

Abstract
Sensory information comprises the substrate from which memories are created. Memories of spatial sensory experience are encoded by means of synaptic plasticity in the hippocampus. Hippocampal dependency on sensory information is highlighted by the fact that sudden and complete loss of a sensory modality results in an impairment of hippocampal function that persists for months. Effects are accompanied by extensive changes in the expression of neurotransmitter receptors in cortex and hippocampus, consistent with a substantial adaptive reorganization of cortical function. Whether gradual sensory loss affects hippocampal function is unclear. Progressive age-dependent hearing loss (presbycusis) is a risk factor for cognitive decline. Here, we scrutinized C57BL/6 mice that experience hereditary and cumulative deafness starting in young adulthood. We observed that 2–4 months postnatally, increases in the cortical and hippocampal expression of GluN2A and GluN2B subunits of the N-methyl-D-aspartate receptor occurred compared to control mice that lack sensory deficits. Furthermore, GABA and metabotropic glutamate receptor expression were significantly altered. Hippocampal synaptic plasticity was profoundly impaired and mice exhibited significant deficits in spatial memory. These data show that during cortical adaptation to cumulative loss of hearing, plasticity-related neurotransmitter expression is extensively altered in the cortex and hippocampus. Furthermore, cumulative sensory loss compromises hippocampal function.
Keywords: C57BL/6, GABA, hearing loss, hippocampus, metabotropic glutamate receptor, NMDA receptor
Introduction
Sensory information processing is a fundamental component of learning, memory, and cognition. Brain structures that are essential for the acquisition and encoding of complex associative memories, such as the hippocampus, use spatial sensory information both to generate metric representation of navigable space (Save et al. 2000; Zhang and Manahan-Vaughan 2015; Draht et al. 2017; Jeffery 2018) and to create robust and long-lasting records of spatial experience (Kemp and Manahan-Vaughan 2007, 2012). The latter is enabled by hippocampal synaptic plasticity (Manahan-Vaughan and Braunewell 1999; Kemp and Manahan-Vaughan 2004; Goh and Manahan-Vaughan 2013), and it has been shown that visuospatial, olfactospatial, and audiospatial experience can be used by the hippocampus to create spatial memories that are recorded by this phenomenon (Kemp and Manahan-Vaughan 2008; André and Manahan-Vaughan 2014; Dietz and Manahan-Vaughan 2017). The mammalian brain is highly adaptable, and there are multiple descriptions of how both humans (Röder et al. 1999; van Boven et al. 2000; Fortin et al. 2008; Fiehler and Rösler 2010) and animals (Lindner et al. 1997; Iura and Udo 2014; Norimoto and Ikegaya 2015) adapt effectively to profound and perpetual sensory loss. Studies of the consequences of loss of visual input and blindness have shown that this adaptation occurs as a consequence of extensive reorganization of the cortex that reflects both changes in the affected primary sensory cortex (Eysel et al. 1999; Goel and Lee 2007) and in other primary and associative sensory areas (Kujala et al. 1995; Sadato et al. 1996; Cohen et al. 1999; Weeks et al. 2000; Merabet et al. 2009; Petrus et al. 2015).
One aspect of this that has received little attention is how the cortex and hippocampus functionally adjust to initial loss of input from a specific sensory modality. Recently, we reported that hereditary blindness that becomes manifest in mice within weeks after birth results in massive and progressive reorganization of neurotransmitter receptor expression in the cortex and hippocampus that persists for months after the onset of blindness (Feldmann et al. 2019). This reorganization is accompanied by a profound impairment of hippocampal long-term potentiation (LTP) and debilitation of hippocampus-dependent spatial learning (Feldmann et al. 2019). These results suggest that cortical and cognitive adaptation to the loss of a sensory modality impacts on the information processing ability of cortical structures, and this compromises, in turn, the ability of the hippocampus to reliably and effectively encode and store sensory experience.
Ultimately, both humans and animals recover from this transitional phase (Röder et al. 1999; van Boven et al. 2000; Fortin et al. 2008; Fiehler and Rösler 2010; Iura and Udo 2014; Norimoto and Ikegaya 2015), and adaptation is ostensibly so effective in rodents that chronically blind mice cannot be distinguished from their “seeing” counterparts on the basis of cued learning behavior (Lindner et al. 1997). Recent studies in human individuals have suggested, however, that the consequences for cognition of gradual sensory loss are insidious. Age-dependent sensorineural hearing loss (presbycusis) comprises a gradual and cumulative loss of hearing sensitivity that first affects high frequencies and slowly extends to mid-frequency ranges (Morton 1991; Gorlin et al. 1995). It is closely associated with cognitive decline (Fortunato et al. 2016; Deal et al. 2017; Golub 2017) and is considered a risk factor for dementia (Teipel et al. 2015; Deal et al. 2017; Lin and Black 2017; Su et al. 2017; Thomson et al. 2017). This risk is not exclusive to the auditory domain (Attems et al. 2015; Fischer et al. 2016) and increases according to the relative number of sensory modalities showing age-related impairments (Brenowitz et al. 2018).
A causal link between cumulative hearing loss and cognitive decline is currently lacking. In the present study, our goal, therefore, was to explore to what extent a gradual loss of hearing sensitivity can result in cortical reorganization and changes in hippocampal function. The C57BL/6 mouse strain develops cumulative deafness that first becomes manifest at the age of 4 weeks with the loss of very high hearing frequencies (Park et al. 2010). With accumulating age, even more frequencies are lost: At 5 months, mice lose their ability to hear socially relevant vocalizations (>32 kHz) (Park et al. 2010), and 24 months postnatally, this mouse strain is completely deaf (Mikaelian 1979). Loss of the ability to discriminate speech is a major debilitatory aspect of presbycusis in human individuals (Morton 1991; Gorlin et al. 1995) and may serve to accelerate cognitive decline. Rodents rely heavily on auditory information in the form of vocalizations for both their social structures and integration, as well as the localization of salient proximal events and features (Brudzynski 2015). Rodent vocalizations predominantly occur outside the range of human hearing and extend into the ultrasound spectrum (Brudzynski 2001, 2015; Brudzynski and Pniak 2002). Thus, the cumulative loss of hearing sensitivity to 32 kHz and higher in adult rodents emulates cumulative loss of speech discrimination in presbycusis in humans. Given the parallels in age-dependent cellular, sensorineural, and perceptual changes in the C57BL/6 mouse strain and presbycusis, the C57BL/6 mouse is therefore considered an effective animal model of age-dependent hearing loss (Kazee et al. 1995; Spongr et al. 1997; Del Campo et al. 2012; Osumi et al. 2012).
We show here that widespread changes in plasticity-related neurotransmitter expression become manifest as early as at 2 months of age in C57BL/6 mice. At 4 months of age, neurotransmitter receptor changes, including GABA, metabotropic glutamate (mGlu) receptors, and GluN2A and GluN2B subunits of the N-methyl-D-aspartate (NMDA) receptor, occur in both primary sensory and association cortices and also extend to the hippocampus. At this time-point, potent impairments in hippocampal LTP and spatial memory become evident. The data indicate that gradual hearing loss is accompanied by extensive adaptive changes in the cortex and hippocampus that hinder effective hippocampal information processing and suggest that progressive hearing loss may be causally linked to cognitive decline.
Materials and Methods
Animals
The present study was carried out in accordance with the European Communities Council Directive of 22 September 2010 (2010/63/EEC) for care of laboratory animals and the requirements of the animal ethics committee of the local government authority (Landesamt fur Arbeitsschutz, Naturschutz, Umweltschutz und Verbraucherschutz [LANUV]). Male C57BL/6 (Charles River) and CBA/CaOlaHsd mice (Harlan [Envigo] Laboratories) were group-housed in a temperature- and humidity-controlled vivarium with a constant 12-h light–dark cycle (lights on from 6 AM to 6 PM) with ad libitum food and water access. The C57BL/6 mouse develops hereditary hearing loss (Mikaelian 1979). It exhibits normal hearing at 1 month of age (Shnerson and Pujol 1983). From this time-point onward, the cochlea begins to degenerate commencing at the cochlear base resulting in high frequencies, being affected first (Mikaelian 1979; Henry and Chole 1980; Park et al. 2010). At 2 years of age, mice are completely deaf (Mikaelian 1979). The CBA/CaOlaHsd mouse strain has no reported deficits in its sensory modalities (Brooks et al. 2004; Walton et al. 2008; Heckman et al. 2017).
Tissue Preparation
Brains were removed when mice were 2 or 4 months old, using isoflurane for inhalational anesthesia, followed by an intraperitoneal injection with sodium pentobarbital. Transcardial perfusion was conducted with cooled, 0.2% heparinized Ringer solution (10 min), followed by 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) (10 min). Brains immersed in PFA for 24 h at 4 °C and subsequently stored in 30% sucrose in PBS. Frozen sections of 30 μm were prepared with a cryostat microtome (Leica) for Nissl staining and immunohistochemistry. Slices from C57BL/6 and CBA/CaOlaHsd mice were processed together to minimize interarray variations between different staining cohorts. Nissl stainings were conducted to verify tissue quality and to choose slices with the correct anatomical features.
Immunohistochemistry
GABAA, GABAB, and GluN2B immunostainings were conducted using an avidin–biotin complex (ABC) method as described previously (Heras et al. 1995; Feldmann et al. 2019). To evaluate the expression of mGlu1, mGlu2/3, and GluN2A, we included streptavidin enhancement (Feldmann et al. 2019). Negative controls comprising tissue incubations with separate primary and secondary antibodies were performed in order to verify that specific binding had occurred.
For the ABC approach, free-floating sections were rinsed in PBS thrice for 10 min. Sections were then placed in 0.3% H2O2 for 20 min to remove endogenous peroxidase activity, thereby ensuring that background staining could be kept to a minimum. Then, they were preincubated with blocking solution containing 20% avidin (avidin–biotin blocking kit, Vector Laboratories), 10% normal serum (Vector Laboratories), and 0.2% Triton X-100 (Tx) for 90 min to reduce nonspecific binding. Sections were subsequently incubated overnight at room temperature with the primary antibody solution, containing 20% biotin (avidin-biotin blocking kit, Vector Laboratories), 1% normal serum, 0.2% Tx, and the relevant primary antibody—GABAA (1:400, monoclonal mouse antibody [AB], MAB341, Merck Millipore), GABAB (1:500, monoclonal mouse AB, ab55051, Abcam), or GluN2B (polyclonal goat AB, sc-1469, Santa Cruz Biotechnology). The secondary antibody was applied for 90 min. A biotinylated horse anti-mouse antibody was used for GABAA and GABAB (1:500, BA-2001, Vector Laboratories) and a biotinylated horse anti-goat antibody was used for GluN2B (1:500, BA-9500, Vector Laboratories). Sections were then immersed in 1:1000 ABC-Elite detection system (Vector Laboratories), 1% normal serum, and 0.1% Tx for 90 min. The staining reaction was then performed using 3,3′-diaminobenzidin (DAB, Sigma-Aldrich) in 0.01% hydrogen peroxide PBS for 10 min.
