

Abstract
Highly pathogenic avian influenza (HPAI) A(H5) viruses belonging to clade 2.3.4.4c of the A/goose/Guangdong/1/96-like (Gs/GD) lineage caused severe global outbreaks in domestic birds from 2014 to 2015, that also represented the first incursions of Gs/GD viruses into Taiwan and the USA. However, few studies have investigated the circulation of clade 2.3.4.4c viruses after 2015. Here, we describe Gs/GD clade 2.3.4.4c and Mexican-like H5N2 viruses that were isolated in Taiwan during active surveillance conducted in chicken farms from February to March 2019. Phylogenetic analysis demonstrated two distinct genome constellations of the clade 2.3.4.4c H5 viruses, with the internal genes of one of the new genotypes closely related to a virus isolated from a pintail (Anas acuta) in Taiwan, providing the first direct evidence that migratory birds play a role in importing viruses into Taiwan. Our study also confirmed the co-circulation of Gs/GD clade 2.3.4.4c and Mexican-like H5 lineage viruses in Taiwan, presenting a rare case where Gs/GD viruses developed sustained transmission alongside another enzootic H5 lineage, raising the possibility that homosubtypic immunity may mask virus transmission, potentially frustrating detection, and the implementation of appropriate control measures. To eradicate H5 viruses from poultry in Taiwan, further studies on the effect of co-circulation in poultry of low pathogenic avian influenza and HPAI viruses are needed. Furthermore, only with continued surveillance efforts globally can we fully discern dispersal patterns and risk factors of virus transmission both to and within Taiwan.
Keywords: zoonotic, pandemic, influenza virus, evolution
1. Introduction
Highly pathogenic avian influenza virus (HPAI) continues to be a public health concern due to its zoonotic potential. A/goose/Guangdong/1/1996-like (Gs/GD) H5N1 viruses first emerged to cause six fatalities in Hong Kong in 1997 (Chan 2002). Since 2003, this virus lineage has been disseminated to countries in Southeast Asia, Middle East, and Europe (Sonnberg, Webby, and Webster 2013). Amplified in poultry populations, primarily chicken and duck, these viruses have occasionally spilled over to wild birds, resulting in long-distance virus transmission (Chen et al. 2006).
The hemagglutinin (HA) genes of Gs/GD H5 viruses have evolved into more than forty distinct clades (Smith et al. 2015), within which a myriad of internal gene constellations have been generated by undergoing reassortment with low pathogenic avian influenza (LPAI) viruses. Since 2008, clade 2.3.4.4 H5 viruses have acquired different neuraminidase (NA) genes from LPAI viruses leading to the emergence of subtypes other than H5N1 including H5N2, H5N6, and H5N8 (Saito et al. 2015; Smith et al. 2015). Clade 2.3.4.4c H5N8 viruses, one of the sub-lineages of clade 2.3.4.4 (WHO 2020), caused severe outbreaks in Korean poultry farms in early 2014 (Lee et al. 2014b; Saito et al. 2015). Before the end of 2014, viruses from the same lineage had spread to Japan, Europe, and unprecedentedly, to Canada, and the USA (Bouwstra et al. 2015; Ip et al. 2015; Kanehira et al. 2015). In North America, these H5 viruses that were likely dispersed between continents by migratory birds, soon reassorted with enzootic LPAI viruses and caused outbreaks in poultry farms (Ip et al. 2015; Saito et al. 2015).
Taiwan reported its first outbreak of clade 2.3.4.4c H5N8 viruses in January 2015 (Lee et al. 2016). By the end of 2015, HPAI viruses were found in 944 farms with 4.15 million birds culled in response to the epidemics (BAPHIQ 2019). As in North America, this is the first time that Gs/GD-like H5 viruses have caused sustained transmission in Taiwan (Huang et al. 2016) (Supplementary Fig. S1). Multiple subtypes, including H5N2, H5N3, and H5N8 viruses, were generated through reassortment (Supplementary Table S1), among which at least five genetic constellations were identified (Huang et al. 2016). Viruses belonging to a Mexican-like H5N2 lineage, which has been circulating in Taiwan since 2003 (Lee et al. 2014a; Li et al. 2017), were also isolated during the 2015 epidemics (Huang et al. 2016). Interestingly, some of these Mexican-like viruses isolated during 2008–14 had more than two basic amino acids at the HA cleavage site, a well-studied virulence marker of avian influenza, and were characterized as potential HPAI (Soda et al. 2011; Lee et al. 2014a; Li et al. 2017). All viruses isolated in 2015, however, had only two basic amino acids and are characterized as LPAI (Li et al. 2017).
