

Abstract
Papillomaviruses (PVs) are non-enveloped icosahedral viruses with a circular double-stranded DNA genome of ∼8,000 base pairs (bp). More than 200 different PV types have been identified to date in humans, which are distributed in five genera, with several strains associated with cancer development. Although widely distributed in vertebrates, Neotropical Primates (NP) PV infection was described for the first time only in 2016. Currently, four complete genomes of NP PVs have been characterized, three from Saimiri sciureus (SscPV1 to SscPV3) and one from Alouatta guariba (AgPV1). In this work, we describe two novel PV strains infecting Callithrix penicillata (provisionally named CpenPV1 and CpenPV2), using anal swab samples from animals residing at the Brasilia Primatology Center and next generation sequencing. The genomes of CpenPV1 (7,288 bp; 41.5% guanine-cytosine content - GC) and CpenPV2 (7,250 bp; 40.7% GC) contain the characteristic open reading frames (ORFs) for the early (E6, E7, E1, E2, and E4) and late (L2 and L1) PV genes. The L1 ORFs, commonly used for phylogenetic identification, share 76 per cent similarity with each other and differ 32 per cent from any other known PV, indicating that these new strains meet the criteria for defining novel species. PV genes phylogenetic variance was analyzed and different degrees of saturation revealed similar levels of topological heterogeneity, ruling out saturation as primary etiological factor for this phenomenon. Interestingly, the two CpenPV strains form a monophyletic clade within the Gammapapillomavirus genus (provisionally named gammapapillomavirus 32). Unlike for other NP PV strains, which grouped into a new sister genus of Alphapapillomavirus, this is the first report of NP PV strains grouping into a genus previously considered to exclusively comprise Old World Primates (OWP) PVs, including human PVs. These findings confirm the existence of a common ancestor for Gammapapillomavirus already infecting primates before the split of OWP and NP at ∼40 million years ago. Finally, our findings are consistent with an ancient within-species diversity model and emphasize the importance of increasing sampling to help understanding the PV-primate codivergence dynamics and pathogenic potential.
Keywords: papillomavirus, neotropical primate, gammapapillomavirus, CpenPV, within-species diversity model
1. Introduction
Papillomaviruses (PVs) are non-enveloped icosahedral particles with circular double-stranded DNA genomes ranging from 5,748 to 8,607 bp (revised in Van Doorslaer et al. 2018). They belong to the Papillomaviridae family that comprises fifty-three genera and more than 100 species. PV infect vertebrates, including mammals, birds, fishes, and reptiles (Van Doorslaer et al. 2018; Willemsen and Bravo 2019). Although more than 200 different PV types have been identified in humans, only 112 have been characterized in other animal species up to 2013 (Rector and Van Ranst 2013). These viruses infect and replicate exclusively in keratinocytes of the skin and mucosae; and can induce non-cancerous wart-like lesions in epithelia as well as mucocutaneous cancer, including squamous cell carcinoma in a wide range of animals (O’Neill et al. 2011; Eleni et al. 2017; Gil da Costa et al. 2017). However, PV are also part of the viral flora of the healthy skin of different mammals, with a prevalence above 75 per cent in primates, including humans, chimpanzees, gorillas, and long-tailed macaques (Antonsson et al. 2000; Antonsson and Hansson 2002).
Due to their wide array of vertebrate hosts, Papillomaviridae may be considered the largest viral family that has infected vertebrates at least 400 million years ago (mya; Van Doorslaer 2013; Lopez-Bueno et al. 2016). The evolutionary history of this group reflects a complex process of manyfold events, like adaptation, tissue tropism specialization, recombination, lineage sorting, and virus–host codivergence (Gottschling et al. 2007, 2011; Bravo and Félez-Sánchez 2015). Unlike other pathogens, PV do not follow Fahrenholz’s rule, which states that phylogenies and divergence times of parasites and their hosts are perfectly matched in a codivergence scenario (Fahrenholz 1913). Differently, PVs are not clustered according to their hosts in phylogenetic trees, and this is the case of non-human primate PVs found to be intermingled within three different human PV (HPV) genera throughout the viral phylogeny (Van Doorslaer et al. 2018).
Formerly, PV cross-species transmissions were considered to be almost non-existent, resulting in abortive infections in non-natural hosts (Shah, Doorbar, and Goldstein 2010). However, multiple cross-species transmission events have been recently reported (García-Pérez et al. 2014; Trewby et al. 2014; Gil da Costa et al. 2017; Canuti et al. 2019) and contributed to changing the paradigm (Gottschling et al. 2007, 2011; Bravo and Félez-Sánchez 2015). Nowadays, different models have been proposed for explaining the genetic heterogeneity of this group. The most plausible hypothesis postulated that specific events of host evolution opened new niches to an ancestral generalist PV that split into several groups of increasingly specialized viruses. These subsequently diversified alongside their hosts (García-Vallvé, Alonso and Bravo 2005; Bravo and Félez-Sánchez 2015; Willemsen and Bravo 2019). Within this scenario, the evolutionary dynamics driving PV diversification was provided by a balance between the availability of new niches and speciation events. This ancient, within-species diversity followed by a codivergence process may explain the extant phylogenetic pattern of the Papillomaviridae tree (Gottschling et al. 2007, 2011; Chen et al. 2018a).
HPVs are the most well-studied PVs to date among the 481 complete PV genomes reported in the PaVE database (https://pave.niaid.nih.gov/). Human strains, accounting for 62 per cent (n = 298) of these reports, are divided into five genera: Alpha-, Beta-, Gamma-, Mu-, and Nupapillomavirus. Only seventeen complete PV genomes infecting Old World Primates (OWP) are presently available, and more recently, four PV genomes infecting Neotropical Primates (NP) have been characterized (Silvestre et al. 2016; Chen et al. 2018b; Long et al. 2018). Phylogenetic analysis of OWP PVs showed that these viruses grouped in minor monophyletic clades with their HPV counterparts within the Alpha-, Beta-, and Gammapapillomavirus genera, with divergence estimates of 14–31 mya (Chan et al. 1997; Chen et al. 2018a). NP PVs are grouped in the Dyomikronpapillomavirus genus, a sister group of Alphapapillomavirus from which they diverged around 40 mya, corresponding to the emergence of NP in the Americas (Chen et al. 2018a).
Given that studies on long-term virus evolution are conditioned by sampling (e.g. Xu et al. 2018), sequencing and analyses of new genomes are expected to further corroborating or rejecting current hypotheses. As more strains of NP PV become characterized, a better understanding of the ancient within-species diversity model can be achieved. In this work, we look forward to bridging this gap through characterization of two novel PV strains infecting the anal mucosa of captive black-tufted marmosets (Callithrix penicillata) from Brazil. Phylogenetic inference revealed that these newly reported viruses belonged to the Gammapapillomavirus, implying that this genus is widespread and older than previously proposed. Overall, our findings are consistent with the ancient within-species diversity model and emphasize the importance of increasing sampling for validating this hypothesis.