GluN2A subunits, as well as mGlu1 and mGlu2/3 receptor expression, were detected by means of streptavidin enhancement. Tris-(hydroxymethyl)-aminomethane-buffered saline was used instead of PBS. The initial protocol elements were the same as those described above. Incubation with the primary antibody specific to mGlu1 (1:200, polyclonal rabbit AB, ab82211, Abcam), mGlu2/3 (1:250, polyclonal rabbit AB, ab5675, Merck Millipore), or GluN2A (1:250, polyclonal rabbit AB, sc-9056, Santa Cruz Biotechnology) lasted for 24 h at room temperature and was followed by secondary antibody incubation with biotinylated goat anti-rabbit antibody (1:500, BA-1000, Vector Laboratories). The sections were then incubated with 1:1000 streptavidin (Cy™3-conjugated Streptavidin, Jackson Laboratories) for 30 min. Anti-streptavidin antibody (1:500, biotinylated goat anti-streptavidin, BA-0500, Vector Laboratories) was applied for another 30 min, followed by DAB staining.
Electrophysiology
Two- or 4-month-old mice were deeply anesthetized with isoflurane and then decapitated. Brains were dissected in cold (4 °C), oxygenated artificial cerebrospinal fluid (aCSF) using 87 mM NaCl, 2.4 mM KCl, 1.3 mM MgSO4, 0.5 mM CaCl2, 26 mM NaHCO3, 1.25 mM NaH2PO4, and 2 mM d-glucose. Following brain dissection, slices (400 μm) were cut using a vibratome and stored on a nylon net in a holding chamber filled with aCSF and glucose (30 °C) for 30 min. The slices were subsequently transferred to an interface recording chamber (Scientific Systems Design Inc) that was continuously percolated with oxygenated aCSF (95% O2, 5% CO2, 125 mM NaCl, 3 mM KCl, 2.5 mM CaCl2, 1.3 mM MgSO4, 1.25 mM NaH2PO4, 13 mM d-glucose, 26 mM NaHCO3) at the rate of 1.5 mL/min. The temperature in the chambers was maintained at 30 ± 2 °C by an automatic temperature controller (PTC03, Scientific Systems Design Inc). The slices acclimatized for 1 h in the recording chamber before recordings were commenced. A bipolar stimulation electrode (Fredrick Haer) was positioned in stratum radiatum of CA1. The recording electrode, which comprised a borosilicate glass-micropipette (1–2 MΩ) filled with aCSF, was placed in the CA1 dendritic area. Biphasic test pulse stimuli (0.025 Hz, duration: 0.2 ms) were applied, and for each time-point, five evoked responses (sample rate: 16 000 Hz) were averaged. An input/output relationship (I/O-curve) was determined by stimulating in a range of 60–600 μA. For experiments, a stimulation intensity that evoked 50% of the I/O maximum was used. Basal synaptic transmission (“baseline”) was recorded for 60 min and then, without changing the stimulus intensity, LTP was induced via high-frequency stimulation (HFS, 3 trains of 100 pulses at 100 Hz, delivered 5 min apart). Evoked responses were followed for 2 h after HFS.
Behavioral Tests
Mice that were 4–5 months of age underwent object recognition (OR) and item–place (IP) tests. In each case, on the day before the acquisition trial, habituation of the mice to the test arena occurred for 10 min. The OR and IP tests corresponded to previously reported paradigms (Goh and Manahan-Vaughan 2013). In brief, for the OR test, mice were presented with two novel objects (in the test arena) for 10 min (acquisition trial). After 24 h, animals were exposed to one of the now familiar objects in its old location and a novel object in the location of the previous object.
In the IP experiment, mice were presented with two novel objects (in the test arena) for 10 min (acquisition trial). After 24 h, animals were re-exposed to the now familiar objects in the same locations as previously. A further 24 h after this, animals were presented with the two objects in a reconfigured spatial location. Each test trial lasted 10 min, during which the total exploration time and the exploration time of the familiar and the novel (or moved) object were measured. Objects and test arenas were cleaned thoroughly between trials to remove olfactory stimuli. The trials were recorded by a camera that was positioned above the test arena to allow offline analysis.
Data Analysis
For the analysis of neurotransmitter receptor distribution, optical density measurements were conducted for selected brain regions using an image-based analysis system (Neurolucida, MBF Bioscience). Protein quantification was conducted by means of a standardized immunoassay (Grube 2004; Feldmann et al. 2019). Cortical and hippocampal areas were examined at the following distances from bregma: 2.2 mm (Fig. 1C, piriform cortex), 0.5 mm (Fig. 1C, somatosensory cortex), −1.8 mm (Fig. 1C, posterior parietal cortex), and −2.5 mm (Fig. 1C, auditory cortex, visual cortex, dentate gyrus, CA1–3). Tissue sections were scrutinized with 2.5-fold magnification using a Leitz Wetzlar Dialux 20 brightfield microscope (Leica). Whole slide images were acquired with a digital camera (QIC-F-CLR-12, QImaging), using the virtual tissue 2D module of Neurolucida. Immunohistochemical background staining was corrected by subtracting density values obtained in the corpus callosum to minimize interarray differences (Dubovyk and Manahan-Vaughan 2018). Luminance information ranging from 0 to 255 was determined for the whole area.
Figure 1.
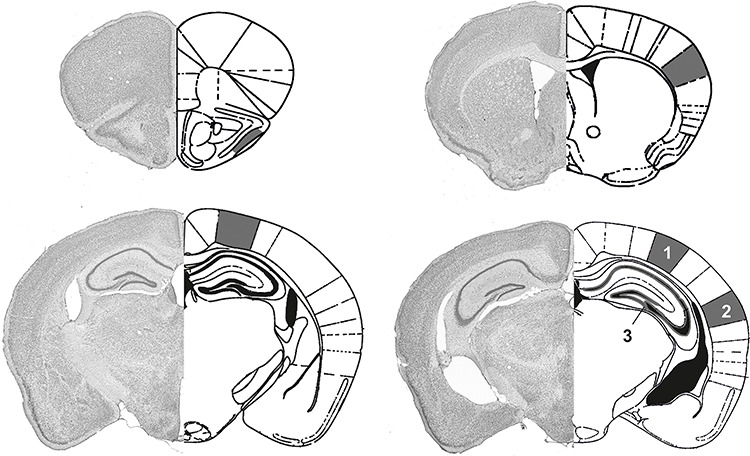
Diagram of areas selected for immunohistochemical analysis. The areas assessed in the mouse cerebral cortex and hippocampus are shown in the histological sections (left side of each example). The specific regions examined are indicated in the schemas on the right side of each example (based on Franklin and Paxinos 2008). Markings in dark gray represent the following areas: Top left: PiC; top right: SC; bottom left: PPC; bottom right: VC (1), AuC (2), and hippocampus (3), including the DG and cornus ammonis subregions (CA1–CA3).
Immunohistochemical data were tested for between-group effects by means of multifactorial analysis of variance (ANOVA) followed by a post hoc Duncan’s test. Between-group factors comprised strain (C57BL/6 vs. CBA/CaOlaHsd) and brain areas (piriform cortex [PiC], somatosensory cortex [SC], posterior parietal cortex [PPC], visual cortex [VC], auditory cortex [AuC], dentate gyrus [DG], CA1, CA2/3, and CA4) and were assessed 2 and 4 months postnatally. Immunohistochemical data from control (CBA/CaOlaHsd) mice were used as a reference in a previous study (Feldmann et al. 2019), in compliance with requirements of the ethics committee of the local government authority (LANUV) to minimize the number of animals used in the experimentation. P values and the mean optical densities determined for the neurotransmitter receptors (region studied at 2 and 4 months) are shown in Table 1 (for significant effects all comparisons).
Table 1.
Statistical comparison of receptor expression in 2- and 4-month-old C57BL6 and CBA/CaOlaHsd mice
| Receptor | Area | C57BL/6 | CBA/CaOlaHsd | P | N |
|---|---|---|---|---|---|
| 2 months | |||||
| GluN2A | PPC | 21.11 ± 1.74 | 17.28 ± 1.01 | <0.001 | 6 |
| VC | 19.77 ± 1.31 | 13.66 ± 1.05 | <0.01 | 6 | |
| AuC | 19.66 ± 1.73 | 13.84 ± 0.87 | <0.01 | 6 | |
| mGlu2/3 | PPC | 33.10 ± 2.51 | 26.09 ± 2.35 | <0.05 | 6 |
| VC | 33.68 ± 2.79 | 24.44 ± 1.77 | <0.01 | 6 | |
| mGlu5 | CA4 | 97.97 ± 2.43 | 83.71 ± 6.17 | <0.05 | 5 |
| GABA-B | AuC | 28.40 ± 1.74 | 20.42 ± 1.55 | <0.05 | 6 |
| CA1 | 31.34 ± 2.04 | 20.72 ± 1.40 | <0.01 | 6 | |
| 4 months | |||||
| GluN2A | PiC | 21.57 ± 1.15 | 16.75 ± 1.52 | <0.05 | 6 |
| SC | 22.52 ± 1.08 | 17.49 ± 1.36 | <0.05 | 6 | |
| GluN2B | PiC | 32.78 ± 2.71 | 12.27 ± 1.82 | <0.001 | 5 |
| SC | 43.31 ± 2.15 | 20.08 ± 1.76 | <0.001 | 5 | |
| PPC | 46.81 ± 4.27 | 26.93 ± 1.94 | <0.001 | 5 | |
| VC | 39.56 ± 3.58 | 23.86 ± 1.35 | <0.001 | 5 | |
| AuC | 37.38 ± 3.17 | 20.03 ± 1.71 | <0.001 | 5 | |
| DG | 23.12 ± 2.19 | 11.97 ± 1.30 | <0.01 | 5 | |
| CA1 | 32.56 ± 2.73 | 23.28 ± 1.28 | <0.05 | 5 | |
| CA3 | 31.64 ± 1.98 | 19.81 ± 1.80 | <0.01 | 5 | |
| CA4 | 21.89 ± 2.52 | 10.25 ± 1.48 | <0.01 | 5 | |
| mGlu1 | SC | 39.68 ± 1.52 | 31.84 ± 1.47 | <0.01 | 5 |
| AuC | 41.96 ± 1.62 | 34.44 ± 2.25 | <0.05 | ||
| mGlu2/3 | PiC | 30.96 ± 1.57 | 24.72 ± 1.24 | <0.01 | 5 |
| AuC | 36.74 ± 1.68 | 31.07 ± 1.51 | <0.05 | 5 | |
| DG | 40.21 ± 2.22 | 34.68 ± 1.41 | <0.05 | 5 | |
| CA1 | 16.54 ± 1.73 | 10.20 ± 0.47 | <0.01 | 5 | |
| CA3 | 19.87 ± 1.91 | 12.31 ± 0.98 | <0.01 | 5 | |
| CA4 | 29.46 ± 3.61 | 21.52 ± 0.86 | <0.001 | 5 | |
| GABA-A | PiC | 64.34 ± 4.59 | 84.58 ± 6.06 | <0.05 | 6 |
| SC | 76.59 ± 5.44 | 104.76 ± 6.06 | <0.01 | 6 | |
| AuC | 78.95 ± 7.88 | 101.36 ± 5.63 | <0.05 | 6 | |
| DG | 79.36 ± 2.87 | 99.29 ± 4.72 | <0.05 | 6 | |
| GABA-B | DG | 28.86 ± 1.76 | 44.06 ± 3.17 | <0.001 | 6 |
| CA3 | 31.80 ± 1.15 | 43.85 ± 3.68 | <0.001 | 6 | |
| CA4 | 32.50 ± 1.64 | 49.87 ± 3.85 | <0.001 | 6 | |
Note: The following regions were examined: AuC, PiC, SC, PPC, VC, DG, CA1 region, CA3 region, and CA4 region. Values shown correspond to the mean optical density ± standard error of the mean. “P” indicates the P values obtained in statistical comparisons between C57BL/6 and CBA/CaOlaHsd mice at 2 months (N = 6 each) and 4 months (N = 6 for C57BL/6, N = 5 for CBA/CaOlaHsd mice). “N” indicates the number of animals used in the comparisons.