There is no evidence that clade 2.3.4.4c H5 viruses persist in Korea, countries in Europe or North America after 2016 (Taubenberger and Morens 2017; WHO 2020). In contrast, during 2016–19, there were 401 farms and 3.67 million birds affected by HPAI viruses in Taiwan, which was mainly the consequence of the 2015 introduction (BAPHIQ 2019). In addition, H5N2 and H5N8 HPAI viruses were continuously detected during 2015–18 based on the government-run surveillance (Supplementary Table S1). However, during the same period there were few studies focusing on clade 2.3.4.4c viruses circulating in Taiwan. Although clades 2.3.4.4b and 2.3.4.4e H5N6 viruses were detected and described in 2017 (Chen et al. 2018; Liu et al. 2018), there is no evidence that these viruses developed sustained transmission in poultry in Taiwan (WHO 2020) (Supplementary Fig. S1 and Table S1). In this study, we aim to understand the current situation of H5 epidemics in Taiwan. By analyzing newly sequenced H5N2 viruses from chicken farms, we confirmed that both clade 2.3.4.4c and Mexican-like H5N2 continued to circulate in 2019. A virus isolated from a pintail (Anas acuta) further demonstrated that migratory birds were able to import genes from the wild bird influenza gene pool, potentially contributing to the H5 reassortants described here.
2. Materials and methods
2.1 Sample collection
Due to the sustained circulation of H5 viruses since its introduction in 2015, active surveillance focusing on the poultry before shipping to the slaughter house was conducted in Yunlin, one of the major agricultural areas in Taiwan. At least twenty cloacal or fresh dropping swabs were sampled from a total of forty-six poultry farms during January and March 2019. There were on average about 18,000 birds in each farm while the survey was conducted. Two chicken farms with avian influenza virus detected in this study were also diagnosed by the government-run surveillance system, and the farms were then subjected to regular control measures including culling all the birds (Lee et al. 2016; Liu et al. 2018). Migratory waterfowls were trapped using funnel trap (Whitworth et al. 2007) in Aogu Wetland Forest Park, Chiayi County, Taiwan, a major wintering site for migratory birds on the west coast. Waterfowls trapped in this study were species migrating through the East Asian–Australasian Flyway from Alaska or Siberia in the winter. Oral and cloacal swabs were collected from trapped waterfowls. A total eighty-nine samples composed of nine species, including pintail, Eurasian wigeon, and garganey, were collected from November 2017 to February 2018. The procedures for wild waterfowls trapping and sample collection were approved by Institutional Animal Care and Use Committee of National Pingtung University of Science and Technology (Approval number: NPUST-106-029).
2.2 Nucleic acid extraction, library preparation, and MinION sequencing
RNA was extracted from the samples using Direct-zol RNA Miniprep Plus (Zymo Research) with DNase I treatment according to manufacturer instructions. Reverse transcription was then conducted with SuperScript III enzyme (Invitrogen) and a mixture of Uni12/Uni12.4 primers (Zhou et al. 2009). Before amplifying whole viral genomes, cDNA derived from chicken samples were screened for influenza A virus by conventional M gene PCR using InfA Forward/Reverse primers (WHO 2018). Samples from wild birds were screened for influenza A viruses by loop-medicated isothermal amplification and further confirmed by conventional RT-PCR as previously described (Wang, Huang, and Chen 2016).Whole viral genomes were subsequently amplified using Phusion HF polymerase (New England Biolabs) and a combination of Uni12/Uni12.4/Uni13 primers as described (Zhou et al. 2009).
PCR products containing whole genomes were purified using KAPA Pure Beads (Roche), and amplicons from different samples was quantified with Qubit dsDNA Assay Kit (Life Technologies) on a Qubit fluorometer prior to library preparation. At least 100 ng DNA was taken from each sample for end-repairing with KAPA Hyper Prep Kit (KAPA), and then barcoded using Native Barcoding Kit (EXP-NBD104, Oxford Nanopore Technologies). Products were further purified as above before pooling equal amounts of each barcoded amplicon. The final sequencing library was constructed from the pooled product using Ligation Sequencing Kit (SQK-LSK109, Oxford Nanopore Technologies). Libraries prepared above consisting of four to six samples were loaded onto a R9.4.1 flow cell (FLO-MIN106, Oxford Nanopore Technologies) on a MinION sequencing device after the priming process. Data were collected after 24–48 h.
2.3 Bioinformatics workflow
Base calling from fast5 files was performed using Guppy v3.15 (Oxford Nanopore Technologies) filtering out reads with a qscore lower than seven. Resulting fastq files were demultiplexed using Porechop v0.24 (https://github.com/rrwick/Porechop), in which only reads containing matched barcodes at both ends were assigned to the new fastq files. Before mapping reads to reference sequences, de novo assembly was conducted using Canu v1.8 (https://github.com/marbl/canu). The generated contigs were used to perform blastn with BLAST+ v2.9 (https://blast.ncbi.nlm.nih.gov) against a local influenza database downloaded from NCBI. Top hits, that is a collections of most similar segments in particular strains, were then scrutinized and selected to prepare a reference genome for each sample. Demultiplexed fastq files were subsequently mapped to respective reference genomes by Minimap2 v2.16 (https://github.com/lh3/minimap2).