2. Material and methods
2.1 NP samples
Anal swab samples (n = 77; Table 1) were collected from captive NP housed at Primate Center of the University of Brasilia (Centro de Primatologia de Brasília – CP/UnB), including C. penicillata (n = 39), Sapajus sp. (n = 36), and Saimiri ustus (n = 2). The Primate Center is an official primate breeding facility for scientific purposes authorized by IBAMA (Instituto Brasileiro do Meio Ambiente e dos Recursos Naturais Renováveis, Brazil under permanent license number 1/53/1999/000006-2), located in a 4,500 ha environmental reserve area (16° 30″ S, 46° 30″ W) belonging to the University of Brasilia. Animals were kept in cages surrounded by natural vegetation and were maintained in couples or in groups according to the rules of IBAMA. Animals were anesthetized and anal samples were collected using sterile cotton swabs with plastic shafts. Swab samples were placed in 1.5 ml centrifuge tubes with 500 μl of PBS and later excised close to the cotton tip using flame-sterilized scissors. Tubes were kept at −20°C until processing. All samples were collected following the national guidelines and provisions of CONCEA (National Council for Animal Experimentation Control, Brazil), which included animal welfare standard operating procedures. This project was approved by Ethics Committee on the Use of Animals of Universidade Federal do Rio de Janeiro (reference number 037/14).
Table 1.
Description of sample distribution, library pools (PV-positive pools indicated in italics), and NGS results.
| Species | Number of cytB PCR-positive samplesa | Pools | Number of samples per pool | Number of reads | Number of filtered readsb | Number of viral reads | Proportion of PV PCR- positive samplesc |
|---|---|---|---|---|---|---|---|
| C. penicillata (n = 39) | 31/39 | P1 | 5 | 1,487,358 | 1,251,893 | 471 | 1/5 |
| P2 | 5 | 3,319,936 | 2,920,471 | 6,369 | 0/5 | ||
| P3 | 5 | 1,723,364 | 1,171,413 | 114 | 0/5 | ||
| P4 | 4 | 4,195,054 | 2,970,257 | 250 | 4/4 | ||
| P7 | 6 | 3,544,820 | 2,420,288 | 288 | 3/6 | ||
| P8 | 6 | 2,205,954 | 1,155,420 | 127 | 0/6 | ||
| Total C. penicillate | 6 | 31 | 16,476,486 | 11,889,742 | 7,619 | 8/31 | |
| Sapajus sp. (n = 36) | 17/36 | P5 | 6 | 2,572,234 | 2,103,728 | 891 | NA |
| P6 | 6 | 2,867,788 | 2,256,673 | 541 | 1/6 | ||
| P9 | 5 | 2,110,732 | 1,518,606 | 122 | NA | ||
| Total Sapajus sp. | 3 | 17 | 7,550,754 | 5,879,007 | 1,554 | 1/6 | |
| S. ustus (n = 2) | 0/2 | NA | 0 | NA | NA | NA | NA |
| Total (n = 77) | 48/77 | 9 | 48 | 24,027,240 | 17,768,749 | 9,173 | 9/37 |
NA, not applicable.
Confirmation of DNA integrity and species identification by polymerase chain reaction (PCR)-amplification of cytB mtDNA sequence.
Trimmed for quality (Q30), length (>50 bp), and host genomes.
Diagnosis PV PCR and/or long specific PV PCR results.
Genomic DNA (gDNA) was isolated using the PureLink® Genomic DNA Mini Kit (Invitrogen, CA, USA) with minor modifications. These consisted of 1, initial addition of 500 μl of RNAlater (Ambion, TX, USA) to each swab sample; 2, use of 400 μl of the mix for gDNA extraction; and 3, collection of final elution volumes of 100 μl of gDNA. Confirmation of gDNA integrity and host species identification was carried out by polymerase chain reaction (PCR)-amplification of 1,260 bp from cytB mitochondrial DNA (mtDNA) using ∼100 ng gDNA as previously described (Irwin, Kocher, and Wilson 1991; Casado et al. 2010). Amplicons were purified, directly sequenced in an ABI 3130XL Genetic Analyzer (Applied Biosystems, Thermo-Fisher Scientific, MA, USA) and manually edited with SeqMan 7.0 (DNASTAR Inc, Madison, USA). All samples with detectable and successfully sequenced cytB were submitted to GenBank BLASTn tool for confirming host species identification and suitability for next generation sequencing (NGS).
2.2 Circular DNA amplification and NGS
Rolling circle amplification (RCA) was used for enriching total circular DNA using the TempliPhi™ Amplification Kit (GE Healthcare Life Sciences, NJ, USA) following the manufacturer’s instructions. Nine sample pools were created for facilitating library preparation (Table 1) with Nextera® DNA Sample Preparation Kit (Illumina®, CA, USA) following the manufacturer’s instructions. Libraries were purified with Agencourt AMPure® XP Beads (Beckman Coulter, IN, USA), and quantified with the Qubit™ dsDNA HS Assay Kit (Thermo-Fisher Scientific) in a Qubit® 2.0 Fluorometer (Thermo-Fisher Scientific). Libraries were mixed in an equimolar solution to a final concentration of 20 pM. NGS by synthesis was carried out in an Illumina Miseq® platform using MiSeq Reagent Kit v2 (2 × 250 paired-end runs—Illumina®).
2.3 Virome analysis
An inhouse pipeline was used to analyze PV diversity in libraries. Briefly, quality analysis of generated sequence reads was conducted with FastQC (Babraham Bioinformatics, Cambridge, UK). Reads of good quality (Phred score > 30 and fragment size ≥ 50 bp) were selected with Sickle-Master (https://github.com/najoshi/sickle). Reads were submitted to host genome filtering using all NP reference genomes (Saimiri boliviensis boliviensis—GCA_000235385.1; Callithrix jacchus—GCA_002754865.1; Aotus nancymaae—GCA_000952055.2; and, Cebus capucinus imitator—GCA_001604975.1) published by Ensembl Project (Zerbino et al. 2018). Filtered reads were BLASTx-aligned against an inhouse, non-redundant viral protein database, with minimal e-value cutoff of 0.01. Reads assigned to viruses were BLASTx-aligned against a total GenBank non-redundant protein database, with minimal e-value cutoff of 1 × 10−5. Viral taxonomic assignment was carried out using the lowest-common ancestor (LCA) algorithm of MEGAN 6.3.5, built 4 (April 2016). The following LCA parameters were used: Min Score 50; Max Expected 1 × 10−5; Top Percent 10; Min Support Percent 75; Min Support 5; Min Complexity 0.3. Sequences without significant hits were designated as ‘unassigned’.
