

Abstract
Purpose
O-GlcNAcylation of cellular proteins contributes to the pathophysiology of diabetes and evidence supports a role for augmented O-GlcNAcylation in diabetic retinopathy. The aim of this study was to investigate the impact of the renin-angiotensin system on retinal protein O-GlcNAcylation.
Methods
Mice fed a high-fat diet were treated chronically with the angiotensin-converting enzyme inhibitor captopril or captopril plus the angiotensin-(1–7) Mas receptor antagonist A779. Western blotting and quantitative polymerase chain reaction were used to analyze retinal homogenates. Similar analyses were performed on lysates from human MIO-M1 retinal Müller cell cultures exposed to media supplemented with angiotensin-(1–7). Culture conditions were manipulated to influence the hexosamine biosynthetic pathway and/or signaling downstream of the Mas receptor.
Results
In the retina of mice fed a high-fat diet, captopril attenuated protein O-GlcNAcylation in a manner dependent on Mas receptor activation. In MIO-M1 cells, angiotensin-(1–7) or adenylate cyclase activation were sufficient to enhance cyclic AMP (cAMP) levels and inhibit O-GlcNAcylation. The repressive effect of cAMP on O-GlcNAcylation was dependent on exchange protein activated by cAMP (EPAC), but not protein kinase A, and was recapitulated by a constitutively active variant of the small GTPase Rap1. We provide evidence that cAMP and angiotensin-(1–7) act to suppress O-GlcNAcylation by inhibition of O-GlcNAc transferase (OGT) activity. In cells exposed to an O-GlcNAcase inhibitor or hyperglycemic culture conditions, mitochondrial superoxide levels were elevated; however, angiotensin-(1–7) signaling prevented the effect.
Conclusions
Angiotensin-(1–7) inhibits retinal protein O-GlcNAcylation via an EPAC/Rap1/OGT signaling axis.
Keywords: O-GlcNAcylation, Ang1–7, EPAC, cAMP, OGT
It is estimated that 16 million Americans will have diabetic retinopathy (DR) by the year 2050.1 As the leading cause of blindness in working-age adults, DR develops because of poor glycemic control and attenuated insulin receptor-mediated signaling. Clinical evidence suggests that dysregulation of the renin-angiotensin system (RAS) may also play an important role in the development of DR, as diabetic patients taking RAS blockers have lower risk for both incidence and progression of DR.2 The specific signaling mechanisms that mediate the positive effects of RAS blockers in retina are not well defined, but are believed to be independent of changes in blood pressure.2 The classic RAS is initiated by cleavage of the pro-peptide angiotensinogen by the enzyme renin to form angiotensin I (AngI), which is subject to further cleavage by angiotensin-converting enzyme (ACE) to generate angiotensin II (AngII). By activating signaling cascades coupled to the G protein-coupled receptor AT1, AngII elevates blood pressure via numerous mechanisms including oxidative stress, vasoconstriction, aldosterone release, and sympathetic activation. Renin, angiotensinogen, ACE, and AT1 have all been identified in the retina of both humans and animals.3–8 Furthermore, levels of AngII in retina are several fold higher than can be expected because of contamination from serum,9 indicating that a local ocular RAS exists independent of circulating levels of the peptide.
A reduction in AngII formation is one of the primary mediators of the beneficial cardiovascular and metabolic effects of ACE inhibitors (ACEi); however, ACEi also increase the counterregulatory angiotensin-(1–7) (Ang1–7) axis of RAS.10–13 Ang1–7 is a degradation product of AngII, generated through cleavage of the peptide by the enzyme ACE2. Additionally, Ang1–7 is formed through the degradation of AngI by endopeptidases such as neprilysin. Ang1–7 binds to the G protein-coupled receptor Mas to produce actions that are generally opposite to those of AngII, such as vasodilation and blood pressure reduction.14 In retina, Ang1–7 localizes to the footplates of Müller glia and extends through Müller cell processes, spanning all layers of the retina.15 Intraocular administration of AAV-Ang1–7 or ACE2 reduces oxidative damage, prevents the formation of acellular capillaries, and normalizes vascular permeability in retina of streptozotocin-induced diabetic rodents.16,17 Similarly, oral delivery of a probiotic expressing ACE2 reduces inflammation and prevents ganglion cell death in the retina of diabetic mice.18 Therefore, methods to increase retinal Ang1–7 signaling may be valuable in treating DR. ACE inhibition is one such method by which retinal levels of Ang1–7 may be increased.15 Indeed, ACE inhibitors correct electroretinogram deficits and attenuate retinal capillary degeneration and inflammation in diabetic mice.19,20 However, whether the beneficial actions of ACE inhibitors in retina are due to reduced AngII versus increased Ang1–7 formation remains unclear.
In the present study, we evaluated the hypothesis that Ang1–7 attenuates retinal protein O-GlcNAcylation via regulation of the hexosamine biosynthetic pathway (HBP). The HBP ultimately converts fructose-6-phosphate into uridine diphosphate β-D-N-acetylglucosamine (UDP-GlcNAc), which serves as the glycosyl donor for the posttranslational modification of proteins by O-GlcNAcylation. Expression and activity of the pathway's rate-limiting enzyme, glutamine:fructose-6-phosphate amidotransferase (GFAT) represent a critical regulatory checkpoint for HBP flux. It is hypothesized hyperglycemic conditions increase flux through the HBP resulting in greater production of UDP-GlcNAc.21,22 In kidney, AngII transcriptionally induces expression of GFAT1,23 indicating the RAS may regulate the rate of HBP flux. Differences in the relative tissue distribution of the GFAT isoforms, however, suggest that GFAT2, rather than GFAT1, is the primary rate-limiting enzyme in the HBP in the retina.24,25 Importantly, steady-state protein O-GlcNAcylation is also determined by the dynamic cycling of GlcNAc residues on and off proteins by O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), respectively.26
Dysregulated O-GlcNAcylation of cellular proteins contributes to the pathophysiology of diabetes27 and evidence supports a role for augmented O-GlcNAc signaling in DR.28 We recently demonstrated that aberrant O-GlcNAc cycling causes mitochondrial reprogramming and excessive generation of reactive oxygen species (ROS) in retina.29 In diabetes, an increase in ROS plays a critical role in the development of retinal dysfunction.30–33 The aim of the present study was to investigate the impact of signaling pathways activated by Ang1–7 on retinal protein O-GlcNAcylation and ROS production. In the retina of mice fed a high-fat diet (HFD), the ACEi captopril attenuated retinal protein O-GlcNAcylation in a manner that was dependent on Mas receptor activation. Surprisingly, the protective effects of Ang1–7 were not consistent with a change in GFAT expression or activity. Rather, the results were consistent with a model wherein Ang1–7 repressed OGT activity by elevating production of cyclic AMP (cAMP), activating exchange protein activated by cAMP (EPAC), and increasing GTP loading of the small GTPase Ras-associated protein 1 (Rap1).
