

Abstract
Purpose
Cone-rod dystrophy (CRD) is a rare hereditary eye disorder that causes progressive degeneration of cone and rod photoreceptors. More than 30 genes, including RAB28, have been associated with CRD; however, only a few RAB28 variants have been reported to be associated with CRD. In this study, we describe two brothers with CRD and a homozygous missense variant, c.55G>A (p.Gly19Arg), in RAB28.
Methods
The missense variant was identified as part of a study investigating underlying genetic defects in a large patient cohort (n = 667) using targeted next-generation sequencing of 125 genes associated with retinal dystrophy. Cellular localization of RAB28 and ciliogenesis in patient fibroblasts were investigated by immunofluorescence microscopy. The effect of the missense variant on RAB28 expression level was investigated by quantitative real-time PCR.
Results
Two brothers of a consanguineous couple presented with CRD, postaxial polydactyly (PAP), and myopia. Both brothers had a homozygous missense RAB28 variant located in the G1 box of the guanosine triphosphate/guanosine diphosphate binding domain of RAB28. This missense variant caused a considerable reduction of RAB28 localized to the cilia, whereas ciliogenesis seemed unaffected.
Conclusions
The missense variant in RAB28 is classified as likely pathogenic with functional effect on protein localization. The combination of retinal dystrophy and PAP are well known from ciliopathies; however, more data are needed to finally conclude that the RAB28 variant described here is the cause of PAP in these brothers.
Keywords: cone-rod dystrophy, RAB28, primary clilium, localization, molecular genetics
Inherited retinal diseases (IRDs) are a major cause of visual impairment and affect approximately 1 in 3500.1 It is a clinically and genetically heterogeneous group of disorders with around 200 associated genes. A common feature is the malfunction of the photoreceptors in the retina. In some cases the cones are predominantly affected; in others, the rods. Both stationary and progressive forms exist, and clinical symptoms and age of onset may vary from severe congenital visual impairment to onset of symptoms in adulthood. IRDs can be isolated or part of a systemic disorder. All modes of Mendelian inheritance as well as mitochondrial inheritance have been observed. Rare cases of digenic inheritance have also been reported.2
Genes associated with IRDs are expressed either in the photoreceptors (rods or cones, or both) or in the supporting retinal pigment epithelium (RPE) cells. The most frequent form of IRD is retinitis pigmentosa (RP), presenting with night blindness and peripheral concentric visual field loss, caused by progressive degeneration of the rod photoreceptors and subsequent degeneration of the cones leading to blindness. Cone-rod dystrophy (CRD) is characterized by an initial loss of cones and symptoms of central vision loss, photophobia, progressive visual field loss, and color vision deficiency. In later stages, the rods also degenerate, typically leading to night blindness. Clinical distinction between RP and CRD can be difficult, especially in the late stages. Several treatment strategies for IRDs are under development; among these, gene therapy has given promising results.
RAB28 (MIM 612994) belongs to the RAB subfamily of the RAS oncogene family of Ras-related small GTPases. RAB GTPases function as molecular switches to control vesicle transport, vesicle budding, and membrane fission, and they regulate vesicle trafficking between organelles. In Caenorhabditis elegans (C. elegans), rab-28 has been linked to an intraflagellar transport (IFT) cargo through the Bardet–Biedl syndrome (BBS) cargo-adaptor protein complex (BBSome).3
RAB28 localizes to 4p15.33 and has two pseudogenes, located on chromosome 9 and X chromosome, respectively. The gene consists of nine exons, and alternative splicing gives rise to three isoforms differing in C-terminus.4 Two of the isoforms contain a C-terminal CAAX farnesylation motif.4,5 RAB28 tissue expression patterns differ among the isoforms, but they are all expressed in the retina.6 In C. elegans, rab-28 is expressed only in ciliated sensory neurons.3 In 2013, sequence changes in RAB28 were identified for the first time as the genetic cause of CORD18.6 To date, only five RAB28 variants are reported in the Human Gene Mutation Database (Fig. 1A).7
Figure 1.