Between-group effects in electrophysiological data were assessed by means of a two-way ANOVA with repeated measures. A post hoc Fisher’s test was used to discriminate significant effects at specific time-points/conditions. In the subsequent text sections, the number of animals is signified by an upper case “N” and the number of slices used is signified by a lower case “n.” All data were shown as mean ± standard error of the mean. All statistical tests were performed using STATISTICA 13 (Statsoft). The level of significance was set at P < 0.05.
Behavioral data were analyzed by determining the exploration time spent at each object within the different trials. Object exploration was determined as touching or sniffing the object or close proximity (<2 cm) of the snout to the object when the snout was pointed directly at the object in question. OR data were expressed as a percentage of the total exploration time for each object per experiment. A discrimination ratio was also calculated, as the difference between the time spent exploring the novel and the familiar objects divided by the total time spent exploring both objects (Goh and Manahan-Vaughan 2013). The IP data were expressed as a percentage of the total exploration time during novel exploration of the objects. The behavioral data were analyzed using ANOVA for assessment of the IP data.
Results
Immunohistochemistry
Brain sections from C57BL/6 and CBA/CaOlaHsd mice were scrutinized for changes in the expression of GABAA and GABAB receptors, mGlu1 and mGlu2/3 receptors, and the GluN2A and GluN2B subunits of the NMDA receptor. The following cortical regions were examined: PiC, SC, PPC, VC, and AuC. In addition the following regions of the dorsal hippocampus were examined: DG, CA1 region, CA3 region, and CA4 region (Fig. 1). Optical density and P values for all receptors, age groups, and brain regions are shown in Table 1, whereas significant values are reported in Table 1.
Expression of the GluN2A subunit of the NMDA receptor is increased in the paoterior parietal cortex, visual cortex and auditory cortex of C57BL/6 mice 2 months postnatally. Four months postnatally, increases are evident in the piriform cortex and somatosensory cortex
In 2-month-old C57BL/6 mice (N = 6), the GluN2A subunit of the NMDA receptor was significantly increased in the cortex compared to control CBA/CaOlaHsd mice (N = 6). This effect was specific to the PPC, VC, and AuC (Fig. 2A; Table 1). Four months postnatally, the levels of GluN2A are significantly increased in C57BL/6 mice (N = 6) in the PiC and SC compared to CBA/CaOlaHsd (N = 6), respectively (Fig. 2B; Table 1).
Figure 2.
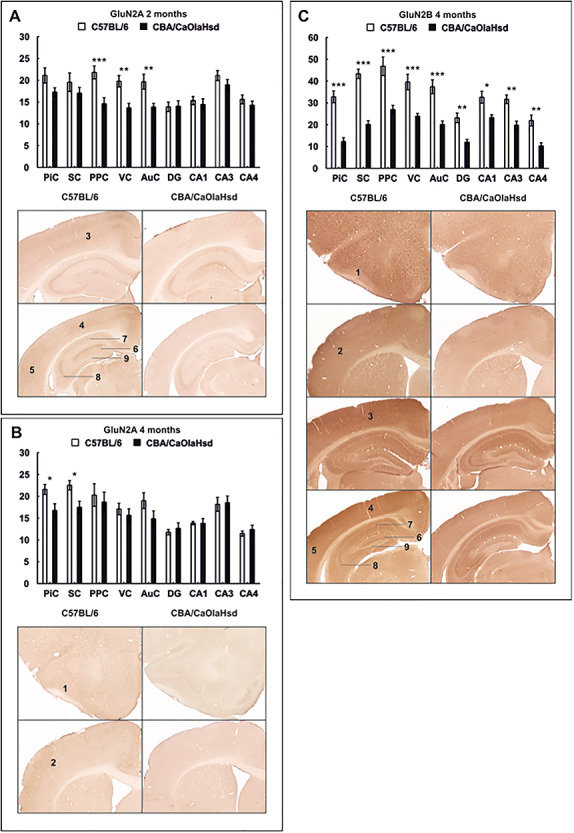
Expression of the GluN2A and GluN2B subunits of the NMDA receptor 2 and 4 months postnatally. Bar charts represent mean optical density of NMDA receptor subunit expression in C57BL/6 and CBA/CaOlaHsd mice 2 and 4 months postnatally. Photomicrographs highlight significantly affected areas as shown in the graphs above. (A) GluN2A expression is significantly increased in the PPC, VC, and AuC of C57Bl/6 mice at the age of 2 months. (B) GluN2A expression is significantly increased in 4-month-old C57BL/6 mice in the PiC and SC. (C) GluN2B expression is globally increased in C57BL/6 mice 4 months postnatally. Data are means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. 1: PiC, 2: SC, 3: PPC, 4: VC, 5: AuC, 6: DG, 7: CA1, 8: CA3, and 9: CA4.
Four months postnatally, the expression of the GluN2B subunit of the NMDA receptor in C57BL/6 mice is increased in all primary sensory cortices and the parietal cortex, as well as in all subfields of the hippocampus
Two months postnatally, no significant differences were evident in the expression of the GluN2B subunit of the NMDA receptor between the two mouse strains (N = 6, each, Table 1).
At the age of 4 months, however, the expression of the GluN2B subunit was increased in C57BL/6 mice (N = 6) compared to controls (N = 6). The increase in GluN2B expression occurred in all cortical areas investigated, as well as in all hippocampal subfields (Fig. 2C; Table 1).
mGlu1 Receptor Expression Is Unchanged at 2 Months and Is Increased in the somatosensory cortex and auditory cortex at 4 Months
In 2-month-old C57BL/6 mice (N = 6), no significant differences in the expression of the metabotropic glutamate receptor mGlu1 were evident compared to CBA/CaOlaHsd mice (N = 6, Table 1).
At the age of 4 months, mGlu1 receptor density was significantly higher in C57BL/6 mice (N = 6) compared to CBA/CaOlaHsd controls (N = 5) in the SC and AuC (Fig. 3A; Table 1).
Figure 3.
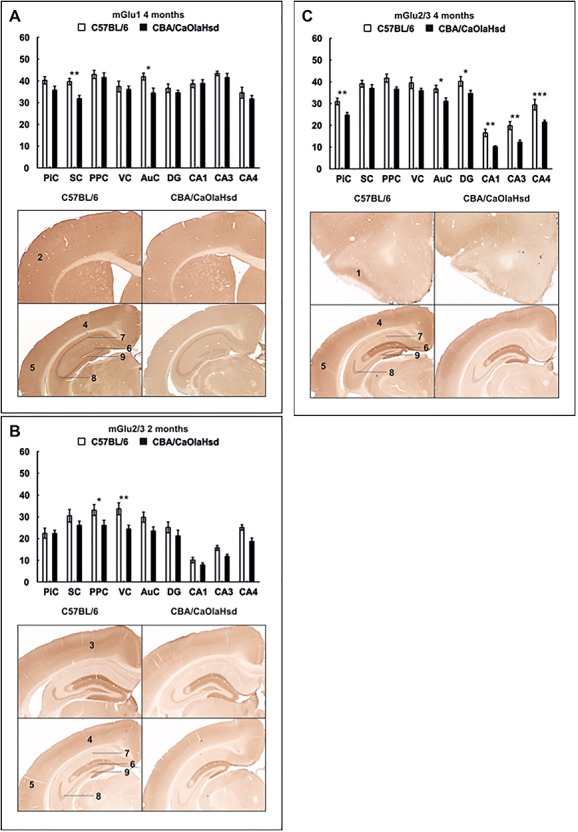
Expression of metabotropic glutamate receptors 2 and 4 months postnatally. Bar charts represent mean optical density of metabotropic glutamate receptor expression in C57BL/6 and CBA/CaOlaHsd mice at the age of 2 and 4 months. Photomicrographs highlight significantly affected areas as shown in the graphs above. (A) mGlu1 expression is significantly increased in 4-month-old C57BL/6 mice in the SC and AuC. (B) Significant increase in mGlu2/3 expression in C57BL/6 mice in the PPC and VC at the age of 2 months. (C) C57BL/6 mice show significantly increased mGlu2/3 expression in the PiC and AuC and in the hippocampus. Data are means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. 1: PiC, 2: SC, 3: PPC, 4: VC, 5: AuC, 6: DG, 7: CA1, 8: CA3, and 9: CA4.
mGlu2/3 Receptor Expression Is Increased in the VC and posterior parietal cortex at 2 Months and in the piriform cortex and auditory cortex of 4-Month-Old C57BL/6 Mice
Two months postnatally, a significant increase in the expression of the mGlu2/3 receptor in the PPC and VC (Fig. 3B; Table 1) of C57BL/6 mice was evident (N = 6) in comparison with CBA/CaOlaHsd mice (N = 6).
Four-month-old C57BL/6 mice (N = 6) showed increased mGlu2/3 levels in both cortical and hippocampal areas compared to CBA/CaOlaHsd (N = 5). This was specific to the PiC and AuC and the hippocampal subfields CA1–4 and the DG (Fig. 3C; Table 1).
GABAA Expression Is Unchanged at 2 Months But Is Increased in the piriform cortex, somatosensory cortex, auditory cortex and dentate gyrus of 4-Month-Old C57BL/6 Mice
In 2-month-old C57BL/6 mice (N = 6), no significant changes of GABAA expression occurred compared to control mice (N = 6, Table 1).
At the age of 4 months, inhibitory GABAA receptors were decreased in the cortex and in the hippocampus of C57BL/6 mice (N = 6) compared to CBA/CaOlaHsd controls (N = 5). This effect was significant in the PiC, SC, and AuC as well as in the DG (Fig. 4A; Table 1).
Figure 4.