Methods
Animals
Three groups of male C57Bl/6J mice (Jackson Laboratory) were placed on a diet containing 60% kcal from fat (HFD) beginning at 5 weeks of age: (1) saline (n = 6 saline infusion plus normal water); (2) captopril (n = 6 saline infusion plus captopril water); and (3) captopril + A779 (n = 5 A779 infusion [400 ng/kg/min] plus captopril water). The mice described here are a subset of those presented in our previous study.34 After 8 weeks of HFD, mice were implanted with a subcutaneous osmotic mini-pump (Alzet Model 2004) for chronic 3-week infusion of saline or the Ang-(1–7) Mas receptor antagonist A779 (Bachem). Immediately following mini-pump implantation, mice received either tap water or water containing the ACEi captopril (50 mg/L; Tocris Bioscience). There were no differences in daily water intake between mice receiving tap water and water containing captopril.34 The dose of captopril was previously demonstrated to be equivalent to captopril doses used for treatment of patients with hypertension.35 Body weight, arterial glucose levels, and plasma AngII and Ang1–7 concentrations were analyzed as previously described.34 Retinas were isolated and flash-frozen in liquid nitrogen, and lysates were prepared as previously described.36 Protein concentrations were assessed by DC Protein Assay (BioRad Laboratories, Inc; Hercules, CA), and supernatants were combined with a 2 × Laemmli buffer, boiled for 5 minutes, and analyzed via Western blotting. Retinal RNA was extracted using TRIzol reagent (Invitrogen) according to the standard manufacturer's protocol. An equal amount of RNA was converted into complementary DNA (cDNA) using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and subjected to quantitative real-time polymerase chain reaction using QuantiTect SYBR Green master mix (Qiagen). Primer sequences can be found in the Table. Changes in messenger RNA (mRNA) expression were normalized to GAPDH mRNA expression using the 2−∆∆CT calculations as previously described.37 All experimental protocols used for the studies described herein were approved by the Institutional Animal Care and Use Committee of Penn State College of Medicine.
Table.
Oligonucleotides Used in qPCR Analysis
| Gene Name | Species | Sequence | |
|---|---|---|---|
| GFAT1 | Mouse | Forward | TAAGGAGATCCAGCGGTGTC |
| Mouse | Reverse | CAGCTGTCTCGCCTGATTGA | |
| GFAT2 | Mouse | Forward | GTCATTCAGCAGTTGGAAGGC |
| Mouse | Reverse | CTTCGTACCCCGATGAGCAA | |
| OGA | Mouse | Forward | AGCGAAGATGGCAGAGGAGT |
| Mouse | Reverse | CCGTGCTCGTAAGGAAGGTA | |
| OGT | Mouse | Forward | GTGCACTGTTCATGGATTACATCATC |
| Mouse | Reverse | TCCATTGTGTATTGTTTGGTGTTG |
qPCR, quantitative polymerase chain reaction.
Cell Culture
MIO-M1 human Müller cells (obtained from the UCL Institute of Ophthalmology, London, UK) were maintained in DMEM (Gibco) containing 5.6 mM glucose, and supplemented with 10% heat inactivated (55˚C, 30 minutes) fetal bovine serum, and 1% penicillin-streptomycin (Invitrogen). For cell culture experiments, Ang1–7 acetate salt hydrate (Sigma Aldrich) was prepared in sterile water and added to culture medium at a final concentration of 10 µM, except where indicated. Thiamet G (TMG) was prepared in dimethylsulfoxide and added to culture medium at a final concentration of 50 nM. TMG and Ang1–7 were added to cell culture wells simultaneously and treatment proceeded for 24 hours. Forskolin (Fsk) was prepared in DMSO and added to cell culture medium at a concentration of 100 µM. The Mas receptor agonist AVE 0991 (Apex Bio) was prepared in DMSO and added to culture medium at a final concentration of 10 µM. Cells were pretreated with Fsk and AVE 0991 for 1 hour or 30 minutes, respectively, before TMG. TMG exposure proceeded for 24 hours. To quantify levels of cAMP, cells were stimulated with Ang1–7 for 30 minutes and then lysed with 0.1 M HCl. cAMP from cell lysates was then quantified via a cAMP competitive enzyme-linked immunosorbent assay (ELISA) kit (Invitrogen) according to manufacturer's instructions. The PKA inhibitor H89 (N-[2-p-bromocinnamylamino-ethyl]-5-isoquinolinesulfonamide) and the EPAC inhibitor ESI-09 (α-[2-(3-Chlorophenyl)hydrazinylidene]-5-(1,1-dimethylethyl)-b-oxo-3-isoxazolepropanenitrile) were prepared in DMSO and added to cell culture medium at final concentrations as indicated in the figures. H89 and ESI-09 were added to cell culture medium for 1 hour before the addition of Fsk for 1 hour, followed by the addition of TMG for 24 hours. D-(+)-Glucosamine hydrochloride (GlcN) was prepared in sterile water and added to culture medium at a final concentration of 30 mM. Where indicated, mannitol (Man) prepared in sterile water was added at a final concentration of 30 mM. The membrane-permeable EPAC agonist 8-pCPT-2′-O-Me-cAMP-AM (8-CPT; Axxora) was prepared in DMSO and added to culture medium at a final concentration of 10 µM. Cells were stimulated with Fsk for 1 hour before addition of GlcN or Man to culture medium for 24 hours. Alternatively, cells were serum starved for 15 minutes before addition of 8-CPT to culture medium. Following 30 minutes of pretreatment, GlcN was added to culture medium for 24 hours. Transfections were performed using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. Plasmids for expression of pAXEF-Rap1-WT (WT) and pAXEF-Rap1-E63(GTP) (Rap1E63) were a gift from Vicki Boussiotis (Addgene plasmids #32697 and #32698; http://n2t.net/addgene:32697; RRID:Addgene_32697; http://n2t.net/addgene:32698; RRID:Addgene_32698). Cells were lysed in 1× Laemmli buffer, boiled for 5 minutes at 100˚C, and analyzed via Western blotting.
Glycosyltransferase Activity Assay
Plasmid for expression of pcDNA2.1HA-OGT was a gift from Gerald W. Hart (University of Georgia). EZview red anti-HA affinity gel beads (Sigma, cat# E6779) were twice washed with lysis buffer (20 mM HEPES, 2 mM EGTA, 50 mM NaF, 100 mM KCl, 0.2 mM EDTA, 50 mM B-glycerophosphate, pH 7.4). After exposure to 10 µM Ang1–7 for 30 minutes, cells were harvested in 1 mL phosphate-buffered saline, spun at 1000g for 5 minutes at 4˚C, and cell pellet was resuspended in 500 µL lysis buffer plus inhibitors (1M DTT, 200 mM benzamidine, 200 mM sodium vanadate, Sigma protease inhibitor cocktail [#P8340]). Clarified cell lysate was added to the prepared anti-HA beads and incubated for 2 hours at 4˚C with gentle rocking. Lysate/bead mix was spun for 30 seconds at 8200g, unbound fraction aspirated, and bead pellet washed three times in 750 µL lysis buffer. OGT activity was assessed using a UDP-Glo glycosyltransferase assay (Promega, catalog #V6961). Bound OGT-HA fusion proteins were resuspended in 50 µL OGT transferase buffer (25 mM HEPES, pH 7.2, 10 mM MnCl2, 1 mM EDTA, and 1 mM PMSF) with 100 µM OGT peptide substrate (AnaSpec, Inc.) and 100 µM UDP-GlcNAc (Promega, catalog #V7071). After 1 hour at room temperature, reactions were quenched with UDP-Glo detection reagent and luminescence was measured using a SpectraMax M5 plate reader (Molecular Devices).
