

Structured Abstract
Purpose of review:
In hematopoiesis, rapid cell fate decisions are necessary for timely responses to environmental stimuli resulting in the production of diverse types of blood cells. Early studies have led to a hierarchical, tree-like view of hematopoiesis with hematopoietic stem cells (HSCs) residing at the apex and serially branching out to give rise to bipotential progenitors with increasingly restricted lineage potential. Recent single cell studies have challenged some aspects of the classical model of hematopoiesis. Here, we review the latest articles on cell fate decision in hematopoietic progenitors, highlighting single cell studies that have questioned previously established concepts and those that have reaffirmed them.
Recent findings:
The hierarchical organisation of hematopoiesis and the importance of transcription factors have been largely validated at the single cell level. In contrast, single cell studies have shown that lineage commitment is progressive rather than switch-like as originally proposed. Furthermore, the reconstruction of cell fate paths suggested the existence of a gradient of hematopoietic progenitors that are in a continuum of changing fate probabilities rather than in a static bipotential state, leading us to reconsider the notion of bipotential progenitors.
Summary:
Single cell transcriptomic and proteomic studies have transformed our view of lineage commitment and offer a drastically different perspective on hematopoiesis.
Keywords: transcription factors, chromatin, lineage bias, fate commitment, fate probability
Introduction
Deciphering the mechanism that underlies cell fate decision is central to understanding normal development and disease. In hematopoiesis, rapid cell fate decisions are necessary for timely responses to environmental stimuli (such as infection or injury) resulting in the production of appropriate amounts of diverse types of blood cells, including red blood cells, megakaryocytes and immune cells [1,2]. Pioneering studies of lineage reprogramming in the 1990s have identified transcription factors (TFs) as the main players underlying cell fate decision. For example, it was shown that ectopic expression of GATA1 at high levels in monocytes drives erythroid and megakaryocytic differentiation whereas low levels of GATA1 convert the same cells into eosinophils [3]. The central role of TFs in cell fate decision has been constantly reaffirmed ever since, through multiple in vitro and in vivo studies (reviewed in [4,5]; also see [6,7]), including a recent demonstration that adult blood cells can be reprogrammed into non-hematopoietic (neuronal) progenitors through manipulating lineage-specifying TFs (LS-TFs) [8*]. Beside TFs, chromatin structure plays a critical role in cell fate decision through restricting or facilitating the binding of TFs (reviewed in [9*] and see [10*]). While chromatin structure is initially established by TFs through the recruitment of chromatin remodeling enzymes, it is at least partially transmissible across cell divisions even in the absence of TFs, and thus can provide a memory of the action of “early” TFs in descendent cells (reviewed in [11*]).
In parallel with the studies described above, experiments combining bone marrow transplant in mice, lineage tracing and colony-forming assays have led to a hierarchical, tree-like view of hematopoiesis with self-renewing hematopoietic stem cells (HSCs) residing at the apex and serially branching out to give rise to progenitors with increasingly restricted lineage potential [12]. While some alternative differentiation paths within the hematopoietic tree have been suggested, including direct conversion of HSCs into megakaryocytes [13], the idea of a hierarchical progression remains central to the classical view of hematopoiesis [2,14*]. The concept of bipotential progenitor originates from this classical model of hematopoiesis whereby most cells (beside HSCs, precursors and terminally differentiated effector cells) are considered as “bipotential progenitors”, that is: cells that transiently accumulate prior to choosing between two fates. In this model, LS-TFs from mutually exclusive lineages are co-expressed at low levels in bipotential progenitors and quantitative changes in their relative levels tilt the balance towards one possible fate or another in a switch-like manner [5].
Recent studies using single cell approaches have challenged some (but not all) aspects of the classical model of hematopoiesis, forcing us to revisit fundamental principles underlying cell fate decision, including the very existence of bipotential progenitors. Here, we review the latest articles on cell fate decision in hematopoietic progenitors, highlighting single cell studies that have questioned previously established concepts and those that have reaffirmed them. Then, we underline an important distinction between cell fate (as measured experimentally) and cell fate potential. Finally, based on recent evidence on the role of progenitors with restricted potential, we propose a revised model of graded cell fate decision in hematopoiesis.
Text of review
1). Recent insights into cell fate decision in hematopoiesis
Recent findings in single cell studies have changed our understanding of the mechanism underlying cell fate decisions in hematopoiesis. This has led to reconsideration of the concept of bipotential progenitors. In this section, we summarize aspects of the classical model of hematopoiesis that have been reaffirmed and those that have been refined by single cell-based assays.
a). The link between cell division, self-renewal and differentiation.
The classical model of hematopoiesis posits that HSCs transition from their multipotent state to a more differentiated state through cell division, and that HSCs are the only cells capable of self-renewal. Challenging this view, fate mapping experiments in unperturbed mouse hematopoiesis showed that multipotent progenitors (MPPs) are also capable of self-renewal [15,16]. Furthermore, murine transplanted HSCs and MPPs can differentiate into restricted progenitors prior to cell division [17*] implying that intermediary progenitor states either can be bypassed, or do not require cell division. While interesting, these findings should be considered carefully in light of other studies suggesting a critical role for the cell cycle in cell fate decision, for instance through regulating the level of the lineage-specifying TF PU.1 in developing macrophages [18] or through synchronizing progenitors for terminal erythroid differentiation [19]. Also interestingly, cell cycle speed has been implicated in cell fate decision [20*]. At this point, the link between self-renewal, cell division and cell fate remains unclear and despite an undeniably important role in controlling the rate of self-renewal or that of transient amplification of progenitors, it remains to be determined whether cell division and/or cell cycle are directly involved in cell fate decision in hematopoietic progenitors.
b). Heterogeneity of the HSC and progenitor compartments, cell fate bias and the hierarchical organization of hematopoiesis.
