

Abstract
Prefrontal cortex (PFC) is highly influenced by the inputs from ventral tegmental area (VTA); however, how the projection from VTA impacts PFC neurons and how the synaptically released dopamine affects PFC activity are largely unclear. Using optogenetics and electrophysiological approaches, we examined the impact of VTA stimulation on PFC principal neurons and parvalbumin-positive (PV+) interneurons and the modulatory role of dopamine. We found that the brief activation of the VTA–PFC circuit immediately induced action potential firing, which was mediated by glutamatergic transmission. However, strong stimulation of VTA gradually induced a marked and prolonged enhancement of the excitability of PFC PV+ interneurons and a modest and short-lived enhancement of the excitability of PFC principal neurons. Blocking dopamine receptors (DARs) shortened the VTA excitation of PFC PV+ interneurons and prolonged the VTA excitation of PFC principal neurons. Blocking GABAA receptors induced a similar effect as DAR antagonists in PFC principal neurons, suggesting that the dopaminergic effect is through influencing the inhibitory transmission system. These results have revealed a role of dopamine in regulating the temporal dynamics of excitation/inhibition balance in VTA–PFC circuit, which provides insights into the functional consequence of activating dopamine system in the mesocortical system.
Keywords: dopamine, optogenetics, parvalbumin, prefrontal cortex, ventral tegmental area
Introduction
The prefrontal cortex (PFC) is a central brain region controlling high-level executive function and goal-directed behaviors (Goldman-Rakic 1994; Miller 2000). In PFC, the balance between excitatory and inhibitory neurotransmission (E/I balance) is essential for the proper execution of PFC-dependent behaviors, and its disruption is involved in a range of psychiatric diseases (Carmichael and Price 1996; Drevets et al. 1997; Mizoguchi et al. 2004; Hoover and Vertes 2007; Foss-Feig et al. 2017; Ferguson and Gao 2018). A major subtype of PFC GABAergic interneurons, parvalbumin-positive (PV+) interneurons, specifically possess synapses innervating the perisomatic region of target cells like PFC pyramidal neurons and form dense gap junctional connectivity with one another (Kawaguchi and Kubota 1993, 1998). This allows PV+ interneurons to control fast feedforward and feedback inhibition, which plays a key role in maintaining the proper E/I balance (El-Boustani and Sur 2014; Hu et al. 2014).
PFC receives many inputs from other brain areas including the ventral tegmental area (VTA). The VTA to PFC circuit plays an important role in addiction, depression, and other mood disorders (Lammel et al. 2012; Chaudhury et al. 2013; Volman et al. 2013; Hauser et al. 2017). Particularly, dopaminergic innervations from VTA to PFC strongly influence the PFC regulation of cognition and emotion (Sawaguchi and Goldman-Rakic 1991; Williams and Goldman-Rakic 1995; Wang et al. 2002).
VTA area contains dopaminergic (~60%), GABAergic (~25%), and glutamatergic neurons (~15%) (Yamaguchi et al. 2007, 2011; Walsh and Han 2014; Yoo et al. 2016). In addition, VTA dopamine neurons could corelease dopamine and other transmitters (Morales and Margolis 2017). Thus the projection from VTA to PFC is expected to elicit complex responses due to the release of multiple transmitters. However, it remains unclear how the E/I balance in PFC is influenced by activing VTA inputs and how the synaptically released dopamine modulates the responses. Here we have used the optogenetic and electrophysiological approaches to reveal the dynamic impact of VTA activation on the excitability of PFC pyramidal projection neurons and PFC PV+ interneurons, as well as the role of dopamine.
Materials and Methods
Animals and Neuron Identification
All experiments were carried out with the approval of the State University of New York at Buffalo Animal Care Committee. Adolescent male rats (3-week-old) were used for virus injection surgeries. Electrophysiology recording experiments were performed 4 weeks later. Neurons of PFC were chosen for recordings. Pyramidal neurons were identified by their large triangular soma and a clear apical dendrite, whereas PV+ interneurons were identified by a small round or oval cell body and the lack of a visible apical dendrite under infrared video microscopy (Kawaguchi and Kubota 1993, 1997, 1998). In addition, when action potentials were elicited by injecting a depolarizing current pulse, PV+ interneurons had fast spikes (base duration = ~ 2 ms) at high frequencies with little frequency adaptation (Kawaguchi and Kubota 1997; Zhong and Yan 2011), while pyramidal neurons had slow spikes (base duration = ~ 4.5 ms) at low frequencies with obvious frequency adaptation (Yang et al. 1996; Zhong and Yan 2011).
Viral Vectors and Stereotaxic Injections
The viral vector, AAV9.CAG.hChR2(H134R)-mCherry.WPRE.SV40, was purchased from Penn Vector Core (Univ. Penn). Rats were anesthetized with ketamine (75 mg/kg) and Xylazine (5 mg/kg). After achieving deep anesthesia, we put animals on a stereotaxic apparatus (KoPF) and bilaterally injected the virus (1 μL) into the VTA region (AP = −5.00 mm, ML = + − 0.7 mm, DV = −7.5 mm) (Paxinos and Watson 1997) using a Hamilton syringe (31 gauge). Injection speed was controlled using a microinjection pump (KD Scientific) (200 nl/min). The injection needle was left in place for 5 min following injection.
