predicted a total of 7999 protein coding genes. The taxonomic placement of S. phasma in Sporothrix is illustrated in Fig. 3. Sporothrix phasma has the smallest genome size when compared to other Sporothrix species for which genome sequences are available. Sprorothrix pallida has the largest genome size (37.8 Mb), followed by S. globosa (33.5 Mb), S. brasiliensis (33.2 Mb) and S. schenckii (32.3 Mb) (D’Alessandro et al. 2016; Huang et al. 2016; Gomez et al. 2018).
Fig. 3.
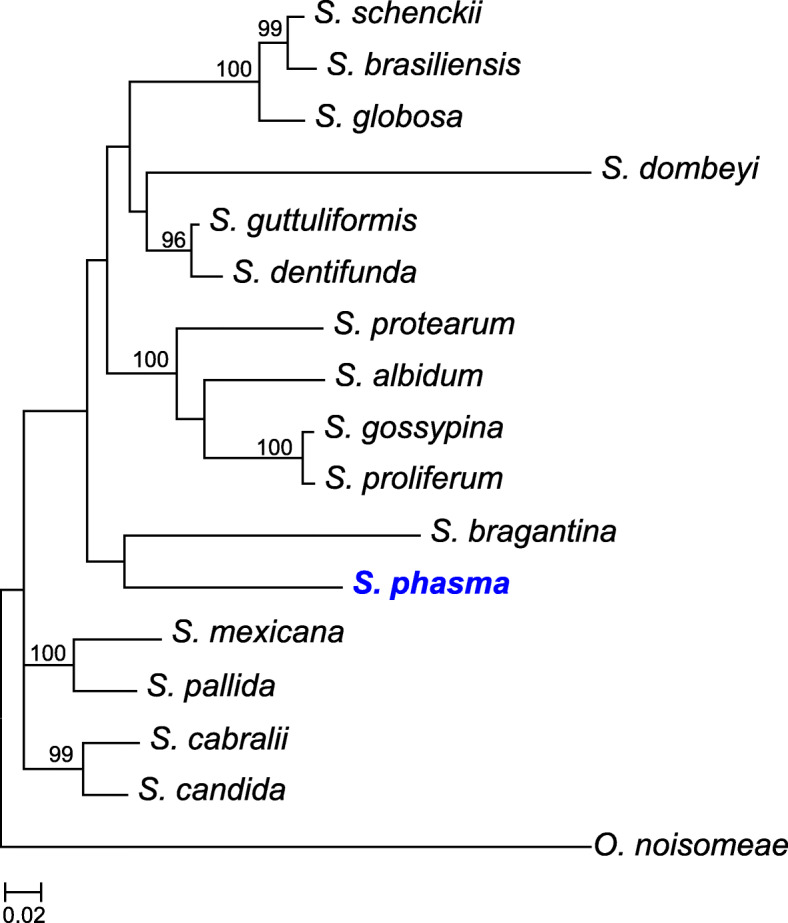
Phylogenetic tree depicting the relationship between genome sequenced S. phasma (in blue and bold) and related species in genus Sporothrix. The tree was constructed from LSU, ITS, BT and CAL gene regions using RaxML. Bootstrap support (1000 replicates) are indicated at the nodes
The unique habitat of S. phasma and its close phylogenetic relationship with various pathogenic taxa in the genus, make the data generated in this study useful for comparative genomics studies. It is intended that these will seek a better understanding of the mechanisms that underpin the ability of Sporothrix species to occupy a remarkable diversity of habitats. Opportunities should also arise to understand why some Sporothrix species have evolved to be animal and plant pathogens.
Authors: R. Chang, M.J. Wingfield, Z.W. De Beer, B.D. Wingfield, T.A. Duong*.
*Contact: tuan.duong@fabi.up.ac.za
IMA GENOME-F 12D
Metagenome sequences of a lichen species Alectoria sarmentosa (Parmeliaceae, Ascomycota).
Introduction
The advent of DNA sequencing has advanced our understanding on biodiversity research (Bik et al. 2012). The analyses of single and mutlilcous datasets in phylogenetic frameworks are frequently used for biodiversity assessment. However, these belong to a small portion of a genome and may bias the accurate species assessment. Therefore a larger portion of genome or whole genome is crucial for a more complete biodiversity assessment. More recently, genome and metagenomic data is being used for biodiversity research. For example, metagenomic data has been shown to be useful for accurate species assessment (including cryptic) especially in mixed sample or multi-lineage assemblages of organisms (‘holobionts’) such as metazoan parasites (Bass et al. 2015), viral pathogens (Bibby 2013) and other microbial communities (Louca et al. 2016).