Consensus sequences were constructed based on previous approaches (Quick et al. 2017; Kafetzopoulou et al. 2019). In brief, Nanopolish v0.11 was employed to detect single nucleotide variants in the aligned reads facilitated by their signal-level data (fast5) (https://github.com/jts/nanopolish). Only variants with a quality score >200 and a minimum support fraction >0.6 were accepted. Details of the bioinformatic scripts can be available at https://github.com/yaotli/minion-influenza. We validated our sequencing pipeline as described in the Supplementary Data. Sequences produced in this study are available from NCBI GenBank database (accession numbers: MN988762-MN988825).
2.4 Phylogenetic analyses
Sequences of avian influenza A viruses were downloaded from the NCBI Influenza Virus Resource (https://www.ncbi.nlm.nih.gov/genomes/FLU/) and GISAID (https://www.gisaid.org). Sequences shorter than 0.95× the lengths of coding region or sequences containing more than 1 per cent ambiguous nucleotides were discarded. Curated sequences were then aligned using MAFFT v7.407 and trimmed to coding regions. Before selecting representative strains, all available sequences of eight segments were separately pooled with sequences generated in our study, and the alignments were subject to maximum likelihood (ML) tree inference using IQ-TREE v1.6.12 (Nguyen et al. 2015) with a general time reversible (GTR) substitution model plus a Gamma-distributed rate. Previous Taiwanese isolates, samples collected in this study and their closely related strains were further selected to generate ML trees inferred by RAxML v8.2.12 (Stamatakis 2014), with a GTR plus gamma substitution model and 1,000 bootstrap iterations. Accession numbers of the sequences analyzed in this study have been provided in Supplementary Table S2.
A Bayesian phylogenetic framework was implemented to explore additional evolutionary and epidemiological information with HA sequences of clade 2.3.4.4c and Mexican-like lineage viruses. TempEst v1.5.3 was used to examine temporal signals in each dataset (Rambaut et al. 2016), taking an ML tree reconstructed by RAxML and times of virus isolation as input. Time-scaled phylogeny was then inferred using BEAST v.1.10.4 (Suchard et al. 2018). We employed a GTR plus gamma substitution model, and an uncorrelated lognormal relaxed molecular clock (Drummond et al. 2006) with a Skygrid demographic prior for each dataset (Gill et al. 2013). Ancestral geographical locations were inferred along the phylogeny by implementing a discrete-state continuous time Markov chain model in BEAST for clade 2.3.4.4c dataset. We employed an asymmetric substitution model with Bayesian stochastic search variable selection for transition parameters (Lemey et al. 2009). Markov Chain Monte Carlo (MCMC) analysis was run for 120 million steps and sampled every 10,000 steps. For each dataset, two parallel runs were conducted and combined with a burnin of 20 million per chain using LogCombiner (Suchard et al. 2018). Parameters logged during the MCMC runs were inspected by Tracer v1.7.1 (Rambaut et al. 2018). Summarized maximum clade credibility (MCC) trees were inferred using TreeAnnotator (Suchard et al. 2018) and subsequently visualized with R package ggtree (Yu et al. 2018).
3. Results
3.1 Detection of avian influenza in chicken farms and wild birds
We detected avian influenza viruses in two separate chicken farms, denoted Farm D and Farm A, in Yunlin, Taiwan in February and March 2019, respectively, during our active surveillance. Five samples tested from Farm A comprised five or six pooled fecal swabs, whereas thirty-two fecal swabs in Farm D were tested individually. Among all the samples, four samples from Farm A (80%) to five samples from Farm D (16%) tested positive for influenza A virus. In addition, we detected influenza in sixteen oropharyngeal or cloacal swabs samples from sixty pintails captured in Aogu Wetlands during the 2017 winter. Whole genomes were generated from six chicken samples to two wild bird samples. Preliminary phylogenetic analysis indicated that viruses from chicken farms were all H5N2 viruses; five of them were clade 2.3.4.4c viruses and one belonged to the Mexican-like H5 lineage (Supplementary Fig. S1). The wild bird genomes were found to be from H6N1 and H1N3 influenza A viruses.