(A) Localization of sequence variants in RAB28 thought to cause CRD. (B) Gly19 is a conserved amino acid through evolution, as are surrounding amino acids.
We report a family with two affected brothers of a consanguineous couple with a homozygous sequence variant, c.55G>A (p.Gly19Arg), located in the conserved guanosine triphosphate (GTP)/guanosine diphosphate (GDP) binding domain of RAB28 (Fig. 1B). Previously reported RAB28 variants have been associated with isolated CRD. Whether the postaxial polydactyly (PAP) observed in the affected brothers is also caused by the RAB28 variant requires more investigation. We provide a thorough ophthalmological description of the affected brothers as well as retrospective data from their medical history.
Methods
The project was performed according to the Declaration of Helsinki and approved by the Regional Ethics Committee. Written informed consent was obtained before the molecular genetic testing.
Clinical Examinations
Retrospective data from the 1970s from the National Eye Clinic for the Visually Impaired in Copenhagen, Denmark (NEC) archive were reviewed. Both brothers (IV-1 and IV-3) were re-examined in 2018 by spectral domain optical coherence tomography (OCT) and by ultra-wide-field pseudocolor and autofluorescence fundus imaging (Optos, Dunfermline, UK). International Society for Clinical Electrophysiology of Vision standard electroretinography (ERG) and Goldmann (III4e) kinetic visual fields were performed on the younger sibling (IV-3) with better preserved visual function. The brothers, IV-1 and IV-3, informed us of other clinical issues in this family.
Molecular Genetic Analysis
Genomic DNA was extracted from peripheral blood using standard protocols. Targeted next-generation sequencing was performed as described previously.8 Briefly, 125 genes, including RAB28, were sequenced using the custom NimbleGen SeqCap Target Enrichment KIT (NimbleGen, Madison, WI, USA) with capture and enrichment of all coding exons, 5′ and 3′ untranslated regions, and 20-bp flanking intronic regions. The enriched DNA libraries were sequenced using the Illumina HiSeq 2000 platform (Illumina, Inc., San Diego, CA, USA). Interpretation and classification of variants was performed according to the American College of Medical Genetics and Genomics (ACMG) guidelines.9 In silico analysis of missense variants was performed using Align GVGD,10,11 SIFT,12 MutationTaster,13 PolyPhen-2,14 and CADD software.15
Cell Studies
Fibroblasts from the patients and one control were obtained by skin biopsy. The cells were cultured in Dulbecco's modified Eagle's medium (DMEM)/F-12 + Gibco GlutaMAX(#31331-028; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with Gibco 10% fetal bovine serum (FBS) (#10270-106) and Gibco 1% penicillin/streptomycin (p/s) (#15140-122). Two different cultures from each patient were established. Cilia were induced by culturing the cells in reduced-serum medium containing 0.5% FBS and 1% p/s for 48 hours unless otherwise specified.
Treatment with siRNA against RAB28 (#SI03024686 and #SI00061705; Qiagen, Hilden, Germany) and with AllStars Negative Control scramble siRNA (#SI03650318; Qiagen) was performed using the DharmaFECTsystem (#T-2001-03; Dharmacon, Lafayette, CO, USA). Twenty-four hours after siRNA transfection, the cells were cultured in reduced-serum medium for 48 hours followed by fixation for immunofluorescence microscopy, or they were used for RNA extraction. For immunofluorescence microscopy, the cells were grown on glass slides.
Total RNA was extracted using the GeneJET RNA Purification Kit (#K0732; Thermo Fisher Scientific), and cDNA was synthesized using the High Capacity cDNA Reverse Transcription Kit (#4368814; Applied Biosystems, Foster City, CA, USA). Real-time quantitative PCR (qRT-PCR) was performed using TaqMan probes against RAB28 (#Hs01017480_m1; Applied Biosystems) and GAPDH (#Hs99999905_m1; Applied Biosystems). The 7500 Fast Real-Time PCR System (Applied Biosystems) was used for qRT-PCR amplification, and the relative standard curve method was used for calculation. RAB28 mRNA expression levels were normalized to GAPDH mRNA.