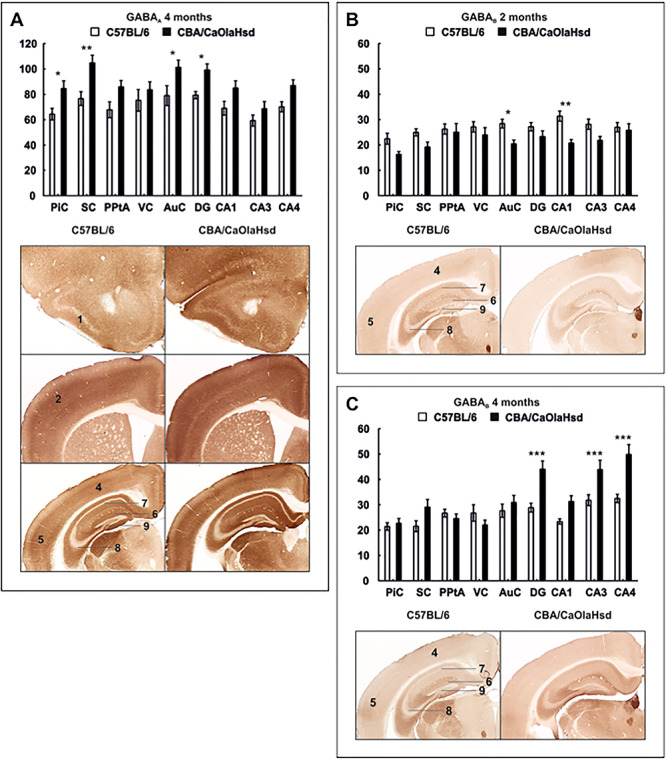
GABA receptor expression differs 2 and 4 months postnatally. Bar charts represent mean optical density of GABAA and GABAB expression in C57BL/6 and CBA/CaOlaHsd mice 2 and 4 months postnatally. Photomicrographs highlight significantly affected areas as shown in the graphs above. (A) Four months postnatally, GABAA is significantly decreased in the PiC, SC, and AuC and in the DG of C57BL/6 mice. (B) Optical density of GABAB receptors is significantly increased in C57BL/6 mice in the AuC and in the hippocampal subfield CA1 2 months postnatally. (C) At the age of 4 months, GABAB expression is significantly lower in the DG and CA3–4 regions of C57BL/6 mice compared with controls. Data are means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001. 1: PiC, 2: SC, 3: PPC, 4: VC, 5: AuC, 6: DG, 7: CA1, 8: CA3, and 9: CA4.
GABAB Expression Is Increased in the auditory cortex and CA1 Region at 2 Months and in the Hippocampus at 4 Months
At the age of 2 months, expression levels of GABAB receptors in C57BL/6 mice (N = 6) were significantly higher in the AuC and in the CA1 region compared with expression levels in CBA/CaOlaHsd mice (N = 5) (Fig. 4B; Table 1).
Four months postnatally, no differences in cortical expression of GABAB were evident. However, C57BL/6 mice (N = 5) show a significantly decreased expression in the hippocampus in the CA3 and CA4 regions as well as in the DG compared to CBA/CaOlaHsd controls (N = 5) (Fig. 4C; Table 1).
The Stimulus–Response Relationship of Hippocampal Field Potentials Is Unaffected in C57BL/6 Mice
The changes in the expression of the plasticity-related receptors described above in the brains of C57Bl/6 mice may have been accompanied by functional alterations at the levels of neural transmission or synaptic plasticity. Given the fact that the hippocampus serves as an integrator of sensory information and a hub for the encoding of associative memory, we explored whether this structure exhibits changes in its stimulus–response relationship in 2- and 4-month-old mice. Here, we observed that the relationship of the fEPSP to stepwise increases in afferent stimulation strength was unaffected in the CA1 region of 2-month-old C57BL/6 mice (N = 6, n = 12) and CBA/CaOlaHsd mice (Fig. 5A; ANOVA: F = (9, 216) = 0.58092, P = 0.81207, N = 6, n = 12.) Similarly, in 4-month-old mice, responses were also equivalent (Fig. 5B; ANOVA: F = (9, 405) = 0.56927, P = 0.82317, N = 10, n = 21).
Figure 5.
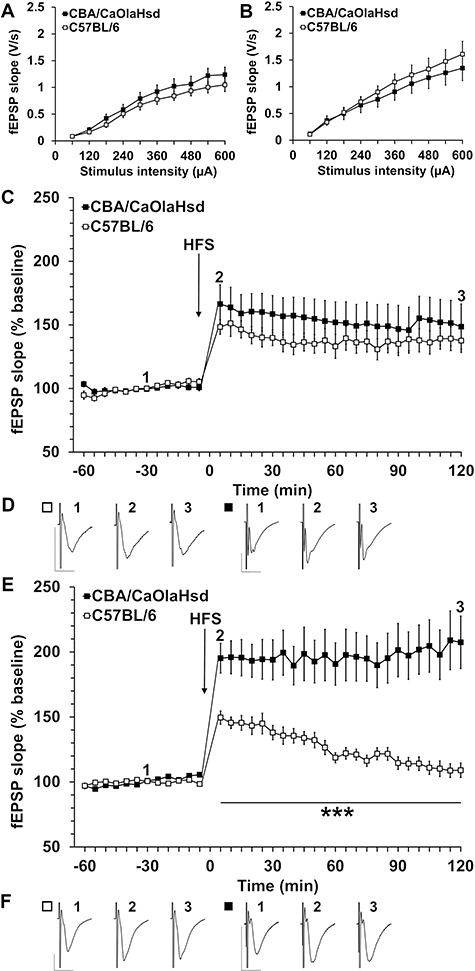
Synaptic plasticity is impaired in C57BL/6 mice. (A) Comparison of input–output relationship in 2-month-old C57BL/6 mice and control (CBA/CaOlaHsd) mice shows no significant differences in fEPSP evoked in the stimulus intensity range of 60–600 μA. (B) Comparison of input–output responses in 4-month-old C57BL/6 and control mice found no significant differences in fEPSP evoked in the stimulus intensity range of 60–600 μA (C) HFS elicits LTP in 2-month-old C57B/6 and control mice, which is not significantly different. (D) Analog examples show fEPSP recorded from 2-month-old C57BL/6 and control mice at the time points indicated in the graph shown in “C.” Vertical scale bar: 1 mV; horizontal scale bar: 10 ms. (E) HFS results in robust LTP in 4-month-old control mice. By contrast, C57BL/6 mice show a significantly impaired response to HFS. The induction and maintenance phases are significantly smaller in magnitude than that elicited in control mice. By 100 min post-HFS, evoked potentials returned to pre-HFS levels. (F) Analog examples show fEPSP recorded from 4-month-old C57BL/6 and control mice at the time points indicated in the graph shown in “E.” Vertical scale bar: 1 mV; horizontal scale bar: 10 ms.
Hippocampal LTP in 4-Month-Old, But Not 2-Month-Old, C57BL/6 Mice Is Significantly Impaired
We then went on to compare LTP in the two mouse strains at the ages of 2 and 4 months. HFS applied to the Schaffer collaterals resulted in robust LTP in C57BL/6 mice that did not differ in its profile from LTP induced in control mice (Fig. 5C,D; ANOVA: F(23, 437) = 0.92878, P = 0.55954, N = 6, n = 9 for each cohort).
At 4 months of age, however, C57BL/6 mice exhibited a very potent debilitation of LTP in comparison with LTP induced in CBA/CaOlaHsd mice (Fig. 5E,F; ANOVA: F(23, 598) = 6.8307, P = 0.0000, N = 10, n = 13).
Item-place Memory Is Impaired, But Object Recognition Memory Is Intact in C57BL/6 Mice
To assess whether the deficits in hippocampal synaptic plasticity that we observed in C57BL/6 mice have functional consequences, we assessed OR memory and IP memory in C57BL/6 (n = 12) and CBA/CaOlaHsd mice (n = 11). OR was calculated as a percentage of the total exploration time of both objects. During the first 10-min exposure to the two test objects, exploration was equally distributed across the objects in both mouse strains (Fig. 6A,B) (A: ANOVA, F(1,20) = 0.66, P = 0.4256; B: ANOVA, F(1,22) = 0.09, P = 0.7607). When the animals were exposed to a familiar object and a novel object (on the same location of the previous object), exploration of the novel object was significantly higher in both CBA/CaOlaHsd (Fig. 6A; ANOVA, F(1,20) = 9.95, P = 0.0049) and C57BL/6 mice (Fig. 6B; ANOVA, F(1,22) = 45.61, P < 0.0001). Assessment of the discrimination ratio for CBA/CaOlaHsd mice (Fig. 6C) and C57BL/6 (Fig. 6D) confirmed that the animals demonstrated effective OR memory (CBA/CaOlaHsd: ANOVA, F(1,20) = 7.60, P = 0.0129; C57BL/6: ANOVA, F(1,22) = 7.51, P = 0.0125).
Figure 6.
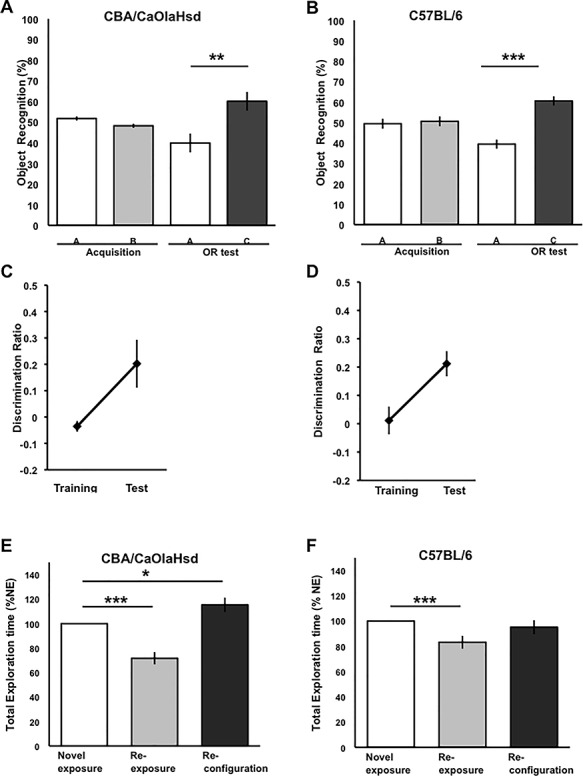
Item-place memory, but not Object Recognition memory, is impaired in C57BL/6 mice. (A and B) Behavioral analysis revealed that CBA/CaOlaHsd mice (n = 11) and C57BL/6 mice (n = 12) performed the OR task successfully with a delay period of 24 h between phases. The mice explored both objects A and B equally during the training phase but explored the novel object C to a significantly greater extent during the test phase (CBA/CaOlaHsd: ANOVA, F(1,20) = 9.99, P = 0.4256; C57BL/6: ANOVA, F(1,22) = 45.61, P < 0.0001). (C, D) Analysis of the discrimination ratios revealed that the mice showed no preference for either object during the training phase but a strong preference for the novel object during the test phase (C: CBA/CaOlaHsd: ANOVA, F(1,20) = 7.60, P = 0.0129; D: C57BL/6: ANOVA, F(1,22) = 7.51, P = 0.0125). (E) CBA/CaOlaHsd mice acquired significant IP memory. Compared to novel exposure, the animals exhibited an habituation effect 24 h later when they were re-exposed to the same objects in the same places F(1,20) = 35.43, P < 0.0001). A further 24 h later, reconfiguration of the objects triggered an increase in exploration (ANOVA, F(1,20) = 6.79, P = 0.0178). By contrast, although C57BL/6 mice (F) exhibited an habituation effect upon object re-exposure (F(1,22) = 44.93, P < 0.0001), they exhibited no significant change in exploration behavior exposure to a spatial reconfiguration of the objects (ANOVA, F(1,22) = 0.52, P = 0.4754). Thus, the C57BL/6 mice failed to generate long-term IP memory.