3.2 Introduction of clade 2.3.4.4c to Taiwan
Phylogenetic analysis of the HA gene suggests that clade 2.3.4.4c viruses entered Korea before 2014, followed by a second wave of dispersal leading to virus circulation in North America, Europe, and Taiwan (Fig. 1A and Supplementary Fig. S2). Estimates of times of the most recent common ancestor (tMRCA) indicate the virus was introduced into Korea in late October 2013, about 1 year before they reached other areas (Table 1). The tMRCA of clade 2.3.4.4c viruses isolated in Taiwan (October 2014) is close to tMRCA of the viruses in North America (September 2014) and Europe (September 2014) with overlapping statistical uncertainty (Table 1 and Fig. 1C). The upper bound of the tMRCA 95 per cent highest posterior density (95% HPD) of viruses in Taiwan is in November 2014, at least 1 month before the first outbreak was reported (Lee et al. 2016). These results illustrate a nearly synchronized invasion of clade 2.3.4.4c virus into different geographical areas in late 2014.
Figure 1.

Establishment and genetic diversity of Gs/GD clade 2.3.4.4c viruses in Taiwan. (A) MCC tree was reconstructed using HA genes of clade 2.3.4.4c viruses. Tips are colored according to their collection location, while branches are colored according to inferred ancestral states. Clade posterior probabilities >0.9 are indicated by white nodes, whereas clade posterior probabilities between 0.9 and 0.7 are shown using filled nodes that match branch color. North Asia here includes isolates from Russia, South Korea, and Japan. (B) Genome constellations of clade 2.3.4.4c viruses in Taiwan are illustrated by a heatmap adjacent to an expanded section of the MCC tree in Fig. 1A (dashed rectangle). Lineages/subtypes are indicated by distinct colors in the heatmap, with three shades of green used to indicate distinct origins within the Eurasian gene pool. Each column represents a gene segment and each row corresponds to an isolate on the MCC tree. Genotypes of the viruses (Huang et al. 2016) are shown to the right of the heatmap, along with the names of viruses sequenced in this study. The green horizontal bar in the MCC tree indicates the 95% HPD of the tMRCA of viruses isolated in 2019. (C) Estimated tMRCAs with 95% HPD of clade 2.3.4.4c viruses from different geographic areas using the same color scheme as panel A. Vertical dashed lines indicate medians of the distributions. Ck, chicken; GP-EA, Eurasian gene pool; GP-NAm, North American gene pool; TW, Taiwan.
Table 1.
tMRCA estimates of H5 clade 2.3.4.4c viruses in different geographical areas.
| Area | Median tMRCAa | Lower 95% HPD | Upper 95% HPD |
|---|---|---|---|
| North Asia | 2013.82 (26 October 2013) | 2013.68 (4 September 2013) | 2013.92 (1 December 2013) |
| North America | 2014.68 (7 September 2014) | 2014.57 (29 July 2014) | 2014.79 (15 October 2014) |
| Europe | 2014.71 (17 September 2014) | 2014.56 (24 July 2014) | 2014.80 (19 October 2014) |
| Taiwan | 2014.76 (3 October 2014) | 2014.62 (15 August 2014) | 2014.86 (10 November 2014) |
tMRCA estimates were calculated using the H5-HA gene.
3.3 New H5N2 reassortants
Both the HA and NA genes of the clade 2.3.4.4c H5N2 viruses isolated in 2019 were most closely related to an H5N2 virus (A/chicken/Taiwan/u7/2016) identified in 2016 (Fig. 2A and Supplementary Fig. S2). Basic amino acids at the HA cleavage site of 2019 viruses (PLRERRRKR/GLF) were also identical to previous clade 2.3.4.4c viruses from Taiwan (Lee et al. 2016). However, by analyzing phylogenetic relationships of all eight genes, we found two novel genotypes each represented by viruses in one farm. We thus assigned Genotypes 7 and 8 to these new isolates (Fig. 1B), following a previous classification (Huang et al. 2016). For Genotype 7, their NP and NS genes were similar to the Gs/GD H5N2 virus found in 2016, which belong to Genotype 2 (Figs 1B and 2B and Supplementary Fig. S3). The PB2, PB1, and PA genes of Genotype 7 did not cluster with other clade 2.3.4.4 viruses identified in Taiwan (Supplementary Fig. S3). Among them, the PB2 and PA originated from the Eurasian gene pool, whereas the PB1 originated from the North America gene pool. This PB1 gene is not closely related to PB1 carried by known HPAI viruses isolated in North America. For Genotype 8, all internal genes except MP were distinct to all known H5 viruses identified in Taiwan, and located within the Eurasian gene pool (Fig. 2B and Supplementary Fig. S3). MP genes of Genotypes 7 and 8 were similar to previous clade 2.3.4.4 viruses in Taiwan that are derived from earlier isolates throughout multiple reassortment events, clustering within a monophyletic group formed by Gs/GD H5 viruses.
Figure 2.