The concept of a hierarchical, tree-like organization of hematopoiesis originates from transplant experiments in mice whereby different cell populations enriched for HSCs [21,22,23,24] or various progenitors [25,26] were shown to drive multilineage, plurilineage or monolineage reconstitution of hematopoiesis in vivo. Furthermore, this model of a structured hierarchy is strongly and independently supported by studies of native hematopoiesis that used genetic barcodes to track the fate of endogenous HSCs or progenitors in vivo [15,27,28,29**] and by single cell RNA sequencing (scRNAseq) experiments that provided snapshots of single hematopoietic stem and progenitor cells transcriptomes [29**,30,31,32*,33**,34,35*,36**,37]. Furthermore, the hierarchical organisation of hematopoiesis has been confirmed using single cell proteomic with temporal barcoding [38*]. While some scRNAseq studies originally failed to detect oligopotent and/or bipotent progenitors [39,40], re-analysis of the same data [39] with different methods [31] as well as more recent studies using either scRNAseq [29**,30,32*,33**,34,35*,36**,37,41,42**] or mass cytometry [38*] approaches have successfully detected oligopotent and bipotent progenitors. Importantly, single cell approaches have also identified alternative paths to the traditional hematopoietic tree suggesting that multiple ontogenies are possible. For example, megakaryocytes have been proposed to arise either directly from HSCs [13,29**,43,44] or from megakaryocyte-erythroid progenitors (MEPs) [33**,38*]. Another example is that of basophils that can originate from granulocyte-macrophage progenitors (GMPs) but also from an erythroid-megakaryocyte lineage branch [33**,35*,38*]. In addition, it has been suggested that monocytes can arise from at least two different routes, a monocyte-dendritic progenitor (MDP) path and a granulocyte-monocyte progenitor (GMP) path [35*,42**]. The existence of these multiple possible hematopoietic trees suggests that progenitors may be more plastic than originally thought and that fate commitment may in fact occur late in differentiation. Moreover, this is consistent with a recent observation that erythroid precursors (CFU-Es) still express low levels of TFs from non-erythroid lineages [45*].
Another interesting observation from scRNAseq studies was that the HSC and progenitors compartments are transcriptionally heterogenous [29**,30,31,32*,33**,34,35*,36**,37,39,40], which seemed to correlate with fate biases detected in single cell transplants [13,23,24,43,46,47,48,49**] and in native hematopoiesis studies [15,16,27,28,29**]. Overall, these findings were taken to suggest that transcriptomic heterogeneity reflects cell fate bias and led to the interpretation that cell fate decisions are made early, in the HSC compartment rather than later in bipotential progenitors, which seems to contradict the classical model of hematopoiesis. However, these studies have not measured cell fate and the transcriptome in the same cells, so the extent to which transcriptomic heterogeneity actually reflects lineage commitment rather than transcriptional stochasticity (that characterizes stem/progenitor cells [50**]) was not clear. To address this question, Weinreb et al. used an approach combining genetic barcoding of heterogeneous progenitor populations with scRNAseq at early and late time-points, which allowed them to correlate gene expression at one time point with fate at a later timepoint [42**]. While the results confirmed that single cell transcriptomics can reflect cell fate, the overall predictive accuracy was only 60% in vitro and 51% in vivo. Furthermore, some fate decision boundaries were not accurately identified [42**]. Thus, one must be careful in interpreting transcriptomic heterogeneity (as measured by current scRNAseq approaches) strictly as lineage bias or commitment. Interestingly, scRNAseq analysis of 60 tissues as part of the establishment of a human atlas revealed that stem and progenitor cells exhibit strong transcriptional stochasticity compared to differentiated cells [50**]. This may explain at least part of the transcriptomic heterogeneity observed in the HSC and progenitor compartments. In addition, while it has been clearly established that cells in the HCS compartment exhibit cell fate biases in specific experimental conditions, it is currently unclear how strongly these biased cells are committed to specific fate(s). For instance, Carrelha et al. found that HSPCs that appear restricted to specific lineage(s) in transplant experiments retain multipotency both in vitro when grown under appropriate conditions and in vivo upon secondary transplant [49**]. Thus, one cannot exclude that in a slightly different in vivo environment (e.g. a different genetic background) or upon external stress or injury, transplanted or endogenous stem and progenitor cells may exhibit different fates or biases. In support of this, a recent study showed that genetic modification of the bone marrow niche (through vascular-specific deletion of Notch delta like ligand DLL4) leads to a profound myeloid bias in HSCs [51**]. Interestingly, the authors also noted that vascular DLL4 in the bone marrow is downregulated upon acute stress [51**]. Thus, cell fate biases appear to depend on the environment, which suggests that HSCs may not be intrinsically committed to specific fates. In summary, even though cells within the HSC or early progenitor compartments exhibit some degree of lineage bias, this is subject to change and the cells do not appear fully committed to specific lineages. This suggests that HSCs and early progenitors remain plastic and that fate commitment may occur later in differentiation, as originally proposed in the classical model of hematopoiesis.
