

Abstract
Objective
Immune checkpoint inhibitors (ICIs) have become the standard of care in various cancers, although the predictive tool is still unknown.
Methods
This study aimed to develop a novel gene panel by selecting DNA damage response (DDR) genes from the Catalogue of Somatic Mutations in Cancer (COSMIC) databank and validating them in previously reported cohorts. This association between DDR gene mutations and tumor mutation burden or microsatellite status was analysed from The Cancer Genome Atlas (TCGA) databank. Furthermore, we made the gene panel clinically accessible and predicted the response in clinical patients receiving ICIs by using cell‐free DNA.
Results
The top 20 mutated DDR genes in various cancers (total 37 genes) were taken from the COSMIC databank, and the DDR genes found to individually predict a response rate > 50% in Van Allen's cohort were selected (Science, 350, 2015 and 207). Eighteen DDR genes were selected as the gene panel. The prevalence and predicted response rate were validated in the other three reported cohorts. Tumor mutational burden‐high was positively associated with mutations of the 18 DDR genes for most cancers. We used cell‐free DNA to test the DDR gene panel and validated by our patients receiving ICIs. This DDR gene panel accounted for approximately 30% of various cancers, achieving a predicted response rate of approximately 60% in patients with a mutated gene panel receiving ICIs.
Conclusion
This gene panel is a novel and reliable tool for predicting the response to ICIs in cancer patients and guides the appropriate administration of ICIs in clinical practice.
Keywords: cell‐free DNA, DNA repair gene, gene panel, immune checkpoint inhibitor, tumor mutational burden
Immune checkpoint inhibitors (ICIs) have been widely used to treat various cancers and have achieved impressive success, resulting in a new era of anticancer therapy. However, only a subset of patients experienced lasting responses; therefore, exploring predictive biomarkers is critical to identify the patients who have benefit from ICIs. This gene panel using liquid biopsy is one of the best predictive biomarkers presently available and can help physicians to predict the response to ICIs and judge the utility of ICIs in clinical practice.

Introduction
Immune checkpoint inhibitors (ICIs) such as anti‐cytotoxic T lymphocyte antigen 4 (CTLA‐4) and anti‐programmed cell death 1 (PD‐1)/programmed cell death‐ligand 1 (PD‐L1) monoclonal antibodies block interactions between cancer cells and the immune system to enhance immune response to the tumor by rebalancing immune surveillance and immune evasion. Ipilimumab, an anti‐CTLA‐4 monoclonal antibody, was the first approved ICI that exhibited activity and efficacy in advanced and metastatic melanoma 1 ; thereafter, nivolumab 2 and pembrolizumab, 3 anti‐PD‐1 antibodies, were approved for melanoma treatment. ICIs are now widely used in various cancers and have achieved impressive success in cancer treatment, resulting in a new era of anticancer therapy. However, only a subset of patients experience lasting responses. Therefore, exploration of predictive biomarkers is critical to optimise the benefits of ICIs in patients.
In 2017, pembrolizumab was the first tissue/site‐agnostic indication approved by U. S. FDA for the patients with microsatellite instability (MSI)‐high or deficient mismatch repair (dMMR) solid tumors. 4 Nivolumab monotherapy and then the combination of nivolumab and ipilimumab demonstrated durable response in MSI‐high or dMMR pretreated colorectal cancer, and the combination was approved by the FDA. 5 , 6 Additionally, high tumor mutational burden (TMB) increases the expression of immunogenic peptides, which influences ICI response in patients with various cancers 7 , 8 such as melanoma, 9 non‐small‐cell lung cancer, 10 small‐cell lung cancer 11 and urothelial carcinoma. 12
Because DNA damage response (DDR) alterations were associated with MSI‐H and high TMB, 13 we hypothesised that a gene panel of DDR alterations could be used to predict the clinical benefit in patients with undergoing ICI treatments. Therefore, in the current study, we aimed to develop a novel gene panel comprised of DDR genes selected from Catalogue of Somatic Mutations in Cancer (COSMIC) databank and validated in previously reported cohorts. Furthermore, we made the gene panel clinically accessible and predicted the response in our patients undergoing ICIs by using cell‐free DNA (cfDNA).
