

Abstract
ADP-ribosylation refers to the post-translational modification of protein substrates with monomers or polymers of the small molecule ADP-ribose. ADP-ribosylation is enzymatically regulated and plays roles in cellular processes including DNA repair, nucleic acid metabolism, cell death, cellular stress responses and antiviral immunity. Recent advances in the field of ADP-ribosylation have led to the development of proteomics approaches to enrich and identify endogenous ADP-ribosylated peptides by liquid chromatography tandem mass spectrometry (LC-MS/MS). A number of these methods rely on reverse-phase solid-phase extraction as a critical step in preparing cellular peptides for further enrichment steps in proteomics workflows. The anionic ion-pairing reagent trifluoroacetic acid (TFA) is typically used during reverse-phase solid-phase extraction to promote retention of tryptic peptides. Here we report that TFA and other carboxylate ion-pairing reagents are inefficient for reverse-phase solid-phase extraction of ADP-ribosylated peptides. Substitution of TFA with cationic ion-pairing reagents, such as triethylammonium acetate (TEAA), improves recovery of ADP-ribosylated peptides. We further demonstrate that substitution of TFA for TEAA in a proteomics workflow specific for identifying ADP-ribosylated peptides increases identification rates of ADP-ribosylated peptides by LC-MS/MS.
Keywords: ADP-ribosylation, LC-MS/MS, site identification, ion-pairing, solid-phase extraction
Introduction
ADP-ribosylation is the post-translational modification (PTM) of protein substrates with monomers, as well as linear and branched polymers of the small molecule adenosine diphosphate ribose (ADP-ribose)1,2. ADP-ribosylation is enzymatically synthesized on substrate proteins by ADP-ribosyltransferases (ARTs), which transfer the ADP-ribose moiety from nicotinamide adenine dinucleotide (NAD+) onto amino acid side chains3. ARTs are divided into a number of different subclasses, with the majority of intracellular ADP-ribosylation being synthesized by diphtheria toxin-like ARTs, commonly known as poly(ADP-ribose)polymerases (PARPs). The human PARP family is comprised of seventeen members that share a conserved PARP domain2–6. Members of the PARP family possess different degrees of ADP-ribosyltransferase activity: PARPs -9 and -13 are enzymatically inactive, -1, -2, -5a, and -5b synthesize poly(ADP-ribose) (PAR) on substrate proteins, and the remaining members are limited to mono(ADP-ribosyl)ation (MARylation)7. As a PTM, ADP-ribosylation is capable of altering the functions of its modified substrates to regulate the enzymatic activity, localization, and protein-protein interactions of target proteins8. ADP-ribosylation plays important roles in a number of cellular processes including signaling, transcription, antiviral defense, and DNA repair8–11.
The expanding scope of ADP-ribosylation biology has led to the development of proteome-wide approaches to identify ADP-ribosylated substrates and sites to investigate the underlying mechanisms. All established methods rely on liquid chromatography tandem mass spectrometry (LC-MS/MS) of modified peptides as the final step to unambiguously identify sites of ADP-ribosylation11–23. Recently, our group has developed a new pipeline that uses enzymatic labeling of terminal ADP-ribose (ELTA) in combination with enzymatic digestion of ADP-ribosylation to phosphoribosylation25 to enrich and identify ADP-ribosylated peptides from cells. In this study, we propose a simple adjustment to a step common in most sample preparation protocols to increase recovery of ADP-ribosylated peptides prior to enrichment. In conjunction with our enrichment workflow, this adjustment enables deeper probing of the ADP-ribosylome.
Reverse-phase chromatography (RPC) is an essential component of MS-based proteomics workflows. RPC enables fractionation of complex peptide mixtures by MS-coupled high-performance liquid chromatography26 (HPLC) as well as concentration, desalting and bulk purification of peptides by solid-phase extraction (SPE)27. RPC relies on hydrophobic interactions between the solid-phase and molecules of interest to promote analyte retention. These hydrophobic interactions can be disrupted when analytes are charged; thus, RPC has limited efficacy for studying highly-charged biological macromolecules. Ion-pairing chromatography (IPC) addresses this issue by supplementing the mobile phase with a small molecule additive, termed ion-pairing reagent (IPR), that (1) carries a charge that is opposite of the analyte of interest to promote an “ion-pair” interaction with the analyte, and (2) possesses hydrophobic properties that increase the retention of the ion-pair on the reverse-phase28. Perfluorinated carboxylic acids, in particular trifluoroacetic acid (TFA), are commonly used as IPRs for HPLC29–31 and SPE27,32 of tryptic peptides due to their dual abilities to stabilize the buffer at a lower pH to shift populations of tryptic peptides to a positive-charge state, and ion-pair in the conjugate base form with the cationic analyte.
After proteolytic digestion of total cellular proteins, SPE of the resulting tryptic peptides is commonly used as a desalting step to allow for downstream processing prior to LC-MS/MS analysis. At this stage, RP-SPE using TFA is a critical buffer exchange step for protocols that enrich for ADP-ribosylated peptides, specifically those that require enzymatic processing steps prior to LC-MS analysis12,22,23,25, because the presence of chaotropic denaturants from the protein digestion step can negatively affect enzyme activity. Though these protocols have been highly effective at identifying hundreds12,25 to thousands22,23 of ADP-ribosylated sites from cells, we were concerned that ADP-ribosylated peptides, particularly those with long PAR chains at sites of modification, may be lost during RP-SPE with TFA. We postulated that the phosphate groups of ADP-ribose polymers would not be fully protonated under these conditions, and that this negative charge would overwhelm the positive charges associated with the basic residues and N-terminus of the tryptic peptide. Therefore, TFA may not act as an effective IPR for this anionic analyte, and significant amounts of endogenous ADP-ribosylated peptides could be lost during this desalting step. We hypothesized that this effect can be mitigated by the substitution of a cationic IPR for TFA as this approach has proven effective for IPC of oligonucleotides33–35.