Lichens represent iconic examples of multi-lineage assemblages of organisms. Lichens form obligate symbiotic association between fungus (exhabitant or host) and microalgae, including cyanobacteria (Hawksworth 2015). In addition to the fungal and photosynthetic partners, a wide variety of non-photosynthetic bacteria, additional fungi as enodlichenic and lichenicolous fungi are regularly found in the lichen symbiosis (Hawksworth 2015; Grube and Wedin 2016; Lumbsch and Rikkinen 2017).
The appearance of cryptic species is a common phenomenon in lichen forming fungi and fungi in general (Crespo and Lumbsch 2010; Lumbsch and Leavitt 2011; Leavitt et al. 2016; Grube et al. 2017). Cryptic biodiversity is an essential component of biodiversity that will be considerably affected by global climate change in the next decades (Bálint et al. 2011). While the molecular studies based on single and multicolus dataset have revolutionized our understanding of species delimitations in lichen-forming fungi, the application of small portion of genome may obscure the accurate cryptic biodiversity assessment.
Here we sequenced metagenome of holobiont of a lichen species Alectoria sarmentosa (Parmeliaceae, Ascomycota) to advance biodiversity research. Our data will also be helpful for accurate assessment of cryptic biodiversity. Alectoria sarmentosa is fruticose lichen belongs to the alectorioid clade of the family Parmeliaceae (Divakar et al. 2015). It is distinguished in having pendent, yellow to greenish grey thallus, pseudocyphellae fusiform, soralia rare and tuberculate (Brodo and Hawksworth 1977). The species is wide spread in Europe, Asia and North America and has been frequently used in bio-monitoring studies especially monitoring forest health (Velmala and Myllys 2011).
Sequenced strains
Alectoria sarmentosa: Norway: Telemark: Close to Notodden village, road E134 at the bus stop called Jepertjónn, 59.36 N 09.25 W, alt., 474 m, in a sparse forest of Pinus sylvestris and Picea abies, on Pinus sylvestris trunk, 20 Aug. 2015, C. G. Boluda, [identified by C. G. Boluda] (MAF-Lich 21,536).
Nucleotide sequence accession numbers
The Alectoria sarmentosa binned metagenome project has been deposited at DDBJ/ENA/GenBank under accession no. PRJNA588068.
Materials and methods
Alectoria sarmentosa was collected from a bark tree in Telemak, Norway and a partial piece of thallus was deposited at the MAF Herbarium (Complutense University of Madrid, Spain) with code MAF-Lich 21,536. The sample was identified using classical procedures as morphology and chemistry; as well as molecular technique with internal transcribed spacer (ITS) sequence of rDNA.
Genomic DNA extraction, sequencing and trimming
Total genomic DNA of the lichen holobiont was extracted from thalli using a commercial kit Quick-DNA™ Fungal/Bacterial Miniprep Kit and following the manufacturers’ instruction. DNA concentration was calculated using the Qubit dsDNA dBR assy kit (Thermo Fisher Scientific, San Diego, CA). Two paired-end libraries (300 bp and 900 bp) were built using a Illumina Tru-Seq library preparation kit. Sequencing was carried out the Unidad de Genómica (Parque Científico de Madrid, Madrid, Spain) with Illumina Miseq platform (250 bp paired-end reads). Raw sequences were downloaded from Illumina BaseSpace application and were quality trimmed and filtered using Trimmomatic-0.36 (Bolger et al. 2014) with following parameters: 4 base long sliding windows, a minimun queality value of 30 and minimun length of 30 bases. (LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36).
Genome assembly, taxonomy assignment and gene prediction
The trimmed paired-end reads were assembled using MetaSPAdes (Nurk et al. 2017) using default parameters and checking the suitability of k-mer (K21, K33, K55 and K77). In order to extract lichen-forming fungal contigs from the metagenome assembly, scaffolds of metagenome were subjected to BLASTX searches using DIAMOND (Buchfink et al. 2015) against a custom database comprising the protein sets of Archaea, Bacteria, Eukaryota, and Viruses of the NCBI nr database (downloaded in August 2018), in addition, 150 complete fungal genomes and 20 algal genomes were added from JGI. Four additional unpublished Parmeliaceae genomes generated from axenic cultures from species within Parmeliaceae [Cetraria islandica, Parmelina carporrhizans, unpublished; Evernia prunastri and Pseudevernia furfuracea (Meiser et al. 2017)] were used as reference genomes for taxonomy assignment, taking only scaffolds belonging to Parmeliaceae. The results of the DIAMOND search were then used as input for MEGAN6 (Huson et al. 2016) for taxonomic assignment (parameters: min-support = 1, min-score = 50, top-hit = 10%, no low complexity filtering).
Contigs belonging to Parmeliaceae were extracted and completeness of Alectoria sarmentosa genome was evaluated with QUAST (Gurevich et al. 2013) and BUSCO approach using Pezizomycotina dataset (Simao et al. 2015). Gene prediction was conducted with MAKER2 (Holt and Yandell 2011), using Augustus v. 3.22 (Stanke et al. 2006) and GeneMarkES (Ter-Hovhannisyan et al. 2008). In order to identify biosynthetic gene clusters, AntiSMASH pipeline v. 4 (Blin et al. 2017) was conducted on contigs with default parameters.