c). Hematopoiesis is a gradual process
One of the main principles underlying the classical model of hematopoiesis is that lineage commitment and differentiation occur in steps whereby stable progenitor populations (defined by specific combinations of cell surface markers) transition from multipotent to oligopotent to unipotent cells in a switch-like manner [2,14*]. The higher resolution offered by single cell approaches measuring either RNAs or proteins revealed that in contrast to this classical view, differentiation is a continuous process with progenitors forming a gradual landscape of cells rather than a succession of stable populations, although some cells clearly accumulate at specific locations along lineages trajectories [29**,33**,34,35*,36**,38*,41,42**,52]. Importantly, time-resolved studies [38*,42**] have confirmed that when placed in a given environment, hematopoietic progenitor cells move in a largely coordinated and unidirectional manner as they differentiate along hierarchically structured paths. Taken together, these findings suggest that loss of fate potential and lineage commitment are progressive rather than switch-like. They also imply the existence of a gradient of hematopoietic progenitors with varying degrees of multi- or oligo-potency spread along differentiation trajectories (Figure 1).
Figure 1. Model of hematopoiesis based on a continuum of changing fate probabilities.
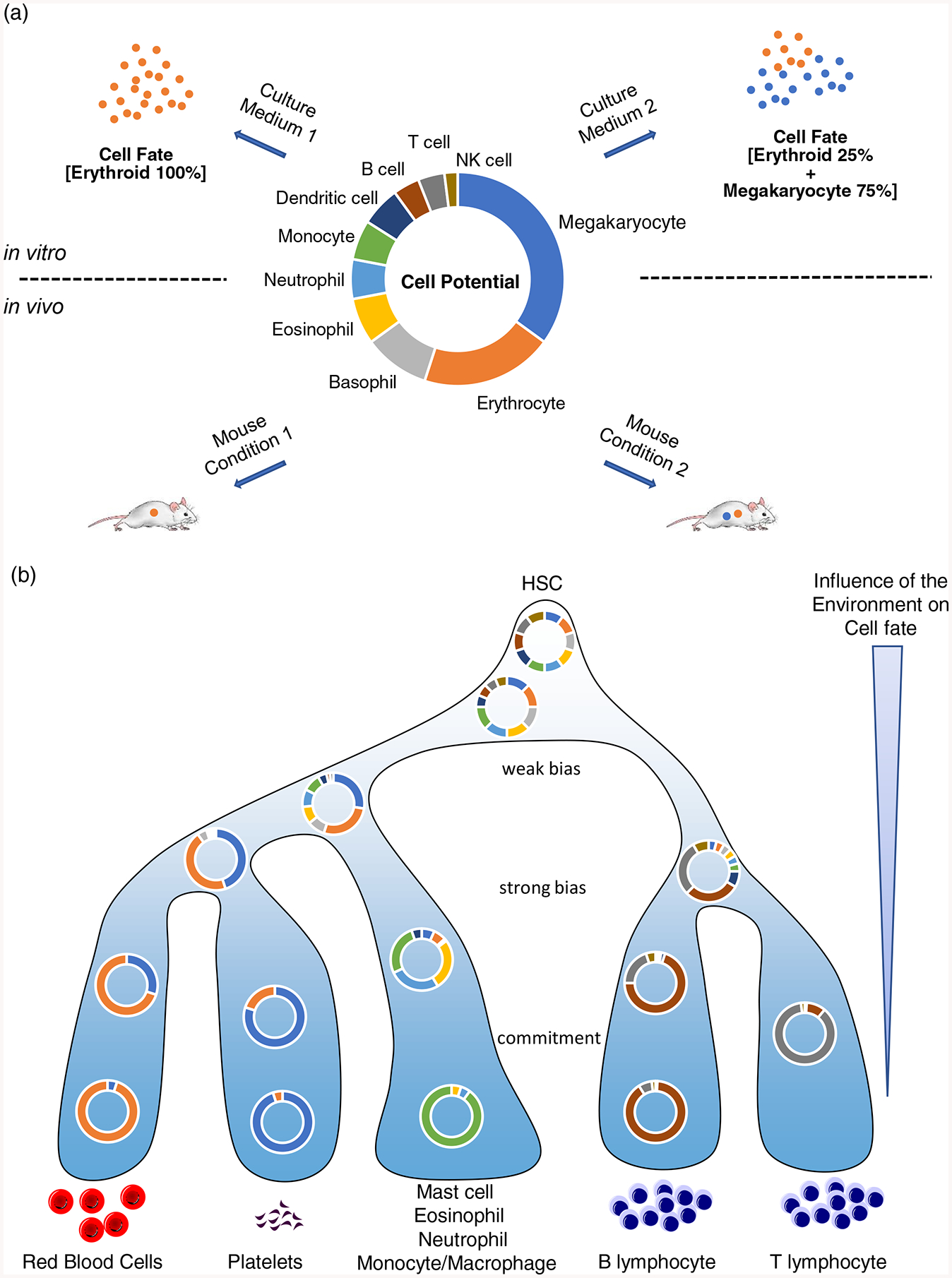
(a) A putative hematopoietic progenitor with specific cell fate potential for various lineages is shown in the middle. Depending on cell culture conditions (top) or in vivo environments (bottom) this same cell can reveal different fates. This illustrates the uncertainty of inferring cell potential from cell fate measurements, both in vitro and in vivo.