Materials and methods
Purification of enzymes
Recombinant PARP-1, hsNudT16a and OAS1 were purified as previously described25.
Generation of 32P-labeled PARylated peptides
PARP-1 was automodified with 32P-NAD+ as previously described36 and digested with trypsin protease at a 1:10 enzyme:substrate ratio at 37 °C overnight.
32P-labeling of MARylated peptide
To label JV-09937 with 32P-α-dAMP, 200 pmol of JV-099 was incubated with 5 μCi of 32P-α-dATP, 2 μg of OAS1, and 2 μg of poly(I:C) (LMW) in OAS1 labeling buffer (20 mM Tris-HCl pH 7.5, 20 mM Mg(OAc)2, 2.5 mM DTT) at 37 °C for 1 hour.
Solid-phase extraction of radioactive peptides
SPE was performed with 1cc 50 mg C18 HyperSep cartridges (Thermo Scientific). Cartridges were conditioned with 80% acetonitrile, and equilibrated, loaded, and washed with either 0.1% trifluoroacetic acid, 0.1% acetic acid, 0.1% formic acid, 100 mM n-butylamine (pH 7.0), 100 mM tetrabutylammonium hydroxide (pH 7.0), or 100 mM triethylammonium acetate (pH 7.0). Radioactive samples were diluted 25-fold in equilibration buffer prior to loading. Samples were eluted with 40% acetonitrile. Loading and wash steps were completed by gravity flow.
Urea polyacrylamide gel electrophoresis
Prior to loading, samples were dried down to completion by vacuum centrifugation and resuspended in Urea-PAGE loading buffer. Samples were loaded on a 15% acrylamide urea gel pre-run to equilibrate at a temperature of 47 °C, and electrophoresed for 2 hours at 1000V. Gels were visualized by autoradiography.
Cell lysate harvesting, protease digestion, and solid-phase extraction
HeLa cells were grown to confluency in DMEM supplemented with 10% FBS. Treated cells were incubated with DMEM supplemented with 10% FBS and 1 mM H2O2 for 10 minutes before being placed on ice and washed with cold 1X PBS to remove excess media. Each 15-cm plate was harvested in 0.5 mL of denaturing buffer (6M guanidine hydrochloride, 100 mM HEPES pH 8.0, 5 mM TCEP, 10 mM CAM) at 95 °C and incubated in the dark for 10 minutes at 95 °C shaking at 1000 RPM. Lysates were diluted 6-fold in 25 mM Tris H-Cl pH 8.5 and supplemented with trypsin protease and LysC endoprotease at a 1:100 enzyme:substrate ratio and incubated at 37° C overnight. Prior to SPE, samples were supplemented with either 10% TFA until the sample reached pH 2, or TEAA (pH 7.0) to a final concentration of 100 mM, and cleared by centrifugation at 5000 G for 30 minutes. SPE was performed with 3 cc 200 mg tC18 Sep-Pak cartridges (Waters). Cartridges were conditioned with 80% acetonitrile, and equilibrated and washed with either 0.1% trifluoroacetic acid or 100 mM triethylammonium acetate (pH 7.0). Samples were eluted with 40% acetonitrile. All steps were completed on a vacuum manifold, with loading and wash steps completed at a low flow rate (~0.5 mL/min). Eluted peptides were dried to completion by vacuum centrifugation and stored at −80 °C until further processing.
Enrichment of ADP-ribosylated peptides
For analysis of total cellular peptides, dried down peptides were resuspended in mQ H2O and quantified by A205 using a nanodrop. Resuspended peptides were cleared by centrifugation at 21,100 G for 10 minutes. To label ADP-ribosylated peptides, each reaction contained 2.5 mg of peptides that were diluted to a concentration of 2.5 mg/mL in a reaction containing N6-azido-hexyl-dATP (Jena Bioscience), 20 μg of OAS1, and 20 μg of poly(I:C) (LMW) in OAS1 labeling buffer (100 mM Tris-HCl pH 7.5, 20 mM Mg(OAc)2, 2.5 mM DTT) at 37 °C for 1 hour. Reactions were supplemented with 100 μL of a 50% slurry of dibenzocyclooctyne-agarose (Click Chemistry Tools) equilibrated in 1X PBS and rotated at room temperature for 1 hour. The agarose was then sequentially washed with 5M NaCl, 20% acetonitrile, 1X PBS, and equilibrated in 200 μL of NudT16 reaction buffer (100 mM HEPES pH 8.0, 15 mM MgCl2). 5 μg of NudT16 was added to the slurry and the reaction was incubated at 37 °C for 2 hours shaking at 1400 RPM. Reactions were stopped by addition of 10% TFA to a final concentration of ~1%, and concentrated and desalted on StageTips38 prior to downstream analysis.