Phylogeny
The complete internal transcribed species region ((ITS1, 5.8S, ITS2; ~ 500 bp), commonly used for species delimitation in Parmeliaceae and the standard DNA barcode for fungi (Schoch et al. 2012) was extracted from Alectoria sarmentosa genome sequence. This was aligned with other ITS sequences of Alectoria species downloaded from NCBI (https://www.ncbi.nlm.nih.gov/). Sequences were aligned using the program MAFFT v. 7 (Katoh and Standley 2013) and the program Gblocks v. 0.91b (Talavera and Castresana 2007) was used to delimit and remove ambiguous alignment nucleotide positions. Maximum likelihood analysis (ML) was conducted with RAxML v. 8.1.11 (Stamatakis 2014), using CIPRES Science Gateway server (http://www.phylo.org/portal2/). Nodal support was evaluated with 1000 bootstrap pseudoreplicates. Phylogenetic trees were drawn using the program FigTree v. 1.4.2 (Rambaut 2009).
Results and discussion
The metassembly of holobiont resulted in 137,274 scaffolds and 161,465,382 bp of length. After taxonomic assignment, we consider all assigned contigs to Parmeliacea belong to Alectoria sarmentosa and yielded the draft genome of 46,540,876 bp (46.5 Mb) assembled into 1788 contigs. Of these, 1046 were longer than 1000 bp and contained 46,193,838 bp (46.1 Mb) of genome. The largest contig was 400,628 bp and the N50 and L50 were 92,863 bp and 140 bp, respectively (Table 2). The genome had a GC content of 40.34% and average coverage of assembly was 53.97. The assessment of genome completeness of our draft genome assembly based on 3156 single-copy orthologos BUSCO genes showed that most of the gene space was covered (96.3%). A. sarmentosa draft genome assembly contained 3041 complete and single-copy BUSCOs, 35 fragmented, 26 duplicated and 80 missing BUSCO genes out of the 3156 BUSCO genes searched (Table 2; Simao et al. 2015).
Table 2.
Assembly metric, genome completeness and number of biosynthetic gene clusters (BGC) of Alectoria sarmentosa binned metagenome
| Assembly Metrics | Genome Completeness | Biosynthetic Gene Clusters | |||
|---|---|---|---|---|---|
| Total length | 46,540,876 bp | Completeness | 96.3% | PKS Type I | 13 |
| Number of contigs | 1788 | Complete (C) | 3041 | PKS Type I-NRPS | 5 |
| Number of contigs (>1000 bp) | 1046 | Single-Copy (SC) | 3015 | PKS Type III | 1 |
| GC content | 40.34% | Duplicated (D) | 26 | PKS Type III -NRPS | 1 |
| N50 contig length | 92,863 bp | Fragmented (F) | 35 | NRPS | 2 |
| Number of genes | 9695 | Missing (M) | 80 | INDOLE | 2 |
| TERPENE | 2 | ||||
| OTHER | 4 | ||||
The gene prediction conducted by MAKER2 (Holt and Yandell 2011) yielded a total of 9695 protein-coding genes (Table 2). Our results are concordant to other recently sequenced genomes of lichen-forming fungi (e.g., Caloplaca flavorubescens: 9695 genes; Park et al. 2013a. Cladonia macilenta: 7322; Park et al. 2013b. Endocarpon pusillum: 9285; Wang et al. 2014. Ramalina intermedia: 8871; Wang et al. 2018b). The analysis of AntiSMASH (Blin et al. 2017) resulted in 61 metabolic gene clusters. The metagenome sequence of A. sarmentosa reported here is the first published genome sequence of the alectorioid clade (Table 2; Fig. 4). The alectorioid clade includes c. Seventy described species distributed in five genera viz.: Alectoria, Bryoria, Bryocaulon, Nodobryoria, and Pseudephebe (Divakar et al. 2015). Phylogenetic analysis of ITS sequence from the sequenced genome confirmed the taxonomic identity as A. sarmentosa (Fig. 4). The draft genome of A. sarmentosa generated in this study will add to the already growing genome database of lichen forming-fungi for future studies of evolutionary biology like speciation or cryptic species discovery, as well as comparative genomic or biosynthetic gene clusters studies. Furthermore, the availability of a genome sequence also provides the opportunity to develop molecular markers, for example species-specific single nucleotide polymorphism (SNPs) markers, or mating types (Alors et al. 2017), which would be important for population studies of this and other closely related taxa.
Fig. 4.