We then assessed IP memory in the mouse strains. Here, 24 h after the initial exposure to the objects, the animals explored the same objects again (in the same locations, i.e., re-exposure). Both groups explored the objects significantly less (Fig. 6E,F) (CBA/CaOlaHsd: F(1,20) = 35.43, P < 0.0001; C57BL/6: F(1,22) = 44.93, P < 0.0001). This reflects an habituation effect, consistent with the animals having generated a memory of the prior exposure to the objects.
A further 24 h later, the same objects underwent a reconfiguration of their location. The CBA/CaOlaHsd exhibited increased exploration behavior that is consistent with their recognition of the novelty of the spatial configuration (Fig. 6E; ANOVA, F(1,20) = 6.79, P = 0.0178). By contrast, the C57BL/6 failed to generate IP memory (Fig. 6F) (ANOVA, F(1,22) = 0.52, P = 0.4754).
Taken together, these data indicate that the C57BL/6 mice were able to engage in OR memory—a low cognitive demand task. By contrast, when the cognitive demands of the task were higher (IP learning), this mouse strain showed impairments compared to CBA/CaOlaHsd mice.
Discussion
The results of this study show that gradual hearing loss in adult mice is accompanied by extensive reorganization of plasticity-related neurotransmitter expression in the cortex and hippocampus, and is accompanied by a potent debilitation of hippocampal synaptic plasticity, as well as memory impairments. Effects are progressive: whereas at 2 months of age mice show no ostensible functional changes in the hippocampus, by 4 months hippocampal LTP is profoundly impaired. In line with this change, spatial memory was also debilitated. Receptor expression changes in the cortex and hippocampus also become more extensive with time. Our data suggest that even at the early stages of progressive hearing loss, the sensory deficit is sufficient to trigger cortical reorganization and to compromise hippocampal function. This finding offers novel insights into the putative causal basis of cognitive decline in age-related hearing loss in humans.
C57BL/6 mice possess the age-related hearing loss (AHL) gene (Johnson et al. 1997). Audiometric studies of C57BL/6 mice revealed that although the sound intensity thresholds required for perception of sound frequencies <10 kHz remain largely unchanged in the first year of life, the thresholds for perception of higher frequencies become progressively higher (Willott 1986; Walton et al. 1995). Deficits begin at 2 months of age (Walton et al. 2008). At 3–5 months of age, sensitivity to acoustic stimuli from 32 to 40 kHz is significantly reduced (Ohlemiller et al. 2000; Park et al. 2010); at 6 months, they exhibit mid-to-high frequency hearing deficits (>16 kHz) (Park et al. 2010). This progressive loss of sensitivity to high frequencies and increase of perceptual intensity thresholds are considered to effectively reflect age-related hearing loss that occurs in mature human individuals (Kazee et al. 1995; Johnson et al. 1997; Park et al. 2010; Bowl and Dawson 2015).
Loss of the ability to discriminate speech is a major debilitatory aspect of presbycusis in human individuals (Morton 1991; Gorlin et al. 1995). Rodents rely heavily on auditory information in the form of vocalizations for both their social structures and integration, as well as the localization of salient proximal events and features (Brudzynski 2015). Rodent vocalizations predominantly occur outside the range of human hearing and extend into the ultrasound spectrum (Brudzynski 2001, 2015; Brudzynski and Pniak 2002). Given the importance of the ultrasonic and high sonic frequency ranges for rodent communication (Brudzynski and Pniak 2002), and previous studies describing cortical and subcortical reorganization following loss of vision (Feldmann et al. 2019), we suggest that the cumulative loss of hearing in C57BL/6 mice may mirror functional changes that occur during presbycusis in the human brain.
The range of human hearing extends from 20 Hz to 20 kHz, with the ear being highly tuned to speech frequencies (ca., 300–3000 Hz). Rodents hear sound frequencies in the range of 200 Hz–90 kHz (Fay 1988; Heffner et al. 2001), but their behaviorally most relevant frequencies occur in the ultrasonic (>20 kHz) range. The loss in perception of sound frequencies in the range of 32–40 kHz that has been reported in 3-month-old C57BL/6 mice was detected at sound pressure levels of 55–65 dB (Park et al. 2010). This is highly relevant on a functional level. Sound frequencies above 20 kHz are vocalized by rodents to communicate and have a sound pressure level of 65–85 dB (van der Poel et al. 1989; Brudzynski 2001; Brudzynski and Pniak 2002; Borta et al. 2005; Wöhr et al. 2005). Thus, a loss of ability to hear frequencies around 32 kHz at a sound pressure of 55–65 dB indicates that the mice can no longer perceive ultrasound vocalizations in this frequency range or higher. Rodent pups vocalize at an average frequency of ca. 40 kHz when isolated from their mothers (Schwarting and Wöhr 2012), 50 kHz vocalizations are emitted during play and tickling (Wöhr et al. 2009; Lukas and Wöhr 2015), as well as social exploration and interactions (Brudzynski and Pniak 2002) and reflect a positive affective state in rodents (Panksepp and Burgdorf 2003; Brudzynski 2015). A loss of ability of the C57BL/6 mice to perceive ultrasonic vocalizations in this range effectively means that they lose their ability to accurately interpret the social behavior of mice they encounter. This may contribute to the more restricted exploration that has been documented in adult C57BL/6 mice (Ohl et al. 2003). It could also explain why a variety of behaviors that are consistent with elevated anxiety are evident in this mouse strain, even though basal levels of corticosterone and its exploration behavior in bright light suggest that this strain is not intrinsically anxious (Trullas and Skolnick 1993).
We detected a significant increase in the expression of GABAB receptors in the auditory cortex in 2-month-old C57BL/6 mice. This change is consistent with a disinhibition of neural circuitry (Chalifoux and Carter 2011), as has been described in neurophysiological studies of the adaptive response of the auditory cortex to loss of sensorineural input strength (Kotak et al. 2005; Mowery et al. 2015). GluN2A subunit expression was increased in the auditory cortex, suggesting a shift in GluN2A/GluN2B ratio that would subserve a reduction in the threshold for induction of synaptic plasticity in this structure. GluN2A and mGlu2/3 receptor expression were increased in the visual and parietal cortex, suggesting that functional reorganization is already underway (Sammons and Keck 2015) at this early and initial states of sensory loss.
At 4 months of age, a time-point where significant deficits in detection of behaviorally salient auditory frequencies are evident (Park et al. 2010); further changes in receptor expression had developed throughout the cortex. Here, elevations in GluN2A were evident in the piriform and somatosensory cortices, whereas the elevations seen in the posterior parietal, auditory, and visual cortices at 2 months were no longer present. In addition, we detected an elevation of mGlu1 receptor expression in the auditory and somatosensory cortices and of mGlu2/3 expression in the piriform and auditory cortices. Strikingly, GluN2B expression was now significantly elevated in all cortical structures. The changes in the primary and parietal cortices are likely to reflect adaptive reorganization: A prolongation of the critical period results in a sustainment of the high expression of GluN2B subunits in the visual cortex (Quinlan et al. 1999), which is a typical characteristic of this period of cortical development (Lee et al. 2015; Isoo et al. 2016). Treatment of rat pups with white noise results in a similar effect (Hogsden and Dringenberg 2009a). Elevations in GluN2B expression or functions in primary visual or auditory cortices in early postnatal development are a characteristic of increased propensity toward synaptic plasticity (Speechley et al. 2007; Hogsden and Dringenberg 2009b) that declines once ocular dominance or tonotopy has become established (Hubel and Wiesel 1970; Kato et al. 1991; Kirkwood et al. 1995; Lehmann and Löwel 2008; Hogsden and Dringenberg 2009a, 2009b; Jang et al. 2009). The elevations of mGlu1 and mGlu2/3 expression in the auditory cortex can be expected to result in changes in excitability: mGlu1 receptors are expressed on neurons and interneurons and modulate both GABA and glutamate release (Cozzi et al. 2002) and mGlu2/3 receptors are autoreceptors for glutamate that negatively modulate its presynaptic release (Mukherjee and Manahan-Vaughan 2013). Thus, the net effect of the changes we detected in the expression of these receptors in the auditory cortex is likely to amount to altered excitatory tonus. Taken together, the changes in receptor expression and in particular the elevation of GluN2B expression throughout the cortex indicate that massive functional reorganization is already underway in 4-month-old C57BL/6 mice. Cortical disinhibition impairs cognitive flexibility in human subjects (Gruber et al. 2010) and is likely to have contributed to the behavioral impairments we detected in hearing-impaired mice.
Receptor expression in the hippocampus was only weakly affected at the 2-month time-point hippocampus, and perhaps unsurprisingly from this perspective, LTP was unaffected. A hint that the hippocampus is already becoming affected by the loss of sensitivity to high auditory frequencies is reflected in the elevation of mGlu5 receptor expression in the CA4 region/hilus, a neural compartment that receives ipsilateral and contralateral synaptic input from the dentate gyrus and extrahippocampal structures such as the septum and is a key beneficiary of hippocampal neurogenesis (Toni and Schinder 2015). Furthermore, GABAB expression is reduced in the CA1 region, although changes in excitation–inhibition balance were not detected during electrophysiological recordings from this subregion.
By the time animals are 4 months old, GluN2B expression is increased in all subfields of the hippocampus and decreased GABAB expression was now evident in the dentate gyrus and CA3 and CA4 regions. At this time-point, very potent deficits in hippocampal synaptic plasticity also became evident. The magnitude and duration of hippocampal LTP are determined by the GluN2A and GluN2B subunits of the NMDA receptor (Ballesteros et al. 2016; Dubovyk and Manahan-Vaughan 2018). Weaker and less persistent forms of LTP are enabled by GluN2A-containing NMDA receptors, whereas stronger and more persistent forms are enabled by GluN2B-containing NMDA receptors (Ballesteros et al. 2016). The mGlu receptors, mGlu1 and mGlu5, are extremely important for the stability and maintenance of multiple forms of hippocampal synaptic plasticity (Mukherjee and Manahan-Vaughan 2013), whereas mGlu2/3 plays an important role in the maintenance of long-term depression (Manahan-Vaughan 1997; Poschel et al. 2005) and in autoreceptor-mediated regulation of presynaptic glutamate release (Mukherjee and Manahan-Vaughan 2013).