LC-MS/MS analysis of enriched peptides
Peptides were eluted from StageTips using 40% acetonitrile/0.1% TFA and dried to completion using vacuum centrifugation. Peptides were resuspended in 0.1% TFA and separated on a Thermo-Dionex RSLCNano UHPLC instrument with a 10-cm long fused silica capillary columns made in-house with a laser puller (Sutter) and packed with 3 μm 120 Å reverse phase C18 beads (Dr. Maisch). The LC gradient was 90 min long with 4-32% B at a flow rate of 300 nL/min. LC solvent A was 0.1% acetic acid and solvent B was 0.1% acetic acid, 80% acetonitrile. MS data was collected with a Thermo Orbitrap Fusion Lumos Tribrid mass spectrometer. Data-dependent analysis was applied using Top10 selection with HCD fragmentation with a normalized collision energy of 25%. Orbitrap MS spectra and MS/MS spectra were acquired at a resolution of 60000 and 30000, respectively.
Database searching of LC-MS/MS data
Raw files were analyzed by MaxQuant39 version 1.5.7.4 using protein, peptide, and site FDRs of 0.01, and a score minimum of 40 for modified peptides and 0 for unmodified peptides, and a delta score minimum of 17 for modified peptides and 0 for unmodified peptides. Sequences from endogenous peptide experiments were searched against a UniProt FASTA file of the human proteome. MaxQuant search parameters were set with variable modifications at acetylation (Protein N-term), oxidation (M) and phosphoribosylation (DEKRSYCTH) and a fixed modification of carbamidomethylation (C). Phosphoribosylation was defined as a modification of C5H9PO7 (212.009). Max-labeled amino acids were 3, max missed cleavages were 2, enzyme was Trypsin/P, max charge was 7, multiplicity was 2. For the purposes of quantifying ion intensities across runs, “Match Between Runs” was enabled with an alignment time window of 20 minutes, and a match time window of 0.7 minutes. Raw files and metadata are deposited into the MassIVE online database under accession number MSV000084446.
Results
Solid-phase extraction with cationic ion-pairing reagents promotes retention and recovery of ADP-ribosylated peptides
To test our hypothesis, we generated 32P-labeled PARylated peptides by automodifying PARP-1 with 32P-NAD+ followed by digestion with trypsin protease, and subjected these peptides to SPE using either 0.1% TFA, 0.1% formic acid (FA) or 0.1% acetic acid (AA) as anionic IPRs, or 100 mM triethylammonium acetate (TEAA) (pH 7), 100 mM n-butylamine (n-but) (pH 7) or 100 mM tetrabutylammonium hydroxide (TBAH) (pH 7) as cationic IPRs. We analyzed the extracted products by urea polyacrylamide gel electrophoresis (Urea-PAGE) and autoradiography. We observed radioactive species, corresponding to the proteolytic products derived from automodified PARP-1, with a range of electrophoretic mobilities that are characteristic of the heterogenous population of PAR generated by PARP-1 automodification in vitro25 (Fig. 1A; Lane 1). Following SPE using FA, AA, or TFA as IPRs, we observed a marked depletion of all of these species in the resulting eluate (Fig. 1A; Lane 2-4). Upon substitution of TEAA (Fig. 1A; Lane 5), n-but, or TBAH (Fig. S1A; Lane 4-5) for these carboxylate IPRs, we observed an increase in recovery of radioactive species across the entire range of the gel. We quantified the recovery of total radioactive signal in the extracted eluates by scintillation counting and calculated the fractional recovery relative to the input sample. We determined the fractions of recovered radioactive signal, when using carboxylates as IPRs, were 5 ± 1% (FA), 6 ± 1% (AA), and 6 ± 2% (TFA) (Fig. 1B). In contrast, the fraction of recovered radioactive signal when using cationic IPRs increased to 26 ± 1% (TEAA) (Fig. 1B), 19 ± 3% (n-but), and 29 ± 2% (TBAH) (Fig. S1B). Notably, increases in retention correlated with increased carbon content of the cationic IPRs, with TBAH (16 carbons) and TEAA (9 carbons) acting more effectively to retain ADP-ribosylated peptides than n-butylamine (4 carbons). This result suggests that cationic IPRs increase recovery of ADP-ribosylated peptides after SPE when compared to anionic IPRs. The remaining experiments were conducted with TFA and TEAA as representative anionic and cationic IPRs, respectively, due to their common usage in proteomics sample preparation.
Figure 1. Solid-phase extraction with triethylammonium acetate promotes retention of ADP-ribosylated peptides.