Optogenetics
Light stimulation of ChR2-expressing terminals in PFC slices was carried out using a UHP-Microscope-LED-460 system (Prizmatix) that provides > 1 W collimated blue light (460 nm peak, 27 nm spectrum half width, 85% peak power at 450 nm). The blue light was transmitted via the microscope objective (Olympus LUMPlanFI/IR, 40X0.80w) and triggered on and off with TTL pulses programmed by pClamp (Molecular Devices) data acquisition software.
Slice Electrophysiology
Rats were anesthetized with isoflurane and decapitated quickly. Coronal slices (350 μm) containing the PFC area were prepared on a vibratome (Leica VT1000s) in the ice-cold sucrose solution (in mM = 234 sucrose, 4 MgSO4, 2.5 KCl, 1 NaH2PO4, 0.1 CaCl2, 15 HEPES, 11 glucose, pH 7.35) and then incubated for 1–4 h at room temperature (22–24 °C) in oxygenated artificial cerebrospinal fluid (ACSF) (in mM = 130 NaCl, 26 NaHCO3, 3 KCl, 5 MgCl2, 1.25 NaH2PO4, 1 CaCl2, 10 Glucose, pH 7.4, and 300 mOsm) before recording. Slice was positioned in a perfusion chamber attached to the fixed stage of an upright microscope (Olympus) and submerged in continuously flowing oxygenated ACSF. Whole-cell patch-clamp experiments were performed with a Multi-Clamp 700A amplifier and Digidata 1322A data acquisition system (Molecular Devices). All recording experiments were conducted at room temperature (22 °C). Neurons were visualized with the infrared differential interference contrast video microscopy. In prelimbic cortex (PrL), layer 5 pyramidal neurons with large triangle-shaped somas and prominent apical dendrites, as well as layer 3–5 PV+ interneurons with small oval-shaped somas and fast-spiking properties, were selected for recordings. Recording electrodes were pulled from borosilicate glass capillaries (1.5/0.86 mm OD/ID) with a micropipette puller (Sutter Instrument Co., model P-97). The resistance of patch electrode was ~ 4.0 MΩ.
To record the spontaneous action potential (sAP), we used a modified ACSF that slightly elevated basal neuronal activity (in mM = 130 NaCl, 26 NaHCO3, 3.5KCl, 0.5 MgCl2, 1.25 NaH2PO4, 1.5 CaCl2, 10 Glucose, pH 7.4, and 300 mOsm), which more closely mimics the ionic composition of brain interstitial fluid in situ (1.0–1.2 mM Ca2+, 1 mM Mg2+, 3.5 mM K+) (Sanchez-Vives and McCormick 2000; Maffei et al. 2004). sAP was recorded in the whole-cell current-clamp mode with the internal solution containing (in mM) 20 KCl, 100 K–gluconate, 10 HEPES, 4 Mg–ATP, 0.5 Na2GTP, and 10 Na–phosphocreatine (Maffei and Turigiano 2008). A small amount of DC current (<50pA) was injected to adjust the interspike potential at −55 to −65 mV (Zhong and Yan 2016). The baseline sAP frequencies were 0.5–1.5 Hz for PFC pyramidal neurons and 0.5–3 Hz for PFC parvalbumin-positive interneurons.
To demonstrate the effect of stimulating VTA inputs on PFC neuronal activity, the sAP frequencies before and after light stimulation at different time points were calculated. Considering the variations on baseline sAP frequencies between cells, mean sAP frequency of each cell before light stimulation was normalized in the group comparison of percent changes induced by VTA stimulation.
Data Analysis
Data were acquired using the software pClamp 9.2 (Molecular Devices). Data sampling frequency was 10 kHz, and filtering frequency was 1 kHz. Data analyses were performed with the Clampfit software (Molecular Devices), Mini Analysis Program (Synaptosoft), and KaleidaGraph software. All data were expressed as the mean ± SEM. We performed statistical analyses using Prism (GraphPad) to attest the normality of the data sets. All data passed Shapiro–Wilk normality test (w > 0.85, P > 0.05), so ANOVA or student t-test (Prism, GraphPad) was used to determine the statistical significance between groups.
Results
Brief Optogenetic Stimulation of VTA Inputs Induces the Immediate Action Potential Firing in PFC Neurons, Which Is Caused by Glutamatergic Transmission
VTA is composed of heterogeneous neuronal types and sends dopaminergic, GABAergic, and glutamatergic projections to other brain areas (Morales and Margolis 2017). To find out the impact of VTA activation on PFC, we injected rats (3-week-old) with the viral vector AAV9.CAG.hChR2(H134R)-mCherry.WPRE.SV40 to VTA (Fig. 1A), which drives the expression of channelrhodopsin-2 (ChR2) in all types of VTA neurons. A representative example about the ChR2 accumulation in synaptic terminals projecting to PFC is shown in Figure 1B.