2.4 PV-specific PCR, library construction and genome assembly
As the virome analysis did not succeed for assembling entire PV genomes, we used the fragments obtained in this step for designing specific primers to characterize full-length genomes. These primers were used for PV-diagnostic PCR and specific PV RCA in individual samples (Supplementary Table S1). Briefly, identification of PV-positive samples was carried out by PV-diagnostic PCR with 50 ng of gDNA mixed with 1× PCR buffer, 0.2 mM deoxyribonucleotide triphosphate (dNTP) mix, 0.4 pmol of each primer, 1.5 mM MgCl2 and 1.25 U GoTaq® DNA Polymerase (Promega). Reactions were carried out in a Veriti® Thermal Cycler (Applied Biosystems, Thermo-Fisher Scientific) with initial denaturation at 95°C for 5 min, followed by thirty-five cycles of denaturation at 95°C for 45 s, annealing at specific primer temperatures (Supplementary Table S1) for 15 s and extension at 72°C at specific times according to fragment length, with a final extension at 72°C for 5 min.
Complete PV genomes were amplified by adding 50 ng of RCA products to a mix containing 1X PCR buffer, 0.2 mM dNTP mix, 0.4 pmol of each primer, 1 mM MgSO4 and 1 U of Platinum™ Taq DNA Polymerase High Fidelity (Thermo-Fisher Scientific). Reactions were carried out in a Veriti® Thermal Cycler with an initial denaturation at 94°C for 2 min, followed by thirty-five cycles of denaturation at 94°C for 15 s, annealing at specific primer temperatures (Supplementary Table S1) for 30 s and extension at 68°C for 9 min. Amplicons were purified and Illumina libraries were prepared, sequenced and analyzed as described above. PV reads were exported and de novo assembled with Geneious® 11.0.5 (Biomatters Ltd, Auckland, NZ, USA), using the option ‘circularize contigs with matching ends’.
2.5 Phylogenetic analyses
In order to re-evaluate the ancient within-species diversity model a set of phylogenetic analyses was performed. First, we assembled a comprehensive dataset of PV genomes available in PaVE database and added the novel genomes herein characterized. We selected four genes present in all genomes (E1, E2, L1, and L2) and carried out individual protein alignments with MAFFT (Katoh and Standley 2013), as reference for aligning their respective nucleotide sequences. Subsequently, each alignment was trimmed with trimAl (Capella-Gutiérrez, Silla-Martínez, and Gabaldón 2009) with the option ‘gappyout’. Concatenated alignments for each kind of data were thereafter constructed. For all datasets, maximum likelihood phylogenies were inferred with IQ-tree (Nguyen et al. 2015) with the GTR + F + I + G4 model (Tavaré 1986; Yang 1994) for nucleotide alignments and the LG + F + I + G4 model (Yang 1994; Le and Gascuel 2008) for amino acid alignments. Both substitution models were evaluated as having the best fit on a formal model selection procedure implemented in IQ-tree (Kalyaanamoorthy et al. 2017). Ultrafast bootstrap (UFBoot; Hoang et al. 2018) and Shimoidara–Hasegawa-like approximate likelihood-ratio test (SH-aLRT; Guindon et al. 2010) measures were used for assessing clade support. Additionally, we formally evaluated whether topological discrepancies between gene trees were statistically significant with the approximately unbiased test (Shimodaira 2002).
2.6 Temporal analysis
Timescales of primate PV groups were inferred with BEAST 1.10.4 (Suchard et al. 2018). At this stage, three different subsets of the concatenated nucleotide alignment were evaluated: one composed of Alphapapillomavirus + Dyoomikronpapillomavirus + PphPV2, another comprising Betapapillomavirus + EePV1, and a third one with Gammapapillomavirus + VvPV1. The Bayesian coalescent skyline tree prior (Drummond et al. 2005) with an uncorrelated relaxed clock log-normal model (Drummond et al. 2006) and the GTR + G4 model of sequence evolution (Tavaré 1986; Yang 1994) were used. Moreover, under the assumption that primate PVs and their hosts followed a codivergence mode of evolution, host divergence times were used for calibrating the viral molecular clock. Calibration time points between OWP PVs and HPVs were set to 28 mya (95% CI = 25–31 mya), coinciding with the divergence of macaque and hominin ancestors (Gibbs et al. 2007; Perez et al. 2013). An exception to this calibration was Pan paniscus Papillomavirus 1 (PpPV1), whose divergence from HPV13 was set at 7 mya (6–8 mya), coinciding with the divergence time between humans and chimpanzees (Patterson et al. 2006). Additionally, calibration points between NP PVs and their closest PVs were set at 49 mya (41–58 mya), coinciding with the time of divergence of NP from OWP (Perez et al. 2013). Importantly, normal priors with soft bounds were used for calibrations, allowing for different divergence time estimates from those specified by the priors.
2.7 Recombination and saturation analysis
As recombination and substitution saturation affect phylogenetic inferences, we analyzed our Gammapapillomavirus dataset for the likely presence of these events. For recombination, we used the standard analysis of the RDP4 software (Martin et al. 2015). With respect to saturation, in addition to generating regular saturation plots for the L1 gene, we developed an exploratory analysis for verifying whether this likely event had any impact on the phylogenetic heterogeneity of Gammapapillomavirus genes (specifically E1, E2, L1, and L2). We hypothesized that substitution saturation could cause long branch attraction artifacts and ultimately lead to incorrect phylogenetic inferences, causing biased topological variance. This analysis is based on the postulation that different codon positions evolve at different rates, thus reaching substitution saturation at different times. We assessed the level of topological variance between gene trees inferred from datasets with combinations of specific codon positions (first and second, only second, and full dataset) and amino acid data. If saturation were the primary cause of heterogeneity of phylogenetic signals, trees inferred from datasets less prone to saturation (e.g. with only second codon positions) were expected to reveal a lower topological variance. We used custom R scripts with functions from APE (Paradis and Schliep 2019) and Phangorn (Schliep, 2011) packages for calculating and plotting Robinson–Foulds (RF) distances (Robinson and Foulds, 1981) between gene trees within each dataset. All codes, alignments and beast xml files used in this work are available in Supplementary Material.