Western Blot Analysis
Lysates were fractionated using Criterion precast 4% to15% or 4% to 20% gels (Bio-Rad). Proteins were transferred to PVDF, blocked in 5% bovine serum albumin in Tris-buffered saline tween 20 for 1 hour, washed, and incubated overnight at 4˚C with the appropriate antibody. Antibodies used included O-GlcNAc (Cell Signaling Technology; catalog #9875), GAPDH (Santa Cruz; catalog #sc-32233), OGA (Novus, catalog #NBP-81244), OGT (Cell Signaling Technology; catalog #24083), and DDK (Origene; catalog #TA5011-100). Incubations with the O-GlcNAc antibody were permitted to incubate for 48 hours. The antigen-antibody interaction was visualized with enhanced chemiluminescence (Clarity Reagent; Bio-Rad Laboratories, Inc.) using a ProteinSimple Fluorochem E imaging system (Santa Clara, CA). Blots were quantified using Image J software (NIH, Bethesda, MD).
ROS Detection
MitoSOX Red mitochondrial superoxide indicator (Invitrogen) was used in MIO-M1 cells to detect mitochondrial superoxide according to the manufacturer's instructions. Briefly, cells were seeded at 200,000 cells per dish in 35 mm, No. 1.5 coverglass, Poly-d-lysine-coated dishes (MatTek Corporation). Cells were then exposed to 50 nM TMG for 24 hours. One milliliter of 5 µM MitoSOX diluted in HBSS/Ca/Mg (Life Technologies) was applied to cells. The dye-loaded cells were incubated for 10 minutes at 37˚C in the dark and then washed with HBSS buffer. Live cells were imaged using an inverted Leica TCS SP8 confocal microscope.
Statistical Analysis
Data are presented as means ± standard error. Analyses were performed using GraphPad Prism (Version 7.0) with P value < 0.05 defined as statistically significant. Means were compared using one-way analysis of variance with Tukey's multiple-comparisons post hoc tests.
Results
ACE Inhibition Attenuates Retinal Protein O-GlcNAcylation via the Mas Receptor
To evaluate the contribution of endogenous Ang1–7 to the effects of ACE inhibition on retinal O-GlcNAcylation, HFD-induced obese mice received chronic treatment with either the ACEi captopril in drinking water or captopril in combination with the Mas receptor antagonist A779. As expected, HFD mice treated with saline vehicle were obese (46 ± 6 g; Fig. 1A) and exhibited mild hyperglycemia following a 4-hour fasting period (129 ± 12 mg/dL; Fig. 1B). Chronic ACE inhibition reduced body mass in HFD mice, and this effect was not reversed by the addition of A779 (Fig. 1A). There was neither an effect of captopril alone nor captopril plus A779 on fasting blood glucose concentrations (Fig. 1B). As expected, ACE inhibition reduced circulating AngII levels (Fig. 1C), and increased circulating Ang1–7 levels (Fig. 1D). Global protein O-GlcNAcylation was attenuated in the retina of mice receiving captopril alone when compared with normal drinking water (Fig. 1E). When mice received captopril in combination with A779, however, the decrease in retinal protein O-GlcNAcylation was reversed (Fig. 1E). Thus, activation of the Mas receptor was necessary for captopril to attenuate retinal protein O-GlcNAcylation in HFD mice. To evaluate the effects of ACE inhibition on expression levels of genes that may directly affect the quantity of O-GlcNAcylated proteins, we evaluated mRNA levels of OGT, OGA, and both isoforms of GFAT expressed in the retina. Expression of the mRNAs encoding OGT, OGA, and GFAT1/2 were not altered in mice receiving captopril alone or captopril plus A779 as compared with control mice (Figs. 1F-I). When evaluated by Western blotting, OGT and OGA protein expression were also similar in retinal lysates (Fig. 1J); however, we were unable to convincingly measure GFAT1 and GFAT2 protein expression by this method.
Figure 1.

Captopril inhibits retinal protein O-GlcNAcylation via a Mas-dependent pathway. Mice were fed a 60% high-fat diet for 11 weeks and treated chronically with saline vehicle (Veh), 50 mg/L of captopril (ACEi), or captopril plus the Ang1-7 Mas receptor antagonist A779 (400 ng/kg/min) during the last 3 weeks of diet. (A–B) Body mass and fasting arterial blood glucose levels were evaluated. (C-D) Plasma Ang II and Ang1-7 levels were measured by radioimmunoassay. (E) Protein O-GlcNAcylation and GAPDH expression were assessed by Western blotting of retinal lysates. (F) GFAT1, (G) GFAT2, (H) OGA, and (I) OGT mRNA expression in whole retina were assessed by PCR. (J) OGT, OGA, and actin protein expression were assessed by Western blotting of retinal lysates. Results are expressed as means + SEM. *P < 0.05 versus Veh; #P < 0.05 versus ACEi alone. Mice are a subset of those previously described.34
Ang1–7 Enhances cAMP Levels and Inhibits Protein O-GlcNAcylation
Because Ang1–7 localizes to Müller cells in the retina,15 we used the human Müller cell line MIO-M1 to investigate direct effects of Ang1–7 on protein O-GlcNAcylation in retinal cells in culture. To enhance O-GlcNAcylation in cell culture, we used the OGA inhibitor TMG. In MIO-M1 cells treated with both Ang1–7 and TMG, protein O-GlcNAcylation was significantly reduced compared to cells treated with TMG alone (Figs. 2A-B). The Ang1–7 Mas receptor is coupled to the Gs G protein in several cell types.38–40 To determine if Ang1–7 also activates Gs-associated signaling pathways in Müller cells, we evaluated cAMP concentrations in cell lysates via ELISA. Ang1–7 significantly elevated cAMP levels in MIO-M1 cells (Fig. 2C). The conversion of ATP to cAMP is catalyzed by adenylate cyclase. Therefore, activating adenylate cyclase can be used to investigate the downstream signaling pathways initiated by the second messenger cAMP. Chang et al.41 previously reported that the adenylate cyclase activator Fsk reduced O-GlcNAc modifications of intracellular proteins in kidney epithelial cells. To determine if Fsk mediated the same effect on O-GlcNAc modified proteins in Müller cells, cells were pretreated with Fsk before inhibiting OGA. Cells exposed to both Fsk and TMG exhibited a reduction in O-GlcNAcylated proteins compared with cells exposed to TMG alone (Fig. 2D). Because TMG is used to block O-GlcNAc removal from proteins in these studies, the data support a mechanism in which activation of pathways generating cAMP suppress the rate of O-GlcNAc addition.