Results
Identification of DNA repair gene panel for predicting clinical responders for checkpoint inhibitor‐based immunotherapies
The top 20 mutated DDR genes in various cancers were taken from the COSMIC databank, and a total of 37 mutated DDR genes were included in the current study. 14 The DNA repair genes found to individually predict a response rate > 50% in a cohort reported by Van Allen et al., 15 who analysed the whole exomes from pretreatment tumor samples in patients with melanoma undergoing treatment with anti‐CTLA‐4, were included. A panel of 18 DNA repair genes were obtained and validated in the following cohorts (Figure 1a and b; Table 1). Moreover, this gene panel was first examined in the cohort in Van Allen et al., and 37 of 110 (33.6%) patients were identified as having mutated genes of this panel, and 18 of 37 (48.6%) patients with a mutated gene panel were responders to anti‐CTLA‐4. Conversely, only nine out of 63 (14.3%) were responders in patients without a mutated gene panel.
Figure 1.
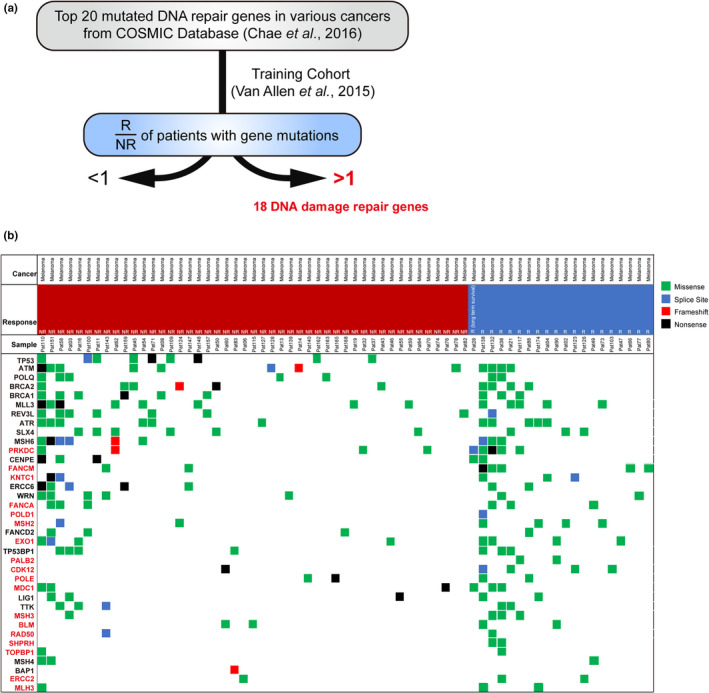
The identification of 18 DNA repair genes. (a) Schema of the identification of 18 DNA repair genes (validation shown in flowing figures). Table 1 presents the information of 18 DNA repair genes. (b) Heatmap of somatic mutations including missense (green), splice site (blue), frameshift (red) and nonsense (black) detected in 110 melanoma patients from Van Allen et al. 15 Left, top 20 mutated DNA repair genes (total 37 genes). Red, the mutation of target gene predict response rate > 50% in the cohort. Block, the mutation of target gene predict response rate < 50% in the cohort. Top, patient phenotype data for cancer type, response and sample ID. NR, No response; R, Response.
Table 1.