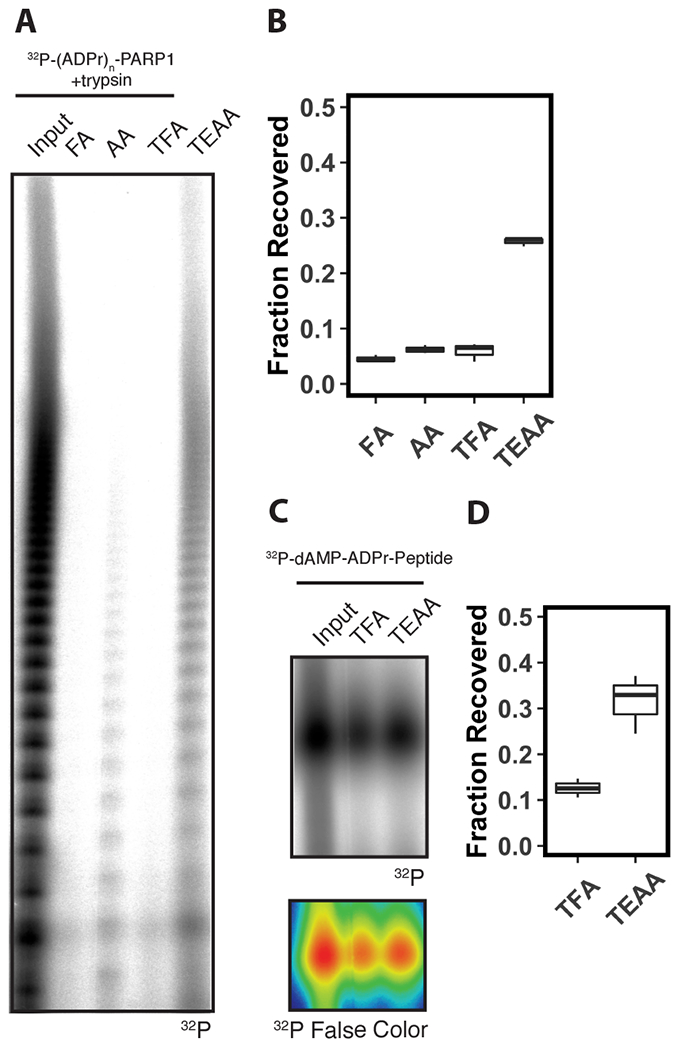
(A) Urea-PAGE analyses of 32P-(ADPr)n-PARP1 digested with trypsin before (Input), and after solid-phase extraction on a C18 cartridge using formic acid (FA), acetic acid (AA) or trifluoroacetic acid (TFA), or triethylammonium acetate (TEAA) as an ion-pairing reagent in the mobile phase. Gel was analyzed by autoradiography. (B) Relative recovery of radioactive species in (A) after solid-phase extraction. Radioactive counts were acquired by scintillation counting, and the relative recovery was calculated as a fraction of the input (n=3). (C) Urea-PAGE analyses of a 32P-labeled synthetic ADPr-peptide before (Input) and after solid-phase extraction on a C18 cartridge using trifluoroacetic acid (TFA) or triethylammonium acetate (TEAA) as an ion-pairing reagent in the mobile phase. Gel was analyzed by autoradiography (upper panel). False color image (lower panel) of autoradiograph with higher intensity pixels represented as red, medium intensity pixels represented as green, and lower intensity pixels represented as blue. (D) Relative recovery of radioactive species in (C). Radioactive counts were acquired by scintillation counting and the relative recovery was calculated as a fraction of the input (n=3).
To determine if the above observations apply to MARylated peptides, we radioactively labeled a synthetic ADP-ribosylated peptide37 with 32P-α-dAMP using ELTA25. The MARylated peptide exhibited improved recovery by SPE when TFA was used as an IPR compared to PARylated peptides (Fig. 1C; Lane 2), but substitution of TEAA as an IPR still improved recovery (Fig. 1C; Lane 3) from 13 ± 2% to 32 ± 7% (Fig. 1D). Collectively, these results demonstrate that recovery of ADP-ribosylated peptides after RP-SPE is more efficient, particularly for PARylated peptides, upon substitution of cationic IPRs for anionic IPRs.
Next, we asked if the reduced recovery of ADP-ribosylated peptides after SPE with TFA is due to lower retention of the peptides on the solid phase. To test this hypothesis, we analyzed the flowthrough and wash fractions during SPE of ADP-ribosylated peptides with TFA or TEAA as IPRs. We analyzed these samples by Urea-PAGE and observed an increased intensity of radioactive species in the flowthrough and wash fractions of TFA extracted samples (Fig. 2A; Lane 2-3), when compared to TEAA extracted samples (Fig. 2A; Lane 5-6). Scintillation counting showed an increase in radiation in both the flowthrough and wash fractions when using TFA as (flowthrough, 19 ± 3%; wash, 22 ± 1%) compared to TEAA (flowthrough, 3 ± 1%; wash, 7 ± 1%) as IPRs (Fig. 2B). Collectively, these data demonstrate that substitution of TFA for TEAA as an IPR during SPE of ADP-ribosylated peptides promotes their retention on the solid-phase to increase recovery.
Figure 2. Solid-phase extraction with triethylammonium acetate promotes retention of ADP-ribosylated peptides on solid phase.