Figure 1.
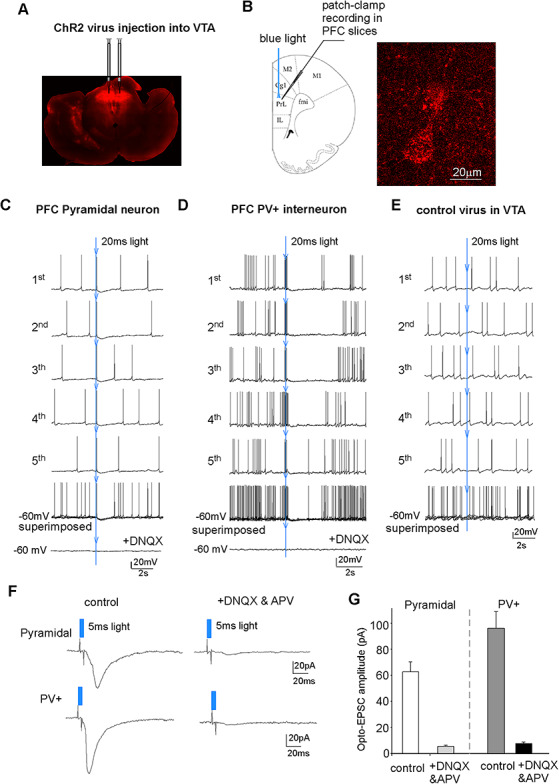
Brief optogenetic stimulation of VTA inputs induced action potentials in PFC neurons, which was mediated by glutamatergic transmission. (A) A confocal image showing the viral expression of ChR2(H134R)-mCherry in VTA at 4 weeks after the injection. (B) A diagram showing the recording condition and a confocal image showing the ChR2-expressing terminals innervating a PFC pyramidal neuron. (C, D, E) Representative sAP recordings in a PFC pyramidal neuron (C) and a PFC PV+ interneuron (D) from animals with ChR2 virus injected into VTA or a PFC pyramidal neuron from an animal with control (no ChR2) virus injected into VTA (E). A 20-ms blue light pulse was delivered at the time indicated by the blue arrow. Individual episodic sAP traces and the superimposed traces were shown. In the end, the AMPAR antagonist DNQX was added, which blocked sAP and the light-induced AP (C, D). (F) Representative traces of excitatory postsynaptic currents (EPSC) evoked by the blue light pulse (5 ms) stimulation in a PFC pyramidal neuron and a PFC PV+ interneuron in the absence (control) or presence of DNQX and APV. (G) Bar graphs showing the amplitude of light-evoked EPSC in PFC pyramidal neurons and PV+ interneurons in the absence (control) or presence of DNQX and APV (control, pyramidal = 62.8 ± 7.7 pA, n = 9; parvalbumin = 96.4 ± 12.9 pA, n = 7).
To determine the impact of optogenetic stimulation of VTA inputs on PFC neuronal activity, we performed whole-cell current-clamp recording to measure the synaptic-driven sAP. To examine the PFC response to VTA stimulation, we first applied single 20-ms light pulses, which were delivered at the fourth second during the episodic 10-second recording. As shown in Figure 1C,D, the 20-ms blue light pulse immediately triggered an action potential in PFC pyramidal neurons (n = 10 cells/3 rats), and a cluster of action potentials in PFC PV+ interneurons (n = 7 cells/4 rats), which occurred almost at the same time point when the light pulse was applied. Application of the AMPAR antagonist DNQX (20 μM) blocked the light-induced AP in both types of neurons, suggesting that the immediate AP firing in PFC neurons to VTA activation is mediated by the direct stimulation of glutamatergic terminals from VTA. Following the immediate triggering of action potentials in PFC neurons by VTA stimulation, there was a short period (~2 s) of membrane hyperpolarization and sAP silencing. This brief inhibition on spontaneous spike could be due to the direct stimulation of GABAergic terminals from VTA or light-induced excitation of PFC GABAergic interneurons (Kabanova et al. 2015).
To confirm that the light-induced alteration of PFC activity is mediated by VTA stimulation, we performed experiments by injecting the control virus without ChR2 (AAV5-hSyn-EGFP) into VTA and delivering blue light pulses in PFC. As shown in Figure 1E, the 20-ms blue light pulses failed to immediately trigger APs and induce the later inhibition of sAP firing in PFC pyramidal neurons (n = 6 cells/2 rats).
To provide a more direct evidence on the glutamatergic response induced by VTA activation, we performed whole-cell voltage-clamp recording to measure synaptic currents in PFC neurons. As shown in Figure 1F,G, a short blue light pulse (5 ms) induced an inward current, which was blocked by application of DNQX (an AMPAR antagonist) and APV (an NMDAR antagonist). It further indicates that brief stimulation of VTA terminals in PFC can evoke glutamatergic excitatory synaptic currents, triggering AP firing.