(b) In this model, hematopoiesis entails gradual changes in cell fate probabilities as progenitors progress along lineage trajectories. The extracellular environment (the niche) can alter cell fate probabilities in all progenitors but its influence gradually decreases during differentiation. In this model there are no bipotential progenitors per se, only progenitors with variable levels of restriction.
d). TFs are main drivers of cell fate decision
As the only molecules capable of reading the DNA code, sequence-specific DNA binding TFs are the main drivers of cell fate decisions [4]. The classical model of hematopoiesis proposed that LS-TFs from competing lineages are co-expressed at low levels in progenitors and that changes in their relative levels produce an imbalance leading to differentiation towards a specific lineage at the expense of another [5]. At the mechanistic level, TFs work through integration of diverse signals from the environment, which results in the establishment of gene expression programs that either maintain multipotency or drive differentiation towards specific paths [1,5,53]. These principles were recently reaffirmed by single cell analyses in embryonic cells and in embryos that have confirmed the key role of the TF TAL1 in coordinating blood, cardiac and endothelial lineages during development [54*,55**].
An important principle that emerged from studying TFs is that their function in establishing cell fate is highly dose-dependent. For example, early studies showed that in monocytes, low levels of GATA1 drive differentiation towards an eosinophilic fate whereas high levels of GATA1 drive differentiation towards an erythroid/megakaryocyte fate [3]. Along the same lines, it was shown that precise stoichiometry of specific TFs combinations is required to drive efficient erythropoiesis [7]. Furthermore, a recent study combining Perturb-seq with CRISP interference to titrate the expression of genes at single cell resolution showed that specific abundance thresholds determine the ability of TFs to modify cell behavior in hematopoietic cells [56*]. Thus, to understand the role of TFs in lineage fate decisions, one must not only determine if a TF is expressed but also measure precisely its level of expression in cells. In this regard, it is important to recognize that there are drastic differences in protein versus mRNA abundances (and dynamics) for master regulators of hematopoiesis [45*]. Furthermore, these differences are exacerbated in hematopoietic stem and progenitor cells with a Pearson correlation coefficient < 0.25 compared to 0.4–0.6 in more differentiated cells [45*]. Thus, approaches that measure proteins (not transcripts) are warranted to decipher the role of TFs in early lineage decisions. Importantly, these methods must be quantitative and provide information on protein stoichiometry to determine relative abundances of lineage-specifying TFs.
With this in mind, we recently used a combination of targeted MS and single cell mass cytometry to examine endogenous TFs in early hematopoiesis and erythropoiesis [38*]. As predicted by the classical model, we found that LS-TFs from distinct hematopoietic lineages are co-expressed in single cell progenitors [38*]. However, in contrast to the original model, TFs abundances change gradually (and not in a switch like manner) during differentiation. Interestingly, ectopic expression of a pro-megakaryocyte TF (FLI1) in early hematopoietic progenitors can overcome strong pro-erythroid environmental signals and redirect cells away from their preferred erythroid path towards a megakaryocytic lineage. This shows that TFs (when expressed above a certain threshold) establish cell fate as opposed to simply reinforce lineage commitment. Thus, even though TFs levels do not change in a switch like manner, quantitative changes in their abundances is the driving force behind cell fate decision, as originally proposed in the classical model of hematopoiesis [57]. Of note, even though TFs abundances are changing gradually during differentiation, these proteins could still function in a switch-like manner in the sense of having a measurable effect on cell fate only upon reaching a specific quantitative threshold above which they would be able to establish a stable gene expression program [56*].
In summary, some of the main aspects of the classical model of hematopoiesis have been validated by single cell studies, including the idea that hematopoiesis is hierarchically organized, and the notion that quantitative changes in LS-TFs determine cell fate in hematopoietic progenitors. On the other hand, the finding that hematopoietic progenitors gradually transition along a continuous landscape of hematopoietic trajectories not only put into question the previously proposed switch-like mechanism for cell fate decisions but also invite us to reconsider how to define hematopoietic progenitors.
2). Cell potential inference and a new model of cell fate decision in hematopoiesis
A commonly used approach to infer potential is to measure cell fate in specific in vitro or in vivo conditions. However, such studies can be misleading because: 1) experimental conditions may not be permissive to all possible fates, leading to inaccurate interpretations of experimental data pertaining to cell fate potential (Figure 1a); and 2) the external environment (i.e. the bone marrow niche or culture medium) strongly influences cell fate probabilities [49**,51**]. Below, we discuss different approaches to infer fate potential from single cell measurements and we propose a unifying model of cell fate decisions based on recent findings.
Figure 2. Proposed mechanism for regulation of cell fate potential at the molecular level.