For immunofluorescence microscopy, the cells were fixed using 4% paraformaldehyde (1000.1000; Hounisen, Skanderborg, Denmark) for 15 minutes at room temperature, permeabilized using 0.2% TritonX-100 (Sigma Aldrich, St. Louis, MO, USA) in PBS for 15 minutes and blocked for 30 minutes in 3% bovine serum albumin. The fixed cells were incubated with the primary antibodies against RAB28 (#PA5-68303; Thermo Fisher Scientific) diluted 1:1000 and Arl13B (#ab136648; Abcam, Cambridge, UK) or anti-acetylated alfa-tubulin antibody (Ac-TUB) (T6793; Sigma-Aldrich) diluted 1:1000, for either 2 hours at room temperature or 16 hours at 4°C. Incubation with secondary antibodies Alexa Flour Goat Anti-Rabbit 488 (#A11008; Life Technologies, Carlsbad, CA, USA) and Alexa Flour Donkey Anti-Mouse 546 (#A10036, Life Technologies) was carried out in the dark for 45 minutes at room temperature. DAPI (#D9542; Sigma-Aldrich) was used to stain the nucleus. For each cell line, at least 100 primary cilia were analyzed per treatment.
Statistical analysis was performed using t-tests or χ2 tests as indicated. Levels of significance were classified as *P < 0.05, **P < 0.005, and ***P < 0.0005.
Results
Clinical features are listed in Table 1, and the family pedigree is shown in Figure 2. Brothers IV-1 and IV-3 had their first visit at NEC in 1971. They both initially presented with high myopia in early childhood. In their teens, both were diagnosed with progressive CRD, and the diagnosis was later confirmed with ERGs revealing undetectable cone responses and moderate to severe reduced rod responses. In adulthood, both developed glaucoma and cataract. At the most recent clinical examination in 2018, IV-1 presented with only light perception, but IV-3 still had useful navigation vision and was able to hold a part-time office job. Both presented with photophobia, absent color vision, and constricted visual fields but only minimal night blindness. The ocular fundus phenotypes in 2018 are shown in Figure 3. Furthermore, both IV-1 and IV-3 had PAP with an extra finger. Only IV-1 and IV-3 had the symptoms of CRD and PAP. Due to the combination of retinal dystrophy and PAP, they were suspected of BBS, and molecular genetic analysis of the BBS genes (BBS1–BBS17) was performed for IV-3; however, the BBS diagnosis could not be confirmed.
Table 1.
Clinical Features
| Individual | Age at Diagnosis (y) | Age at Last Visit (y) | Refractive Error at Diagnosis | Visual Acuity at Diagnosis | Visual Acuity at Final Visit | Color Vision at Presentation | ERG | Goldmann Visual Field IV4e | Other |
|---|---|---|---|---|---|---|---|---|---|
| IV:1 | 20 | 67 | –12D BE | RE, 3/60 LE, 6/60 | RE, LP – P LE, LP + P | Red–green color vision deficit | Age 20: severely reduced scotopic responses; photopic responses undetectable | Age 20: normal peripheral visual field | Polydactyly Age 26: glaucoma Age 40: bilateral cataract surgery Age 60: prostate cancer |
| IV:3 | 14 | 61 | –11D BE | RE, 6/18 LE, 6/24 | RE, 6/60 LE, 6/75 | Protan-type color vision deficit | Age 29: moderately reduced scotopic responses; absent light-adapted single flash and flicker cone responses, Age 61: recordable but severely reduced scotopic ERG; unrecordable photopic responses | Age 14: mild concentric constriction of peripheral visual field Age 61: eccentric fixation; two paracentral islands of preserved visual field of 20° and 10° diameter, respectively | Polydactyly Age 39: bilateral cataract surgery Age 41: glaucoma Age 54: prostate cancer Age 59: skin melanoma |
RE, right eye, LE, left eye, BE, both eyes; LP + P and LP – P, light perception visual acuity with or without projection, respectively.