ML phylogeny of the N2 and NP genes of H5N2 viruses isolated in this study. Viruses isolated in this study are labeled in red, whereas tip labels in blue and brown represent previously identified Gs/GD and Mexican-like H5 viruses from Taiwan, respectively. Genotypes in clade 2.3.4.4c (1–8) and major lineages are also labeled. Bootstrap supporting values >70 are highlighted by black circles at nodes. The long branches at the tree root are shown as shorter dashed branches. Both scale bars represent 0.001 substitutions per site. BHG, bar headed goose; Ck, chicken; Dk, duck; GFc, gyrfalcon; Gs, goose; GWteal, green winged teal; HMn, hill myna, JPWe, Japanese white eye; Korea, South Korea; Mall, mallard; Pt, pintail; Sb, spoonbill; teal, common teal; TW, Taiwan; WSw, whooper swan.
The NP genes in Genotype 8 viruses were closely related to that of the H1N3 virus (A/pintail/Taiwan/WB2478/2017) isolated from a pintail in Taiwan sequenced as part of this study (Fig. 2B). The NP gene A/pintail/Taiwan/WB2478/2017 is also the most similar sequence to Genotype 8 among all available NP sequences (Supplementary Fig. S4). Other internal genes in A/pintail/Taiwan/WB2478/2017 were most closely related to genes of viruses identified in Japan (Supplementary Fig. S3). This finding provides evidence that migratory birds have acted as vectors introducing influenza virus gene segments into the local H5 genetic sink.
3.4 Mexican-like H5N2 lineage
We also sequenced a Mexican-like H5N2 virus (A/chicken/Taiwan/A1/2019) isolated in Farm A where Genotype 8 viruses were also found. The HA phylogeny further indicates A/chicken/Taiwan/A1/2019 was descended from a subclade of Group B H5N2 viruses (described in Lee et al. 2014a) that possessed two basic amino acids at the HA cleavage site (as shown by Ck/TW/CY/A2628/2012 in Fig. 3). In comparison, all of the viruses in Group A and some viruses in Group B during 2008–14 possessed three or more basic amino acid residues at the cleavage site. Furthermore, A/chicken/Taiwan/A1/2019 had a unique glycine residue at position −3 (Fig. 3). We confirmed this residue by another library containing at least 4,000× coverage at this codon (Supplementary Fig. S6).
Figure 3.

Distribution of basic amino acid residues at the HA cleavage site in Mexican-like H5N2 virus from Taiwan. Tips on the MCC tree are aligned with their corresponding sequences with basic amino acids immediately preceding the cleavage site highlighted in red. The virus isolated in this study is labeled in red. Clade posterior probabilities >0.9 are indicated by white circles at nodes on the MCC tree, whereas clade posterior probabilities between 0.9 and 0.7 are shown with black circles at nodes. Ck, chicken; TW, Taiwan.
The HA sequence of this new virus, plus other Taiwanese Mexican-like H5N2 viruses isolated during 2003–15 displayed a strong clock signal (Supplementary Fig. S5), and the regression line of genetic distance to time of the Mexican-like H5N2 viruses in Taiwan is also similar to that of viruses isolated in the Americas from 1994 to 2009. Taken together, these results suggest the enzootic circulation of Mexican-like H5N2 viruses in Taiwan. Phylogenetic analyses show this H5N2 virus possessed a classical Mexican-like H5N2 genotype as its HA and NA were most similar to H5N2 viruses isolated from Taiwan in 2015, whereas internal genes clustered with enzootic H6N1 viruses (Figs 2 and 3 and Supplementary Fig. S3).
3.5 Diversity of the two H5N2 populations in Taiwan
Isolation of Gs/GD and Mexican-like H5 viruses indicated both lineages were circulating in Taiwan at the time we sampled. With limited genetic data from either lineage after 2015 (Figs 1 and 3), it is difficult to assess viral population sizes directly. Interestingly, the clade 2.3.4.4 HA phylogeny showed that viruses from Farm A to Farm D formed distinct monophyletic groups (Fig. 1B). The tMRCA of viruses from the two farms was estimated as late 2016 to mid-2017 (median, 2017.02; 95% HPD, 2016.62–2017.48), 2 years prior to detection, and suggesting the presence of a much higher underlying HA diversity in poultry (Fig. 1B).
Since the two viruses isolated during 2016 diverged roughly at the time clade 2.3.4.4 viruses were introduced into Taiwan (i.e. about 1.5–2 years before their isolation), from a population genetics perspective (Ho and Shapiro 2011), similar coalescent intervals imply population diversity in 2019 was no less than the level of diversity in 2016 (Fig. 1B). Direct comparison between sequences shows HA genetic diversity in 2019 is more than five times of viruses identified in the 1st year of introduction (Fig. 4). For the Mexican-like H5N2 lineage, their HA diversities reached a peak in 2013 and then declined to the similar level as Gs/GD in 2015 (Fig. 4). This observation is in agreement with the phylogenetic tree that no Group A virus was found after 2013.
Figure 4.