Details of 18 DNA repair genes
| Official symbol | Official full name | Locus |
|---|---|---|
| BLM | BLM RecQ like helicase | 15q26.1 |
| CDK12 | Cyclin‐dependent kinase 12 | 17q12 |
| ERCC2 | ERCC excision repair 2, TFIIH core complex helicase subunit | 19q13.32 |
| EXO1 | Exonuclease 1 | 1q43 |
| FANCA | FA complementation group A | 16q24.3 |
| FANCM | FA complementation group M | 14q21.2 |
| KNTC1 | Kinetochore associated 1 | 12q24.31 |
| MDC1 | Mediator of DNA damage checkpoint 1 | 6p21.33 |
| MLH3 | mutL homolog 3 | 14q24.3 |
| MSH2 | mutS homolog 2 | 2p21‐p16.3 |
| MSH3 | mutS homolog 3 | 5q14.1 |
| PALB2 | Partner and localizer of BRCA2 | 16p12.2 |
| POLD1 | DNA polymerase delta 1, catalytic subunit | 19q13.33 |
| POLE | DNA polymerase epsilon, catalytic subunit | 12q24.33 |
| PRKDC | Protein kinase, DNA‐activated, catalytic subunit | 8q11.21 |
| RAD50 | RAD50 double‐strand break repair protein | 5q31.1 |
| SHPRH | SNF2 histone linker PHD RING helicase | 6q24.3 |
| TOPBP1 | DNA topoisomerase II binding protein 1 | 3q22.1 |
Validation of established DNA repair gene panel in other cohorts
The first validation cohort was collected from patients with metastatic melanoma treated with anti‐PD‐1 and anti‐CTLA‐4 reported by Hugo et al. 16 (n = 38) and Snyder et al. 9 (n = 64), respectively. In 102 patients with melanoma, 34 (33.3%) patients had a mutated gene panel and 22 (64.7%) were responsive to ICI (Figure 2a). Moreover, patients with a mutated gene panel had significantly longer progression‐free survival (PFS; P = 0.0097) and overall survival (OS; P = 0.0086) than patients with a wild‐type gene panel (Figure 2c).
Figure 2.
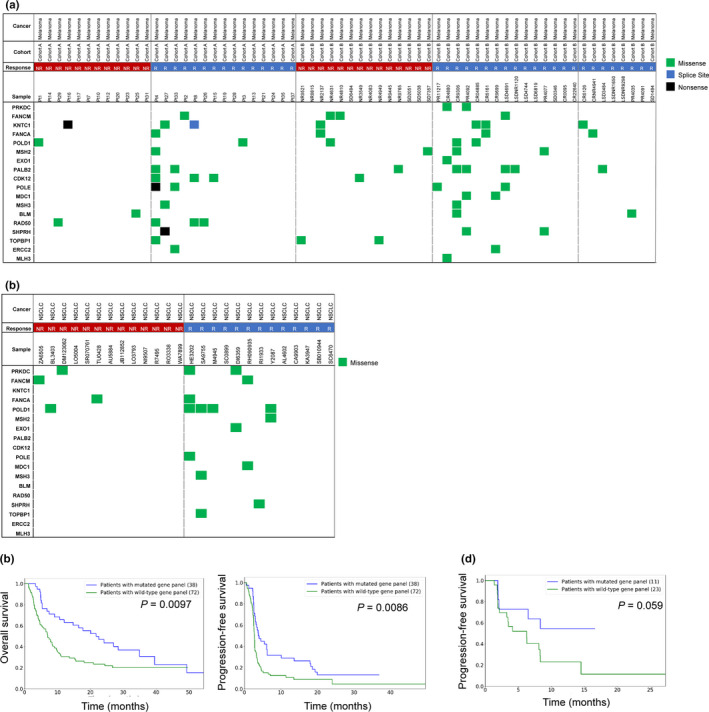
The validation of 18 DNA repair genes for prediction of ICI response. (a) Heatmap of somatic mutations including missense (green), splice site (blue), and nonsense (black) detected in 38 melanoma patients (cohort A) from Snyder et al., 9 and 64 melanoma patients (cohort B) from Hugo et al. 16 Left, 18 DNA repair genes identified in Figure 1. Top, patient phenotype data for cancer type, response, cohort and sample ID. NR, No response; R, Response. (b) Heatmap of missense (green) detected in 31 non‐small‐cell lung cancer patients from Rizvi et al. 10 Left, 18 DNA repair genes identified in Figure 1. Top, patient phenotype data for cancer type, response and sample ID. (c) Kaplan–Meier survival curves are plotted for overall survival and progression‐free survival comparing melanoma patients (Van Allen et al. 15 ) with mutated and wild‐type DDR genes. The P‐value calculated using the log‐rank test is shown. NR, No response; NSCLC, Non‐small‐cell lung cancer; R, Response. (d) Kaplan–Meier survival curves are plotted for progression‐free survival comparing non‐small‐cell lung cancer patients (Rizvi et al. 10 ) with mutated and wild‐type DDR genes. The P‐value was calculated using the log‐rank test and is shown in the panel.