Strong Optogenetic Stimulation of VTA Inputs Induces a Prolonged and Large Increase of PFC PV+ Interneuron Activity, Which is Turned into a Short-Lived Effect by Blocking Dopamine Receptors
Since VTA dopaminergic neurons often have tonic firing (~5 Hz, Ellwood et al. 2017), we applied a 60-pulse train of light (20 ms, 3 Hz) to induce the strong activation of PFC-projecting terminals from VTA and examined the impact on PFC neuron activity in a longer time frame (at least 30 min). We first recorded PV+ fast-spiking interneurons. As shown in Figure 2A–C, the blue light train (60 pulses) stimulation induced a fast decrease of sAP firing within 30 s, followed by a large and long-lasting increase of sAP frequency in PV+ interneurons. The light-induced increase of sAP frequency in PFC PV+ interneurons took 6–10 min to reach the maximal level. At the 10th minute, the frequency of sAP was increased by ~ 450% (Fig. 2D). The huge increase of sAP frequency lasted for a long time. At the 30th minute after light stimulation, the frequency of sAP was still increased by ~310% (Fig. 2D). No significant change was observed on sAP frequency without light stimulation (Fig. 2A,B, n = 6 cells/3 rats).
Figure 2.
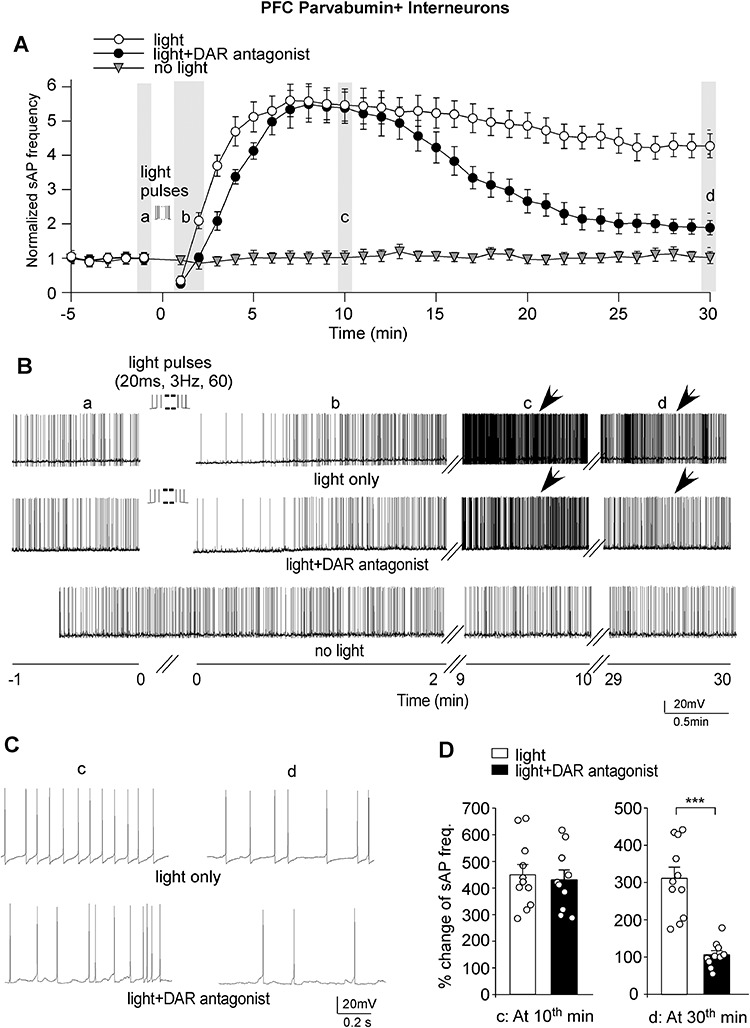
Strong optogenetic stimulation of VTA inputs induced a prolonged and large increase of PFC PV+ interneuron excitability, which was switched to a transient effect by blocking DARs. (A) Plots showing the averaged (mean ± SEM) sAP frequency before and after the 60-pulse train of light (20 ms, 3 Hz) stimulation in parvalbumin interneurons in the absence or presence of the DAR antagonists (SCH23390, sulpiride, and L-745870). A control PV+ interneuron without light stimulation was also shown. The mean sAP frequency at the baseline (before light stimulation) of each group was normalized. (B) Representative traces of sAP in parvalbumin interneurons at different time frames in different conditions. (C) The expanded view of sAP traces at the time points (indicated by arrows in B). (D) Bar graphs showing the percent changes of sAP frequencies in PFC PV+ interneurons at the 10th or 30th minute after light (20 ms, 3 Hz, 60 pulses) stimulation in the absence or presence of DAR antagonists (10th minute, light = 449.8% ± 38.2%, n = 11 cells/5 rats; light+DAR antagonist = 429.3% ± 37.5%, n = 10 cells/5 rats; 30th minute, light = 311.5% ± 29.8%, light+DAR antagonist = 105.7% ± 10.9%). ***P < 0.001, t-test.