Figure 2.
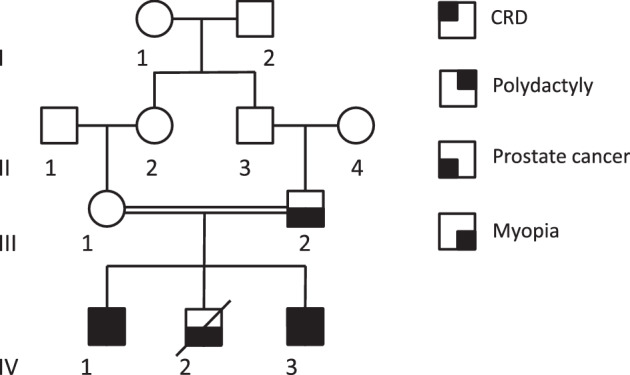
Pedigree of the family. For simplicity not all family members mentioned in the text are shown in the figure. Open circles and squares are unaffected females and males, respectively. Filled squares indicate a symptom depending on location in the square.
Figure 3.

Ultra-wide-field fundus image (A) and blue-light autofluorescence (C) image of IV-3 shows peripapillary atrophy and well-circumscribed chorioretinal atrophy in the macula; furthermore, two to three rows of multiple nummular atrophic and heavily pigmented chorioretinal areas are noted. Severely attenuated retinal vessels, especially arteries, are observed. (B) OCT shows an ellipsoid zone and outer limiting membrane preserved temporal to fovea (white arrow) and intraretinal cysts and lamellar retinal hole formation in the fovea (asterisk). (D) Ultra-wide-field fundus image of IV-1 demonstrates extensive atrophy of the retina and the choroid around the disk and nasal midperiphery and well-demarcated retina and choroid atrophy in the macula. As for IV-3, severely attenuated retinal vessels, especially arteries, are observed. The far-peripheral retina is relatively spared in IV-1. (E, F) Progression of retinal degeneration over time. Right-eye color fundus photographs of younger sibling in 1993 (E) and in 2018 (F). Central chorioretinal atrophy with hyperpigmentation has developed in the macula. Peripapillary and nasal midperipheral atrophy has significantly progressed. Images were taken with different cameras and different angle lenses; therefore, direct comparison of vascular changes was complicated. However, vessels seem to have straightened slightly over time.
Several other clinical findings were described in the family. Deuteranomaly (green) was found in IV-2 and in the brother of III-1 (not shown on the pedigree). Furthermore, a dominant form of myopia was observed in III-2, IV-1, IV-2, and IV-3 and in the daughter of IV-2 (not shown on the pedigree). For IV-1 and IV-3, it could not be determined whether their myopia was related to the retinal dystrophy or if they inherited the apparently dominant myopia segregating in the family, or both. Some of the family members also had prostate cancer (III-2, IV-1, IV-2, and IV-3).
Genetic Studies
Both brothers were homozygous for c.55G>A (p.Gly19Arg). Gly19 is a conserved amino acid located in the G1 box of the G domain of RAB28 (Fig. 1B). Five in silico prediction programs predict p.Gly19Arg to be pathogenic. The variant is reported in 2 out of 257.380 alleles in the gnomAD database.16 p.Gly19Arg segregates with disease in this family in an autosomal recessive inheritance pattern; however, the family is small, and brother IV-2 was not available for analysis because he died from prostate cancer. This study shows that p.Gly19Arg alters the function of RAB28. Consequently, following the ACMG guidelines for interpretation of variants,9 c.55G>A is classified as likely pathogenic.
Cell Studies
Previous studies have found rab-28 to be localized in neuro-cilia in C. elegans3 and in the RPE cells and outer segments of photoreceptors in mice.17 Jensen et al.3 also linked rab-28 as an IFT cargo through the BBSome. To validate the pathogenicity of the human p.Gly19Arg RAB28 variant, the expression level of RAB28 mRNA and the cellular localization of the RAB28 protein were investigated in fibroblast cells obtained from the two affected brothers and control cells for comparison.