In another cohort of 31 patients with non‐small‐cell lung cancer undergoing anti‐PD‐1 reported by Rizvi et al., 10 11 (35.5%) had a mutated gene panel and 22 (64.7%) of them were responsive to ICI (Figure 2b). Moreover, patients with a mutated gene panel had a trend of superior PFS to patients with a wild‐type gene panel (P = 0.059; Figure 2d).
Association between mutation status in the DDR gene panel and TMB
We further examined the association between mutation status in the gene panel and TMB in various cancers on the basis of somatic mutation data from the TCGA database. In most cancers (27/33), the mutation of these genes was significantly associated with high TMB (Figure 3), indicating that the samples with a mutated DDR gene panel tend to have higher TMB. Because few patients with cholangiocarcinoma, diffuse large B‐cell lymphoma, mesothelioma, testicular germ cell tumors, thymoma, and uveal melanoma had a mutated DDR gene panel, this gene panel had no significant association with TMB.
Figure 3.
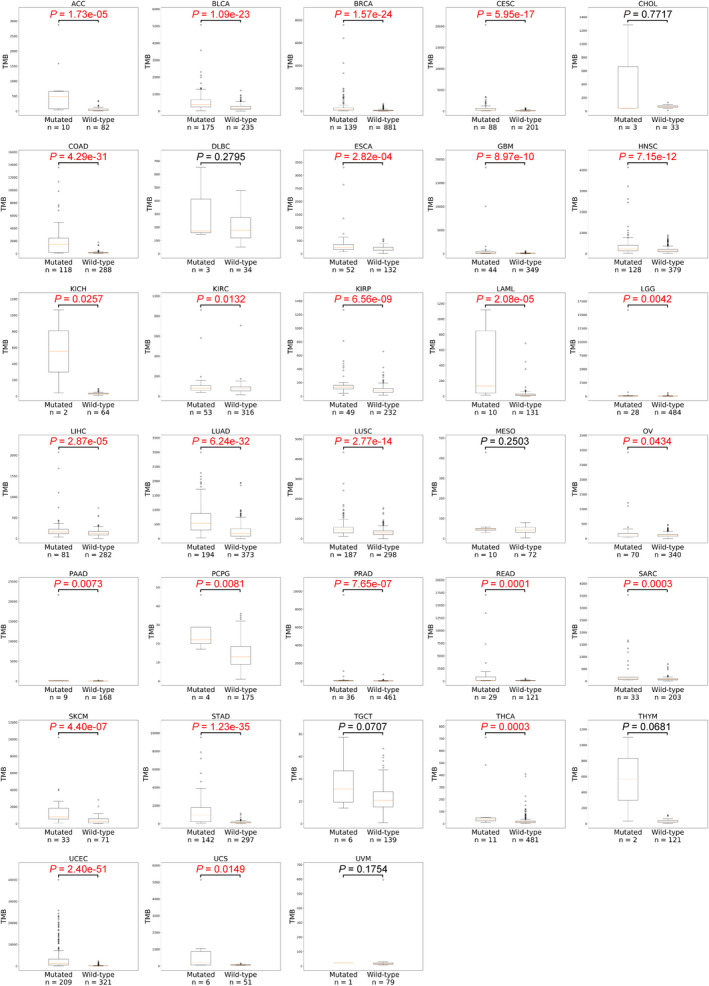
The association of tumor mutation burden and mutations of 18 DDR genes in 33 cancers. TMB (tumor mutational burden) was analysed with (mutated) or without (wild‐type) mutations of 18 DDR genes in 33 cancer from TCGA. Box‐and‐whisker plots depict the distribution of mutation counts. The P‐value was calculated using the Wilcoxon rank‐sum test. The P‐value and sample number in each group are shown. ACC, Adrenocortical carcinoma; BLCA, Bladder urothelial carcinoma; BRCA, Breast invasive carcinoma; CESC, Cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, Cholangiocarcinoma; COAD, Colon adenocarcinoma; DLBC, Lymphoid neoplasm diffuse large B‐cell lymphoma; ESCA, Esophageal carcinoma; GBM, Glioblastoma multiforme; HNSC, Head and neck squamous cell carcinoma; KICH, Kidney chromophobe; KIRC, Kidney renal clear cell carcinoma; KIRP, Kidney renal papillary cell carcinoma; LAML, Acute myeloid leukaemia; LGG, Brain Lower Grade Glioma; LIHC, Liver hepatocellular carcinoma; LUAD, Lung adenocarcinoma; LUSC, Lung squamous cell carcinoma; MESO, Mesothelioma; OV, Ovarian serous cystadenocarcinoma; PAAD, Pancreatic adenocarcinoma; PCPG, Pheochromocytoma and paraganglioma; PRAD, Prostate adenocarcinoma; READ, Rectum adenocarcinoma; SARC, Sarcoma; SKCM, Skin cutaneous melanoma; STAD, Stomach adenocarcinoma; TGCT, Testicular germ cell tumors; THCA, Thyroid carcinoma; THYM, Thymoma; UCEC, Uterine corpus endometrial carcinoma; UCS, Uterine carcinosarcoma; UVM, Uveal melanoma.