Figure 2.

Ang1-7 inhibits O-GlcNAcylation and promotes cAMP levels in retinal cells in culture. MIO-M1 cells were cultured in medium containing 5 mM glucose supplemented with 10% FBS. (A) Culture medium was supplemented with the O-GlcNAcase inhibitor Thiamet G (TMG) in the presence or absence of Ang1-7 for 24 hours. Protein O-GlcNAcylation in cell lysates was assessed by Western blotting. (B) MIO-M1 cells were exposed to culture medium containing TMG in the presence or absence of 10−5 M Ang1-7 for 24 hours. Protein O-GlcNAcylation is expressed relative to GAPDH. (C) Relative cAMP levels were assessed in lysates from cells exposed to culture medium containing 10−5 M Ang1-7 for 30 minutes by ELISA. (D) Cells were exposed to culture medium containing TMG in the presence or absence of the adenylate cyclase activator forskolin (Fsk). Results are expressed as means + SEM. *P < 0.05 versus Veh; #P < 0.05 versus TMG alone.
cAMP-mediated Attenuation of O-GlcNAcylation is EPAC-dependent
cAMP classically regulates cellular physiology via activation of protein kinase A (PKA), but can also activate EPAC. EPAC is involved in activation of the Ras family member Rap1 by promoting GDP/GTP exchange. To determine if these downstream effector proteins are required for cAMP-induced attenuation of O-GlcNAcylation, either PKA or EPAC were inhibited and cells were subsequently exposed to Fsk and TMG. In cells exposed to the PKA inhibitor H89, protein O-GlcNAcylation was attenuated by adenylate cyclase activation (Fig. 3A, C). In contrast, EPAC inhibition with ESI-09 prevented the suppressive effect of adenylate cyclase activation on protein O-GlcNAcylation (Figs. 3B-C). These results support a role for cAMP-dependent activation of EPAC, rather than PKA, in suppressing the rate of O-GlcNAc addition in Müller cells. To confirm that the effect of ESI-09 on O-GlcNAcylation was via EPAC inhibition, wild-type and constitutively active variants of the EPAC downstream effector Rap1 were expressed in MIO-M1 cells. Rap1E63 expression decreased protein O-GlcNAcylation in response to TMG compared with expression of WT Rap1 (Figs. 3D-E). Taken together, these results support a model wherein activation of the EPAC/Rap1 signaling axis suppresses protein O-GlcNAcylation.
Figure 3.

EPAC and the GTPase Rap1 inhibit O-GlcNAcylation in retinal cells in culture. MIO-M1 cells were cultured in medium containing 5 mM glucose supplemented with 10% FBS. (A) TMG was added to culture medium in the presence or absence of Fsk, as well as the PKA inhibitor H89 or (B) the EPAC inhibitor ESI-09. Protein O-GlcNAcylation and GAPDH expression were assessed by Western blotting. (C) Protein O-GlcNAcylation in (A) and (B) were quantified and expressed relative to GAPDH. Quantification in (C) is for 10−5 and 10−4 M H89 and ESI-09, respectively. Cells were transfected with plasmids for expression of either wild-type (WT) Rap1 or a constitutively active Rap1 variant (E63). Protein O-GlcNAcylation in (D) was quantified in (E). Total protein levels were evaluated by staining. Results are expressed as means + SEM. *P < 0.05 versus Veh; #P < 0.05 versus TMG or WT in (C) and (E), respectively; $P < 0.05 versus Fsk alone.
Ang1–7 Acts Downstream of GFAT by Inhibiting OGT
The phosphorylation of glucosamine (GlcN) produces glucosamine-6-phosphate (GlcN-6-P), which may be metabolized further by enzymes in the HBP. Importantly, GlcN-6-P enters the HBP downstream of the rate-limiting enzyme GFAT. The cAMP-mediated attenuation of retinal protein O-GlcNAcylation is likely due to either a restricted pool of substrate (UDP-GlcNAc) available for OGT, or reduced activity of OGT itself. In cells exposed to culture medium supplemented with GlcN, protein O-GlcNAcylation was enhanced (Fig. 4A). However, in cells pretreated with Fsk before addition of GlcN to cell culture medium, O-GlcNAcylation was significantly reduced as compared with cells exposed to GlcN alone (Figs. 4B-C). No significant change in O-GlcNAc-modified proteins was observed in cells exposed to the osmotic control mannitol (Figs. 4B-C). To determine if activation of EPAC could similarly attenuate GlcN-induced protein O-GlcNAcylation, cells were pretreated with the EPAC activator 8-CPT. In cells pretreated with 8-CPT before addition of GlcN to cell culture medium, O-GlcNAcylation was significantly reduced as compared to cells exposed to GlcN alone (Figs. 4D-E). If Fsk or 8-CPT limits the pool of available UDP-GlcNAc via negative regulation of GFAT activity, it is expected that Fsk and 8-CPT would have no effect on O-GlcNAcylation induced by GlcN. However, the results described here support the alternate hypothesis that EPAC-specific signaling pathways negatively regulate O-GlcNAcylation downstream of GFAT. To determine if Ang1–7 inhibited OGT activity, HA-tagged OGT was expressed in and immunoprecipitated from cells exposed to Ang1–7. HA-tagged OGT was then assayed for glycosyltransferase activity. The glycosyltransferase activity of OGT isolated from cells exposed to Ang1–7 was attenuated compared with OGT from cells exposed to vehicle (Fig. 4F).
Figure 4.

Ang1-7 inhibits OGT activity in retinal cells in culture. MIO-M1 cells were cultured in medium containing 5 mM glucose supplemented with 10% FBS. (A) Cells were exposed to culture medium containing glucosamine (GlcN) for 24 hours. (B–E) Cells were exposed to culture medium containing 30 mM glucosamine (GlcN), 30 mM mannitol (Man), and/or Fsk or 8-CPT. Protein O-GlcNAcylation was assessed by Western blotting. Total protein levels were evaluated by staining. O-GlcNAcylation in (B) and (D) was quantified in (C) and (E). (F) HA-tagged OGT was expressed in MIO-M1 cells in culture. Forty-eight hours after transfection, cells were exposed to Ang1-7 for 30 minutes, HA-tagged OGT was immunoprecipitated from cell lysates, and in vitro OGT activity was evaluated by glycosyltransferase assay. Results are expressed as means + SEM. *P < 0.05 versus no treatment (NT) or Veh; #P < 0.05 versus GlcN or Veh.