Briefly, cells from IV-1, IV-3, and a control individual were grown under standard growth conditions, or under reduced-serum conditions to induce formation of the primary cilium (referred to as cilium hereafter).
Investigation of the RAB28 expression level by qRT-PCR revealed that RAB28 mRNA was expressed in both control and patient cells, indicating that the variant did not affect the expression level; furthermore, the expression level was significantly increased under reduced-serum conditions (Fig. 4A). Investigation of the cellular localization of the RAB28 protein in control cells by immunofluorescence microscopy showed that 73% of the cilia stained positive for RAB28 (Table 2), consistent with ciliary localization of RAB28 in human cells (Fig. 4B). Interesting, the patient cell lines showed a significant reduction in the number of RAB28-positive cilia (36% RAB28-positive cilia each) (Table 2).
Figure 4.

(A) Serum reduction leads to increased expression of RAB28. Expression profiles of RAB28 mRNA were normalized to the amount of endogenous GADPH mRNA. A significant increase in the level of RAB28 was observed for all samples under reduced-serum conditions compared to standard growth conditions (**P = 0.001815 for control; *P = 0.007181 for IV-1; **P = 0.003329 for IV-3). Three independent experiments were performed with similar results. Statistical analysis was performed using t-tests. (B) Ciliary localization of RAB28 is significantly reduced in patient cell lines. IFM analysis of control and patient cells grown under reduced-serum conditions. Cilia were labeled with anti-acetylated alfa-tubulin (AC-TUB) antibody (red). RAB28 protein (green) can be observed in the cilia in the two control cells (white arrows), whereas none of the three displayed patient cells (IV-1) shows ciliary localization of RAB28. Nuclei were visualized with DAPI staining (blue). The ciliary localization of RAB28 was significantly reduced in both patient cell lines compared to control (see Table 2). Scale bar, 10 µm. (C) siRNA-mediated knock down of RAB28 leads to a reduction of ciliary RAB28. Normalized expression profile of RAB28 mRNA expression in control cells treated with siRNA is compared with RAB28 (siRAB28) and a negative control (siScramble) grown under serum reduced conditions. Expression profiles of RAB28 were normalized to the amount of endogenous GAPDH mRNA. The expression level in control cells grown under standard conditions treated with siScramble is set to 1. Treatment with siRab28 decreases RAB28 expression by 75.76 ± 2% under reduced-serum conditions (see Table 2). GADPH, glyceraldehyde 3-phosphate dehydrogenase; AC-TUB, anti-acetylated alfa-tubulin; IFM, immunofluorescence microscopy.
Table 2.
Investigation of Cilia in Patient Versus Control Fibroblasts
| Number of Cells | Number of Cilia | RAB28-Positive Cilia (%) | ||
|---|---|---|---|---|
| 1 | Control cells | 156 | 124 | 91 (73%) |
| 2 | Patient IV-1 cells | 165 | 142 | 51 (36%)** |
| 3 | Patient IV-3 cells | 223 | 179 | 65 (36%)*** |
| 4 | Control cells + siScramble | 166 | 128 | 94 (73%) |
| 5 | Control cells + siRAB28 | 155 | 104 | 33 (28%)*** |
Rows 1–3: The numbers represent the sum of the five cultures investigated in two independent experiments. Cilia were labeled with antibody against Arl13B or acetylated alfa-tubulin and the RAB28 protein was labeled with specific RAB28 antibody. Nuclei were visualized with DAPI staining. The ciliary localization of RAB28 was significantly reduced in both patients’ cell lines compared to the control (patient IV-1/control, **P = 0.000746; patient IV-3/control, ***P = 0.000408), whereas no significant difference was observed in the number of cilia (ciliogenesis: patient IV-1/control, P = 0.630227; patient IV-3/control, P = 0.950036). Statistical analysis was performed using the χ2 test. Rows 4 and 5: Control cell cultures were treated with siRNA against RAB28 or with a negative control siRNA (siScramble). The numbers represent the sum from two independent experiments. Cilia were labeled with anti-Arl13B or anti-acetylated alpha-tubulin antibody and the RAB28 protein was labeled with specific RAB28 antibody; all cells were grown under serum-restricted conditions. Nuclei were visualized with DAPI staining. The ciliary localization of RAB28 was significantly reduced in the control cells treated with siRNA against RAB28 compared with cells treated with siScramble (siRAB28/siScramble, ***P = 0.000429). No significant difference was observed in the number of cilia (ciliogenesis: siRAB28/siScramble, P = 0.377439). Statistical analysis was performed using the χ2 test.