Association between mutation status in the DDR gene panel and MSI status
Because MSI status is also a potential biomarker for immunotherapy, we investigated the association between mutation status in the gene panel and MSI status. With the somatic mutation data available, the MSI status for every sample in TCGA dataset can be predicted using the MSIseq tool 17 with high accuracy. Among 33 cancers, eight cancer types had no MSI‐H samples predicted by MSIseq (Figure 4a). In 25 cancer types with MSI‐H samples, the mutated DNA repair gene panel was significantly associated with MSI‐H in 17 cancer types (Figure 4b; Supplementary table 1), indicating that the samples with a mutated DDR gene panel tend to be MSI‐H in most cancers. For those MSS samples, we also investigated the association between mutation status in the gene panel and TMB. The results indicated that in 27/33 cancers, the mutation of these genes was significantly associated with high TMB, demonstrating that the MSS samples with a mutated DDR gene panel also tend to have higher TMB (Supplementary figure 1). In summary, the samples with a mutation in the gene panel tend to be MSI‐H or have higher TMB in most cancers, implying that they may be more responsive to ICIs. Therefore, the identified DDR gene panel can serve as a predictive biomarker for ICIs in different cancers.
Figure 4.
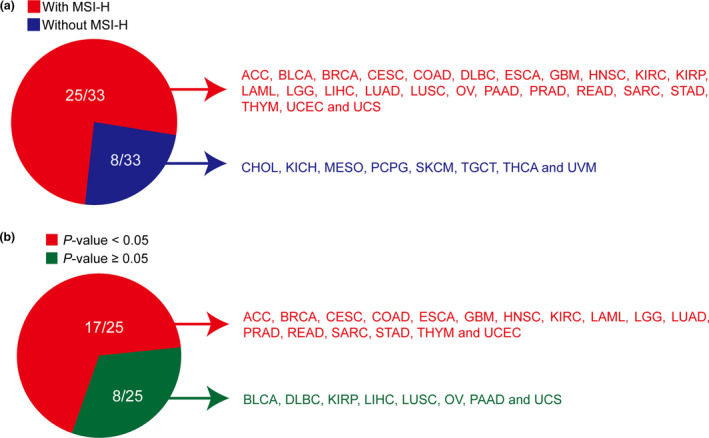
The association between mutation status in DDR gene panel and microsatellite instability status. (a) The microsatellite instability (MSI) status for 33 cancers in the TCGA MC3 dataset was predicted by the MSIseq tool. Red (n = 25): with MSI‐H (microsatellite instability‐high); Blue (n = 8): without MSI‐H. (b) Association between mutation status in DDR gene panel and MSI status in 25 MSI‐H cancers described in a. The P‐value was calculated using Fisher's exact test and is shown here and in Supplementary table 1. Red, P < 0.05 (n = 17); green, P ≥ 0.05 (n = 8).