A maximum likelihood (ML) tree based on ITS sequences of Alectoria species including the genome sequence of Alectoria sarmentosa reported here. The ML tree obtained with RAxML is shown. The isolate from which the genome was sequenced is indicated in blue and bold. ML bootstrap values ≥75% are indicated at the branches. Two species of Nodobryoria were used as out-group (Divakar et al. 2015)
Authors: D. Pizarro, A. Crespo, P.K. Divakar *.
*Contact: pdivakar@farm.ucm.es
Acknowledgements
The authors of Calonectria genome project thank QianLi Liu, JieQiong Li, and QuanChao Wang from the CERC (China Eucalypt Research Centre) - FABI (Forestry and Agricultural Biotechnology Institute) Tree Protection Programme (CFTPP) for their assistance with sample preparation.
The authors of the Celoporthe dispersa and Sporothrix phasma studies thank the University of Pretoria, the Tree Protection Co-operative Programme (TPCP), the DST-NRF Centre of Excellence in Tree Health Biotechnology, the National Research Foundation (NRF), and the DST-NRF SARChI chair in Fungal Genomics.
The authors of Alectoria sarmentosa genome project thank C. G. Boluda for providing fresh materials for metagenome sequencing.
Adherence to national and international regulations
Not applicable to the specific isolates used in this manuscript. All isolates are maintained in culture collections as per government regulations and quarantine specifications.
Authors’ contributions
The manuscript is comprised of four sections, 12A, 12B, 12C, 12D. Each are authored separately and there is a specific contact person. In each case the contact person has taken the lead in writing and compiling the manuscript. The other authors have been involved in the idea behind the manuscript, editing and analysis of the data. All authors read and approved the final manuscript. 12A Draft genome sequences of five Calonectria species from Eucalyptus plantations in China. FFL, SFC, BDW, MJW, TAD. Contact: shuaifei.chen@gmail.com. 12B Draft genome sequences of Celoporthe dispersa, a causal agent of canker diseases. BDW, TAD, MS, MAF, AMK. Contact: brenda.wingfield@fabi.up.ac.za. 12C Draft genome sequence of Sporothrix phasma. RC, MJWingfield, ZWdeB, BDW, TAD. Contact: tuan.duong@fabi.up.ac.za. 12D Metagenome sequences of a lichen species Alectoria sarmentosa (Parmeliaceae, Ascomycota). DP, AC, PKD. Contact: pdivakar@farm.ucm.es
Authors’ information
No additional information provided.
Funding
The Calonectria genome project was financed by the projects of the Fundamental Research Funds for the Central Non-Profit Research Institution of CAF (Project No. CAFYBB2018QC003), the National Natural Science Foundation of China (NSFC) (project numbers: 31622019), Top Young Talents Program in National Special Support Program for High-level Talents (Ten-thousand Talents Program) (project no. W03070115) and Top Young Talents Program in Science and Technology of Guangdong Special Support Program (project no. 2017TQ04N764), the Tree Protection Co-operative Programme (TPCP), and the National Research Foundation (NRF, Grant 95875), South Africa.
The authors of the Celoporthe dispersa and Sporothrix phasma studies acknowledge the National Research Foundation (NRF) and Department of Science and Technology (DST) of South Africa for providing scholarship funding for students as well as members of the Tree Protection Cooperative Programme at the Forestry and Agricultural Biotechnology Institute for providing financial support for this project. These projects from part of the DST/NRF Centre of Excellence in Tree Health Biotechnology, and the SARChI in Fungal Genomics.
The authors of Alectoria sarmentosa genome project acknowledge the Spanish Ministerio de Ciencia e Innovacion (CGL2013–42498-P) and the Santander-Universidad Complutense de Madrid (PR75/18–21605 and G/6400100/3000).
Availability of data and materials
All data and material are available the relevant details (data banks, culture collections and herbaria) are given in the manuscript.