To verify the specificity of the RAB28 antibody we performed siRNA-mediated knock down of RAB28 in the control fibroblasts. Treatment with siRNA against RAB28 reduced the number of RAB28-positive cilia significantly (from 73% to 28% RAB28-positive cilia), whereas treatment with a negative control siRNA (siScramble) did not, confirming the specificity of the RAB28 antibody (Table 2).
We subsequently investigated whether ciliogenesis was affected by the RAB28 variant by assessing the percentage of ciliated cells. Cilia were identified on 79% (124/156) of the control cells grown under reduced-serum conditions (Table 2). Cells from IV-1 had 80% ciliated cells (142/165), whereas cells from IV-3 had 86% ciliated cells (179/223). Also, control cells treated with siRNA against RAB28 mRNA had a percentage of ciliated cells similar to that found for control cells treated with siScramble: 77% (128/166) versus 69% (104/155) (Table 2). Thus, no significant difference in ciliogenesis was observed.
Investigation of the expression level of RAB28 mRNA in siRNA-treated cells confirms the specificity of siRNA against RAB28. The treatment leads to a significant reduction (75.76 ± 2%) in RAB28 expression, although it did not completely ablate it (Fig. 4C).
In conclusion, these data underline the pathogenicity of the RAB28 variant by showing reduced ciliary localization of RAB28 protein. In contrast, the RAB28 variant did not affect ciliogenesis.
Discussion
We have described a family with several clinical features including CRD, PAP, high myopia, at least two forms of dyschromatopsia, and prostate cancer. Brothers IV-1 and IV-3 had all of the above-mentioned clinical features, and the third brother (IV-2) had myopia, deuteranomaly (green), and prostate cancer. Because the father (III-2) also suffered from prostate cancer and myopia, we consider these symptoms to be inherited independent of the CRD. Apart from IV-2, a maternal uncle also had deuteranomaly (green), suggesting the presence of an X-linked form of deuteranomaly in these two individuals. The finding of a homozygous missense variant in RAB28 affecting one of the invariable amino acids in the G motif makes this a plausible explanation for the CRD.
PAP has not previously been reported as a clinical symptom in individuals with RAB28 variants. We speculate that the RAB28 variant is the cause of PAP in the brothers presented here because RAB28 locates to the cilia like many products of genes associated with PAP, another small GTPase (Rab34) has been shown to be associated with PAP in mice,18 and, finally, IFT27 (which belongs to the RAB family of genes) is associated with BBS presenting with PAP.19 We cannot, however, rule out that another recessive gene is causing the PAP, especially as the parents are consanguineous.
Five other disease-causing RAB28 variants have been reported previously: two missense variants, p.Ser23Phe in a female of Korean descent20 and p.Cys217Trp in a female of Spanish descent21; two nonsense variants: p.Glu189* and p.Arg137* in a German and a Moroccan Jewish family, respectively6; and, finally, a splice variant c.172+1G>C in a Spanish family21 (Table 3). Classification according to the ACMG guidelines9 renders the two missense variants as variants of unknown significance, and the two nonsense variants and the splice site variant are classified as likely pathogenic. The ophthalmological findings are very similar with early onset of macula dystrophy, dyschromatopsia, progressive visual loss, (high) myopia, and lack of night blindness in all affected individuals. Thus, it seems that different types of RAB28 variants (missense, nonsense, and splice) are a cause of CRD in different ethnic groups.