To investigate the role of dopamine in the PFC response to VTA stimulation, we applied antagonists to block dopamine receptors (DARs), including the D1R antagonist SCH23390 (10 μM), D2/3R antagonist sulpiride (10 μM), and D4R antagonist L-745870 (5 μM). Slices were exposed to these antagonists for at least 5 min before sAP was recorded. As shown in Figure 2A–C, when DA receptors were blocked, the blue light train (60 pulses) stimulation still induced a short decrease of sAP firing followed by a large increase of sAP frequency (~430%) in PFC PV+ interneurons at the early stage (Fig. 2D). However, the duration of the increasing effect was significantly shortened. At the 30th minute after light stimulation, the increase in sAP frequency was dropped to ~110% (Fig. 2D), which was significantly (P < 0.001, t-test) different from the light-induced effect on sAP in the absence of DAR antagonists.
To quantify the temporal dynamics of the effect of VTA stimulation on PFC interneuron activity, we fitted the curves (Fig. 2A) with exponential equations to obtain decay time constants. We found that blocking DA receptors significantly shortened the increasing effect of VTA stimulation on sAP frequency in PV+ interneurons (decay time constants, light = 73.2 min; light+DAR antagonist = 16.7 min). These data suggest that the strong stimulation of VTA inputs gradually induces a large increase of PFC PV+ interneuron activity, which is sustained by DAR activation.
Strong Optogenetic Stimulation of VTA Inputs Induces a Short and Modest Increase of PFC Pyramidal Neuron Activity, Which Is Turned into a Long-Lasting Effect by Blocking DARs
Next, we examined the impact of the strong activation of terminals from VTA on PFC pyramidal neurons. As shown in Figure 3A–C, in response to the 60-pulse train of light (20 ms, 3 Hz) stimulation, sAP frequency in PFC pyramidal neurons was quickly decreased within 1 min and then started to increase and reached the maximal level at ~5 min. At the 10th minute, the frequency of sAP was increased by ~ 130% (Fig. 3D). However, the increasing effect did not last long and returned to the control level within 25 min. At the 30th minute after light stimulation, the frequency of sAP was even slightly decreased (Fig. 3D). The sAP frequency in PFC pyramidal neurons did not show significant changes without light stimulation (Fig. 3A,B, n = 7 cells/3 rats).
Figure 3.
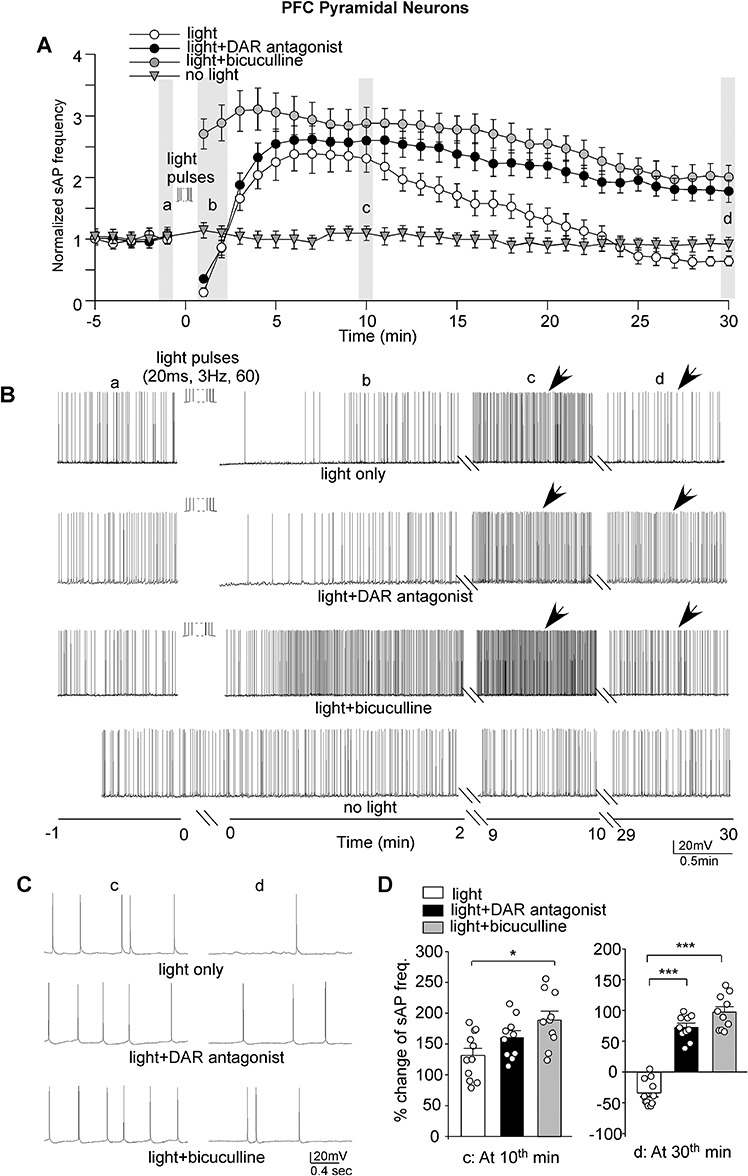
Strong optogenetic stimulation of VTA inputs induced a transient and modest increase of PFC pyramidal neuron excitability, which was turned into a long-lasting effect by blocking DARs or GABAA receptors. (A) Plots showing the averaged (mean ± SEM) sAP frequency before and after the 60-pulse train of light (20 ms, 3 Hz) stimulation in PFC pyramidal neurons in the absence or presence of the DAR antagonists (SCH23390, sulpiride, and L-745870) or the GABAA receptor antagonist bicuculline. A control neuron without light stimulation was also shown. The mean sAP frequency at the baseline (before light stimulation) of each group was normalized. (B) Representative traces of sAP in PFC pyramidal neurons at different time frames in different conditions. (C) The expanded view of sAP traces at the time points (indicated by arrows in B). (D) Bar graphs showing the percent changes of sAP frequencies in PFC pyramidal neurons at the 10th or 30th minute after light (20 ms, 3 Hz, 60 pulses) stimulation in the absence or presence of DAR antagonists or GABAA receptor antagonist bicuculline (10th minute, light = 131.5% ± 11.5%, n = 11 cells/4 rats; light+DAR antagonist = 159.8% ± 10.8%, n = 10 cells/4 rats; light+bicuculline = 188.9% ± 14.8%, n = 10 cells/4 rats); 30th minute, light = −33.6% ± 6.4%; light+DAR antagonist = 72.1% ± 6.9%; light+bicuculline = 97.2% ± 8.8%). ***P < 0.001, ANOVA.