Ethics approval and consent to participate
Not applicable, no humans, human subjects nor data were used in this manuscript.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Afgan E, Baker D, Van den Beek M, Blankenberg D, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Eberhard C, Grüning B. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Research. 2016;44:W3–W10. doi: 10.1093/nar/gkw343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfenas R, Pereira O, Ferreira M, Jorge V, Crous P, Alfenas A. Calonectria metrosideri, a highly aggressive pathogen causing leaf blight, root rot, and wilt of Metrosideros spp. in Brazil. Forest Pathology. 2013;43:257–265. doi: 10.1111/efp.12035. [DOI] [Google Scholar]
- Alfenas R, Pereira OL, Jorge VL, Crous PW, Alfenas AC. A new species of Calonectria causing leaf blight and cutting rot of three forest tree species in Brazil. Tropical Plant Pathology. 2013;38:513–521. doi: 10.1590/S1982-56762013000600007. [DOI] [Google Scholar]
- Alors D, Dal Grande F, Cubas P, Crespo A, Schmitt I, Molina MC, Divakar PK. Panmixia and dispersal from the Mediterranean Basin to Macaronesian Islands of a macrolichen species. Scientific Reports. 2017;7:40879. doi: 10.1038/srep40879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bálint M, Domisch S, Engelhardt CHM, Haase P, Lehrian S, Sauer J, Theissinger K, Pauls SU, Nowak C. Cryptic biodiversity loss linked to global climate change. Nature Climate Change. 2011;1:313–318. doi: 10.1038/nclimate1191. [DOI] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV. SPAdes: a new genome assembly algorithm and its applications to single–cell sequencing. Journal of Computational Biology. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass D, Stentiford GD, Littlewood DTJ, Hartikainen H. Diverse applications of environmental DNA methods in parasitology. Trends in Parasitology. 2015;31:499–513. doi: 10.1016/j.pt.2015.06.013. [DOI] [PubMed] [Google Scholar]
- Bibby K. Metagenomic identification of viral pathogens. Trends in Biotechnology. 2013;31:275–279. doi: 10.1016/j.tibtech.2013.01.016. [DOI] [PubMed] [Google Scholar]
- Bik HM, Porazinska DL, Creer S, Caporaso GJ, Knight R, Thomas WK. Sequencing our way towards understanding global eukaryotic. Trends in Ecology & Evolution. 2012;27:233–243. doi: 10.1016/j.tree.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K, Wolf T, Chevrette MG, Lu X, Schwalen CJ, Kautsar SA, Suarez Duran HG, de Los Santos EL, Kim HU, Nave M, Dickschat JS. antiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Research. 2017;45:W36–W41. doi: 10.1093/nar/gkx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre–assembled contigs using SSPACE. Bioinformatics. 2011;27:578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- Boetzer M, Pirovano W. Toward almost closed genomes with GapFiller. Genome Biology. 2012;13:R56. doi: 10.1186/gb-2012-13-6-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodo IM, Hawksworth DL. Alectoria and allied genera in North America. Opera Botanica. 1977;42:1–164. [Google Scholar]
- Buchfink B, Chao X, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nature Methods. 2015;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- Chen SF, Gryzenhout M, Roux J, Xie YJ, Wingfield MJ, Zhou XD. Novel species of Celoporthe from Eucalyptus and Syzygium trees in China and Indonesia. Mycologia. 2011;103:1384–1410. doi: 10.3852/11-006. [DOI] [PubMed] [Google Scholar]
- Chen SF, Lombard L, Roux J, Xie YJ, Wingfield MJ, Zhou XD. Novel species of Calonectria associated with Eucalyptus leaf blight in Southeast China. Persoonia. 2011;26:1–12. doi: 10.3767/003158511X555236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowling RM, Richardson DM. Fynbos: South Africa's unique floral kingdom. Cape Town: Fernwood Press; 1995. [Google Scholar]
- Crespo A, Lumbsch HT. Cryptic species in lichen forming fungi. IMA Fungus. 2010;1:167–170. doi: 10.5598/imafungus.2010.01.02.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crous PW. Taxonomy and pathology of Cylindrocladium (Calonectria) and allied genera. St Paul, MN: American Phytopathological Society Press; 2002. [Google Scholar]
- D’Alessandro E, Giosa D, Huang L, Zhang J, Gao W, Brankovics B, Oliveira MME, Scordino F, Lo Passo C, Criseo G, van Diepeningen AD, Huang H, de Hoog GS, Romeo O. Draft genome sequence of the dimorphic fungus Sporothrix pallida, a nonpathogenic species belonging to Sporothrix, a genus containing agents of human and feline sporotrichosis. Genome Announcements. 2016;4:e00184–e00116. doi: 10.1128/genomeA.00184-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Beer ZW, Duong TA, Wingfield MJ. The divorce of Sporothrix and Ophiostoma: solution to a problematic relationship. Studies in Mycology. 2016;83:165–191. doi: 10.1016/j.simyco.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Beer ZW, Marincowitz S, Duong TA, Kim JJ, Rodrigues A, Wingfield MJ. Hawksworthiomyces gen. Nov. (Ophiostomatales), illustrates the urgency for a decision on how to name novel taxa known only from environmental nucleic acid sequences (ENAS) Fungal Biology. 2016;120:1323–1340. doi: 10.1016/j.funbio.2016.07.004. [DOI] [PubMed] [Google Scholar]