3. Results
3.1 Virome description
Following pool screening, more than 24 million reads (average 2,669,693; min. 1,487,358 to max. 4,195,054) were obtained, and over 17 million reads (74%) were kept after trimming for quality (Q30), length (>50 bp) and host genome filtering (Table 1). Despite that ≤ 0.2 per cent of filtered reads per pool (totaling 9,173; average 1,019, min. 114 to max. 6,369) were classified into viral families, 7/9 pools (78%) were PV-positive. Infection was confirmed by specific PV-diagnostic PCR in 4/7 pools (57%) corresponding to 9/37 samples (24.3%), one Sapajus sp. and eight C. penicillata. With long specific PV PCR from RCA products, four complete PV genomes (SWA17, SWA18, SWA19, and SWA53), corresponding to two different PV types (SWA17/SWA18/SWA19 Type 1 and SWA53 Type 2) of ∼7 kb from C. penicillata, were successfully amplified, sequenced, and assembled (Fig. 1 and Table 2). The remaining samples did not provide data for amplifying complete PV genomes (unpublished data).
Figure 1.

Complete gene maps of the C. penicillata papillomavirus Types 1 and 2 (provisionally named CpenPV1 and CpenPV2) genomes. Gene annotations are represented in green. Both genomes contained five putative early genes (E6, E7, E1, E2, and E4) and two late genes (L2 and L1). All positions were annotated (in bp) from the first nucleotide of the E6 gene. The 3′UTR was located between the L1 stop codon and the E6 start codon. The green ring represents the A + T content and the blue ring indicates C + G content. The location of the primers used for the specific PV RCA is represented by arrowheads.
Table 2.
Genome characteristics of the two novel CpenPV genomes characterized.
| Type | Genome length (nt) | % GC | Nucleotide positiona and size (amino acids) of ORF |
Most closely related PV typeb | % L1 sequence identityb | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E6 | E7 | E1 | E2 | E4 | L2 | L1 | 3′UTR | |||||
| CpenPV1 | 7,288 | 41.5 | 1–429 (142) | 432–719 (95) | 709–2,496 (595) | 2,438–3,568 (376) | 2,925–3,326 (133) | 3,576–5,171 (531) | 5,184–6,716 (510) | 6,717–7,288 (572c) | HPV65 | 66 |
| CpenPV2 | 7,250 | 40.7 | 1–429 (142) | 431–721 (96) | 711–2,498 (595) | 2,440–3,564 (374) | 2,924–3,325 (133) | 3,573–5,165 (530) | 5,179–6,705 (508) | 6,706–7,250 (545c) | HPV-mSK_065 | 68 |
3′UTR, 3′ untranslated region.
Nucleotide positions in the genome are relative to the starting ATG codon of the E6 gene (Position 1).
Results are based on L1 ORFs NCBI Blast search. HPV65, Human papillomavirus type 65. HPV-mSK_065, Human papillomavirus isolate HPV-mSK_065.
Nucleotide size of 3′UTR.
3.2 Novel PV strains characterization
The two novel genomes were submitted to the Animal Papillomavirus Reference Center. The recommendation was to report them as novel types that have not yet been classified and provisionally named C. penicillata papillomavirus Types 1 and 2 (CpenPV1 and CpenPV2). The CpenPV1 genome was predicted to be 7,288 bp long, with a guanine-cytosine (GC) content of 41.5 per cent (Table 2), whereas the CpenPV2 genome was predicted to be 7,250 bp long, with a GC content of 40.7 per cent. Both genomes contain five putative early genes (E6, E7, E1, E2, and E4) and two late genes (L2 and L1; Table 2). The complete L1 open reading frames (ORFs) of these two types shared 76 per cent of nucleotide identity and differed by ≥ 32 per cent from any other known Gammapapillomavirus (HPV65 being the most similar to CpenPV1 and HPV-mSK_065 to CpenPV2), indicating that these isolates filled the criteria to be classified as new types of Gammapapillomavirus in a novel species, provisionally named gammapapillomavirus 32. Analysis of predicted CpenPV genes revealed an E2–E4 splicing event, a reason why the E4 gene was annotated in complete overlap with the E2 gene. The CpenPV1 genome contained six polyadenylation sites (AATAAA) at positions 753, 2,329, 3,663, 5,556, 6,498, and 6,849. The CpenPV2 genome contained eight polyadenylation sites at positions 1,170, 1,793, 1,913, 2,331, 2,638, 4,370, 5,551, and 6,833. All positions were annotated from the first nucleotide of the E6 gene.
Both predicted E6 ORFs from CpenPV1 and CpenPV2 contained two putative conserved zinc-binding domain [CxxC(x)30CxxC/CxxC(x)32CxxC and CxxC(x)30CxxC/CxxC(x)29CxxC] separated by 33 and 36 amino acids, respectively. Both predicted E7 ORFs contained a single putative conserved zinc-binding domain [CxxC(x)29CxxC]. The retinoblastoma protein binding site (LXCXE) was not found. The predicted E1 ORFs of CpenPV1 contained two Cyclin A interaction motifs (RXL) and CpenPV2 with six, which might account for an efficient PV replication (Ma et al. 1999). Moreover, both types showed a single adenosine triphosphate (ATP)-dependent helicase GX4GK(T/S) domain. The E4 ORFs were predicted to encode a proline-rich (14 proline residues of 133 amino acids for both types) peptide containing four Cyclin A interaction motifs in CpenPV1 and three in CpenPV2. The CpenPV1 3′ untranslated region (3′UTR), also known as long control region (LCR), was 572-bp long and the CpenPV2 3′UTR was 545-bp long, located between the L1 stop codon and the E6 start codon. Four LCR E2 binding sites (ACC-X6-GGT) were identified in both CpenPV1 and CpenPV2 3′UTR. LCR E1 binding sites (ATTGTT-X3-AACAAT) were not found. The CpenPV1 LCR contained three putative TATA boxes (TATAA), while CpenPV2 showed only one.
3.3 Phylogenetic inferences
Phylogenetic reconstruction performed with the concatenated nucleotide dataset recovered the previously described structure of the Papillomaviridae tree, with all genera being monophyletic and with high support (Fig. 2). As expected, PV strains infecting OWP grouped within the above mentioned five genera (Alpha-, Beta-, Gamma-, Mu-, and Nupapillomavirus), without a strictly defined monophyly of host species. As expected, previously described NP PVs clustered within the new Dyoomikronpapillomavirus genus, closely related to the Alphapapillomavirus genus that include non-human OWP PV and HPV strains. Conversely, the novel viruses herein described, CpenPV1 and CpenPV2, grouped in a monophyletic clade within Gammapapillomavirus. More specifically, this new clade, provisionally named gammapapillomavirus 32, was a sister group of another containing all congeneric species, except for gammapapillomavirus 6 and gammapapillomavirus 7 (UFBoot = 100; SH-aLRT = 100). This phylogenetic arrangement suggests that the Gammapapillomavirus genus comprises two distinct major clades, herein named A and B, with gamapapillomavirus 32 being the first lineage to diverge within Clade A (Fig. 2). The tree inferred from the concatenated amino acid dataset showed that gammapapillomavirus 32 grouped in a sister clade with gammapapillomavirus 10 and gammapapillomavirus 30, although this relationship was not strongly supported (UFBoot = 32.3; SH-aLRT = 87).