To find out the role of dopamine, we applied DAR antagonists SCH23390 (10 μM), sulpiride (10 μM), and L-745870 (5 μM). As shown in Figure 3A–C, when DA receptors were blocked, the blue light train (60 pulses) stimulation still induced a short decrease of sAP firing followed by a modest increase of sAP frequency (~160%) in PFC pyramidal neurons at the early stage (Fig. 3D). However, the duration of the increasing effect was significantly prolonged. At the 30th minute after light stimulation, the frequency of sAP was still increased by ~72% (Fig. 3D), which was significantly (P < 0.001, one-way ANOVA) different from the light-induced effect on sAP in the absence of DAR antagonists. Quantification of the temporal dynamics of the effect of VTA stimulation on PFC pyramidal neuron activity indicated that blocking DA receptors significantly prolonged the increasing effect of VTA stimulation on sAP frequency (decay time constants, light = 17.9 min; light+D1R antagonist = 54.5 min). These data suggest that the strong stimulation of VTA inputs gradually induces a modest increase of PFC pyramidal neuron activity, which is terminated by DAR activation.
The Modulatory Effect of Dopamine on PFC Pyramidal Neuron Activity Involves GABAergic Transmission
How does the optogenetic stimulation of dopaminergic terminals affect the duration of the changes in PFC pyramidal neuron activity? One possibility is via the modification of GABAergic transmission. To test this, we applied the GABAAR antagonist bicuculline (5 μM). Bicuculline application increased the sAP frequency and maintained it at a higher but stable level (125.7% ± 15.7% increase at the 7th minute; 74.7% ± 18.5% increase at the 10th minute, n = 8). Brain slices were perfused with bicuculline-containing ACSF for > 10 min to get a stable baseline sAP before light stimulation. As shown in Figure 3A–C, when synaptic inhibition was blocked, the blue light train (60 pulses) no longer induced the fast decrease of sAP firing but instead induced a fast increase of sAP frequency (Fig. 3D). Similar to what was observed when DARs were blocked, the light-induced increasing effect on sAP in the presence of bicuculline sustained for a long time. At the 30th minute, the frequency of sAP was still increased by ~ 97% (Fig. 3D, decay time constant, light+bicuculline = 58.1 min), which was significantly (P < 0.001, one-way ANOVA) different from the effect in the absence of the GABAAR antagonist. It suggests that dopamine may terminate the increasing effect on pyramidal neuron excitability by activating GABAergic inhibition.
Discussion
VTA–PFC circuit function is crucial for mood regulation (Walsh and Han 2014). Stimulation of VTA neurons would simultaneously activate the glutamatergic, GABAergic, and dopaminergic pathways (Kabanova et al. 2015; Yoo et al. 2016; Ellwood et al. 2017). Here we have found that a brief optogenetic stimulation of VTA terminals induces the immediate action potential firing and the excitatory synaptic currents in PFC neurons, which is mediated by glutamatergic synaptic transmission. Consistently, it has been found that once the dopaminergic projection from VTA to PFC is activated, it elicits a transient (lasting for a few milliseconds) excitatory effect on PFC pyramidal neurons and interneurons (Kabanova et al. 2015; Ellwood et al. 2017), which is caused by the coreleased glutamate from VTA dopaminergic neurons (Ellwood et al. 2017). More importantly, in this study, we have revealed the differential responses of PFC PV+ interneurons and pyramidal neurons to a strong optogenetic stimulation of VTA terminals and the role of dopamine in modulating these delayed responses.