Validation of DNA repair gene panel in our cohort by fresh, formalin‐fixed paraffin‐embedded and cell‐free DNA in plasma
To investigate the correlation between tissue and liquid biopsies, NGS from ten paired tissue (fresh and paraffin‐embedded) and cell‐free DNA (cfDNA; Figure 5a; Table 2; Supplementary table 2) were collected and analysed. The highly significant correlation of mutation events from either fresh or paraffin‐embedded tissue and mutation events of cfDNA via liquid biopsy was found (Figure 5b).
Figure 5.
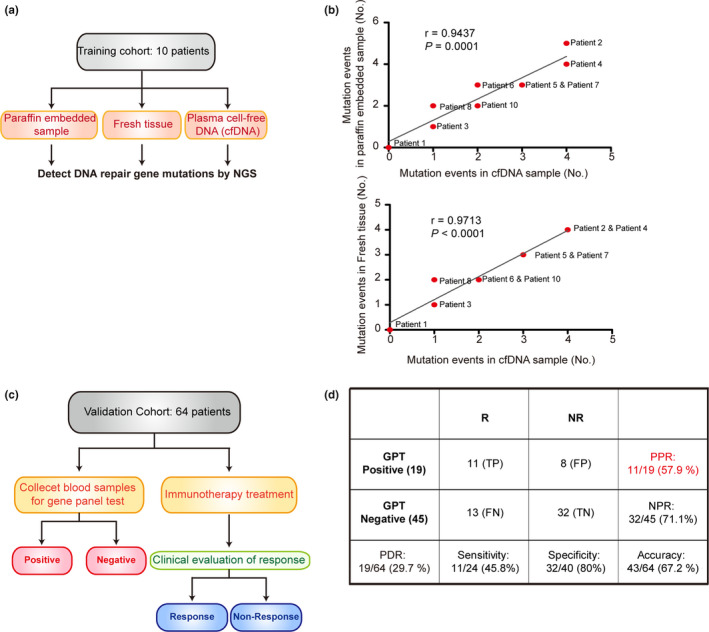
The validation of DNA repair gene panel by cfDNA. (a) Schema for experimental design for b. (b) Association between mutation events in paraffin‐embedded sample, fresh tissue and cell‐free DNA (cfDNA) in 10 patients with cancer. The P‐value was calculated using the Student's t‐test. The correlation coefficients r and P‐values are shown in each panel. See Table 2 for DNA repair gene mutations in fresh tissues, cfDNA and paraffin‐embedded samples. Data are from a single experiment. (c) Schema for experimental design for panel d. (d) A table for showing the validation of DNA repair gene panel using cfDNA. Data are from a single experiment. FN, False negative; FP, False positive; GPT, Gene panel test; NPR, Negative predictive rate; NR, No response; PDR, Positive detection rate; PPR, Positive predictive rate; R, Response; TN, True negative; TP, True positive.
Table 2.