Figure 2.

Phylogenetic tree based on complete genome dataset of the Papillomaviridae family. The Papillomavirus (PV) strains infecting OWP have tips highlighted in red and NP in green. The three main PV genera are highlighted in pink, blue, and purple corresponding to Alpha-, Beta-, and Gammapapillomavirus, respectively. The Mu- and Nupapillomavirus genera are highlighted in gray. The previously described Dyoomikronpapillomavirus genus is highlighted in orange. The novel viruses herein described are provisionally named CpenPV1 and CpenPV2 (asterisks), and formed a monophyletic group within Gammapapillomavirus. The Gammapapillomavirus are divided in two clades, herein named A and B, novel NP viruses being the first lineage to diverge in Clade A.
Gene-wise phylogenetic analyses confirmed the previously described topological incongruence across Gammapapillomavirus genes (Fig. 3). Importantly, the approximately unbiased tests revealed that these topological discrepancies were statistically significant (P < 0.05), suggesting that these phylogenetic patterns were produced by consistent phylogenetic signals, rather than resulting from random stochastic errors affecting gene tree inferences. In fact, gammapapillomavirus 32 showed variable phylogenetic arrangements between the genes herein analyzed (Fig. 3B–E). In the E1 tree (Fig. 3B), these viruses grouped as a sister group of a clade containing all gammapapillomaviruses, except for gammapapillomavirus 6, gammapapillomavirus 7 (including HPVs and MmPV5 strains), and gammapapillomavirus 30 (UFBoot = 78; SH-aLRT = 96). Differently, in the E2 tree (Fig. 3C), the CpenPV clade was a sister group of gammapapillomavirus 10 (UFBoot = 26.1; SH-aLRT = 39). In a rather different topology, the L1 tree (Fig. 3D) grouped these new viruses in sister clade containing gammapapillomavirus 31 (the new MmPV7 strain; Long et al. 2018) and gammapapillomavirus 19 (UFBoot = 93.2; SH-aLRT = 76). The L2 tree (Fig. 3E) revealed that gammapapillomavirus 32 was a sister group of a clade containing several congeneric species.
Figure 3.

Gene-wise phylogenetic analyses showing topological incongruence across Gammapapillomavirus genes. (A) Distributions of RF distances between gene trees: first and second codon Positions (1–2), only second codon Positions (2), full codon (Full), and amino acids (Protein). Phylogenetic trees based on E1 (B), E2 (C), L1 (D), and L2 (E) nucleotide sequences of all Gammapapillomavirus species, including the new provisionally named gammapapillomavirus 32, grouping CpenPV1 and CpenPV2 herein described. PV strains infecting OWP and NP are shown in red and green, respectively. Only nodes with UFBoot and SH-aLRT values above 70 are represented, indicated by the full black circles (empty black circles highlight nodes with higher than 70 estimated by only one algorithm).
Recombination events involving gammapapilomavirus 32 or other non-human primate PVs were not detected, while only HPV recombination received moderate support (see RDP4 output file in Supplementary Material). Moreover, while saturation plots revealed a considerable amount of sequence saturation (Supplementary Fig. S1A–C), this event did not affect gene tree topologies. Irrespective of datasets (with full, first, and second codon positions, only second codon positions, and amino acids), the distribution of RF distances between gene trees did not show significant differences (P > 0.05, Analysis of Variance - ANOVA - test), suggesting that saturation was not a primary factor of phylogenetic heterogeneity (Fig. 3A). We therefore focused downstream analysis using the full nucleotide dataset that contained the longest alignments, and with reduced stochastic errors.
3.4 Temporal analyses
Timescale estimates of the three different datasets of primate PVs were fairly consistent (Supplementary Fig. S2 and Fig. 4). Overall, our findings indicated that OWP PVs diverged from HPVs between 19.91 and 34.34 mya, which accounted, respectively, for the minimum and maximum bounds of 95 per cent highest posterior densities (HPDs) across these estimates. PpPV1 was exceptional, in diverging from HPV13 around 6.61 mya (95% HPD = 6.12–7.67). With respect to NP PVs, the divergence time estimate between Alphapapillomavirus and Dyoomikronpapillomavirus was ∼51 mya (95% HPD = 46.21–53.73; Supplementary Fig. S2A), while the CpenPV clade diverged from its sister group within Gammapapillomavirus some 41.98 mya (95% HPD = 39.15–46.99; Fig. 4). Noticeably, CpenPV strains diverged from each other between 8 and 17 mya (95% HPD = 7.93–16.82), an interval including several divergence times between HPV types. Finally, the Betapapillomavirus diverged from its PV sister group at ∼37.58 mya (95% HPD = 34.02–39.82) (Supplementary Fig. S2B).
Figure 4.

Divergence time estimates for Gammapapillomavirus based on host divergence times used for calibrating the viral molecular clock. Calibration points are highlighted with black circles. Branch lengths are proportional to divergence times and purple bars represent the 95 per cent HPD interval of divergence times. PV strains infecting OWP and NP are shown in red and green, respectively. The outlier group is highlighted in gray.
4. Discussion
In this work, we sequenced and characterized two novel NP PV genomes and reanalyzed the ancestral within-species diversity model for Papillomaviridae diversification (García-Vallvé, Alonso and Bravo 2005; Bravo and Félez-Sánchez 2015; Willemsen and Bravo 2019). This reanalysis contemplated the cross-species transmission and the observed phylogenetic patterns that remarkably deviated from Fahrenholz’s rule. For this reason, our findings were analyzed following the proposition that PV diversity emerged consequently to niche specialization in an ancestral host. This diversity was subsequently inherited and codiverged along descendant host lineages.
The phylogenetic placement and divergence time of the novel strains, CpenPV1 and CpenPV2, were critical for this analysis. According to this hypothesis, these viruses should have grouped near other primate PV strains, with divergence times compatible with the splitting interval of host species. However, if CpenPVs shared a most recent common ancestor (MRCA) with viruses infecting more distantly related hosts—for example, a laurasiatherian mammal—and with much older divergence time estimates, the current model would be questionable. Moreover, this model would not be compatible with more recent divergence times between CpenPVs and other primate PVs. Our findings ruled out these two alternative scenarios, supporting the ancestral within-species diversity model followed by codivergence (Figs 2 and 4).