Ang1–7 Inhibits O-GlcNAcylation-induced Mitochondrial Superoxide
We recently reported that increasing retinal protein O-GlcNAcylation elevates levels of mitochondrial superoxide.29 To determine if Ang1–7 signaling could normalize the effect of O-GlcNAcylation on superoxide levels, MitoSox staining was evaluated in live MIO-M1 cells. OGA inhibition increased superoxide in MIO-M1 cells; however, Ang1–7 (Figs. 5A-B) or the Mas receptor agonist AVE 0991 (Figs. 5C-D) reversed the TMG effect on superoxide levels. Similarly, in MIO-M1 cells exposed to hyperglycemic culture conditions, superoxide levels were found to be elevated, and Ang1–7 addition to culture medium was sufficient to normalize superoxide levels in cells exposed to hyperglycemic conditions (Figs. 5E-F). These results support the hypothesis that Ang1–7 signaling is sufficient to prevent increased mitochondrial superoxide production in response to stimuli that enhance O-GlcNAcylation.
Figure 5.

Ang1-7 inhibits O-GlcNAcylation-induced mitochondrial superoxide in retinal cells in culture. MIO-M1 cells were cultured in medium containing 5 mM glucose supplemented with 10% FBS. (A–D) Cells were exposed to culture medium containing TMG in the presence or absence of Ang1-7 or the Mas receptor agonist AVE 0991. (E–F) Cells were exposed to culture medium containing the 30 mM glucose (HG) in the presence or absence of Ang1-7. Mitochondrial superoxide was assessed in live cells by MitoSox Red. MitoSox fluorescence was visualized by confocal laser microscopy (A, C, E) and quantified (B, D, F, respectively). Representative MitoSox and bright-field (BF) images are shown. Results are expressed as means + SEM. *P < 0.05 versus no treatment (NT) or low glucose (LG); #P <.05 versus TMG or HG alone. RFU, relative fluorescent units.
Discussion
In the present study, we evaluated the impact of pathways activated by Ang1–7 on O-GlcNAcylation of retinal proteins. Within the eye, O-GlcNAcylated proteins have been described in the lens, cornea, retinal pigment epithelium, neural retina, and retinal vasculature.28,42–44 Previous studies report increased O-GlcNAcylation of retinal proteins in diabetic mice and during the active neovascularization phase of oxygen-induced retinopathy.28,43 More recently, we demonstrated that in the retina of mice fed a HFD protein O-GlcNAcylation is enhanced concomitant with transcriptional upregulation of GFAT2.25 GFAT expression and activity represent a critical regulatory checkpoint in HBP flux. In this study, we found that in the retina of HFD mice, captopril attenuated retinal protein O-GlcNAcylation, and the effect was prevented by Mas-receptor inhibition. This finding provides mechanistic insight into the role for elevated Ang1–7 levels in the beneficial effects of ACE inhibition in retina, and suggests that direct targeting of this hormone may be advantageous. Surprisingly, the repressive effect of captopril was not explained by a reduction in the expression of GFAT2, or other enzymes linked to O-GlcNAcylation (i.e., GFAT1, OGT, OGA). Rather, the results here are consistent with a model wherein Mas receptor activation by Ang1–7 inhibits OGT activity via activation of EPAC/Rap1 (Fig. 6).
Figure 6.
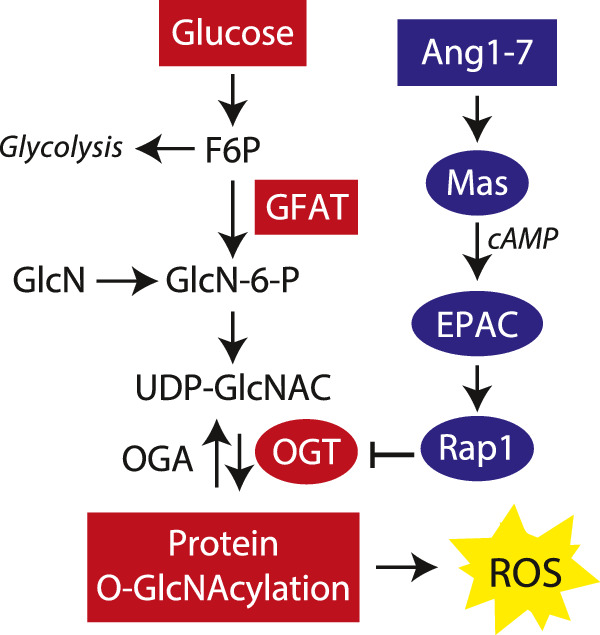
Working model for inhibition of retinal protein O-GlcNAcylation by Ang1-7. The results support a model wherein Ang1-7 inhibits O-GlcNAcylation-induced reactive oxygen species (ROS) via a novel Mas/EPAC/Rap1/OGT signaling axis. Ang1-7 activates the Mas receptor to enhance cAMP levels and activate EPAC. In turn, EPAC promotes Rap1 GDP release and GTP binding. Activation of this signaling axis suppresses protein O-GlcNAcylation downstream of GFAT by inhibiting OGT activity.
This study extends upon previous seminal work in the field demonstrating that retinal Müller cells are an important source of AngII and its metabolite Ang1–7, and that the ACEi captopril can cross the blood-retinal barrier to increase levels of Ang1–7.15,45 The existence of an ocular RAS including components of the Ang1–7-ACE2-Mas receptor axis has been well documented,3,7,9,16,46–49 but the signaling cascades activated downstream of the Mas receptor in retinal Müller cells are not well defined. Herein, we found cAMP levels were elevated in MIO-M1 retinal cells following exposure to culture medium containing Ang1–7. An increase in cellular cAMP concentrations following exposure to Ang1–7 has been previously described in other cell types50 and found to be dependent on activation of the Mas receptor and adenylate cyclase.39 Increasing intracellular cAMP concentrations via stimulation of adenylate cyclase has also been reported to decrease protein O-GlcNAcylation in kidney cell cultures exposed to hyperglycemic conditions.41 Notably, a reduction in O-GlcNAc levels following adenylate cyclase stimulation in kidney cell cultures was overcome by supplementation with GlcN.41 GlcN enters the HBP upon direct phosphorylation to GlcN-6-P, and thus bypasses the rate-limiting GFAT enzymes. Coupled with the finding that PKA inhibits GFAT1 by phosphorylating the enzyme on S205 in vitro,41 the preceding observations led us to initially hypothesize that the Ang1–7-initiated signaling cascade inhibits O-GlcNAcylation by repressing GFAT1 activity. However, we subsequently discovered that in both the retina of mice fed an HFD and in Müller cells in culture, GFAT2 is the predominant isoform, rather than GFAT1.25 Unlike GFAT1, the enzymatic activity of GFAT2 is enhanced by PKA-dependent phosphorylation.51 Thus, a repressive effect of PKA activation on O-GlcNAcylation, mediated by phosphorylation of the GFAT isoforms seemed unlikely. Indeed, PKA inhibition was not sufficient to prevent the repressive effect of adenylate cyclase activation on O-GlcNAcylation in Müller cells.