PFC interneurons act as an important brake on excitatory signaling and control the output of pyramidal neurons (Ferguson and Gao 2018). A single fast-spiking PV+ interneuron contacts nearly every local pyramidal neuron (Packer and Yuste 2011), which allows for potent feedforward and feedback inhibition (Hu et al. 2014). In this study, we have found that PV+ interneuron excitability is markedly increased by optogenetic stimulation of VTA terminals, and this huge effect (4-fold increase) lasts for a long time (at least 30 min). In contrast, the same optogenetic stimulation of VTA terminals only induces a modest (1.5-fold increase) and transient (<25 min) enhancement of PFC pyramidal neuron excitability. It suggests that VTA activation will alter E/I balance in PFC, causing prolonged strengthening of the PFC inhibitory circuit.
PFC functions are strongly influenced by dopamine signaling (Sawaguchi and Goldman-Rakic 1991; Williams and Goldman-Rakic 1995; Lavin et al. 2005). Both PFC pyramidal neurons and PV+ interneurons are regulated by exogenously applied dopamine or DAR agonists (Zhou and Hablitz 1999; Lavin et al. 2005; Zhong and Yan 2016). In this study, we have found that blocking DARs do not affect the early enhancement of PFC neuron excitability induced by the strong stimulation of VTA terminals, but shortens the effect duration in PV+ interneurons and prolongs the effect duration in pyramidal neurons. It suggests that synaptically released dopamine controls the timing of PFC activity changes in response to VTA stimulation, resulting in the long-term potentiation of PFC PV+ interneuron excitability and short-term potentiation of PFC pyramidal neuron excitability.
Why does dopamine have opposing effects in PFC pyramidal neurons versus PV+ interneurons? We have found that the GABAAR antagonist elicits a similar effect as DAR antagonists in PFC pyramidal neurons—preventing the termination of the light-induced enhancing effect on sAP. It suggests that the effect of dopamine may be through the activation of GABAergic inhibition. Consistent with this, dopamine prolongs the large enhancement of PV+ interneuron excitability in response to optogenetic stimulation of VTA terminals. Thus, dopamine released from VTA terminals may directly act on PFC PV+ interneurons, which then affect the excitability of PFC pyramidal neurons. These results have revealed the role of dopamine in regulating the temporal dynamics of E/I balance in VTA–PFC circuit, providing insights into the functional consequence of activating dopamine system in the mesocortical system.
Funding
National Institutes of Health (grants MH108842, AG056060 to Z.Y.).
Notes
P.Z. carried out optogenetics and electrophysiological experiments and analyzed data. L.Q. took confocal images. Z.Y. designed the project and wrote the paper.
References
- Carmichael ST, Price JL. 1996. Connectional networks within the orbital and medial prefrontal cortex of macaque monkeys. J Comp Neurol. 371:179–207. [DOI] [PubMed] [Google Scholar]
- Chaudhury D, Walsh JJ, Friedman AK, Juarez B, Ku SM, Koo JW, Ferguson D, Tsai HC, Pomeranz L, Christoffel DJ, et al. 2013. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature. 493:532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC, Price JL, Simpson JR, Todd RD, Reich T. 1997. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 386:824–827. [DOI] [PubMed] [Google Scholar]
- El-Boustani S, Sur M. 2014. Response-dependent dynamics of cell-specific inhibition in cortical networks in vivo. Nat Commun. 5:5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellwood IT, Patel T, Wadia V, Lee AT, Liptak AT, Bender KJ, Sohal VS. 2017. Tonic or phasic stimulation of dopaminergic projections to prefrontal cortex causes mice to maintain or deviate from previously learned behavioral strategies. J Neurosci. 37:8315–8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson BR, Gao W-J. 2018. PV interneurons: critical regulators of E/I balance for prefrontal cortex-dependent behavior and psychiatric disorders. Front Neural Circuits. 12:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foss-Feig JH, Adkinson BD, Ji JL, Yang G, Srihari VH, McPartland JC. 2017. Searching for cross-diagnostic convergence: neural mechanisms governing excitation and inhibition balance in schizophrenia and autism spectrum disorders. Biol Psychiatry. 81:848–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman-Rakic PS. 1994. Working memory dysfunction in schizophrenia. J Neuropsychiatry Clin Neurosci. 6:348–357. [DOI] [PubMed] [Google Scholar]
- Hauser TU, Eldar E, Dolan RJ. 2017. Separate mesocortical and mesolimbic pathways encode effort and reward learning signals. Proc Natl Acad Sci. 114:E7395–E7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover WB, Vertes RP. 2007. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct Funct. 212:149–179. [DOI] [PubMed] [Google Scholar]
- Hu H, Gan J, Jonas P. 2014. Interneurons fast-spiking, parvalbumin GABAergic interneurons: from cellular design to microcircuit function. Science. 345:1255263. [DOI] [PubMed] [Google Scholar]