The novel viruses herein described grouped within the Gammapapillomavirus, mainly as the first lineage to diverge within Clade A. This accounted for the first report of NP PV strains grouping in a genus formerly considered to exclusively comprise OWP PVs, including HPVs (Fig. 2), a finding that may result from sampling bias. Previously described NP PVs were included in a single genus, Dyoomykronpapillomavirus, a sister clade of Alphapapillomavirus (Silvestre et al. 2016; Chen et al. 2018a,b). Our divergence time estimates were coincident with the proposed model (Shah, Doorbar and Goldstein 2010; Van Doorslaer and McBride 2016), since CpenPVs diverged from their sister PV clade ∼42 mya, consistently with the time of NP and OWP divergence (Perez et al. 2013; Schrago et al. 2014). In fact, analyses of these novel strains reassessed the divergence time estimate of the Gammapapillomavirus MRCA to ∼45 mya (95% HPD = 42.21–51.08), older than the one inferred by Chen et al. (2018a), but more in consonance with other studies (Shah, Doorbar and Goldstein 2010; Van Doorslaer and McBride 2016). Our estimate on the origin of Alphapapillomavirus was 44.83 mya (95% HPD = 39.8–45.43) and 37.58 mya (95% HPD = 34.02–39.82) for Betapapillomavirus, the only main primate PV genus not presently related to any other NP PV strain. The identification of new PVs following more extensive sampling might provide valuable data for an eventual reassessment of the phylogeny and the time estimates of the emergence of Betapapillomavirus (Fig. 5).
Figure 5.
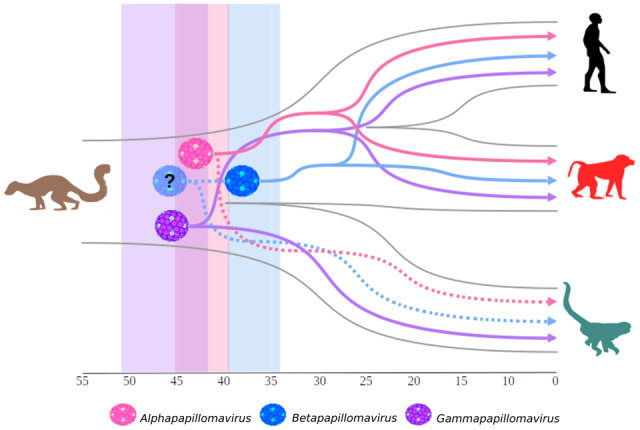
Schematic model of ancient within-species diversity model followed by codivergence. Representation of the divergence time estimate for the three main PV genera: Alpha- (pink virus; 44.83 mya), Beta- (blue virus; 37.58 mya), and Gammapapillomavirus (purple virus; 45 mya) ancestors; plotted in the Primates topology. OWP (red silhouette), including Homo (black silhouette) split around 25 mya, and NP (green silhouette) diverged around 40 mya. The Plesiadapis genus (brown silhouette) is the oldest known mammalian fossil related to Primates, which existed around 56 mya (Gingerich 1975). In this model, the divergence time estimates for Alphapapillomavirus (pink shadow; 95% HPD: 39.8–45.43 mya) and Gammapapillomavirus (purple shadow; 95% HPD: 42.21–51.08 mya) ancestors are compatible with the divergence of host species. Continuous lines represent PV strains already sampled and dashed lines represent putative strains that might explain more likely ancient ancestors. For instance, the Betapapillomavirus (blue shadow; 95% HPD: 34.02–39.82 mya) has a more recent ancestor than that predicted by the splitting of OWP from NP. We therefore hypothesize that the currently unknown presence of Betapapillomavirus strains circulating in NP (dashed line) would indicate an older ancestor of this genus, represented by the blue shadow virus with a question mark.
Overall, our results were consistent with the presence of an ancient within-species diversity followed by codivergence. This model has been confirmed in other viruses, like herpesviruses (McGeoch, Rixon, and Davison 2006), polyomaviruses (Buck et al. 2016), and some genera of retroviruses (Niewiadomska and Gifford 2013). Except for retroviruses, these viruses contain double-stranded DNA genomes and use host cell polymerases with proofreading capability during replication. This reduces the emergence of novel mutations—and in consonance with other epidemiological factors—inhibit their capacity of establishing efficient infections in novel hosts and their rapid adaptation to new environments (Holmes 2008). On the other hand, as these viruses cause benign, mostly unapparent, persistent infections, they seem to be capable of slowly adapting to different niches within a single host whenever they became available during host evolution (e.g. in the emergence of sweat glands; Bernard, Chan, and Delius 1994). These more specialized viruses keep evolving alongside descendant host lineages, further specializing and diversifying. In this respect, the mechanism of PV replication someway modulates their long-term evolution, as observed in other viruses (Geoghegan, Duchêne and Holmes 2017).
The differences of phylogenetic signals between PV genes (Gottschling et al. 2007; Murahwa et al. 2019; Shah, Doorbar and Goldstein 2010) were herein revised. As expected, our estimates suggest phylogenies inferred from E1, E2, L1, and L2 were statistically different at a significant level, supporting the idea that Gammapapillomavirus genes had different evolutionary histories (Fig. 3). Despite being a general trend for the whole family, the topological heterogeneity of the L1 and L2 genes of Gammapapillomavirus was remarkable and not entirely explained by ancient recombination events (Murahwa et al. 2019). As this variation in evolutionary histories could not be fully explained by this event, we analyzed whether the extent of phylogenetic heterogeneity could have resulted from substitution saturation, a process known to affect the inference of viral evolution (see Holmes and Duchêne 2019). In fact, saturation plots revealed considerable amounts of substitution saturation (Supplementary Fig. S1). Notwithstanding, gene tree inferences performed on datasets affected by different degrees of saturation revealed similar levels of topological heterogeneity, ruling out saturation as its primary etiological factor (Fig. 3 and Supplementary Fig. S1). Some caution needs to be taken while considering this conclusion. As RF distances are not sensitive to gene tree uncertainty, this could in principle lead to overestimation of topological differences between gene trees, questioning this proposition. However, in view that our topological tests revealed consistent heterogeneity in phylogenetic signals, the majority of differences between phylogenies probably resulted from variation in the evolutionary history of genes rather than statistical artifacts. Finally, as commented elsewhere (Murahwa et al. 2019), this conundrum of phylogenetic variance is most likely a reflex of the limitations of current methods for tracing ancient recombination.
The findings herein reported reinforced the current hypothesis on PV evolution based on two novel genomes, comprising two new PV types (CpenPV1 and CpenPV2; named gammapapilomavirus 32 species). They emphasized the importance of NGS and increasing host sampling for obtaining a more accurate depiction of the evolutionary history of Papillomaviridae (Fig. 5). The Gammapapillomavirus MRCA was dated back to the common ancestor of OWP and NP consequently to the finding of these two novel PV types. This might also be the case of the Betapapillomavirus MRCA if further strains of this genus were eventually found in NP. Moreover, we have shown, for the first time that substitution saturation did not affect phylogenetic incongruence among different PV genes. Further work with increased sampling of unexplored hosts should provide an even broader perspective of the evolution of this group.