The signaling events initiated by cAMP are classically thought of as being mediated by PKA; however, EPAC is another key signaling molecule that is sensitive to this second messenger.52 EPAC functions as the guanine nucleotide exchange factor for the small GTPase Rap1. EPAC1 and EPAC2 protein isoforms are differentially expressed in retina.53 With immunostaining, both EPAC isoforms colocalize in Müller cell bodies within the inner nuclear layer of the retina.53 In the present study, EPAC inhibition prevented the repressive effect of adenylate cyclase activation on protein O-GlcNAcylation in Müller cells in culture. Moreover, expression of a constitutively active Rap1 variant was sufficient to repress O-GlcNAcylation in Müller cells following OGA inhibition. We initially suspected that cAMP/EPAC/Rap1 signaling may have acted to repress O-GlcNAcylation via a reduction in GFAT2 enzymatic activity, and thus functioned in a manner analogous to PKA-dependent GFAT1 inhibition in other cell types. However, unlike the previous observation in kidney cell cultures,41 adenylate cyclase activation was, in fact, sufficient to inhibit GlcN-induced protein O-GlcNAcylation in Müller cells in culture. Thus, the results are consistent with a model wherein EPAC and Rap1 act to inhibit O-GlcNAcylation downstream of GFAT.
Herein, we provided evidence that Ang1–7 exerts an inhibitory effect on OGT activity via EPAC activation. Although one study has identified a novel interaction between the glycolytic enzyme pyruvate kinase and soluble adenylate cyclase that results in EPAC/Rap1 activation,54 there are currently no studies demonstrating that EPAC interacts with the metabolic enzymes of the HBP. The results here support a model wherein EPAC activation inhibits O-GlcNAcylation downstream of GFAT. Importantly, other enzymes upstream of OGT and downstream of GFAT may also be impacted. A possible mechanism by which the Ang1–7/Mas/EPAC/Rap1 axis may act to repress OGT activity is via activation of a tyrosine phosphatase. Insulin-stimulated phosphorylation of tyrosine residues on OGT enhances the enzyme's catalytic activity.55 In vitro, OGT is directly phosphorylated by Src tyrosine kinase, as it contains well over a dozen predicted Src tyrosine phosphorylation sites.55 This is consistent with a stimulatory effect of tyrosine phosphatase inhibitors on OGT activity.55 In proximal tubular cells, Ang1–7 acts via the Mas receptor to promote Src homology 2-containing protein-tyrosine phosphatase-1 (SHP-1) activity56 and Rap1b-deficient cells have impaired SHP-1 activity.57 It is therefore tempting to speculate that pathways initiating a cAMP second messenger response may suppress OGT activity by promoting EPAC/Rap1-dependent activation of SHP-1 and OGT dephosphorylation. Unfortunately, we were unable to consistently detect OGT tyrosine phosphorylation by Western blotting following immunoprecipitation of the protein.
Mitochondrial dysfunction and increased ROS production are central to the development of diabetic retinopathy.30–33 As a critical regulator of mitochondrial function,58,59 the O-GlcNAc modification posttranslationally modifies several mitochondrial proteins directly in response to diabetes and hyperglycemic conditions.60,61 We recently reported that enhanced O-GlcNAcylation of retinal proteins promotes 4E-BP1-dependent mitochondrial dysfunction and oxidative stress.29 In that study, the OGA inhibitor TMG enhanced O-GlcNAcylation and increased reactive oxygen species in the retina. Here, Müller cells exposed to TMG exhibited an increase in mitochondrial superoxide production. However, Ang1–7 addition to culture medium was not only sufficient to prevent an increase in O-GlcNAcylation, but also normalized mitochondrial superoxide levels following OGA inhibition. This supports the observation that diabetes-induced oxidative stress is normalized in the retina of diabetic rodents receiving intraocular AAV-Ang1–7/ACE2.16,17 The finding that Ang1–7 activates the EPAC/Rap1 signaling axis and attenuates mitochondrial superoxide production supports previous studies indicating EPAC activation suppresses ROS production.62–64 Thus, strategies to enhance Ang1–7-mediated EPAC activation in retina could serve to prevent the increase in oxidative stress that is associated with diabetes-induced O-GlcNAcylation.
The present study identified a novel molecular link between Ang1–7, protein O-GlcNAcylation, and ROS production in retina. Ang1–7 signaling reduced protein O-GlcNAcylation in the retina of HFD mice. Similarly, Ang1–7 attenuated O-GlcNAcylation in human Müller cells by increased production of cAMP, with this effect dependent on EPAC activation. Specifically, Ang1–7 inhibited OGT activity. Thus, although further studies are needed, targeted activation of this newly identified Ang1–7/EPAC/OGT signaling axis may be beneficial in correcting the abnormally elevated protein O-GlcNAcylation that is associated with diabetic retinal complications.
Acknowledgments
The authors thank Gerald W. Hart (University of Georgia) for generously providing the plasmid encoding HA-tagged OGT and Scot R. Kimball (Penn State College of Medicine) for critically evaluating the manuscript. The authors thank Joseph Giordano and Sarah Stenske for assistance with Western blotting.
Parts of this study were presented in abstract form at the 78th Scientific Session of the American Diabetes Association, Orlando, FL, June 2018.
Supported by the American Diabetes Association Pathway to Stop Diabetes Grant 1-14-INI-04, R01 EY029702 (MDD), and R00 HL122507 (ACA).