- Divakar PK, Crespo A, Wedin M, Leavitt SD, Hawksworth DL, Myllys L, McCune B, Randlane T, Bjerke JW, Ohmura Y, Schmitt I. Evolution of complex symbiotic relationships in a morphologically derived family of lichen-forming fungi. The New Phytologist. 2015;208:1217–1226. doi: 10.1111/nph.13553. [DOI] [PubMed] [Google Scholar]
- Duong TA, De Beer ZW, Wingfield BD, Wingfield MJ. Characterization of the mating-type genes in Leptographium procerum and Leptographium profanum. Fungal Biology. 2013;117:411–421. doi: 10.1016/j.funbio.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Gomez OM, Alvarez LC, Muñoz JF, Misas E, Gallo JE, Jimenez MDP, Arango M, McEwen JG, Hernandez O, Clay OK. Draft genome sequences of two Sporothrix schenckii clinical isolates associated with human sporotrichosis in Colombia. Genome Announcements. 2018;6:e00495–e00418. doi: 10.1128/genomeA.00495-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grube M, Gaya E, Kauserud H, Smith AM, Avery SV, Fernstad SJ, Muggia L, Martin MD, Eivindsen T, Koljalg U, Bendiksby M. The next generation fungal diversity researcher. Fungal Biology Reviews. 2017;31:124–130. doi: 10.1016/j.fbr.2017.02.001. [DOI] [Google Scholar]
- Grube M, Wedin M. Lichenized Fungi and the Evolution of Symbiotic Organization. Microbiology Spectrum. 2016;4:1. doi: 10.1128/microbiolspec.FUNK-0011-2016. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology. 2003;59:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assem-blies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawksworth DL. Lichenization: the origins of a fungal life style. In: Upreti DK, Divakar PK, Shukla V, Bajpai R, editors. Recent Advances in Lichenology. New Delhi: Springer; 2015. pp. 1–10. [Google Scholar]
- Holt C, Yandell M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics. 2011;12:491. doi: 10.1186/1471-2105-12-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Gao W, Giosa D, Criseo G, Zhang J, He T, Huang X, Sun J, Sun Y, Huang J, Zhang Y, Brankovics B, Scordino F, D'Alessandro E, van Diepeningen A, de Hoog S, Huang H, Romeo O. Whole-genome sequencing and in silico analysis of two strains of Sporothrix globosa. Genome Biology and Evolution. 2016;8:3292–3296. doi: 10.1093/gbe/evw230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Beier S, Flade I, Górska A, El-Hadidi M, Mitra S, Ruscheweyh HJ, Tappu R. MEGAN community edition – interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Computational Biology. 2016;12:e1004957. doi: 10.1371/journal.pcbi.1004957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Istace B, Friedrich A, d'Agata L, Faye S, Payen E, Beluche O, Caradec C, Davidas S, Cruaud C, Liti G, Lemainque A. Denovo assembly and population genomic survey of natural yeast isolates with the Oxford Nanopore MinION sequencer. Gigascience. 2017;6:giw018. doi: 10.1093/gigascience/giw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, Malla S. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology. 2018;36:338. doi: 10.1038/nbt.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Research. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavitt SD, Divakar PK, Crespo A, Lumbsch HT. A matter of time – understanding the limits of the power of molecular data for delimiting species boundaries. Herzogia. 2016;29:479–492. doi: 10.13158/heia.29.2.2016.479. [DOI] [Google Scholar]
- Li JQ, Wingfield MJ, Liu QL, Barnes I, Roux J, Lombard L, Crous PW, Chen S. Calonectria species isolated from Eucalyptus plantations and nurseries in South China. IMA Fungus. 2017;8:259–294. doi: 10.5598/imafungus.2017.08.02.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QL, Chen SF. Two novel species of Calonectria isolated from soil in a natural forest in China. MycoKeys. 2017;26:25–60. doi: 10.3897/mycokeys.26.14688. [DOI] [Google Scholar]