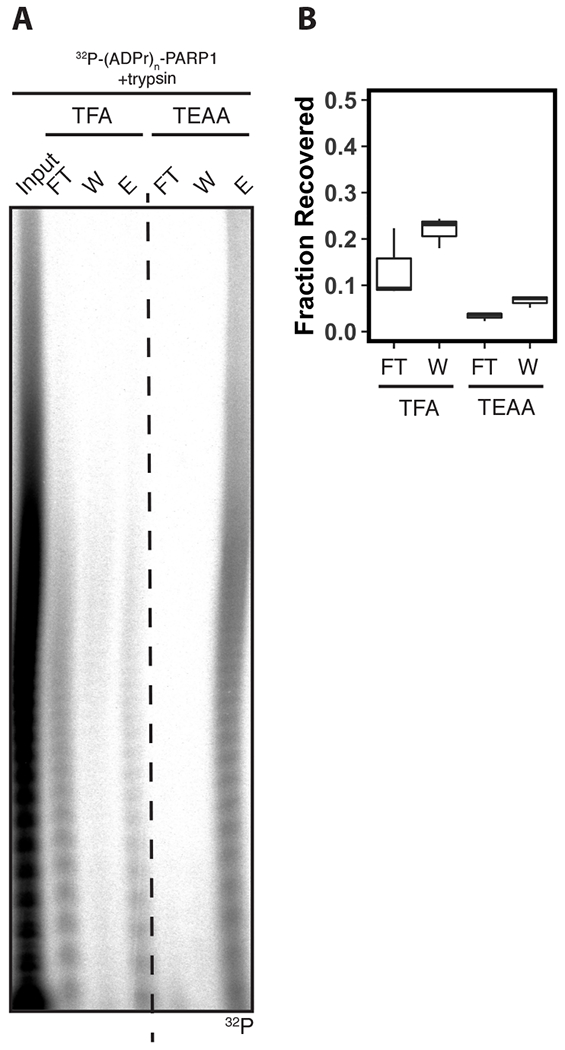
(A) Urea-PAGE analyses of 32P-(ADPr)n-PARP1 digested with trypsin before (Input), and after solid-phase extraction on a C18 cartridge using trifluoroacetic acid (TFA) or triethylammonium acetate (TEAA). Flowthrough (FT), wash (W), and elution (E) fractions are included in separate lanes. Gel was analyzed by autoradiography. (B) Relative recovery of radioactive species in (A). Radioactive counts were acquired by scintillation counting, and the relative recovery was calculated as a fraction of the input (n=3).
Solid-phase extraction of endogenous ADP-ribosylated peptides using triethylammonium acetate increases ADP-ribosylome coverage
Next, we asked if this principle could be incorporated into a proteomics sample preparation protocol that enriches for ADP-ribosylated peptides to increase the depth of enrichment of the endogenous ADP-ribosylome and improve identification rates of ADP-ribosylated peptides by LC-MS/MS. To test this hypothesis, we treated HeLa cells with 1 mM H2O2 for 10 minutes to induce ADP-ribosylation of a known set of endogenous substrates12,22–25. We harvested the cells using the chaotropic denaturing reagent guanidine-hydrochloride, and digested total cellular proteins with trypsin and LysC proteases after reduction and alkylation. Tryptic peptides were then subjected to RP-SPE using either 0.1% TFA or 100 mM TEAA (pH 7) as an IPR in the equilibration, loading, and washing buffers.
Extracted ADP-ribosylated peptides were enriched using our previously developed pipeline25 that consists of three steps: (1) ELTA-based labeling of ADP-ribosylated peptides with the enzyme oligoadenylate synthetase 1 (OAS1) and N6-(N-azido)hexyl-dATP, (2) conjugation of azido-functionalized peptides to dibenzocyclooctyne (DBCO)-agarose through copper-free click chemistry40, and (3) treatment of agarose-conjugated peptides with hsNudT16 phosphodiesterase16,17 to release peptides for LC-MS/MS analysis. Enriched peptides were analyzed on an Orbitrap Fusion Lumos Tribrid mass spectrometer using higher-collisional energy dissociation fragmentation41 to maximize identification rates, and raw data were searched using Andromeda on the MaxQuant interface39.
Using TFA as an IPR for SPE, we identified >200 modified peptides per run (Fig. 3A). Substituting TEAA as an IPR for TFA resulted in a ~3-fold increase in the identification rate of modified peptides (Fig. 3A). A portion of this increase can be attributed to repeat MS/MS scans of peptides identified in the TEAA extracted sample due to the increased enrichment and subsequent peak intensity of these ions (Fig. 3B [grey dots], Fig. 3C). However, the majority of the increase in the identification rate was due to the presence of unique modified peptides (Fig. 3D) from the TEAA extracted sample that were not identified in the TFA extracted sample (Fig. 3B [black dots, y-axis]). The increased identification rate resulted in a ~3-fold increase in the number of unique ADP-ribosylated peptides (Fig. 3D) and a corresponding ~2-fold increase in unique ADP-ribosylated proteins per run (Fig. 3E, Table S1). There were no observable differences in the distribution of MaxQuant scores for modified peptides identified in TFA vs TEAA extracted samples (Fig. 3F).
Figure 3. Solid-phase extraction of tryptic peptides with triethylammonium acetate improves enrichment of ADP-ribosylated peptides.