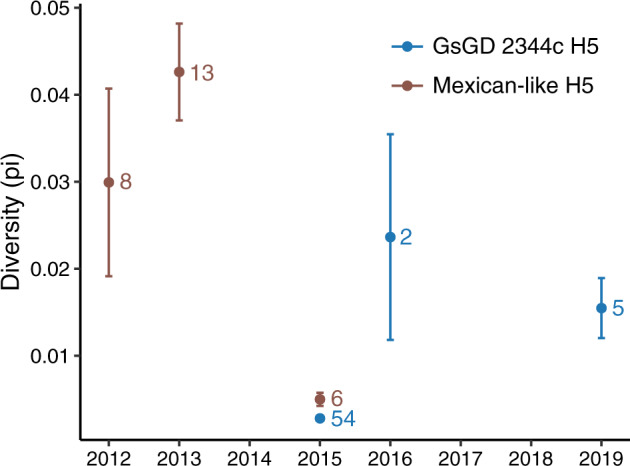
Temporal changes in genetic diversities of Taiwanese clade 2.3.4.4c and Mexico-like lineage viruses in Taiwan. All available HA genes were used to calculate the genetic diversity (π) detected in each year. Sample sizes are indicated next to the estimated values. Error bars represent SD.
4. Discussion
In this study, we analyzed newly isolated H5 viruses belonging to two major lineages in Taiwan—clade 2.3.4.4c H5N2 viruses from the HPAI Gs/GD lineage and a Mexican-like LPAI H5N2 virus in a farm where Gs/GD viruses were also identified. Our results indicate that multiple clade 2.3.4.4c reassortant viruses continued to circulate after the 2015 outbreaks. Consistent with previous studies (Huang et al. 2016; Lee et al. 2016) our data show that the gene segments incorporated during these reassortment events were related to viruses identified in eastern Asia, that is the Eurasian gene pool, within 5 years.
As Taiwan is an island, it is intuitive to speculate that migratory birds play a role in bridging the gap between the Eurasian gene pool and local influenza virus transmission, but a direct evidence has been lacking. Liu et al. (2018) reported a clade 2.3.4.4b H5N6 virus recovered from a dead spoonbill in Taiwan, however, there was no evidence of genetic exchange between this H5N6 virus (or a similar strain) and locally transmitted viruses in Taiwan (Supplementary Fig. S3). Here, we showed a pintail captured in Taiwan carried a virus which potentially contributed NP genes to the chicken infecting viruses, providing a direct example of migratory birds transmitting influenza genes into the Taiwan influenza gene pool. Migratory birds, including pintails (A.acuta), fly from Northeast Asia to Taiwan to overwinter each year (Cheng et al. 2010; Huang et al. 2016). Taiwan is located at the middle of the East Asia and Australian flyways (Cheng et al. 2010), and more intense surveillance in Taiwan would be crucial to better understand the interaction between these gene pools.
Identification of a Mexican-like H5N2 virus in this study indicates that this lineage has been circulating in Taiwan since it was first detected in 2003 (Lee et al. 2014a; Li et al. 2017). We further found that this virus had a low pathogenic marker, with two basic amino acids at the HA cleavage site. Although we were unable to directly examine its pathogenicity in chickens, closely related H5N2 viruses isolated in 2015 also with two basic amino acids were all low pathogenic (Li et al. 2017). Li et al. (2017) proposed that current control measures, which mandates culling in farms diagnosed with viruses having HPAI but not farms diagnosed with LPAI viruses, allowed Mexican-like H5N2 viruses to persist in poultry populations. Corroborating this hypothesis, our results show that the 2019 H5N2 strain originated from a subgroup that has maintained low pathogenic markers since 2012. As far as we are aware, the acquisition and then loss of basic amino acids, and the associated phenotype, in these viruses has not been previously recorded. Other hypotheses, for example, competitive exclusion with Gs/GD-like viruses leading to the replacement of HPAI Mexican-like H5N2 viruses cannot be discounted.
Although co-circulation of Gs/GD and Mexican-like H5 viruses has been described in previous studies (Huang et al. 2016; Li et al. 2017), the detailed distribution and dispersal of these two lineages in Taiwan was unknown. We found evidence for the first time that viruses of these two lineages could co-circulate within a single farm. Although only a single Mexican-like virus was detected, repeated sequencing ascertained that the sequence was not a contaminant (Supplementary Data). Co-circulation of these two lineages in poultry farms may further complicate disease control efforts for two reasons. First, pre-exposure to one of the virus strains may confer homosubtypic immunity against other H5 stains, resulting in milder symptoms or lower mortality thereby masking virus transmission. Second, even though sick birds are detected, the farm may be exempted from further control measures if solely LPAI sequences are detected. Further studies on how pre-exposure to LPAI Mexican-like viruses may mediate pathogenicity of Gs/GD HPAI virus in poultry are needed.