- Lombard L, Chen SF, Mou X, Zhou XD, Crous PW, Wingfield MJ. New species, hyper-diversity and potential importance of Calonectria spp. from Eucalyptus in South China. Studies in Mycology. 2015;80:151–188. doi: 10.1016/j.simyco.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard L, Crous PW, Wingfield BD, Wingfield MJ. Multigene phylogeny and mating tests reveal three cryptic species related to Calonectria pauciramosa. Studies in Mycology. 2010;66:15–30. doi: 10.3114/sim.2010.66.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard L, Crous PW, Wingfield BD, Wingfield MJ. Phylogeny and systematics of the genus Calonectria. Studies in Mycology. 2010;66:31–69. doi: 10.3114/sim.2010.66.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard L, Crous PW, Wingfield BD, Wingfield MJ. Species concepts in Calonectria (Cylindrocladium) Studies in Mycology. 2010;66:1–14. doi: 10.3114/sim.2010.66.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard L, Wingfield MJ, Alfenas AC, Crous PW. The forgotten Calonectria collection: pouring old wine into new bags. Studies in Mycology. 2016;85:159–198. doi: 10.1016/j.simyco.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes-Bezerra LM, Nino-Vega G, Mora-Montes HM, Zhang Y, Rodrigues AM, de Camargo ZP, de Hoog S. Sporotrichosis between 1898 and 2017: the evolution of knowledge on a changeable disease and on emerging etiological agents. Medical Mycology. 2018;56:S126–S143. doi: 10.1093/mmy/myx103. [DOI] [PubMed] [Google Scholar]
- Louca S, Jacques SMS, Pires APF, Leal JS, Srivastava DS, Parfrey LW, Farjalla VF, Doebeli M. High taxonomic variability despite stable functional structure across microbial communities. Nature Ecology Evolution. 2016;1:0015. doi: 10.1038/s41559-016-0015. [DOI] [PubMed] [Google Scholar]
- Lumbsch HT, Leavitt SD. Goodbye morphology? A paradigm shift in the delimitation of species in lichenized fungi. Fungal Diversity. 2011;50:59–72. doi: 10.1007/s13225-011-0123-z. [DOI] [Google Scholar]
- Lumbsch HT, Rikkinen J. Chapter 4 evolution of lichens. In: Dighton J, White JF, editors. The Fungal Community CRC Press Taylor & Francis Group, Boca Raton, FL 33487-2742. 2017. pp. 53–62. [Google Scholar]
- Meiser A, Otte J, Schmitt I, Grande FD. Sequencing genomes from mixed DNA samples—evaluating the metagenome skimming approach in lichenized fungi. Scientific Reports. 2017;7:14881. doi: 10.1038/s41598-017-14576-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES science gateway for inference of large phylogenetic trees. New Orleans, LA: Institute of Electrical and Electronics Engineers; 2010. pp. 1–8. [Google Scholar]
- Nakabonge G, Roux J, Gryzenhout M, Wingfield MJ. Distribution of Chrysoporthe canker pathogens on Eucalyptus and Syzygium spp. in eastern and southern Africa. Plant Disease Reporter. 2006;90:734–740. doi: 10.1094/PD-90-0734. [DOI] [PubMed] [Google Scholar]
- Ngubane N, Dreyer L, Oberlander K, Roets F. Two new Sporothrix species from Protea flower heads in south African grassland and savanna. Antonie Van Leeuwenhoek. 2018;111:965–979. doi: 10.1007/s10482-017-0995-3. [DOI] [PubMed] [Google Scholar]
- Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. MetaSPAdes: anew versatile metagenomic assembler. Genome Research. 2017;27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-Y, Choi J, Kim JA, Jeong M-H, Kim S, Lee Y-H, Hur J-S. Draft genome sequence of Cladonia macilenta KoLRI003786, a lichen-forming fungus producing biruloquinone. Genome Announcements. 2013;1:e00695–e00613. doi: 10.1128/genomeA.00695-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-Y, Choi J, Kim JA, Yu N-H, Kim S, Kondratyuk SY, Lee Y-H, Hur J-S. Draft genome sequence of lichen-forming fungus Caloplaca flavorubescens strain KoLRI002931. Genome Announcements. 2013;1:e00678–e00613. doi: 10.1128/genomeA.00678-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham NQ, Barnes I, Chen S, Liu F, Dang QN, Pham TQ, Lombard L, Crous PW, Wingfield MJ. Ten new species of Calonectria from Indonesia and Vietnam. Mycologia. 2019;111:78–102. doi: 10.1080/00275514.2018.1522179. [DOI] [PubMed] [Google Scholar]
- Rambaut A. FigTree 1.4.2. 2009. [Google Scholar]
- Roets F, Crous PW, Wingfield MJ, Dreyer LL. Mite-mediated hyperphoretic dispersal of Ophiostoma spp. from the infructescences of south African Protea spp. Environmental Entomology. 2009;38:143–152. doi: 10.1603/022.038.0118. [DOI] [PubMed] [Google Scholar]
- Roets F, De Beer ZW, Dreyer LL, Zipfel R, Crous PW, Wingfield MJ. Multi-gene phylogeny for Ophiostoma spp. reveals two new species from Protea infructescences. Studies in Mycology. 2006;55:199–212. doi: 10.3114/sim.55.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roets F, De Beer ZW, Wingfield MJ, Crous PW, Dreyer LL. Ophiostoma gemellus and Sporothrix variecibatus from mites infesting Protea infructescences in South Africa. Mycologia. 2008;100:496–510. doi: 10.3852/07-181R. [DOI] [PubMed] [Google Scholar]
- Roets F, Wingfield BD, De Beer ZW, Wingfield MJ, Dreyer LL. Two new Ophiostoma species from Protea caffra in Zambia. Persoonia. 2010;24:18–28. doi: 10.3767/003158510X490392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roets F, Wingfield MJ, Crous PW, Dreyer LL. Discovery of fungus-mite mutualism in a unique niche. Environmental Entomology. 2007;36:1226–1237. doi: 10.1093/ee/36.5.1226. [DOI] [PubMed] [Google Scholar]