Data availability
Sequencing data files are available in the SRA database under BioProject accession PRJNA524802 (BioSample accession: SAMN11040671–SAMN11040674). Accession numbers for the CpenPV1 and CpenPV2 complete genome sequences determined in this study are available in GenBank under the accession numbers MN535763 and MN535764, respectively.
Supplementary Material
Acknowledgments
This work was supported in part by the Conselho Nacional de Desenvolvimento Científico e Tecnológico/CNPq (grant 312903/2017-0 to A.F.A.S.) and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro/FAPERJ (grants E-26/112.647/2012 to M.A.S. and E26/202.738/2018 to A.F.A.S.) for funding this study. We thank to all the workers of the Centro de Primatologia de Brasília, Universidade de Brasília (Distrito Federal, Brazil) for assistance with sample collection. We are also grateful to Carolina Furtado and Nicole Scherer from Instituto Nacional de Câncer Jośe de Alencar (Rio de Janeiro, Brazil) for assistance with next generation sequencing and analyses.
Conflict of interest: None declared.
References
- Antonsson A. et al. (2000) ‘The Ubiquity and Impressive Genomic Diversity of Human Skin Papillomaviruses Suggest a Commensalic Nature of These Viruses’, Journal of Virology, 74: 11636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonsson A., Hansson B. G. (2002) ‘Healthy Skin of Many Animal Species Harbors Papillomaviruses Which Are Closely Related to Their Human Counterparts’, Journal of Virology, 76: 12537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard H. U., Chan S. Y., Delius H. (1994) ‘Evolution of Papillomaviruses’, Current Topics in Microbiology and Immunology, 186: 33–54. [DOI] [PubMed] [Google Scholar]
- Bravo I. G., Félez-Sánchez M. (2015) ‘Papillomaviruses: Viral Evolution, Cancer and Evolutionary Medicine’, Evolution, Medicine, and Public Health, 2015: 32–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck C. B. et al. (2016) ‘The Ancient Evolutionary History of Polyomaviruses’, PLoS Pathogens, 12: e1005574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canuti M. et al. (2019) ‘New Insight into Avian Papillomavirus Ecology and Evolution from Characterization of Novel Wild Bird Papillomaviruses’, Frontiers in Microbiology, 10: 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutiérrez S., Silla-Martínez J. M., Gabaldón T. (2009) ‘trimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses’, Bioinformatics (Oxford, England), 25: 1972–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado F. et al. (2010) ‘Mitochondrial Divergence between 2 Populations of the Hooded Capuchin, Cebus (Sapajus) Cay (Platyrrhini, Primates)’, Journal of Heredity, 101: 261–9. [DOI] [PubMed] [Google Scholar]
- Chan S. Y. et al. (1997) ‘Genomic Diversity and Evolution of Papillomaviruses in Rhesus Monkeys’, Journal of Virology, 71: 4938–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. et al. (2018. a) ‘Niche Adaptation and Viral Transmission of Human Papillomaviruses from Archaic Hominins to Modern Humans’, PLoS Pathogens, 14: e1007352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. et al. (2018. b) ‘Complete Genome Sequences of Three Novel Saimiri sciureus Papillomavirus Types Isolated from the Cervicovaginal Region of Squirrel Monkeys’, Genome Announcements, 6: e01400–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J. et al. (2005) ‘Bayesian Coalescent Inference of past Population Dynamics from Molecular Sequences’, Molecular Biology and Evolution, 22: 1185–92. [DOI] [PubMed] [Google Scholar]
- Drummond A. J. et al. (2006) ‘Relaxed Phylogenetics and Dating with Confidence’, PLoS Biology, 4: e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eleni C. et al. (2017) ‘Detection of Papillomavirus DNA in Cutaneous Squamous Cell Carcinoma and Multiple Papillomas in Captive Reptiles’, Journal of Comparative Pathology, 157: 23–6. [DOI] [PubMed] [Google Scholar]
- Fahrenholz H. (1913) ‘Ectoparasiten und Abstammungslehre’, Zoologischer Anzeiger, 41: 371–4. [Google Scholar]
- García-Pérez R. et al. (2014) ‘Novel Papillomaviruses in Free-Ranging Iberian Bats: No Virus–Host Co-Evolution, No Strict Host Specificity, and Hints for Recombination’, Genome Biology and Evolution, 6: 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Vallvé S., Alonso A., Bravo I. G. (2005) ‘Papillomaviruses: Different Genes Have Different Histories’, Trends in Microbiology, 13: 514–21. [DOI] [PubMed] [Google Scholar]
- Geoghegan J. L., Duchêne S., Holmes E. C. (2017) ‘Comparative Analysis Estimates the Relative Frequencies of co-Divergence and Cross-Species Transmission within Viral Families’, PLoS Pathogens, 13: e1006215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs R. A. et al. ; Rhesus Macaque Genome Sequencing and Analysis Consortium. (2007) ‘Evolutionary and Biomedical Insights from the Rhesus Macaque Genome’, Science, 316: 222–34. [DOI] [PubMed] [Google Scholar]
- Gil da Costa R. M. et al. (2017) ‘An Update on Canine, Feline and Bovine Papillomaviruses’, Transboundary and Emerging Diseases, 64: 1371–9. [DOI] [PubMed] [Google Scholar]
- Gingerich P. D. (1975) ‘New North American Plesiadapidae (Mammalia, Primates) and a Biostratigraphic Zonation of the Middle and Upper Paleocene’, Contributions from the Museum of Paleontology the University of Michigan, 24: 135–48. [Google Scholar]
- Gottschling M. et al. (2007) ‘Multiple Evolutionary Mechanisms Drive Papillomavirus Diversification’, Molecular Biology and Evolution, 24: 1242–58. [DOI] [PubMed] [Google Scholar]
- Gottschling M. et al. (2011) ‘Quantifying the Phylodynamic Forces Driving Papillomavirus Evolution’, Molecular Biology and Evolution, 28: 2101–13. [DOI] [PubMed] [Google Scholar]
- Guindon S. et al. (2010) ‘New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0’, Systematic Biology, 59: 307–21. [DOI] [PubMed] [Google Scholar]
- Hoang D. T. et al. (2018) ‘UFBoot2: Improving the Ultrafast Bootstrap Approximation’, Molecular Biology and Evolution, 35: 518–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E. C. (2008) ‘Evolutionary History and Phylogeography of Human Viruses’, Annual Review of Microbiology, 62: 307–28. [DOI] [PubMed] [Google Scholar]
- Holmes E. C., Duchêne S. (2019) ‘Can Sequence Phylogenies Safely Infer the Origin of the Global Virome?’, mBio, 10: 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin D. M., Kocher T. D., Wilson A. C. (1991) ‘Evolution of the Cytochrome b Gene of Mammals’, Journal of Molecular Evolution, 32: 128–44. [DOI] [PubMed] [Google Scholar]