The limited sampling conducted in this study, only two farms in one county, determine that inferences on population diversity are less likely reflect that of the total viral population in Taiwan. Other analyses, including the timing introduction of clade 2.3.4.4 viruses and their genome constellations are not affected by sample size. Therefore, our data should be interpreted as the ‘lower bound’ estimates of genetic diversity in the area. In fact, a new subtype of clade 2.3.4.4c, H5N5, along with clade H5N2 outbreaks were reported during the time of our investigation (OIE 2019). In this regard, we urge for comprehensive surveillance of poultry farms in Taiwan to identify sources and patterns of local transmission in the island to inform control efforts and halt current enzootic transmission.
Advances in both sequencing and phylogenetic techniques have been increased their utility as powerful tools in facilitating infectious disease control (Quick et al. 2016). Our study provides a pipeline for local governments in Taiwan to rapidly generate sequences without virus culture and to implement real-time sequence analyses during an epidemic. However, only with sufficient sequences collected consistently through time and across a broad geographic area, could future studies discern dispersal patterns and risk factors of virus transmission within Taiwan.
Data availability
Accession numbers of the sequences analyzed in this study have been provided in the Supplementary Data. Sequences produced in this study are available from NCBI GenBank database.
Funding
The work is funded by Bureau of Animal and Plant Health Inspection and Quarantine (108AS-21.3.2-BQ-B1). The funding source of this study had no role in the study design, data collection, data analysis, data interpretation, or writing of the report. G.J.D.S. is supported by the Duke-NUS Signature Research Programme funded by the Ministry of Health, Singapore and by contract HHSN272201400006C from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, USA.
Conflict of interest: None declared.
Supplementary Material
References
- BAPHIQ. (2019) Epidemic Situation of Avian Influenza <https://twai.baphiq.gov.tw/AI/> accessed 31 Dec 2019.
- Bouwstra R. et al. (2015) ‘Full-genome Sequence of Influenza A(H5N8) Virus in Poultry Linked to Sequences of Strains from Asia, the Netherlands, 2014’, Emerging Infectious Diseases, 21: 872–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P. K. (2002) ‘Outbreak of Avian Influenza A(H5N1) Virus Infection in Hong Kong in 1997’, Clinical Infectious Diseases, 34: S58–64. [DOI] [PubMed] [Google Scholar]
- Chen H. et al. (2006) ‘Establishment of Multiple Sublineages of H5N1 Influenza Virus in Asia: Implications for Pandemic Control’, Proceedings of the National Academy of Sciences, 103: 2845–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. H. et al. (2018) ‘Reassortant Clade 2.3.4.4 of Highly Pathogenic Avian Influenza A(H5N6) Virus, Taiwan, 2017’, Emerging Infectious Diseases, 24: 1147–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M. C. et al. (2010) ‘Avian Influenza Monitoring in Migrating Birds in Taiwan During 1998–2007’, Avian Diseases, 54: 109–14. [DOI] [PubMed] [Google Scholar]
- Drummond A. J. et al. (2006) ‘Relaxed Phylogenetics and Dating with Confidence’, PLoS Biology, 4: e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill M. S. et al. (2013) ‘Improving Bayesian Population Dynamics Inference: A Coalescent-based Model for Multiple Loci’, Molecular Biology and Evolution, 30: 713–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S. Y., Shapiro B. (2011) ‘Skyline-plot Methods for Estimating Demographic History from Nucleotide Sequences’, Molecular Ecology Resources, 11: 423–34. [DOI] [PubMed] [Google Scholar]
- Huang P. Y. et al. (2016) ‘Genetic Characterization of Highly Pathogenic H5 Influenza Viruses from Poultry in Taiwan, 2015’, Infection, Genetics and Evolution, 38: 96–100. [DOI] [PubMed] [Google Scholar]
- Ip H. S. et al. (2015) ‘Novel Eurasian Highly Pathogenic Avian Influenza a H5 Viruses in Wild Birds, Washington, USA, 2014’, Emerging Infectious Diseases, 21: 886–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafetzopoulou L. E. et al. (2019) ‘Metagenomic Sequencing at the Epicenter of the Nigeria 2018 Lassa Fever Outbreak’, Science, 363: 74–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehira K. et al. (2015) ‘Characterization of an H5N8 Influenza a Virus Isolated from Chickens during an Outbreak of Severe Avian Influenza in Japan in April 2014’, Archives of Virology, 160: 1629–43. [DOI] [PubMed] [Google Scholar]
- Lee C. C. et al. (2014. a) ‘Emergence and Evolution of Avian H5N2 Influenza Viruses in Chickens in Taiwan’, Journal of Virology, 88: 5677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. S. et al. (2016) ‘Highly Pathogenic Avian Influenza Viruses H5N2, H5N3, and H5N8 in Taiwan in 2015’, Veterinary Microbiology, 187: 50–7. [DOI] [PubMed] [Google Scholar]
- Lee Y. J. et al. (2014. b) ‘Novel Reassortant Influenza A(H5N8) Viruses, South Korea, 2014’, Emerging Infectious Diseases, 20: 1087–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P. et al. (2009) ‘Bayesian Phylogeography Finds Its Roots’, PLoS Computational Biology, 5: e1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K. P. et al. (2017) ‘Sequence Diversity and Associated Pathogenicity of the Hemagglutinin Cleavage Site of H5N2 Avian Influenza Viruses Isolated from Chickens in Taiwan during 2013–2015’, Journal of Veterinary Medical Science, 79: 108–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. P. et al. (2018) ‘Detection of Reassortant H5N6 Clade 2.3.4.4 Highly Pathogenic Avian Influenza Virus in a Black-faced Spoonbill (Platalea minor) Found Dead, Taiwan, 2017’, Infection, Genetics and Evolution, 62: 275–8. [DOI] [PubMed] [Google Scholar]
- Nguyen L. T. et al. (2015) ‘IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-likelihood Phylogenies’, Molecular Biology and Evolution, 32: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OIE. (2019) EMPRES-i <http://empres-i.fao.org/empres-i> accessed 31 Dec 2019.