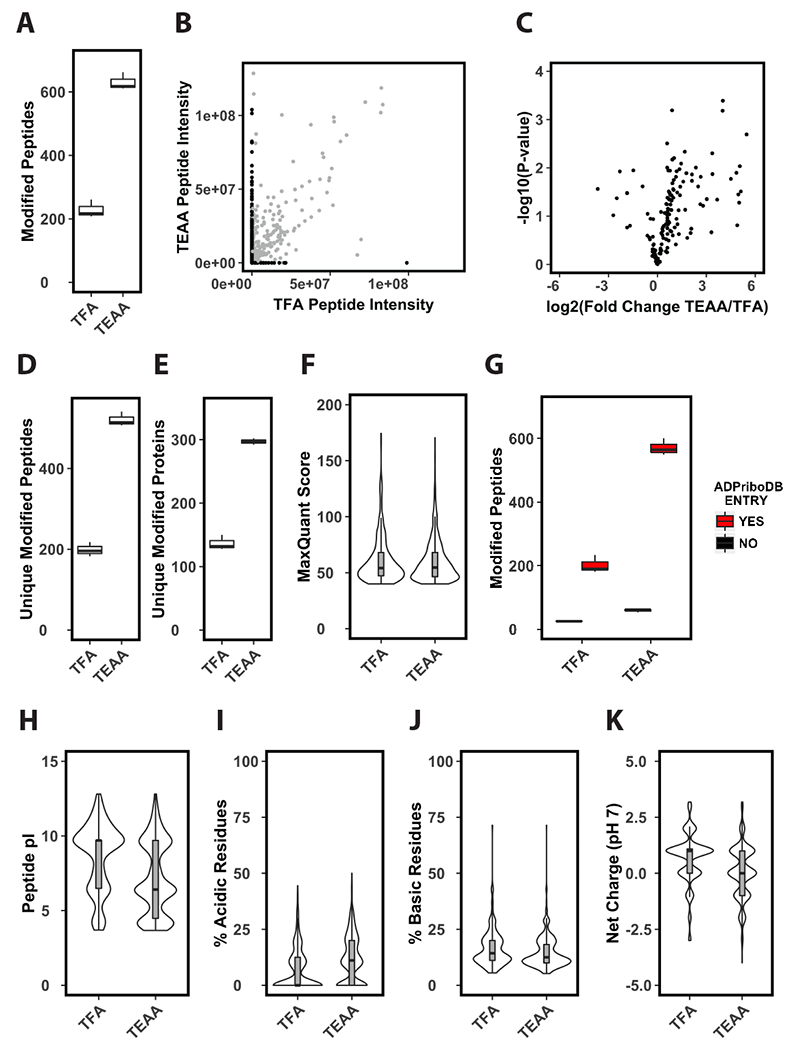
(A) Identifications per LC-MS/MS run of enriched phosphoribosylated peptides. Prior to enrichment tryptic cellular peptides were extracted by solid-phase using either TFA or TEAA as an ion-pairing reagent (n=3). (B) Average ion current intensity of enriched phosphoribosylated peptides. Peptides identified by both methods shown in grey, peptides identified by only one method are shown in black (n=3). (C) Volcano plot analysis of enriched phosphoribosylated peptides identified in both TFA and TEAA samples (n=3). (D-E) Unique identifications per LC-MS/MS run of enriched phosphoribosylated peptides (D) or proteins (E) (n=3). (F) Distribution of MaxQuant scores of phosphoribosylated peptides identified in TFA or TEAA extracted samples. (G) Identified phosphoribosylated peptides per LC-MS/MS runs in TFA or TEAA extracted samples that come from previously identified ADP-ribosylated substrates (n=3). This criterion is based on the presence (YES) or absence (NO) of the substrate from the curated ADP-ribosylated substrate database ADPriboDB. (H-K) Distribution of predicted pI values (H), percentage of acidic (I) or basic (J) residues, and average predicted net charge at pH 7 (K) of phosphoribosylated peptides identified in TFA or TEAA extracted samples.
We sought to determine if the increased identification rate of modified peptides in TEAA extracted samples resulted in the identification of previously uncharacterized ADP-ribosylated substrates. We cross-compared the genes of the identified peptides from TFA or TEAA extracted samples with ADPriboDB42, a curated database of known ADP-ribosylated substrates. We found that the majority of identified peptides came from protein substrates present in ADPriboDB in both the TFA and TEAA extracted samples (Fig. 3G). These data suggest that usage of TEAA as an IPR during SPE does not bias extraction towards or against previously uncharacterized ADP-ribosylated substrates but can be used to specifically increase the efficiency of SPE prior to enrichment.
Notably, there were a number of modified peptides that were uniquely identified in samples extracted with TFA (Fig. 3B [black dots, x-axis]), but these ions were significantly lower in number and intensity compared to the population that were uniquely identified in the TEAA extracted sample. However, the absence of these peptides in the TEAA extracted sample may suggest that anionic IPRs, such as TFA, may be favorable for the SPE of ADP-ribosylated peptides with certain physicochemical properties. Differences were observed in the distribution of pI values for the backbone sequence of the identified modified peptides from TFA vs TEAA extracted samples (Fig. 3H). This difference could be due to a specific decrease in the percentage of acidic residues in peptides from TFA extracted samples (Fig. 3I). Additionally, we observed a slight increase in the percentage of basic residues of peptides from TFA extracted samples (Fig. 3J). As expected, these changes shifted the distribution of the predicted net charge of TFA extracted peptides at pH 7 from a median value of ~0 to an approximate value of +1 (Fig. 3K). Notably, TFA, as an anionic IPR, should be more efficient at extracting peptides with a net positive charge. However, the net charge values above are calculated for the backbone sequence of the modified peptides, and do not take into account the negative charge contribution from the ADP-ribose modification. This negative charge should outweigh the charge contributions from acidic and basic residues on the peptide backbone – especially if the peptide is modified by poly(ADP-ribose). Therefore, it is unclear if the physicochemical differences of the peptide backbone sequences described above would significantly contribute to the differences in efficiency of extraction of ADP-ribosylated peptides in vitro (Fig. 1–2), or identification rates of endogenous ADP-ribosylated peptides that we observe between TFA and TEAA extracted samples (Fig. 3). However, these observations emphasize that diverse chromatographic approaches should be considered when conducting comprehensive proteomics analyses of complex PTMs.
Conclusion
In summary, we have shown that the cationic ion-pairing reagent TEAA, when compared to anionic IPRs, increases retention of ADP-ribosylated peptides on a reverse-phase cartridge for the purpose of solid-phase extraction. The substitution of TEAA for carboxylic acid IPRs allows for greater recovery after extraction of ADP-ribosylated peptides (Fig. 1–2) and, when used in conjunction with a proteomics enrichment pipeline, leads to higher identification rates of endogenous ADP-ribosylated peptides from cells (Fig. 3). This strategy can be applied to alternate proteomics enrichment pipelines to boost identification rates of ADP-ribosylated peptides as well as other protocols that seek to extract ADP-ribosylated substrates for the purposes of purification and analysis.