Disclosure: S.K. Dierschke, None; A.L. Toro, None; A.J. Barber, None; A.C. Arnold, None; M.D. Dennis, None
References
- 1. Saaddine JB, Honeycutt AA, Narayan KM, Zhang X, Klein R, Boyle JP. Projection of diabetic retinopathy and other major eye diseases among people with diabetes mellitus: United States, 2005-2050. Arch Ophthalmol. 2008; 126: 1740–1747. [DOI] [PubMed] [Google Scholar]
- 2. Wang B, Wang F, Zhang Y, et al.. Effects of RAS inhibitors on diabetic retinopathy: a systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2015; 3: 263–274. [DOI] [PubMed] [Google Scholar]
- 3. Danser AH, van den Dorpel MA, Deinum J, et al.. Renin, prorenin, and immunoreactive renin in vitreous fluid from eyes with and without diabetic retinopathy. J Clin Endocrinol Metab. 1989; 68: 160–167. [DOI] [PubMed] [Google Scholar]
- 4. Deinum J, Derkx FH, Danser AH, Schalekamp MA. Identification and quantification of renin and prorenin in the bovine eye. Endocrinology. 1990; 126: 1673–1682. [DOI] [PubMed] [Google Scholar]
- 5. Sramek SJ, Wallow IH, Tewksbury DA, Brandt CR, Poulsen GL. An ocular renin-angiotensin system. Immunohistochemistry of angiotensinogen. Invest Ophthalmol Vis Sci. 1992; 33: 1627–1632. [PubMed] [Google Scholar]
- 6. Feman SS, Mericle RA, Reed GW, May JM, Workman RJ. Serum angiotensin converting enzyme in diabetic patients. Am J Med Sci. 1993; 305: 280–284. [DOI] [PubMed] [Google Scholar]
- 7. Wagner J, Jan Danser AH, Derkx FH, et al.. Demonstration of renin mRNA, angiotensinogen mRNA, and angiotensin converting enzyme mRNA expression in the human eye: evidence for an intraocular renin-angiotensin system. Br J Ophthalmol. 1996; 80: 159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murata M, Nakagawa M, Takahashi S. Expression and localization of angiotensin II type 1 receptor mRNA in rat ocular tissues. Ophthalmologica. 1997; 211: 384–386. [DOI] [PubMed] [Google Scholar]
- 9. Danser AH, Derkx FH, Admiraal PJ, Deinum J, de Jong PT, Schalekamp MA. Angiotensin levels in the eye. Invest Ophthalmol Vis Sci. 1994; 35: 1008–1018. [PubMed] [Google Scholar]
- 10. Campbell DJ, Lawrence AC, Towrie A, Kladis A, Valentijn AJ. Differential regulation of angiotensin peptide levels in plasma and kidney of the rat. Hypertension. 1991; 18: 763–773. [DOI] [PubMed] [Google Scholar]
- 11. Kohara K, Brosnihan KB, Ferrario CM. Angiotensin(1-7) in the spontaneously hypertensive rat. Peptides. 1993; 14: 883–891. [DOI] [PubMed] [Google Scholar]
- 12. Kucharewicz I, Pawlak R, Matys T, Pawlak D, Buczko W. Antithrombotic effect of captopril and losartan is mediated by angiotensin-(1-7). Hypertension. 2002; 40: 774–779. [DOI] [PubMed] [Google Scholar]
- 13. Ishiyama Y, Gallagher PE, Averill DB, Tallant EA, Brosnihan KB, Ferrario CM. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension. 2004; 43: 970–976. [DOI] [PubMed] [Google Scholar]
- 14. Santos RA, Simoes e Silva AC, Maric C, et al.. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA. 2003; 100: 8258–8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Senanayake PD, Bonilha VL, J WP, et al.. Retinal angiotensin II and angiotensin-(1-7) response to hyperglycemia and an intervention with captopril. J Renin Angiotensin Aldosterone Syst. 2018; 19:1470320318789323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Verma A, Shan Z, Lei B, et al.. ACE2 and Ang-(1-7) confer protection against development of diabetic retinopathy. Mol Ther. 2012; 20: 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dominguez JM, Hu P, Caballero S, et al.. Adeno-associated virus overexpression of angiotensin-converting enzyme-2 reverses diabetic retinopathy in type 1 diabetes in mice. Am J Pathol. 2016; 186: 1688–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Verma A, Xu K, Du T, et al.. Expression of human ACE2 in Lactobacillus and beneficial effects in diabetic retinopathy in mice. Mol Ther Methods Clin Dev. 2019; 14: 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bui BV, Armitage JA, Tolcos M, Cooper ME, Vingrys AJ. ACE inhibition salvages the visual loss caused by diabetes. Diabetologia. 2003; 46: 401–408. [DOI] [PubMed] [Google Scholar]
- 20. Zhang JZ, Xi X, Gao L, Kern TS. Captopril inhibits capillary degeneration in the early stages of diabetic retinopathy. Curr Eye Res. 2007; 32: 883–889. [DOI] [PubMed] [Google Scholar]
- 21. McClain DA. Hexosamines as mediators of nutrient sensing and regulation in diabetes. J Diabetes Complications. 2002; 16: 72–80. [DOI] [PubMed] [Google Scholar]
- 22. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001; 414: 813–820. [DOI] [PubMed] [Google Scholar]
- 23. James LR, Ingram A, Ly H, Thai K, Cai L, Scholey JW. Angiotensin II activates the GFAT promoter in mesangial cells. Renal Physiol. 2001; 281: F151–F162. [DOI] [PubMed] [Google Scholar]
- 24. Oki T, Yamazaki K, Kuromitsu J, Okada M, Tanaka I. cDNA cloning and mapping of a novel subtype of glutamine:fructose-6-phosphate amidotransferase (GFAT2) in human and mouse. Genomics. 1999; 57: 227–234. [DOI] [PubMed] [Google Scholar]
- 25. Dai W, Dierschke SK, Toro AL, Dennis MD. Consumption of a high fat diet promotes protein O-GlcNAcylation in mouse retina via NR4A1-dependent GFAT2 expression. Biochim Biophys Acta Mol Basis Dis. 2018; 1864: 3568–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007; 446: 1017–1022. [DOI] [PubMed] [Google Scholar]
- 27. Issad T, Masson E, Pagesy P. O-GlcNAc modification, insulin signaling and diabetic complications. Diabetes Metab. 2010; 36: 423–435. [DOI] [PubMed] [Google Scholar]
- 28. Semba RD, Huang H, Lutty GA, Van Eyk JE, Hart GW. The role of O-GlcNAc signaling in the pathogenesis of diabetic retinopathy. Proteomics Clin Appl. 2014; 8: 218–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dierschke SK, Miller WP, Favate JS, et al.. O-GlcNAcylation alters the selection of mRNAs for translation and promotes 4E-BP1-dependent mitochondrial dysfunction in the retina. J Biol Chem. 2019; 294: 5508–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kowluru RA, Kern TS, Engerman RL, Armstrong D. Abnormalities of retinal metabolism in diabetes or experimental galactosemia. III. Effects of antioxidants. Diabetes. 1996; 45: 1233–1237. [DOI] [PubMed] [Google Scholar]
- 31. Nishikawa T, Edelstein D, Du XL, et al.. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000; 404: 787–790. [DOI] [PubMed] [Google Scholar]
- 32. Kowluru RA, Kowluru V, Xiong Y, Ho YS. Overexpression of mitochondrial superoxide dismutase in mice protects the retina from diabetes-induced oxidative stress. Free Radic Biol Med. 2006; 41: 1191–1196. [DOI] [PubMed] [Google Scholar]
- 33. Kanwar M, Chan PS, Kern TS, Kowluru RA. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci. 2007; 48: 3805–3811. [DOI] [PubMed] [Google Scholar]