- Quick J. et al. (2016) ‘Real-time, Portable Genome Sequencing for Ebola Surveillance’, Nature, 530: 228–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick J. et al. (2017) ‘Multiplex PCR Method for MinION and Illumina Sequencing of Zika and Other Virus Genomes Directly from Clinical Samples’, Nature Protocols, 12: 1261–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2016) ‘Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen)’, Virus Evolution, 2: vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. et al. (2018) ‘Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7’, Systematic Biology, 67: 901–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T. et al. (2015) ‘Intracontinental and Intercontinental Dissemination of Asian H5 Highly Pathogenic Avian Influenza Virus (Clade 2.3.4.4) in the Winter of 2014–2015’, Reviews in Medical Virology, 25: 388–405. [DOI] [PubMed] [Google Scholar]
- Smith G. J. et al. (2015) ‘Nomenclature Updates Resulting from the Evolution of Avian Influenza A(H5) Virus Clades 2.1.3.2a, 2.2.1, and 2.3.4 during 2013–2014’, Influenza and Other Respiratory Viruses, 9: 271–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soda K. et al. (2011) ‘A Low Pathogenic H5N2 Influenza Virus Isolated in Taiwan Acquired High Pathogenicity by Consecutive Passages in Chickens’, Journal of Veterinary Medical Science, 73: 767–72. [DOI] [PubMed] [Google Scholar]
- Sonnberg S., Webby R. J., Webster R. G. (2013) ‘Natural History of Highly Pathogenic Avian Influenza H5N1’, Virus Research, 178: 63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. (2014) ‘RAxML Version 8: A Tool for Phylogenetic Analysis and Post-analysis of Large Phylogenies’, Bioinformatics, 30: 1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard M. A. et al. (2018) ‘Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10’, Virus Evolution, 4: vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger J. K., Morens D. M. (2017) ‘H5Nx Panzootic Bird Flu—Influenza’s Newest Worldwide Evolutionary Tour’, Emerging Infectious Diseases, 23: 340–2. [Google Scholar]
- Wang L. C., Huang D., Chen H. W. (2016) ‘Simultaneous Subtyping and Pathotyping of Avian Influenza Viruses in Chickens in Taiwan Using Reverse Transcription Loop-Mediated Isothermal Amplification and Microarray’, Journal of Veterinary Medical Science, 78: 1223–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth D. et al. (2007) ‘Wild Birds and Avian Influenza: An Introduction to Applied Field Research and Disease Sampling Techniques’. FAO Animal Production and Health Manual, 5
- WHO. (2018) WHO Information for the Molecular Detection of Influenza Viruses (November 2018) <https://www.who.int/influenza/gisrs_laboratory/molecular_diagnosis/>.
- WHO. (2020) Antigenic and Genetic Characteristics of Zoonotic Influenza Viruses and Candidate Vaccine Viruses Developed for Potential Use in Human Vaccines <https://www.who.int/influenza/vaccines/virus/characteristics_virus_vaccines> accessed 31 Mar 2020.
- Yu G. et al. (2018) ‘Two Methods for Mapping and Visualizing Associated Data on Phylogeny Using ggtree’, Molecular Biology and Evolution, 35: 3041–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B. et al. (2009) ‘Single-reaction Genomic Amplification Accelerates Sequencing and Vaccine Production for Classical and Swine Origin Human Influenza a Viruses’, Journal of Virology, 83: 10309–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Accession numbers of the sequences analyzed in this study have been provided in the Supplementary Data. Sequences produced in this study are available from NCBI GenBank database.