GABAA and GABAB receptors contribute importantly to the regulation of excitation–inhibition balance (Fritschy 2008), information transfer by means of neuronal oscillations (Pelkey et al. 2017), and the dynamic control of the magnitude and direction of change of synaptic strength (Lehmann et al. 2012). The decrease in GABAB expression, at 4 months postnatally, is likely to cause a decrease in basal excitatory tonus through disinhibition of GABAergic terminals (Cozzi et al. 2002). But the voltage-dependent Mg2+-block of GluN2B-containing NMDA receptors requires a strong membrane depolarization (Erreger et al. 2005; Clarke et al. 2013) and correspondingly, the threshold for inducing GluN2B-dependent LTP is higher (Berberich et al. 2005, 2007; Köhr et al. 2003; Ballesteros et al. 2016). The decrease in GABAB expression may thus explain why LTP is impaired in 4–month-old C57BL/6 mice despite elevations in expression of GluN2B.
Spatial learning deficits have been reported in C57BL/6 mice (Ohl et al. 2003; Park et al. 2016). The deficits in hippocampal synaptic plasticity that we observed in the present study can be expected to contribute to these deficits, given the tight interconnection between hippocampal synaptic plasticity and hippocampus-dependent learning (Kemp and Manahan-Vaughan 2007, 2008). Although learning about spatially configured auditory information supported hippocampal encoding of spatial experience through synaptic plasticity (Dietz and Manahan-Vaughan 2017), the hippocampus can use the visual and olfactory modalities to encode space (Kemp and Manahan-Vaughan 2008; André and Manahan-Vaughan 2014). Thus, the loss of auditory input per se is not likely to be the primary reason why hippocampal synaptic plasticity or spatial learning is impaired in the C57BL/6 mice. It seems more likely that cortical reorganization and the associated changes in neurotransmitter receptor expression drive the deficits. Here, C57BL/6 mice were capable of learning the cognitively demanding task (object recognition) but were impaired at 4 months in acquiring item-place memory, a task that is cognitively more challenging.
Presbycusis is associated with cognitive impairment and dementia (Fortunato et al. 2016; Golub 2017; Thomson et al. 2017). To what extent effects are causal is unclear, although studies of spatial learning in noise-challenged C57BL/6 mice indicate that hearing loss may lead to cognitive impairments (Park et al. 2016). Our data indicate that cortical adaptation to the loss of the ability of the C57BL/6 mice to perceive high sound frequencies even at a relatively young age is massively disruptive to hippocampal function. We propose that an impoverishment of functional encoding of learning experience by means of synaptic plasticity that is brought about by hearing loss serves to amplify or indeed accelerate emerging deficits in cognition that are triggered by neurodegeneration or age-related cognitive decline.
Digressing briefly from the topic of presbycusis and its functional basis, one important point that one should mention here, is that this issue is not only highly pertinent to our understanding of how the brain copes with and adapts to sensory loss but also highly pertinent for studies of the mechanisms underlying memory and cognition, given the widespread establishment of the C57BL/6 mouse as the “substrain” for transgenic mouse lines and the inbred strain for studies of the cellular and behavioral basis of memory encoding in mice. This mouse strain is not alone in developing cumulative hearing deficits: 129/J (Zheng et al. 1999), A/J (Zheng et al. 1999), BALB/c (Erway et al. 1993; Willott et al. 1998), CBA/CaJ (May et al. 2006), DBA/2J (Erway et al. 1993; Liu et al. 2019), and LP/J (Steel et al. 1987) mice all exhibit age-dependent loss of hearing, and the CBA/J mouse that has been used for decades in multiple studies as a health control for scrutiny of hearing loss (Henry and McGinn 1992; Henry et al. 2004) is blind (Feldmann et al. 2019). This hereditary form of blindness, which becomes manifest soon after birth, also results in massive changes in receptor expression across the brain and impairs hippocampal synaptic plasticity (Feldmann et al. 2019). CBA/J mice also become deaf in old age (Henry et al. 2004; Sha et al. 2008). The results of the present study indicate that studies of hippocampal synaptic plasticity in C57BL/6 and other strains of hearing-impaired mice may generate an outcome that is confounded by impoverished hippocampal function.
In conclusion, this study shows that progressive hearing loss triggers extensive reorganization of both sensory and association cortices at the level of expression of plasticity-related neurotransmitter receptors and demonstrates that, in parallel, hippocampal function is compromised. This finding is in line with previous reports that the first months of adaptation to blindness profoundly affect hippocampal synaptic plasticity and hippocampus-dependent learning ability (Feldmann et al. 2019). This study offers three very important take-home messages: Firstly, our results, taken together with the results of the abovementioned study (Feldmann et al. 2019), indicate how vulnerable the hippocampus is to changes in function or in sensory information processing at the cortical level. Secondly, from the perspective of basic research, the C57BL/6 mouse, or indeed any mouse strain that exhibits cumulative deafness or another kind of sensory deficit, is not an ideal subject for long-term studies of brain function, given that deafness-related changes in sensory and hippocampal information processing can be expected to confound the interpretation of results. Thirdly, given that cumulative loss of hearing is a typical feature of aging in humans (Morton 1991; Gorlin et al. 1995), our results suggest that age-related hearing loss may be a contributor to loss of function of the hippocampus that occurs with age.
Funding
German Research Foundation (DFG) (grant SFB 874/B1, project no. 122679504 to D.M.V.).
Notes
We are very grateful to Ute Neubacher, Jens Colitti-Klausnitzer, and Beate Krenzek for technical support and to Nadine Kollosch and Petra Küsener for animal care. Conflict of Interest: None declared.
Author Contributions
D.M.-V. developed the concept and strategy of the study, interpreted the data, and wrote the paper. Immunohistochemistry was conducted and analyzed by M.F. Electrophysiology were conducted and analyzed by D.B. and O.S. Behavioral experiments were conducted and analyzed by O.S. Figures were prepared by M.F., D.B., and O.S.
References
- André ME, Manahan-Vaughan D. 2014. Spatial olfactory learning facilitates long-term depression in the hippocampus. Hippocampus. 23:963–968. [DOI] [PubMed] [Google Scholar]
- Attems J, Walker L, Jellinger KA. 2015. Olfaction and aging: a mini-review. Gerontology. 61(6):485–490. [DOI] [PubMed] [Google Scholar]
- Ballesteros JJ, Buschler A, Köhr G, Manahan-Vaughan D. 2016. Afferent input selects NMDA receptor subtype to determine the persistency of hippocampal LTP in freely behaving mice. Front Behav Neurosci. 8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berberich S, Jensen V, Hvalby O, Seeburg PH, Köhr G. 2007. The role of NMDAR subtypes and charge transfer during hippocampal LTP induction. Neuropharmacology. 52:77–86. [DOI] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby O, Köhr G. 2005. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci. 25:6907–6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borta A, Wöhr M, Schwarting RKW. 2005. Rat ultrasonic vocalization in aversively motivated situations and the role of individual differences in anxiety-related behavior. Behav Brain Res. 166:271–280. [DOI] [PubMed] [Google Scholar]
- Bowl MR, Dawson SJ. 2015. The mouse as a model for age-related hearing loss-a mini-review. Gerontology. 61(2):149–157. [DOI] [PubMed] [Google Scholar]
- Brenowitz WD, Han F, Kukull WA, Nelson PT. 2018. Treated hypothyroidism is associated with cerebrovascular disease but not Alzheimer’s disease pathology in older adults. Neurobiol Aging. 62:64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks SP, Pask T, Jones L, Dunnett SB. 2004. Behavioural profiles of inbred mouse strains used as transgenic backgrounds. I: motor tests. Genes Brain Behav. 3:206–215. [DOI] [PubMed] [Google Scholar]
- Brudzynski SM. 2001. Pharmacological and behavioral characteristics of 22 kHz alarm calls in rats. Neurosci Biobehav Rev. 25:611–617. [DOI] [PubMed] [Google Scholar]
- Brudzynski SM. 2015. Pharmacology of ultrasonic vocalizations in adult rats: significance, call classification and neural substrate. Curr Neuropharmacol. 13(2):180–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudzynski SM, Pniak A. 2002. Social contacts and production of 50-kHz short ultrasonic calls in adult rats. J Comp Psychol. 116:73–82. [DOI] [PubMed] [Google Scholar]
- Chalifoux JR, Carter AG. 2011. GABAB receptor modulation of synaptic function. Curr Opin Neurobiol. 21(2):339–344. doi: 10.1016/j.conb.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke RJ, Johnson JW. 2013. NMDA receptor NR2 subunit dependence of the slow component of magnesium unblock. J Neurosci. 26:5825–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LG, Weeks RA, Sadato N, Celnik P, Ishii K, Hallett M. 1999. Period of susceptibility for cross-modal plasticity in the blind. Ann Neurol. 45:451–460. [DOI] [PubMed] [Google Scholar]
- Cozzi A, Meli E, Carlà V, Pellicciari R, Moroni F, Pellegrini-Gimpietro DE. 2002. Metabotropic glutamate 1 (mGlu1) receptor antagonists enhance GABAergic neurotransmission: a mechanism for the attenuation of post-ischemic injury and epileptiform activity? Neuropharmacology. 43(2):119–130. [DOI] [PubMed] [Google Scholar]
- Deal JA, Albert MS, Arnold M, Bangdiwala SI, Chisolm T, Davis S, Eddins A, Glynn NW, Goman AM, Minotti M, et al. . 2017. A randomized feasibility pilot trial of hearing treatment for reducing cognitive decline: results from the aging and cognitive health evaluation in elders pilot study. Alzheimers Dement (N Y). 3(3):410–415. doi: 10.1016/j.trci.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Campo HM, Measor KR, Razak KA. 2012. Parvalbumin immunoreactivity in the auditory cortex of a mouse model of presbycusis. Hear Res. 294(1–2):31–39. [DOI] [PubMed] [Google Scholar]