Cell potential in hematopoietic stem/progenitor cells is defined by a combination of several factors: 1) TFs that establish gene transcription programs and introduce epigenetic marks on chromatin; 2) chromatin structure that influences TFs by preventing or promoting their binding to specific genomic locations; 3) External signals from the environment that influence TFs and chromatin structure to allow fate plasticity.
a). De novo prediction of bipotential progenitors using single cell data
The high-resolution transcriptome maps of homeostatic cells offer the possibility of reconstructing fate paths de novo. Several methods have been put forward to infer the fate of collections of cells (FateID [58], PBA [59] and Palantir [60]). These methods build on the idea that the single cell measurements represent a sampling of the path from stem cells to mature cells and therefore can be used to infer fate maps. Each of the methods works from different starting points, for instance FateID takes user-specified mature cell populations and using random forests classifiers, works iteratively backwards to identify common progenitors and fate probabilities. On the other hand, Palantir works forward from a user specified starting cell, calculates trajectories and end points and constructs a markov chain to derive the fate probabilities. PBA utilises both starting points (stem cells) and end points (mature cells) to model the cells as part of a stationary stochastic dynamical system where cells are produced at the sources (stem cells) and removed at the sinks (mature cells). Each of these methods predicts a probability map that in itself makes the idea of bipotential progenitor less clear. Indeed, within this probabilistic framework cells are in a continuum of changing fate probabilities rather than in areas with clear cutoffs (Figure 1b). So, we propose that it may be more accurate to refer to those cells as “fate restricted” progenitors rather than “bipotential” progenitors. Additionally, there is the possibility that the environment/niche can shape this landscape and the probabilities within it (Figure 1b).
While cell potential can be predicted from scRNA seq measurement, transcriptomic data alone is unlikely to encompass all the information necessary to define cell fate probabilities in progenitors. Indeed, several studies have suggested that chromatin structure and epigenetic marks (i.e. DNA methylation and histone modifications) hold key information pertaining to cell fate potential. For example, transplant experiments using color-tagged HSCs combined with measures of DNA methylation and chromatin accessibility (ATAC seq) identified stereotypical intra-clonal behaviours whereby HSPCs fate correlates well with chromatin modifications [61]. Similarly, a recent scRNA seq study that tracked ascendant-descendent cell relationships suggested some inheritance of fate potential within cell “families” [62], a feature that characterizes epigenetic mechanisms [63]. Most interestingly, using transgenic mice, Izzo et al. [64**] found that deletion of the DNA methyltransferase Dnmt3a biases the HSCs towards an erythroid fate at the expense of a monocyte/macrophage fate whereas deletion of an enzyme with the opposite function (i.e. Tet2 that mediates removal of the DNA methylation mark) has the opposite effect (i.e. biasing of HSCs towards a monocyte/macrophage fate at the expense of an erythroid fate). Taken together, these findings demonstrate that DNA methylation, an epigenetically transmitted mark, directly regulates cell fate potential. It remains to be determined whether similar effects on fate probability occur with histone modifications.
b). What is the mechanism of cell fate decision in hematopoietic progenitors?
TFs are the main drivers of cell fate decision. Mechanistically, they regulate transcription both directly through the recruitment of RNA polymerase II, and indirectly through the recruitment of chromatin remodeling enzymes that shape the chromatin landscape. Notably, chromatin structure strongly influences the function of TFs through either limiting or stabilizing TFs binding to specific genomic locations [65]. As chromatin structure is (at least partly) heritable, TFs can have a delayed effect on cell fate through establishing chromatin accessibility of descendent cells.
Based on recent evidence described above, we propose that fate potential in hematopoietic stem/progenitor cells is defined by a combination of several factors that influence each others’ dynamically: 1) TFs that establish gene transcription programs and introduce epigenetic marks on chromatin; 2) chromatin structure (partly inherited) that strongly influences TFs by preventing or promoting their binding to specific genomic locations; 3) External signals from the environment that influence TFs (for example through post-translational modifications) and chromatin structure to allow some variation in fate probabilities in response to stress or injury (Figure 2). In this model, we also propose that cell fate “decisions” occur through gradual acquisition of biases in progenitors which entail quantitative changes in LS-TFs abundances and chromatin structure modification for the establishment of increasingly restricted gene expression programs. Furthermore, the reversibility of epigenetic reactions provides an opportunity for extracellular signals to reshape the chromatin landscape at various time-points and, providing the right TFs are available at sufficient abundances, alter cell fate decisions. Furthermore, it is likely that the influence of the extracellular environment decreases as chromatin structure is becoming increasingly constrained when progenitors progress along differentiation trajectories (Figure 1b). Thus, our model is compatible both with the concept that the environment (the niche) has an important role in shaping cell fate and with the idea that early cell fate biases can be preserved (to some extent) through epigenetic memory. Again, the model implies that the influence of the environment on cell fate gradually decreases during differentiation with differentiating cells progressively losing their fate plasticity (Figure 1b).
Conclusion
In the past couple of years, single cell transcriptomic and proteomic studies have transformed our view of lineage commitment and brought a drastically different perspective on hematopoietic progenitors and their function in lineage commitment. Yet, we still know very little on the molecular mechanisms that control these cellular behaviours in single cells. And while we have begun to understand the central role of chromatin (mostly DNA methylation) in this process, the mechanism through which competing LS-TFs gradually establish and reinforce mutually exclusive gene expression programs in single progenitor cells remains mostly unknown. Quantitative approaches that measure proteins abundances in single cells combined with single cell analyses of histones and TFs genomic binding [66,67] will be invaluable to answer these fundamental questions in the near future.