- Roets F, Wingfield MJ, Crous PW, Dreyer LL. Fungal radiation in the cape floristic region: an analysis based on Gondwanamyces and Ophiostoma. Mol. Phylogenetics and Evolution. 2009;51:111–119. doi: 10.1016/j.ympev.2008.05.041. [DOI] [PubMed] [Google Scholar]
- Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. PNAS. 2012;109:6241–6246. doi: 10.1073/pnas.1117018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert KA, De Beer ZW, Wingfield MJ. The Ophiostomatoid Fungi: expanding Frontiers. 2013. [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- Soares TPF, Ferreira MA, Mafia RG, Oliveira LSS, Hodges CS, Alfenas AC. Canker disease caused by Chrysoporthe doradensis and C. cubensis on Eucalyptus sp. and Tibouchina spp. in Brazil. Tropical Plant Pathology. 2018;43:314–322. doi: 10.1007/s40858-018-0238-9. [DOI] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research. 2006;34:W435–W439. doi: 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Morgenstern B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Research. 2005;33:W465–W467. doi: 10.1093/nar/gki458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systematic Biology. 2007;56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira M, Almeida L, Kubitschek-Barreira P, Alves FL, Kioshima ES, Abadio AK, Fernandes L, Derengowski LS, Ferreira KS, Souza RC, Ruiz JC, Curty de Andrade N, C Paes H, Nicola A, Albuquerque P, Gerber A, Martins V, Df Peconick L, Viggiano Neto A, Felipe MS. Comparative genomics of the major fungal agents of human and animal Sporotrichosis: Sporothrix schenckii and Sporothrix brasiliensis. BMC Genomics. 2014;15:943. doi: 10.1186/1471-2164-15-943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Research. 2008;18:1979–1990. doi: 10.1101/gr.081612.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theron-De Bruin N, Dreyer LL, Ueckermann EA, Wingfield MJ, Roets F. Birds mediate a fungus-mite mutualism. Microbial Ecology. 2018;75:863–874. doi: 10.1007/s00248-017-1093-9. [DOI] [PubMed] [Google Scholar]
- Velmala S, Myllys L. Alectora. In: Thell A, Moberg R, editors. Parmeliaceae. Nordic lichen flora. 2011. pp. 14–16. [Google Scholar]
- Wang W, Liu Q, Li G, Liu F, Chen S. Phylogeny and pathogenicity of Celoporthe species from plantation Eucalyptus in southern China. Plant Disease. 2018;102:1915–1927. doi: 10.1094/PDIS-12-17-2002-RE. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yuan X, Chen L, Wang X, Li C. Draft genome sequence of the lichen-forming fungus Ramalina intermedia strain YAF0013. Genome Announcements. 2018;6:e00478–e00418. doi: 10.1128/genomeA.00478-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y-Y, Liu B, Zhang X-Y, Zhou Q-M, Zhang T, Li H, Yu Y-F, Zhang X-L, Hao X-Y, Wang M, Wang L, Wei J-C. Genome characteristics reveal the impact of lichenization on lichen-forming fungus Endocarpon pusillum Hedwig (Verrucariales, Ascomycota) BMC Genomics. 2014;15:34. doi: 10.1186/1471-2164-15-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield BD, Ades PK, Al-Naemi FA, Beirn LA, Bihon W, Crouch JA, De Beer ZW, De Vos L, Duong TA, Fields CJ, Fourie G. IMA genome-F 4: draft genome sequences of Chrysoporthe austroafricana, Diplodia scrobiculata, Fusarium nygamai, Leptographium lundbergii, Limonomyces culmigenus, Stagonosporopsis tanaceti, and Thielaviopsis punctulata. IMA Fungus. 2015;6:233–248. doi: 10.5598/imafungus.2015.06.01.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield BD, Barnes I, De Beer ZW, De Vos L, Duong TA, Kanzi AM, Naidoo K, Nguyen HD, Santana QC, Sayari M, Seifert KA. IMA genome-F 5: draft genome sequences of Ceratocystis eucalypticola, Chrysoporthe cubensis, C. deuterocubensis, Davidsoniella virescens, Fusarium temperatum, Graphilbum fragrans, Penicillium nordicum, and Thielaviopsis musarum. IMA Fungus. 2015;6:493–506. doi: 10.5598/imafungus.2015.06.02.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JJ, Qin SY, Hao YY, Ren J, Tan P, Ali HB, Kevin DH, Wang Y. A new species of Calonectria causing leaf disease of water lily in China. Mycotaxon. 2012;122:177–185. doi: 10.5248/122.177. [DOI] [Google Scholar]
- Ye X, Zhong Z, Liu H, Lin L, Guo M, Guo W, Wang Z, Zhang Q, Feng L, Lu G, Zhang F. Whole genome and transcriptome analysis reveal adaptive strategies and pathogenesis of Calonectria pseudoreteaudii to Eucalyptus. BMC Genomics. 2018;19:358. doi: 10.1186/s12864-018-4739-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data and material are available the relevant details (data banks, culture collections and herbaria) are given in the manuscript.