- 34. Loloi J, Miller AJ, Bingaman SS, Silberman Y, Arnold AC. Angiotensin-(1-7) contributes to insulin-sensitizing effects of angiotensin-converting enzyme inhibition in obese mice. Am J Physiol Endocrinol Metab. 2018; 315: E1204–e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Premaratna SD, Manickam E, Begg DP, et al.. Angiotensin-converting enzyme inhibition reverses diet-induced obesity, insulin resistance and inflammation in C57BL/6J mice. Int J Obes (Lond). 2012; 36: 233–243. [DOI] [PubMed] [Google Scholar]
- 36. Dennis MD, Kimball SR, Fort PE, Jefferson LS. Regulated in development and DNA damage 1 is necessary for hyperglycemia-induced vascular endothelial growth factor expression in the retina of diabetic rodents. J Biol Chem. 2015; 290: 3865–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 38. Sahr A, Wolke C, Maczewsky J, et al.. The angiotensin-(1-7)/Mas axis improves pancreatic beta-cell function in vitro and in vivo. Endocrinology. 2016; 157: 4677–4690. [DOI] [PubMed] [Google Scholar]
- 39. Tetzner A, Gebolys K, Meinert C, et al.. G-protein-coupled receptor MrgD is a receptor for angiotensin-(1-7) involving adenylyl cyclase, cAMP, and phosphokinase A. Hypertension. 2016; 68: 185–194. [DOI] [PubMed] [Google Scholar]
- 40. Liu GC, Oudit GY, Fang F, Zhou J, Scholey JW. Angiotensin-(1-7)-induced activation of ERK1/2 is cAMP/protein kinase A-dependent in glomerular mesangial cells. Am J Physiol Renal Physiol. 2012; 302: F784–790. [DOI] [PubMed] [Google Scholar]
- 41. Chang Q, Su K, Baker JR, Yang X, Paterson AJ, Kudlow JE. Phosphorylation of human glutamine:fructose-6-phosphate amidotransferase by cAMP-dependent protein kinase at serine 205 blocks the enzyme activity. J Biol Chem. 2000; 275: 21981–21987. [DOI] [PubMed] [Google Scholar]
- 42. Xu C, Liu GD, Feng L, Zhang CH, Wang F. Identification of O-GlcNAcylation modification in diabetic retinopathy and crosstalk with phosphorylation of STAT3 in retina vascular endothelium cells. Cell Physiol Biochem. 2018; 49: 1389–1402. [DOI] [PubMed] [Google Scholar]
- 43. Gurel Z, Sieg KM, Shallow KD, Sorenson CM, Sheibani N. Retinal O-linked N-acetylglucosamine protein modifications: implications for postnatal retinal vascularization and the pathogenesis of diabetic retinopathy. Mol Vis. 2013; 19: 1047–1059. [PMC free article] [PubMed] [Google Scholar]
- 44. Xu C, Liu G, Liu X, Wang F. O-GlcNAcylation under hypoxic conditions and its effects on the blood-retinal barrier in diabetic retinopathy. Int J Mol Med. 2014; 33: 624–632. [DOI] [PubMed] [Google Scholar]
- 45. Senanayake P, Drazba J, Shadrach K, et al.. Angiotensin II and its receptor subtypes in the human retina. Invest Ophthalmol Vis Sci. 2007; 48: 3301–3311. [DOI] [PubMed] [Google Scholar]
- 46. Berka JL, Stubbs AJ, Wang DZ, et al.. Renin-containing Muller cells of the retina display endocrine features. Invest Ophthalmol Vis Sci. 1995; 36: 1450–1458. [PubMed] [Google Scholar]
- 47. Brandt CR, Pumfery AM, Micales B, et al.. Renin mRNA is synthesized locally in rat ocular tissues. Curr Eye Res. 1994; 13: 755–763. [DOI] [PubMed] [Google Scholar]
- 48. Kida T, Ikeda T, Nishimura M, et al.. Renin-angiotensin system in proliferative diabetic retinopathy and its gene expression in cultured human muller cells. Jpn J Ophthalmol. 2003; 47: 36–41. [DOI] [PubMed] [Google Scholar]
- 49. Foureaux G, Nogueira BS, Coutinho DCO, Raizada MK, Nogueira JC, Ferreira AJ. Activation of endogenous angiotensin converting enzyme 2 prevents early injuries induced by hyperglycemia in rat retina. Braz J Med Biol Res. 2015; 48: 1109–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tallant EA, Clark MA. Molecular mechanisms of inhibition of vascular growth by angiotensin-(1-7). Hypertension. 2003; 42: 574–579. [DOI] [PubMed] [Google Scholar]
- 51. Hu Y, Riesland L, Paterson AJ, Kudlow JE. Phosphorylation of mouse glutamine-fructose-6-phosphate amidotransferase 2 (GFAT2) by cAMP-dependent protein kinase increases the enzyme activity. J Biol Chem. 2004; 279: 29988–29993. [DOI] [PubMed] [Google Scholar]
- 52. de Rooij J, Zwartkruis FJ, Verheijen MH, et al.. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998; 396: 474–477. [DOI] [PubMed] [Google Scholar]
- 53. Whitaker CM, Cooper NG. Differential distribution of exchange proteins directly activated by cyclic AMP within the adult rat retina. Neuroscience. 2010; 165: 955–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Onodera Y, Nam JM, Bissell MJ. Increased sugar uptake promotes oncogenesis via EPAC/RAP1 and O-GlcNAc pathways. J Clin Invest. 2014; 124: 367–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Whelan SA, Lane MD, Hart GW. Regulation of the O-linked beta-N-acetylglucosamine transferase by insulin signaling. J Biol Chem. 2008; 283: 21411–21417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gava E, Samad-Zadeh A, Zimpelmann J, et al.. Angiotensin-(1-7) activates a tyrosine phosphatase and inhibits glucose-induced signalling in proximal tubular cells. Nephrol Dial Transplant. 2009; 24: 1766–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kumar S, Xu J, Kumar RS, et al.. The small GTPase Rap1b negatively regulates neutrophil chemotaxis and transcellular diapedesis by inhibiting Akt activation. J Exp Med. 2014; 211: 1741–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tan EP, Villar MT, E L, et al.. Altering O-linked beta-N-acetylglucosamine cycling disrupts mitochondrial function. J Biol Chem. 2014; 289: 14719–14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tan EP, McGreal SR, Graw S, et al.. Sustained O-GlcNAcylation reprograms mitochondrial function to regulate energy metabolism. J Biol Chem. 2017; 292: 14940–14962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ma J, Liu T, Wei AC, Banerjee P, O'Rourke B, Hart GW. O-GlcNAcomic profiling identifies widespread O-linked beta-N-acetylglucosamine modification (O-GlcNAcylation) in oxidative phosphorylation system regulating cardiac mitochondrial function. J Biol Chem. 2015; 290: 29141–29153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ma J, Banerjee P, Whelan SA, et al.. Comparative proteomics reveals dysregulated mitochondrial O-GlcNAcylation in diabetic hearts. J Proteome Res. 2016; 15: 2254–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yang Z, Kirton HM, Al-Owais M, et al.. Epac2-Rap1 signaling regulates reactive oxygen species production and susceptibility to cardiac arrhythmias. Antioxid Redox Signal. 2017; 27: 117–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mukai E, Fujimoto S, Sato H, et al.. Exendin-4 suppresses SRC activation and reactive oxygen species production in diabetic Goto-Kakizaki rat islets in an Epac-dependent manner. Diabetes. 2011; 60: 218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stokman G, Qin Y, Booij TH, et al.. Epac-Rap signaling reduces oxidative stress in the tubular epithelium. J Am Soc Nephrol. 2014; 25: 1474–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]