Key points:
Single cell approaches give a high-resolution view of cell fate, complementing classical studies
Hematopoietic progenitors are in a continuum of changing fate probabilities suggesting it may be more accurate to refer to those cells as “fate restricted” progenitors rather than “bipotential” progenitors
Cell fate potential in progenitors is highly influenced by the extracellular environment (the niche) suggesting multiple hematopoietic trees are possible
Cell fate potential is encoded by the interplay between lineage-specifying transcription factors (LS-TFs) and chromatin structure
Cell fate “decisions” occur through gradual acquisition of biases in progenitors which entail quantitative changes in LS-TFs abundances and chromatin structure modification for the establishment of increasingly restricted gene expression programs
Acknowledgements
None.
Financial support and sponsorship
The work was supported by NIH DK098449 (M.B.), CIHR MOP89834, MOP343603 and CEEHRC Initiative (M.B.), Canadian Cancer Research Society (M.B.), and MRC Computational Biology Fellowship MC_UU_12025 (E.M.) and MRC Strategic Alliance Funding (E.M.)
Footnotes
Conflicts of interest
There are no conflicts of interest.
References
- 1.Orkin SH, Zon LI: Hematopoiesis: an evolving paradigm for stem cell biology. Cell 2008, 132:631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eaves CJ: Hematopoietic stem cells: concepts, definitions, and the new reality. Blood 2015, 125:2605–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kulessa H, Frampton J, Graf T: GATA-1 reprograms avian myelomonocytic cell lines into eosinophils, thromboblasts, and erythroblasts. Genes Dev 1995, 9:1250–1262. [DOI] [PubMed] [Google Scholar]
- 4.Orkin SH: Diversification of haematopoietic stem cells to specific lineages. Nat Rev Genet 2000, 1:57–64. [DOI] [PubMed] [Google Scholar]
- 5.Graf T, Enver T: Forcing cells to change lineages. Nature 2009, 462:587–594. [DOI] [PubMed] [Google Scholar]
- 6.Org T, Duan D, Ferrari R, et al. : Scl binds to primed enhancers in mesoderm to regulate hematopoietic and cardiac fate divergence. EMBO J 2015, 34:759–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Capellera-Garcia S, Pulecio J, Dhulipala K, et al. : Defining the Minimal Factors Required for Erythropoiesis through Direct Lineage Conversion. Cell Rep 2016, 15:2550–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8*.Thier MC, Hommerding O, Panten J, et al. : Identification of Embryonic Neural Plate Border Stem Cells and Their Generation by Direct Reprogramming from Adult Human Blood Cells. Cell Stem Cell 2019, 24:166–182 e113. [DOI] [PubMed] [Google Scholar]; This study demonstrates that TFs can reprogram adult peripheral blood cells into neural stem cells, illustrating the importance of TFs in cell fate plasticity.
- 9*.Klemm SL, Shipony Z, Greenleaf WJ: Chromatin accessibility and the regulatory epigenome. Nat Rev Genet 2019, 20:207–220. [DOI] [PubMed] [Google Scholar]; This review provides molecular insights into how TFs regulate chromatin accessibility to establish and maintain cell identity.
- 10*.Ludwig LS, Lareau CA, Bao EL, et al. : Transcriptional States and Chromatin Accessibility Underlying Human Erythropoiesis. Cell Rep 2019, 27:3228–3240 e3227. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses chromatin accessibility profiling coupled with transcriptome analysis to provide insights into human erythropoiesis.
- 11*.Brand M, Nakka K, Zhu J, et al. : Polycomb/Trithorax Antagonism: Cellular Memory in Stem Cell Fate and Function. Cell Stem Cell 2019, 24:518–533. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review examines the molecular mechanism of cell fate memory in stem cells.
- 12.Weissman IL, Anderson DJ, Gage F: Stem and progenitor cells: origins, phenotypes, lineage commitments, and transdifferentiations. Annu Rev Cell Dev Biol 2001, 17:387–403. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto R, Morita Y, Ooehara J, et al. : Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 2013, 154:1112–1126. [DOI] [PubMed] [Google Scholar]
- 14.Laurenti E, Gottgens B: From haematopoietic stem cells to complex differentiation landscapes. Nature 2018, 553:418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun J, Ramos A, Chapman B, et al. : Clonal dynamics of native haematopoiesis. Nature 2014, 514:322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Busch K, Klapproth K, Barile M, et al. : Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 2015, 518:542–546. [DOI] [PubMed] [Google Scholar]
- 17*.Grinenko T, Eugster A, Thielecke L, et al. : Hematopoietic stem cells can differentiate into restricted myeloid progenitors before cell division in mice. Nat Commun 2018, 9:1898. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that HSCs can differentiate into restricted progenitors without cell division and before entering the S phase of the cell cycle.
- 18.Kueh HY, Champhekar A, Nutt SL, et al. : Positive feedback between PU.1 and the cell cycle controls myeloid differentiation. Science 2013, 341:670–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hwang Y, Futran M, Hidalgo D, et al. : Global increase in replication fork speed during a p57(KIP2)-regulated erythroid cell fate switch. Sci Adv 2017, 3:e1700298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20*.Lu YC, Sanada C, Xavier-Ferrucio J, et al. : The Molecular Signature of Megakaryocyte-Erythroid Progenitors Reveals a Role for the Cell Cycle in Fate Specification. Cell Rep 2018, 25:2083–2093 e2084. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identifies cell cycle speed as a possible mechanism involved in cell fate specification in hematopoietic progenitors.