Table 3.
Sequence Variants in RAB28
| Exon/Intron | cDNA (NM_004249.3) | Predicted Protein Change | Domain/Putative Functional Consequence | Status (ho/he) | Phenotype | rs Number/MAF gnomAD | ACMG Classification | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | c.55G>A | p.Gly19Arg | G1 domain G domain GXXXXGKS/T | Ho | CRD, polydactyly | 0,00000771 only heterozygous alleles (1) | LP (PM1, PM2, PP3, PS3) | This study |
| 1 | c.68C>T | p.Ser23Phe | G1 domain G domain GXXXXGKS/T | Ho | CRD | rs769199865 gnomAD 0,00003193 only heterozygous alleles (7) | VUS (PM2, PP3) | Lee et al. (2017)20 |
| Intron 2 | c.172+1G>C | p.? splicing defect | Skipping of exon 2 | Ho | CRD | rs875989778, NP | LP (PVS1, PM2) | Riveiro-Álvarez et al. (2015)21 |
| 5 | c.409C>T | p.Arg137* | — | Ho | CRD | rs398123044 0.000008148 only heterozygous alleles (2) | LP (PVS1, PM2) | Roosing et al. (2013)6 |
| 6 | c.565C>T | p.Gln189* | — | Ho | CRD | rs786200944, NP | LP (PVS1, PM2) | Roosing et al. (2013)6 |
| 8 | c.651T>G | p.Cys217Trp | — | Ho | CRD | rs751163782 0.000004076 only heterozygous alleles (1) | VUS (PM2, PP3) | Riveiro-Álvarez et al. (2015)21 |
Sequence variations reported in RAB28. PM1, PM2, PP1, PP3, and PVS1 refer to ACMG classification according to Richards et al.9 Ho, homozygous; He, heterozygous; VUS, variant of unknown significance; NP, not present; LP, likely pathogenic.
As mentioned above, the RAB28 missense variant found in this study, p.Gly19Arg, reduced the amount of RAB28 localized to the cilium but did not seem to alter ciliogenesis in human dermal fibroblasts. The observed increase in RAB28 mRNA expression in the human fibroblasts after serum starvation is consistent with localization of RAB28 in the cilium. Our observations are in agreement with the results from Jensen et al.,3 who showed that rab-28 in C. elegans is not a core component of the BBSome or the IFT pathway. Rather, it could be speculated that RAB28 associates with the BBSome and the IFT in order to be transported into the cilia, where it exerts its function. The BBSome is a highly conserved protein complex essential for IFT in both humans and mice.22–24 In mice, RAB28 functions in the cilia and is required for phagocytosis of the discs in the outer segments of the photoreceptors.17 The same mechanism may be present in humans.
As the variant described in this study does not affect the ability of a cell to form a cilium, further experiments are necessary to verify whether the ultrastructure of the cilium is normal in the patient cell lines.
In conclusion, RAB28 variants are a rare cause of CRD in various ethnic groups. These variants cause a comparable ophthalmological phenotype and furthermore could also be a cause of PAP.
Acknowledgments
We thank the families who participated in this study. We thank Pia Skovgaard, Bodil Olsen, Judy Rasmussen, and Anne Obling Madsen for technical assistance and Jette Bune Rasmussen for help preparing the figures.
Supported by Velux Foundation Grant 32700.