- Kabanova A, Pabst M, Lorkowski M, Braganza O, Boehlen A, Nikbakht N, Pothmann L, Vaswani A, Musgrove R, Monte DA, et al. 2015. Function and developmental origin of a mesocortical inhibitory circuit. Nat Neurosci. 18:872–882. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y. 1993. Correlation of physiological subgroupings of on pyramidal cells with parvalbumin- and calbindinD28k-immunoreactive neurons in layer V of rat frontal cortex. J Neurophysiol. 70:387–396. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y. 1997. GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Creb Cortex. 7:476–486. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y. 1998. Neurochemical features and synaptic connections of large physiologically-identified GABAergic cells in the rat frontal cortex. Neuroscience. 85:677–701. [DOI] [PubMed] [Google Scholar]
- Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM, Deisseroth K, Malenka RC. 2012. Input-specific control of reward and aversion in the ventral tegmental area. Nature. 491:212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin A, Nogueira L, Lapish CC, Wightman RM, Phillips PE, Seamans JK. 2005. Mesocortical dopamine neurons operate in distinct temporal domains using multimodal signaling. J Neurosci. 25:5013–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A, Nelson SB, Turigiano GG. 2004. Selective reconfiguration of layer 4 visual cortical circuitry by visual deprivation. Nat Neurosci. 7:1353–1359. [DOI] [PubMed] [Google Scholar]
- Maffei A, Turigiano GG. 2008. Multiple modes of network homeostasis in visual cortical layer 2/3. J Neurosci. 28:4377–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EK. 2000. The prefrontal cortex and cognitive control. Nat Rev Neurosci. 1:59–65. [DOI] [PubMed] [Google Scholar]
- Mizoguchi K, Ishige A, Takeda S, Aburada M, Tabira T. 2004. Endogenous glucocorticoids are essential for maintaining prefrontal cortical cognitive function. J Neurosci. 24:5492–5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, Margolis EB. 2017. Ventral tegmental area: cellular heterogeneity, connectivity and behaviour. Nat Rev Neurosci. 18:73–85. [DOI] [PubMed] [Google Scholar]
- Packer AM, Yuste R. 2011. Dense, unspecific connectivity of neocortical parvalbumin-positive interneurons: a canonical microcircuit for inhibition? J Neurosci. 31:13260–13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. 1997. The rat brain in stereotaxic coordinates. San Diego (CA): Academic. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vives MV, McCormick DA. 2000. Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat Neurosci. 3(10):1027–1034. [DOI] [PubMed] [Google Scholar]
- Sawaguchi T, Goldman-Rakic PS. 1991. D1 dopamine receptors in prefrontal cortex: involvement in working memory. Science. 251:947–950. [DOI] [PubMed] [Google Scholar]
- Volman SF, Lammel S, Margolis EB, Kim Y, Richard JM, Roitman MF, Lobo MK. 2013. New insights into the specificity and plasticity of reward and aversion encoding in the mesolimbic system. Neuroscience. 33:17569–17576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh JJ, Han MH. 2014. The heterogeneity of ventral tegmental area neurons: projection functions in a mood-related context. Neuroscience. 282:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhong P, Yan Z. 2002. Dopamine D4 receptors modulate GABAergic signaling in pyramidal neurons of prefrontal cortex. J Neurosci. 22:9185–9193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GV, Goldman-Rakic PS. 1995. Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature. 376:572–575. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Sheen W, Morales M. 2007. Glutamatergic neurons are present in the rat ventral tegmental area. Eur J Neurosci. 25:106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T, Wang HL, Li X, Ng TH, Morales M. 2011. Mesocorticolimbic glutamatergic pathway. J Neurosci. 31:8476–8490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CR, Seamans JK, Gorlova N. 1996. Electrophysiological and morphological properties of layers V-VI principal cells in rat prefrontal cortex in vitro. J Neurosci. 16:1904–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo JH, Zell V, Gutierrez-Reed N, Wu J, Ressler R, Shenasa MA, Johnson AB, Fife KH, Faget L, Hnasko TS. 2016. Ventral tegmental area glutamate neurons co-release GABA and promote positive reinforcement. Nat Commun. 7:13697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong P, Yan Z. 2011. Differential regulation of the excitability of prefrontal cortical fast-spiking interneurons and pyramidal neurons by serotonin and fluoxetine. PLoS One. 6(2):e16970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong P, Yan Z. 2016. Distinct physiological effects of dopamine D4 receptors on prefrontal cortical pyramidal neurons and fast-spiking interneurons. Cereb Cortex. 26:180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F-M, Hablitz JJ. 1999. Dopamine modulation of membrane and synaptic properties of interneurons in rat cerebral cortex. J Neurophysiol. 81:967–976. [DOI] [PubMed] [Google Scholar]