Supplementary Material
Acknowledgements
We thank members of the Leung lab for their critiques on the manuscript. We thank Jim Voorneveld and Dmitri Filippov for the ADP-ribosylated peptide JV-099, and Elad Elkayam for recombinant OAS1. We thank Casey Daniels, Robert O’ Malley, and Robert Cole for their insights on RP-SPE. This work was supported by R01GM104135 (A.K.L.L), T32CA009110 (R.L.M), and R01AR065459 (S.-E.O) from the NIH, as well as Research Scholar Award (RSG-16-062-01-RMC) (A.K.L.L) from American Cancer Society. This work utilized an EASY-nLC1200 UHPLC and Thermo Scientific Orbitrap Fusion Lumos mass spectrometer purchased with funding from an NIH SIG grant S10OD021502 (S.-E.O.).
Footnotes
Declaration of Interests
A patent of the ELTA technique was filed in April 2019.
References
- 1.Gupte R, Liu Z, Kraus WL. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev 2017;31:101–26. 10.1101/gad.291518.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palazzo L, Mikoč A, Ahel I. ADP-RIBOSYLATION: new facets of an ancient modification. FEBS J 2017. 10.1111/febs.14078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hottiger MO, Hassa PO, Lüscher B, Schüler H, Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci 2010;35:208–19. 10.1016/j.tibs.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Cohen MS, Chang P. Insights into the biogenesis, function, and regulation of ADP-ribosylation. Nat Chem Biol 2018;14:236–43. 10.1038/nchembio.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pascal JM, Ellenberger T. The rise and fall of poly(ADP-ribose): An enzymatic perspective. DNA Repair (Amst) 2015. 10.1016/j.dnarep.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amé J-C, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays 2004;26:882–93. 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 7.Vyas S, Matic I, Uchima L, Rood J, Zaja R, Hay RT, et al. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat Commun 2014;5:4426 10.1038/ncomms5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lüscher B, Bütepage M, Eckei L, Krieg S, Verheugd P, Shilton BH. ADP-Ribosylation, a Multifaceted Posttranslational Modification Involved in the Control of Cell Physiology in Health and Disease. Chem Rev 2018;118:1092–136. 10.1021/acs.chemrev.7b00122. [DOI] [PubMed] [Google Scholar]
- 9.Bock FJ, Todorova TT, Chang P. RNA Regulation by Poly(ADP-Ribose) Polymerases. Mol Cell 2015;58:959–69. 10.1016/j.molcel.2015.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leung AKL. Poly(ADP-ribose): An organizer of cellular architecture. J Cell Biol 2014;205:613–9. 10.1083/jcb.201402114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hottiger MO. Nuclear ADP-Ribosylation and Its Role in Chromatin Plasticity, Cell Differentiation, and Epigenetics. Annu Rev Biochem 2015;84:227–63. 10.1146/annurev-biochem-060614-034506. [DOI] [PubMed] [Google Scholar]
- 12.Martello R, Leutert M, Jungmichel S, Bilan V, Larsen SC, Young C, et al. ADP-ribosylome of mammalian cells and tissue. Nat Commun 2016;7:1–13. 10.1038/ncomms12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leslie Pedrioli DM, Leutert M, Bilan V, Nowak K, Gunasekera K, Ferrari E, et al. Comprehensive ADP-ribosylome analysis identifies tyrosine as an ADP-ribose acceptor site. EMBO Rep 2018;19:1–11. 10.15252/embr.201745310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chapman JD, Gagné JP, Poirier GG, Goodlett DR. Mapping PARP-1 auto-ADP-ribosylation sites by liquid chromatography-tandem mass spectrometry. J Proteome Res 2013;12:1868–80. 10.1021/pr301219h. [DOI] [PubMed] [Google Scholar]
- 15.Daniels CM, Ong SE, Leung AKL. Phosphoproteomic approach to characterize protein mono- and poly(ADP-ribosyl)ation sites from cells. J Proteome Res 2014;13:3510–22. 10.1021/pr401032q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daniels CM, Thirawatananond P, Ong S-E, Gabelli SB, Leung AKL. Nudix hydrolases degrade protein-conjugated ADP-ribose. Sci Rep 2015;5:18271 10.1038/srep18271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palazzo L, Thomas B, Jemth A-S, Colby T, Leidecker O, Feijs KLH, et al. Processing of protein ADP-ribosylation by Nudix hydrolases. Biochem J 2015;468:293–301. 10.1042/BJ20141554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palazzo L, Daniels CM, Nettleship JE, Rahman N, McPherson RL, Ong S-E, et al. ENPP1 processes protein ADP-ribosylation in vitro. FEBS J 2016;283:3371–88. 10.1111/febs.13811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Wang J, Ding M, Yu Y. Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat Methods 2013;10:981–4. 10.1038/nmeth.2603. [DOI] [PubMed] [Google Scholar]