- Kalyaanamoorthy S. et al. (2017) ‘ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates’, Nature Methods, 14: 587–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le S. Q., Gascuel O. (2008) ‘An Improved General Amino Acid Replacement Matrix’, Molecular Biology and Evolution, 25: 1307–20. [DOI] [PubMed] [Google Scholar]
- Long T. et al. (2018) ‘Complete Genome Sequences of Six Novel Macaca mulatta Papillomavirus Types Isolated from Genital Sites of Rhesus Monkeys in Hong Kong SAR, China’, Microbiology Resource Announcements, 7: e01414–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bueno A. et al. (2016) ‘Concurrence of Iridovirus, Polyomavirus, and a Unique Member of a New Group of Fish Papillomaviruses in Lymphocystis Disease-Affected Gilthead Sea Bream’, Journal of Virology, 90: 8768–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T. et al. (1999) ‘Interaction between Cyclin-Dependent Kinases and Human Papillomavirus Replication-Initiation Protein E1 is Required for Efficient Viral Replication’, Proceedings of the National Academy of Sciences of the United States of America, 96: 382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D. P. et al. (2015) ‘RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes’, Virus Evolution, 1: vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch D. J., Rixon F. J., Davison A. J. (2006) ‘Topics in Herpesvirus Genomics and Evolution’, Virus Research, 117: 90–104. [DOI] [PubMed] [Google Scholar]
- Murahwa A. T. et al. (2019) ‘Evolutionary Dynamics of Ten Novel Gamma-PVs: Insights from Phylogenetic Incongruence, Recombination and Phylodynamic Analyses’, BMC Genomics, 20: 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L. T. et al. (2015) ‘IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies’, Molecular Biology and Evolution, 32: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewiadomska A. M., Gifford R. J. (2013) ‘The Extraordinary Evolutionary History of the Reticuloendotheliosis Viruses’, PLoS Biology, 11: e1001642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill S. H. et al. (2011) ‘Detection of Human Papillomavirus DNA in Feline Premalignant and Invasive Squamous Cell Carcinoma’, Veterinary Dermatology, 22: 68–74. [DOI] [PubMed] [Google Scholar]
- Paradis E., Schliep K. (2019) ‘APE 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R’, Bioinformatics, 35: 526–8. [DOI] [PubMed] [Google Scholar]
- Patterson N. et al. (2006) ‘Genetic Evidence for Complex Speciation of Humans and Chimpanzees’, Nature, 441: 1103–8. [DOI] [PubMed] [Google Scholar]
- Perez S. I. et al. (2013) ‘Divergence Times and the Evolutionary Radiation of New World Monkeys (Platyrrhini, Primates): an Analysis of Fossil and Molecular Data’, PLoS One, 8: e68029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rector A., Van Ranst M. (2013) ‘Animal Papillomaviruses’, Virology, 445: 213–23. [DOI] [PubMed] [Google Scholar]
- Robinson D. F., Foulds L. R. (1981) ‘Comparison of Phylogenetic Trees’, Mathematical Biosciences, 53: 131–47. [Google Scholar]
- Schliep K. P. (2011) ‘Phangorn: Phylogenetic Analysis in R’, Bioinformatics, 27: 592–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrago C. G. et al. (2014) ‘Multispecies Coalescent Analysis of the Early Diversification of Neotropical Primates: Phylogenetic Inference under Strong Gene Trees/Species Tree Conflict’, Genome Biology and Evolution, 6: 3105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S. D., Doorbar J., Goldstein R. A. (2010) ‘Analysis of Host-Parasite Incongruence in Papillomavirus Evolution Using Importance Sampling’, Molecular Biology and Evolution, 27: 1301–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimodaira H. (2002) ‘An Approximately Unbiased Test of Phylogenetic Tree Selection’, Systematic Biology, 51: 492–508. [DOI] [PubMed] [Google Scholar]
- Silvestre R. V. et al. (2016) ‘First New World Primate Papillomavirus Identification in the Atlantic Forest, Brazil: Alouatta guariba Papillomavirus’, Genome Announcement, 4: e00725–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard M. A. et al. (2018) ‘Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1’, Virus Evolution, 4: vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaré S. (1986) ‘Some Probabilistic and Statistical Problems in the Analysis of DNA Sequences’, in Miura R. M. (ed.) Lectures on Mathematics in the Life Sciences, Vol. 17, pp. 57–86. Providence, RI, USA: American Mathematical Society. [Google Scholar]
- Trewby H. et al. (2014) ‘Analysis of the Long Control Region of Bovine Papillomavirus Type 1 Associated with Sarcoids in Equine Hosts Indicates Multiple Cross-Species Transmission Events and Phylogeographical Structure’, Journal of General Virology, 95: 2748–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Doorslaer K. (2013) ‘Evolution of the Papillomaviridae’, Virology, 445: 11–20. [DOI] [PubMed] [Google Scholar]
- Van Doorslaer K. et al. ; ICTV Report Consortium. (2018) ICTV Virus Taxonomy Profile: Papillomaviridae’, Journal of General Virology, 99: 989–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Doorslaer K., McBride A. A. (2016) ‘Molecular Archeological Evidence in Support of the Repeated Loss of a Papillomavirus Gene’, Scientific Reports, 6:33028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen A., Bravo I. G. (2019) ‘Origin and Evolution of Papillomavirus (Onco) Genes and Genomes’, Philosophical Transactions of the Royal Society B: Biological Sciences, 374: 20180303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X. et al. (2018) ‘Endogenous Retroviruses of Non-Avian/Mammalian Vertebrates Illuminate Diversity and Deep History of Retroviruses’, PLoS Pathogens, 14: e1007072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. (1994) ‘Maximum Likelihood Phylogenetic Estimation from DNA Sequences with Variable Rates over Sites: Approximate Methods’, Journal of Molecular Evolution, 39: 306–14. [DOI] [PubMed] [Google Scholar]
- Zerbino D. R. et al. (2018) ‘Ensembl 2018’, Nucleic Acids Research, 46: D754–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data files are available in the SRA database under BioProject accession PRJNA524802 (BioSample accession: SAMN11040671–SAMN11040674). Accession numbers for the CpenPV1 and CpenPV2 complete genome sequences determined in this study are available in GenBank under the accession numbers MN535763 and MN535764, respectively.