- Dietz B, Manahan-Vaughan D. 2017. Hippocampal long-term depression is facilitated by the acquisition and updating of memory of spatial auditory content and requires mGlu5 activation. Neuropharmacology. 115:30–41. doi: 10.1016/j.neuropharm.2016.02.026. [DOI] [PubMed] [Google Scholar]
- Draht F, Zhang S, Rayan A, Schönfeld F, Wiskott L, Manahan-Vaughan D. 2017. Directionality determines place cell activity as a function of learning. Front Behav Neurosci. 11:92. doi: 10.3389/fnbeh.2017.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubovyk V, Manahan-Vaughan D. 2018. Gradient in expression of synaptic plasticity and plasticity-related neurotransmitter receptors along the hippocampal dorso-ventral axis. Hippocampus. 28:136–150. doi: 10.1002/hipo.22816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erreger K, Dravid SM, Banke TG, Wyllie DJA, Traynelis SF. 2005. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol. 563:345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erway LC, Willott JF, Archer JR, Harrison DE. 1993. Genetics of age-related hearing loss in mice: I. Inbred and F1 hybrid strains. Hear Res. 65(1–2):125–132. [DOI] [PubMed] [Google Scholar]
- Eysel UT, Schweigart G, Mittmann T, Eyding D, Qu Y, Vandesande F, Orban G, Arckens L. 1999. Reorganization in the visual cortex after retinal and cortical damage. Restor Neurol Neurosci. 15(2, 3):153–164. [PubMed] [Google Scholar]
- Fay RR. 1988. Comparative psychoacoustics. Hear Res. 34(3):295–305. [DOI] [PubMed] [Google Scholar]
- Feldmann M, Beckmann D, Eysel U, Manahan-Vaughan D. 2019. Early loss of vision results in extensive reorganisation of plasticity-related receptors and alterations in hippocampal function. Cereb Cortex. 29(2):892–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehler K, Rösler F. 2010. Plasticity of multisensory dorsal stream functions: evidence from congenitally blind and sighted adults. Restor Neurol Neurosci. 28(2):193–205. [DOI] [PubMed] [Google Scholar]
- Fischer ME, Cruickshanks KJ, Schubert CR, Pinto AA, Carlsson CM, Klein BE, Klein R, Tweed TS. 2016. Age-related sensory impairments and risk of cognitive impairment. J Am Geriatr Soc. 64:1981–1987. doi: 10.1111/jgs.14308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin M, Voss P, Lord C, Lassonde M, Pruessner J, Saint-Amour D, Rainville C, Lepore F. 2008. Wayfinding in the blind: larger hippocampal volume and supranormal spatial navigation. Brain. 131(Pt 11):2995–2005. [DOI] [PubMed] [Google Scholar]
- Fortunato S, Forli F, Gugliemli V, De Corso E, Paludetti G, Berrettini S, Fetoni AR. 2016. A review of new insights on the association between hearing loss and cognitive decline in ageing. Acta Otorhinolaryngol Ital. 36(3):155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. 2008. The mouse brain in stereotaxic coordinates. San Diego (CA):Academic Press; ISBN-13: 978-0123742445. [Google Scholar]
- Fritschy JM. 2008. Epilepsy, E/I balance and GABA(A) receptor plasticity. Front Mol Neurosci. 1:5. doi: 10.3389/neuro.02.005.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel A, Lee HK. 2007. Persistence of experience-induced homeostatic synaptic plasticity through adulthood in superficial layers of mouse visual cortex. J Neurosci. 27(25):6692–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh JJ, Manahan-Vaughan D. 2013. Endogenous hippocampal LTD that is enabled by spatial object recognition requires activation of NMDA receptors and the metabotropic glutamate receptor, mGlu5. Hippocampus. 23(2):129–138. [DOI] [PubMed] [Google Scholar]
- Golub JS. 2017. Brain changes associated with age-related hearing loss. Curr Opin Otolaryngol Head Neck Surg. 25(5):347–352. [DOI] [PubMed] [Google Scholar]
- Gorlin RJ, Toriello HV, Cohen MM. 1995. In: Motulsky AG, Bobrow M, Harper PS, Scriver C, editors. Hereditary Hearing Loss and Its Syndromes. Vol 28 New York (NY): Oxford University Press. [Google Scholar]
- Grube D. 2004. Constants and variables in immunohistochemistry. Arch Histol Cytol. 67:115–134. [DOI] [PubMed] [Google Scholar]
- Gruber AJ, Calhoon GG, Shusterman I, Schoenbaum G, Roesch MR, O'Donnell P. 2010. More is less: a disinhibited prefrontal cortex impairs cognitive flexibility. J Neurosci. 30(50):17102–17110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckman JJ, Proville R, Heckman GJ, Azarfar A, Celikel T, Englitz B. 2017. High-precision spatial localization of mouse vocalizations during social interaction. Sci Rep. 7:3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffner RS, Koay G, Heffner HE. 2001. Audiograms of five species of rodents: implications for the evolution of hearing and the perception of pitch. Hear Res. 157(1–2):138–152. [DOI] [PubMed] [Google Scholar]
- Henry J, Petrides M, St-Laurent M, Sziklas V. 2004. Spatial conditional associative learning: effects of thalamo-hippocampal disconnection in rats. Neuroreport. 15(15):2427–2431. [DOI] [PubMed] [Google Scholar]
- Henry KR, Chole RA. 1980. Genotypic differences in behavioral, physiological and anatomical expressions of age-related hearing loss in the laboratory mouse: original papers travaux originaux. Audiology. 19(5):369–383. [DOI] [PubMed] [Google Scholar]
- Henry KR, McGinn MD. 1992. The mouse as a model for human audition. Audiology. 31(4):181–189. [DOI] [PubMed] [Google Scholar]
- Heras A, Roach C, Key M. 1995. Enhanced polymer detection system for immunohistochemistry. Mod Pathol. 8:165A.7777478 [Google Scholar]
- Hogsden JL, Dringenberg HC. 2009a. NR2B subunit-dependent long-term potentiation enhancement in the rat cortical auditory system in vivo following masking of patterned auditory input by white noise exposure during early postnatal life. Eur J Neurosci. 30(3):376–384. [DOI] [PubMed] [Google Scholar]
- Hogsden JL, Dringenberg HC. 2009b. Decline of long-term potentiation (LTP) in the rat auditory cortex in vivo during postnatal life: involvement of NR2B subunits. Brain Res. 1283:25–33. [DOI] [PubMed] [Google Scholar]
- Hubel DH, Wiesel TN. 1970. The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J Physiol. 206(2):419–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoo N, Ohno T, Isowaki M, Fukuda S, Murabe N, Mizukami H, Ozawa K, Mishina M, Sakurai M. 2016. The decline in synaptic GluN2B and rise in inhibitory neurotransmission determine the end of a critical period. Sci Rep. 6:34196. doi: 10.1038/srep34196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iura Y, Udo H. 2014. Behavioral analyses of visually impaired Crx knockout mice revealed sensory compensation in exploratory activities on elevated platforms. Behav Brain Res. 258:1–7. doi: 10.1016/j.bbr.2013.10.020. [DOI] [PubMed] [Google Scholar]
- Jang HJ, Cho KH, Kim HS, Hahn SJ, Kim MS, Rhie DJ. 2009. Age-dependent decline in supragranular long-term synaptic plasticity by increased inhibition during the critical period in the rat primary visual cortex. J Neurophysiol. 101:269–275. [DOI] [PubMed] [Google Scholar]
- Jeffery KJ. 2018. The hippocampus: from memory, to map to memory map. Trends Neurosci. 41(2):64–66. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Erway LC, Cook SA, Willott JF, Zheng QY. 1997. A major gene affecting age-related hearing loss in C57BL/6J mice. Hear Res. 114(1–2):83–92. [DOI] [PubMed] [Google Scholar]
- Kato N, Artola A, Singer W. 1991. Developmental changes in the susceptibility to long-term potentiation of neurones in rat visual cortex slices. Dev Brain Res. 60:43–50. [DOI] [PubMed] [Google Scholar]
- Kazee AM, Han LY, Spongr VP, Walton JP, Salvi RJ, Flood DG. 1995. Synaptic loss in the central nucleus of the inferior colliculus correlates with sensorineural hearing loss in the C57BL/6 mouse model of presbycusis. Hear Res. 89(1–2):109–120. [DOI] [PubMed] [Google Scholar]
- Kemp A, Manahan-Vaughan D. 2004. Hippocampal long-term depression and long-term potentiation encode different aspects of novelty acquisition. Proc Natl Acad Sci USA. 101:8192–8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp A, Manahan-Vaughan D. 2007. Hippocampal long-term depression: master or minion of declarative memory processes. Trends Neurosci. 30:111–118. [DOI] [PubMed] [Google Scholar]
- Kemp A, Manahan-Vaughan D. 2008. The hippocampal CA1 region and dentate gyrus differentiate between environmental and spatial feature encoding through long-term depression. Cereb Cortex. 18:968–977. [DOI] [PubMed] [Google Scholar]
- Kemp A, Manahan-Vaughan D. 2012. Passive spatial perception facilitates the expression of pesistent hippocampal long-term depression. Cereb Cortex. 22:1614–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood A, Lee HK, Bear MF. 1995. Co-regulation of long-term potentiation and experience-dependent synaptic plasticity in visual cortex by age and experience. Nature. 375:328–331. [DOI] [PubMed] [Google Scholar]
- Köhr G, Jensen V, Koester HJ, Mihaljevic AL, Utvik JK, Kvello A, Ottersen OP, Seeburg PH, Sprengel R, Hvalby O. 2003. Intracellular domains of NMDA receptor subtypes are determinants for long-term potentiation induction. J Neurosci. 23:10791–10799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotak VC, Fujisawa S, Lee FA, Karthikeyan O, Aoki C, Sanes DH. 2005. Hearing loss raises excitability in the auditory cortex. J Neurosci. 25:3908–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujala T, Alho K, Kekoni J, Hämäläinen H, Reinikainen K, Salonen O, Standertskjöld-Nordenstam CG, Näätänen R. 1995. Auditory and somatosensory event-related brain potentials in early blind humans. Exp Brain Res. 104(3):519–526. [DOI] [PubMed] [Google Scholar]
- Lee C, Joo K, Kim MJ, Rhie DJ, Jang HJ. 2015. GluN2B-containing N-methyl-D-aspartate receptors compensate for the inhibitory control of synaptic plasticity during the early critical period in the rat visual cortex. J Neurosci Res. 93(9):1405–1412. doi: 10.1002/jnr.23604. [DOI] [PubMed] [Google Scholar]
- Lehmann K, Löwel S. 2008. Age-dependent ocular dominance plasticity in adult mice. PLoS One. 3:e3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann K, Steinecke A, Bolz J. 2012. GABA through the ages: regulation of cortical function and plasticity by inhibitory interneurons. Neural Plast. 2012:892784. doi: 10.1155/2012/892784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin VYW, Black SE. 2017. Linking deafness and dementia: challenges and opportunities. Otol Neurotol. 38(8):e237–e239. doi: 10.1097/MAO.0000000000001408. [DOI] [PubMed] [Google Scholar]
- Lindner MD, Plone MA, Schallert T, Emerich DF. 1997. Blind rats are not profoundly impaired in the reference memory Morris water maze and cannot be clearly discriminated from rats with cognitive deficits in the cued platform task. Brain Res Cogn Brain Res. 5(4):329–333. [DOI] [PubMed] [Google Scholar]