Disclosure: C. Jespersgaard, None; A.B. Hey, None; T. Ilginis, None; T.D. Hjortshøj, None; M. Fang, None; M. Bertelsen, None; N. Bech, None; H. Jensen, None; L.J. Larsen, None; Z. Tümer, None; T. Rosenberg, None; K. Brøndum-Nielsen, None; L.B. Møller, None; K. Grønskov, None
References
- 1. Bertelsen M, Jensen H, Bregnhoj JF, Rosenberg T. Prevalence of generalized retinal dystrophy in Denmark. Ophthalmic Epidemiol. 2014; 21: 217–223. [DOI] [PubMed] [Google Scholar]
- 2. Kajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science. 1994; 264: 1604–1608. [DOI] [PubMed] [Google Scholar]
- 3. Jensen VL, Carter S, Sanders AA, et al.. Whole-organism developmental expression profiling identifies RAB-28 as a novel ciliary GTPase associated with the BBSome and intraflagellar transport. PLoS Genet. 2016; 12: e1006469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brauers A, Schurmann A, Massmann S, et al.. Alternative mRNA splicing of the novel GTPase Rab28 generates isoforms with different C-termini. Eur J Biochem. 1996; 237: 833–840. [DOI] [PubMed] [Google Scholar]
- 5. Lee SH, Baek K, Dominguez R. Large nucleotide-dependent conformational change in Rab28. FEBS Lett. 2008; 582: 4107–4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roosing S, Rohrschneider K, Beryozkin A, et al.. Mutations in RAB28, encoding a farnesylated small GTPase, are associated with autosomal-recessive cone-rod dystrophy. Am J Hum Genet. 2013; 93: 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stenson PD, Ball E, Howells K, Phillips A, Mort M, Cooper DN. Human Gene Mutation Database: towards a comprehensive central mutation database. J Med Genet. 2008; 45: 124–126. [DOI] [PubMed] [Google Scholar]
- 8. Jespersgaard C, Fang M, Bertelsen M, et al.. Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy. Sci Rep. 2019; 9: 1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Richards S, Aziz N, Bale S, et al.. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 2006; 34: 1317–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tavtigian SV, Deffenbaugh AM, Yin L, et al.. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006; 43: 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016; 11: 1–9. [DOI] [PubMed] [Google Scholar]
- 13. Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010; 7: 575–576. [DOI] [PubMed] [Google Scholar]
- 14. Adzhubei IA, Schmidt S, Peshkin L, et al.. A method and server for predicting damaging missense mutations. Nat Methods. 2010; 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019; 47: D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Karczewski KJ, Weisburd B, Thomas B, et al.. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017; 45: D840–D845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ying G, Boldt K, Ueffing M, Gerstner CD, Frederick JM, Baehr W. The small GTPase RAB28 is required for phagocytosis of cone outer segments by the murine retinal pigmented epithelium. J Biol Chem. 2018; 293: 17546–17558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dickinson ME, Flenniken AM, Ji X, et al.. High-throughput discovery of novel developmental phenotypes. Nature. 2016; 537: 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schaefer E, Delvallee C, Mary L, et al.. Identification and characterization of known biallelic mutations in the IFT27 (BBS19) gene in a novel family with Bardet-Biedl syndrome. Front Genet. 2019; 10: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee GI, Lee C, Subramanian S, et al.. A novel likely pathogenic variant in the RAB28 gene in a Korean patient with cone-rod dystrophy. Ophthalmic Genet. 2017; 38: 587–589. [DOI] [PubMed] [Google Scholar]
- 21. Riveiro-Alvarez R, Xie YA, Lopez-Martinez MA, et al.. New mutations in the RAB28 gene in 2 Spanish families with cone-rod dystrophy. JAMA Ophthalmol. 2015; 133: 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mourao A, Nager AR, Nachury MV, Lorentzen E. Structural basis for membrane targeting of the BBSome by ARL6. Nat Struct Mol Biol. 2014; 21: 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakayama K, Katoh Y.. Ciliary protein trafficking mediated by IFT and BBSome complexes with the aid of kinesin-2 and dynein-2 motors. J Biochem. 2018; 163: 155–164. [DOI] [PubMed] [Google Scholar]
- 24. Wei Q, Zhang Y, Li Y, Zhang Q, Ling K, Hu J. The BBSome controls IFT assembly and turnaround in cilia. Nat Cell Biol. 2012; 14: 950–957. [DOI] [PMC free article] [PubMed] [Google Scholar]