- 21.Smith LG, Weissman IL, Heimfeld S: Clonal analysis of hematopoietic stem-cell differentiation in vivo. Proc Natl Acad Sci U S A 1991, 88:2788–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Osawa M, Hanada K, Hamada H, et al. : Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science 1996, 273:242–245. [DOI] [PubMed] [Google Scholar]
- 23.Muller-Sieburg CE, Cho RH, Thoman M, et al. : Deterministic regulation of hematopoietic stem cell self-renewal and differentiation. Blood 2002, 100:1302–1309. [PubMed] [Google Scholar]
- 24.Dykstra B, Kent D, Bowie M, et al. : Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell 2007, 1:218–229. [DOI] [PubMed] [Google Scholar]
- 25.Kondo M, Weissman IL, Akashi K: Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell 1997, 91:661–672. [DOI] [PubMed] [Google Scholar]
- 26.Akashi K, Traver D, Miyamoto T, et al. : A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 2000, 404:193–197. [DOI] [PubMed] [Google Scholar]
- 27.Perie L, Duffy KR, Kok L, et al. : The Branching Point in Erythro-Myeloid Differentiation. Cell 2015, 163:1655–1662. [DOI] [PubMed] [Google Scholar]
- 28.Pei W, Feyerabend TB, Rossler J, et al. : Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature 2017, 548:456–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29**.Rodriguez-Fraticelli AE, Wolock SL, Weinreb CS, et al. : Clonal analysis of lineage fate in native haematopoiesis. Nature 2018, 553:212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses transposon tagging to trace the fate of hematopoietic stem and progenitor cells in unperturbed hematopoiesis
- 30.Olsson A, Venkatasubramanian M, Chaudhri VK, et al. : Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature 2016, 537:698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haghverdi L, Buttner M, Wolf FA, et al. : Diffusion pseudotime robustly reconstructs lineage branching. Nat Methods 2016, 13:845–848. [DOI] [PubMed] [Google Scholar]
- 32.Karamitros D, Stoilova B, Aboukhalil Z, et al. : Single-cell analysis reveals the continuum of human lympho-myeloid progenitor cells. Nat Immunol 2018, 19:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tusi BK, Wolock SL, Weinreb C, et al. : Population snapshots predict early haematopoietic and erythroid hierarchies. Nature 2018, 555:54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng S, Papalexi E, Butler A, et al. : Molecular transitions in early progenitors during human cord blood hematopoiesis. Mol Syst Biol 2018, 14:e8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pellin D, Loperfido M, Baricordi C, et al. : A comprehensive single cell transcriptional landscape of human hematopoietic progenitors. Nat Commun 2019, 10:2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36**.Popescu DM, Botting RA, Stephenson E, et al. : Decoding human fetal liver haematopoiesis. Nature 2019, 574:365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses scRNA seq to study the development of human fetal liver. The authors identify a change in the differentiation potential of HSCs and MPPs during gestation.
- 37.Drissen R, Thongjuea S, Theilgaard-Monch K, et al. : Identification of two distinct pathways of human myelopoiesis. Sci Immunol 2019, 4. [DOI] [PubMed] [Google Scholar]
- 38*.Palii CG, Cheng Q, Gillespie MA, et al. : Single-Cell Proteomics Reveal that Quantitative Changes in Co-expressed Lineage-Specific Transcription Factors Determine Cell Fate. Cell Stem Cell 2019, 24:812–820 e815. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses single cell mass cytometry to show that antagonist LS-TFs are co-expressed in hematopoietic progenitors and that quantitative changes in TFs levels drive cell fate.
- 39.Paul F, Arkin Y, Giladi A, et al. : Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell 2016, 164:325. [DOI] [PubMed] [Google Scholar]
- 40.Velten L, Haas SF, Raffel S, et al. : Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol 2017, 19:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dahlin JS, Hamey FK, Pijuan-Sala B, et al. : A single-cell hematopoietic landscape resolves 8 lineage trajectories and defects in Kit mutant mice. Blood 2018, 131:e1–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42**.Weinreb C, Rodriguez-Fraticelli A, Camargo FD, et al. : Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 2020, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses an approach combining genetic barcoding of heterogeneous progenitor populations with scRNAseq at early and late time-points, which allowed the authors to trace simultaneously the cells’ transcriptome and clonal relationships over time during differentiation in vitro and upon transplantation. The results confirmed that single cell transcriptomics can reflect cell fate but also highlight that mRNA levels alone (as detected by scRNA-Seq) do not encode the full heterogeneity of cell states
- 43.Sanjuan-Pla A, Macaulay IC, Jensen CT, et al. : Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature 2013, 502:232–236. [DOI] [PubMed] [Google Scholar]
- 44.Notta F, Zandi S, Takayama N, et al. : Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016, 351:aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45*.Gillespie MA, Palii CG, Sanchez-Taltavull D, et al. : Absolute Quantification of Transcription Factors Reveals Principles of Gene Regulation in Erythhropoiesis. Molecular Cell 2020, In press. 10.1016/j.molcel.2020.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]; Using targeted mass spectrometry and RNAseq, this study highlights drastic discrepancies in protein versus mRNA levels for master regulators of hematopoiesis in HSPCs.