- 20.Zhen Y, Zhang Y, Yu Y. A Cell-Line-Specific Atlas of PARP-Mediated Protein Asp/Glu-ADP-Ribosylation in Breast Cancer. Cell Rep 2017;21:2326–37. 10.1016/j.celrep.2017.10.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gagné J-P, Langelier M-F, Pascal JM, Poirier GG. Hydrofluoric Acid-Based Derivatization Strategy To Profile PARP-1 ADP-Ribosylation by LC–MS/MS. J Proteome Res 2018;17:2542–51. 10.1021/acs.jproteome.8b00146. [DOI] [PubMed] [Google Scholar]
- 22.Larsen SC, Hendriks IA, Lyon D, Jensen LJ, Nielsen ML, Larsen SC, et al. Systems-wide Analysis of Serine ADP-Ribosylation Reveals Widespread Occurrence and Site-Specific Overlap with Phosphorylation Resource Systems-wide Analysis of Serine ADP-Ribosylation Reveals Widespread Occurrence and Site-Specific Overlap with Phosphoryl. CellReports 2018;24:2493–2505. e4 10.1016/j.celrep.2018.07.083. [DOI] [PubMed] [Google Scholar]
- 23.Hendriks IA, Larsen SC, Nielsen ML. An advanced strategy for comprehensive profiling of ADP-ribosylation sites using mass spectrometry-based proteomics. Mol Cell Proteomics 2019;18:1010–24. 10.1074/mcp.TIR119.001315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bilan V, Selevsek N, Kistemaker HAV, Abplanalp J, Feurer R, Filippov DV., et al. New quantitative mass spectrometry approaches reveal different ADP-ribosylation phases dependent on the levels of oxidative stress. Mol Cell Proteomics 2017;16:949–58. 10.1074/mcp.O116.065623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ando Y, Elkayam E, McPherson RL, Dasovich M, Cheng S-J, Voorneveld J, et al. ELTA: Enzymatic Labeling of Terminal ADP-Ribose. Mol Cell 2019;73:845–856. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Premstaller A, Oberacher H, Walcher W, Timperio AM, Zolla L, Chervet JP, et al. High-performance liquid chromatography - Electrospray ionization mass spectrometry using monolithic capillary columns for proteomic studies. Anal Chem 2001;73:2390–6. [DOI] [PubMed] [Google Scholar]
- 27.Kamysz W, Okrój M, Łempicka E, Ossowski T, Łukasiak J. Fast and efficient purification of synthetic peptides by solid-phase extraction. Acta Chromatogr 2004:180–6. [Google Scholar]
- 28.Cecchi T. Ion pairing chromatography. Crit Rev Anal Chem 2008;38:161–213. [DOI] [PubMed] [Google Scholar]
- 29.Bennett HPJ, Browne CA, Solomon S. The Use of Perfluorinated Carboxylic Acids in the Reversed-Phase HPLC of Peptides. J Liq Chromatogr 1980;3:1353–65. [Google Scholar]
- 30.Chen Y, Mehok AR, Mant CT, Hodges RS. Optimum concentration of trifluoroacetic acid for reversed-phase liquid chromatography of peptides revisited. J Chromatogr A 2004;1043:9–18. [DOI] [PubMed] [Google Scholar]
- 31.Åsberg D, Langborg Weinmann A, Leek T, Lewis RJ, Klarqvist M, Leśko M, et al. The importance of ion-pairing in peptide purification by reversed-phase liquid chromatography. J Chromatogr A 2017;1496:80–91. [DOI] [PubMed] [Google Scholar]
- 32.Herraiz T, Casal V. Evaluation of solid-phase extraction procedures in peptide analysis. J Chromatogr A 1995;708:209–21. [DOI] [PubMed] [Google Scholar]
- 33.Fountain KJ, Gilar M, Gebler JC. Analysis of native and chemically modified oligonucleotides by tandem ion-pair reversed-phase high-performance liquid chromatography/electrospray ionization mass spectrometry. Rapid Commun Mass Spectrom 2003;17:646–53. [DOI] [PubMed] [Google Scholar]
- 34.Hölzl G, Oberacher H, Pitsch S, Stutz A, Huber CG. Analysis of biological and synthetic ribonucleic acids by liquid chromatography-mass spectrometry using monolithic capillary columns. Anal Chem 2005;77:673–80. [DOI] [PubMed] [Google Scholar]
- 35.Dickman MJ, Hornby DP. Enrichment and analysis of RNA centered on ion pair reverse phase methodology. Rna 2006;12:691–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thirawatananond P, McPherson RL, Malhi J, Nathan S, Lambrecht MJ, Brichacek M, et al. Structural analyses of NudT16–ADP-ribose complexes direct rational design of mutants with improved processing of poly(ADP-ribosyl)ated proteins. Sci Rep 2019;9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Voorneveld J, Rack JGM, Ahel I, Overkleeft HS, Van Der Marel GA, Filippov DV. Synthetic α- And β-Ser-ADP-ribosylated Peptides Reveal α-Ser-ADPr as the Native Epimer. Org Lett 2018;20:4140–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rappsilber J, Ishihama Y, Mann M. Stop And Go Extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 2003;75:663–70. [DOI] [PubMed] [Google Scholar]
- 39.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV., Mann M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J Proteome Res 2011;10:1794–805. [DOI] [PubMed] [Google Scholar]
- 40.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, et al. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci U S A 2007;104:16793–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods 2007;4:709–12. [DOI] [PubMed] [Google Scholar]
- 42.Vivelo CA, Wat R, Agrawal C, Tee HY, Leung AKL. ADPriboDB: The database of ADP-ribosylated proteins. Nucleic Acids Res 2017;45:D204–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.