- Liu X, Xie Y, Huang S, Xu A, Zhao M, Kang X, Yan A, Li P, Jin C, Han F. 2019. Characterization of the early pathology of cochlear stereocilia in four inbred mouse strains with progressive hearing loss. Histol Histopathol. 34(7):811–820. doi: 10.14670/HH-18-086. [DOI] [PubMed] [Google Scholar]
- Lukas M, Wöhr M. 2015. Endogenous vasopressin, innate anxiety, and the emission of pro-social 50-kHz ultrasonic vocalizations during social play behavior in juvenile rats. Psychoneuroendocrinology. 56:35–44. [DOI] [PubMed] [Google Scholar]
- Manahan-Vaughan D. 1997. Group 1 and 2 metabotropic glutamate receptors play differential roles in hippocampal long-term depression and long-term potentiation in freely moving rats. J Neurosci. 17:3293–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manahan-Vaughan D, Braunewell K-H. 1999. Novelty acquisition is associated with induction of hippocampal long-term depression. Proc Natl Acad Sci USA. 96:8739–8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May BJ, Kimar S, Prosen CA. 2006. Auditory filter shapes of CBA/CaJ mice: behavioral assessments. J Acoust Soc Am. 120(1):321–330. [DOI] [PubMed] [Google Scholar]
- Merabet LB, Battelli L, Obretenova S, Maguire S, Meijer P, Pascual-Leone A. 2009. Functional recruitment of visual cortex for sound encoded object identification in the blind. Neuroreport. 20(2):132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikaelian DO. 1979. Development and degeneration of hearing in the C57/b16 mouse: relation of electrophysiologic responses from the round window and cochlear nucleus to cochlear anatomy and behavioral responses. Laryngoscope. 89(1):1–15. [DOI] [PubMed] [Google Scholar]
- Morton NE. 1991. Genetic epidemiology of hearing impairment. Ann N Y Acad Sci. 630:16–31. [DOI] [PubMed] [Google Scholar]
- Mowery TM, Kotak VC, Sanes DH. 2015. Transient hearing loss within a critical period causes persistent changes to cellular properties in adult auditory cortex. Cereb Cortex. 25:2083–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Manahan-Vaughan D. 2013. Role of metabotropic glutamate receptors in persistent forms of hippocampal plasticity and learning. Neuropharmacology. 66:65–81. [DOI] [PubMed] [Google Scholar]
- Norimoto H, Ikegaya Y. 2015. Visual cortical prosthesis with a geomagnetic compass restores spatial navigation in blind rats. Curr Biol. 25(8):1091–1095. [DOI] [PubMed] [Google Scholar]
- Ohl FW, Deliano M, Scheich H, Freeman WJ. 2003. Analysis of evoked and emergent patterns of stimulus-related auditory cortical activity. Rev Neurosci. 14(1–2):35–42. [DOI] [PubMed] [Google Scholar]
- Ohlemiller KK, Wright JS, Heidbreder AF. 2000. Vulnerability to noise-induced hearing loss in ‘middle-aged’ and young adult mice: a dose–response approach in CBA, C57BL, and BALB inbred strains. Hear Res. 149(1–2):239–247. [DOI] [PubMed] [Google Scholar]
- Osumi Y, Shibata SB, Kanda S, Yagi M, Ooka H, Shimano T, Asako M, Kawamoto K, Kuriyama H, Inoue T, et al. . 2012. Downregulation of N-methyl-D-aspartate receptor ζ1 subunit (GluN1) gene in inferior colliculus with aging. Brain Res. 1454:23–32. [DOI] [PubMed] [Google Scholar]
- Panksepp J, Burgdorf J. 2003. "Laughing" rats and the evolutionary antecedents of human joy? Physiol Behav. 79:533–547. [DOI] [PubMed] [Google Scholar]
- Park SN, Back SA, Park KH, Kim DK, Park SY, Oh JH, Park YS, Yeo SW. 2010. Comparison of cochlear morphology and apoptosis in mouse models of presbycusis. Clin Exp Otorhinolaryngol. 3(3):126–135. doi: 10.3342/ceo.2010.3.3.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Kim MJ, Sikandaner H, Kim DK, Yeo SW, Park SN. 2016. A causal relationship between hearing loss and cognitive impairment. Acta Otolaryngol. 136(5):480–483. doi: 10.3109/00016489.2015.1130857. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, McBain CJ. 2017. Hippocampal GABAergic inhibitory interneurons. Physiol Rev. 97(4):1619–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrus E, Rodriguez G, Patterson R, Connor B, Kanold PO, Lee HK. 2015. Vision loss shifts the balance of feedforward and intracortical circuits in opposite directions in mouse primary auditory and visual cortices. J Neurosci. 35(23):8790–8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poschel B, Wroblewska B, Heinemann U, Manahan-Vaughan D. 2005. The metabotropic glutamate receptor mGluR3 is critically required for hippocampal long-term depression and modulates long-term potentiation in the dentate gyrus of freely moving rats. Cereb Cortex. 15:1414–1423. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Olstein DH, Bear MF. 1999. Bidirectional, experience dependent regulation of N-methyl-d-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc Natl Acad Sci USA. 96:12876–12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röder B, Teder-Sälejärvi W, Sterr A, Rösler F, Hillyard SA, Neville HJ. 1999. Improved auditory spatial tuning in blind humans. Nature. 400(6740):162. [DOI] [PubMed] [Google Scholar]
- Sadato N, Pascual-Leone A, Grafman J, Ibañez V, Deiber MP, Dold G, Hallett M. 1996. Activation of the primary visual cortex by braille reading in blind subjects. Nature. 380(6574):526. [DOI] [PubMed] [Google Scholar]
- Sammons RP, Keck T. 2015. Adult plasticity and cortical reorganization after peripheral lesions. Curr Opin Neurobiol. 35:136–141. [DOI] [PubMed] [Google Scholar]
- Save E, Nerad L, Poucet B. 2000. Contribution of multiple sensory information to place field stability in hippocampal place cells. Hippocampus. 10:64–76. [DOI] [PubMed] [Google Scholar]
- Schwarting RKW, Wöhr M. 2012. On the relationships between ultrasonic calling and anxiety-related behavior in rats. Braz J Med Biol Res. 45(4):337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha SH, Kanicki A, Dootz G, Talaska AE, Halsey K, Dolan D, Altschuler R, Schacht J. 2008. Age-related auditory pathology in the CBA/J mouse. Hear Res. 243(1–2):87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shnerson A, Pujol R. 1983. Development: anatomy, electrophysiology and behavior In: Willott JF, editor. The Auditory Psychobiology of the Mouse. Springfield (IL): Thomas, pp. 395–425. [Google Scholar]
- Speechley WJ, Hogsden JL, Dringenberg HC. 2007. Continuous white noise exposure during and after auditory critical period differentially alters bidirectional thalamocortical plasticity in rat auditory cortex in vivo. Eur J Neurosci. 26(9):2576–2584. [DOI] [PubMed] [Google Scholar]
- Spongr VP, Flood DG, Frisina RD, Salvi RJ. 1997. Quantitative measures of hair cell loss in CBA and C57BL/6 mice throughout their life spans. J Acoust Soc Am. 101(6):3546–3553. [DOI] [PubMed] [Google Scholar]
- Steel KP, Moorjani P, Bock GR. 1987. Mixed conductive and sensorineural hearing loss in LP/J mice. Hear Res. 28(2–3):227–236. [DOI] [PubMed] [Google Scholar]
- Su P, Hsu CC, Lin HC, Huang WS, Yang TL, Hsu WT, Lin CL, Hsu CY, Chang KH, Hsu YC. 2017. Age-related hearing loss and dementia: a 10-year national population-based study. Eur Arch Otorhinolaryngol. 274(5):2327–2334. doi: 10.1007/s00405-017-4471-5. [DOI] [PubMed] [Google Scholar]
- Teipel S, Drzezga A, Grothe MJ, Barthel H, Chételat G, Schuff N, Skudlarski P, Cavedo E, Frisoni GB, Hoffmann W, et al. . 2015. Multimodal imaging in Alzheimer's disease: validity and usefulness for early detection. Lancet Neurol. 14(10):1037–1053. [DOI] [PubMed] [Google Scholar]
- Thomson RS, Auduong P, Miller AT, Gurgel RK. 2017. Hearing loss as a risk factor for dementia: a systematic review. Laryngoscope Investig Otolaryngol. 2:69–79. doi: 10.1002/lio2.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toni N, Schinder AF. 2015. Maturation and functional integration of new granule cells into the adult hippocampus. Cold Spring Harb Perspect Biol. 8(1):a018903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trullas R, Skolnick P. 1993. Differences in fear motivated behaviors among inbred mouse strains. Psychopharmacology. 111(3):323–331. [DOI] [PubMed] [Google Scholar]
- Van Boven RW, Hamilton RH, Kauffman T, Keenan JP, Pascual–Leone A. 2000. Tactile spatial resolution in blind braille readers. Neurology. 54(12):2230–2236. [DOI] [PubMed] [Google Scholar]
- Van der Poel AM, Noach EJK, Miczek KA. 1989. Temporal patterning of ultrasonic distress calls in the adult rat: effects of morphine and benzodiazepines. Psychopharmacology. 97:147–148. [DOI] [PubMed] [Google Scholar]
- Walton JP, Frisina RD, Meierhans LR. 1995. Sensorineural hearing loss alters recovery from short-term adaptation in the C57BL/6 mouse. Hear Res. 88(1–2):19–26. [DOI] [PubMed] [Google Scholar]
- Walton JP, Barsz K, Wilson WW. 2008. Sensorineural hearing loss and neural correlates of temporal acuity in the inferior colliculus of the C57BL/6 mouse. J Assoc Res Otolaryngol. 9(1):90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks R, Horwitz B, Aziz-Sultan A, Tian B, Wessinger CM, Cohen LG, Hallett M, Rauschecker JP. 2000. A positron emission tomographic study of auditory localization in the congenitally blind. J Neurosci. 20(7):2664–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willott JF. 1986. Effects of aging, hearing loss, and anatomical location on thresholds of inferior colliculus neurons in C57BL/6 and CBA mice. J Neurophysiol. 56(2):391–408. [DOI] [PubMed] [Google Scholar]
- Willott JF, Turner JG, Carlson S, Ding D, Bross LS, Falls WA. 1998. The BALB/c mouse as an animal model for progressive sensorineural hearing loss. Hear Res. 115(1–2):162–174. [DOI] [PubMed] [Google Scholar]
- Wöhr M, Borta A, Schwarting RKW. 2005. Overt behavior and ultrasonic vocalization in a fear conditioning paradigm: a dose-response study in the rat. Neurobiol Learn Mem. 84:228–240. [DOI] [PubMed] [Google Scholar]
- Wöhr M, Kehl M, Borta A, Schänzer A, Schwarting RKW, Höglinger GU. 2009. New insights into the relationship of neurogenesis and affect: tickling induces hippocampal cell proliferation in rats emitting appetitive 50-kHz ultrasonic vocalizations. Neuroscience. 163:1024–1030. [DOI] [PubMed] [Google Scholar]
- Zhang S, Manahan-Vaughan D. 2015. Spatial olfactory learning contributes to place field formation in the hippocampus. Cereb Cortex. 25(2):423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR, Erway LC. 1999. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res. 130(1–2):94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]