- 46.Challen GA, Boles NC, Chambers SM, et al. : Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell 2010, 6:265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morita Y, Ema H, Nakauchi H: Heterogeneity and hierarchy within the most primitive hematopoietic stem cell compartment. J Exp Med 2010, 207:1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilson NK, Kent DG, Buettner F, et al. : Combined Single-Cell Functional and Gene Expression Analysis Resolves Heterogeneity within Stem Cell Populations. Cell Stem Cell 2015, 16:712–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49**.Carrelha J, Meng Y, Kettyle LM, et al. : Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature 2018, 554:106–111. [DOI] [PubMed] [Google Scholar]; This study uses transplant experiments to show that HSPCs that appear restricted to specific lineage(s) in transplant experiments retain multipotency both in vitro when grown under appropriate conditions and in vivo upon secondary transplant.
- 50**.Han X, Zhou Z, Fei L, et al. : Construction of a human cell landscape at single-cell level. Nature 2020. [DOI] [PubMed] [Google Scholar]; This study uses scRNA seq of 60 human tissues to demonstrate that stem and progenitor cells exhibit strong transcriptional stochasticity compared to differentiated cells
- 51**.Tikhonova AN, Dolgalev I, Hu H, et al. : The bone marrow microenvironment at single-cell resolution. Nature 2019, 569:222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses genetic modification of the bone marrow niche (through vascular-specific deletion of Notch delta like ligand DLL4) to show a profound myeloid bias in HSCs. This shows that cell fate biases depend on the environment, which suggests that HSCs may not be intrinsically committed to specific fates.
- 52.Nestorowa S, Hamey FK, Pijuan Sala B, et al. : A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 2016, 128:e20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trompouki E, Bowman TV, Lawton LN, et al. : Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration. Cell 2011, 147:577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54*.Chagraoui H, Kristiansen MS, Ruiz JP, et al. : SCL/TAL1 cooperates with Polycomb RYBP-PRC1 to suppress alternative lineages in blood-fated cells. Nat Commun 2018, 9:5375. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study implicates the recruitment of chromatin-regulatory factors by TFs to regulate cell fate decisions in embryonic hematopoiesis.
- 55**.Pijuan-Sala B, Griffiths JA, Guibentif C, et al. : A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 2019, 566:490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses scRNA seq in mouse embryos at sequential time points of development to build a map of differentiation. Furthermore, through the use of chimeric Tal1−/− embryos they confirm the key role of the TF TAL1 in coordinating blood, cardiac and endothelial lineages during development
- 56*.Jost M, Santos DA, Saunders RA, et al. : Titrating gene expression using libraries of systematically attenuated CRISPR guide RNAs. Nat Biotechnol 2020, 38:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that specific abundance thresholds determine the ability of TFs to modify cell behavior
- 57.Graf T: Transcription Factor Stoichiometry Drives Cell Fate: Single-Cell Proteomics to the Rescue. Cell Stem Cell 2019, 24:673–674. [DOI] [PubMed] [Google Scholar]
- 58.Herman JS, Sagar Grun D: FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data. Nat Methods 2018, 15:379–386. [DOI] [PubMed] [Google Scholar]
- 59.Weinreb C, Wolock S, Tusi BK, et al. : Fundamental limits on dynamic inference from single-cell snapshots. Proc Natl Acad Sci U S A 2018, 115:E2467–E2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Setty M, Kiseliovas V, Levine J, et al. : Characterization of cell fate probabilities in single-cell data with Palantir. Nat Biotechnol 2019, 37:451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu VWC, Yusuf RZ, Oki T, et al. : Epigenetic Memory Underlies Cell-Autonomous Heterogeneous Behavior of Hematopoietic Stem Cells. Cell 2017, 168:944–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tak T, Prevedello G, Simon G, et al. : Simultaneous tracking of division and differentiation from individual hematopoietic stem and progenitor cells reveals within-family homogeneity despite population heterogeneity. bioRxiv (preprint) 2020. [Google Scholar]
- 63.Cavalli G, Heard E: Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571:489–499. [DOI] [PubMed] [Google Scholar]
- 64**.Izzo F, Lee SC, Poran A, et al. : DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat Genet 2020, 52:378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses scRNA seq in transgenic mice with deletion of enzymes involved in DNA methylation and demethylation to demonstrate differences in HSPCs cell fate biases. This shows that DNA methylation, an epigenetically transmitted mark, directly regulates cell fate potential
- 65.Zaret KS, Mango SE: Pioneer transcription factors, chromatin dynamics, and cell fate control. Curr Opin Genet Dev 2016, 37:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Q, Xiong H, Ai S, et al. : CoBATCH for High-Throughput Single-Cell Epigenomic Profiling. Mol Cell 2019, 76:206–216 e207. [DOI] [PubMed] [Google Scholar]
- 67.Moudgil A, Wilkinson MN, Chen X, et al. : Self-reporting transposons enable simultaneous readout of gene expression and transcription factor binding in single cells. 2019. [Google Scholar]