Detection of DNA repair gene mutations in fresh tissues, cell‐free DNAs (cfDNAs) and paraffin‐embedded samples from nine patients by next‐generation sequencing
| Individual | DNA | Gene | Variant | Chr | Type | Consequence | Amino acids | Codons |
|---|---|---|---|---|---|---|---|---|
| 1 | Tissue | ND | ||||||
| 1 | cfDNA | ND | ||||||
| 1 | Paraffin embedded | ND | ||||||
| 2 | Tissue | PALB2 | C>C/G | 16 | snv | missense_variant | E/D | gaG/gaC |
| 2 | cfDNA | PALB2 | C>C/G | 16 | snv | missense_variant | E/D | gaG/gaC |
| 2 | Paraffin embedded | PALB2 | C>C/G | 16 | snv | missense_variant | E/D | gaG/gaC |
| 2 | Paraffin embedded | FANCA | G>G/A | 16 | snv | synonymous_variant | Y | taC/taT |
| 2 | Tissue | KNTC1 | A>A/G | 12 | snv | missense_variant | I/V | Ata/Gta |
| 2 | cfDNA | KNTC1 | A>A/G | 12 | snv | missense_variant | I/V | Ata/Gta |
| 2 | Paraffin embedded | KNTC1 | A>A/G | 12 | snv | missense_variant | I/V | Ata/Gta |
| 2 | Tissue | RAD50 | A>A/G | 5 | snv | synonymous_variant | E | gaA/gaG |
| 2 | cfDNA | RAD50 | A>A/G | 5 | snv | synonymous_variant | E | gaA/gaG |
| 2 | Paraffin embedded | RAD50 | A>A/G | 5 | snv | synonymous_variant | E | gaA/gaG |
| 2 | Tissue | POLE | T>T/C | 12 | snv | missense_variant | ||
| 2 | cfDNA | POLE | T>T/C | 12 | snv | missense_variant | ||
| 2 | Paraffin embedded | POLE | T>T/C | 12 | snv | downstream_gene_variant | ||
| 3 | Tissue | TOPBP1 | T>T/C | 3 | snv | missense_variant | D/G | gAt/gGt |
| 3 | cfDNA | TOPBP1 | T>T/C | 3 | snv | missense_variant | D/G | gAt/gGt |
| 3 | Paraffin embedded | TOPBP1 | T>T/C | 3 | snv | missense_variant | D/G | gAt/gGt |
| 4 | Tissue | PALB2 | C>C/A | 16 | snv | missense_variant | D/Y | Gac/Tac |
| 4 | cfDNA | PALB2 | C>C/A | 16 | snv | missense_variant | D/Y | Gac/Tac |
| 4 | Paraffin embedded | PALB2 | C>C/A | 16 | snv | missense_variant | D/Y | Gac/Tac |
| 4 | Tissue | PRKDC | A>A/G | 8 | snv | missense_variant | L/P | cTg/cCg |
| 4 | cfDNA | PRKDC | A>A/G | 8 | snv | missense_variant | L/P | cTg/cCg |
| 4 | Paraffin embedded | PRKDC | A>A/G | 8 | snv | missense_variant | L/P | cTg/cCg |
| 4 | Tissue | PRKDC | G>G/A | 8 | snv | synonymous_variant | Y | taC/taT |
| 4 | cfDNA | PRKDC | G>G/A | 8 | snv | synonymous_variant | Y | taC/taT |
| 4 | Paraffin embedded | PRKDC | G>G/A | 8 | snv | synonymous_variant | Y | taC/taT |
| 4 | Tissue | FANCA | C>C/T | 16 | snv | synonymous_variant | ||
| 4 | cfDNA | FANCA | C>C/T | 16 | snv | synonymous_variant | ||
| 4 | Paraffin embedded | FANCA | C>C/T | 16 | snv | downstream_gene_variant | ||
| 5 | Tissue | FANCA | G>G/T | 16 | snv | synonymous_variant | A | gcC/gcA |
| 5 | cfDNA | FANCA | G>G/T | 16 | snv | synonymous_variant | A | gcC/gcA |
| 5 | Paraffin embedded | FANCA | G>G/T | 16 | snv | synonymous_variant | A | gcC/gcA |
| 5 | Tissue | SHPRH | T>T/C | 6 | snv | missense_variant | T/A | Act/Gct |
| 5 | cfDNA | SHPRH | T>T/C | 6 | snv | missense_variant | T/A | Act/Gct |
| 5 | Paraffin embedded | SHPRH | T>T/C | 6 | snv | missense_variant | T/A | Act/Gct |
| 5 | Tissue | SHPRH | C>C/A | 6 | snv | synonymous_variant | P | ccG/ccT |
| 5 | cfDNA | SHPRH | C>C/A | 6 | snv | synonymous_variant | P | ccG/ccT |
| 5 | Paraffin embedded | SHPRH | C>C/A | 6 | snv | synonymous_variant | P | ccG/ccT |
| 6 | Tissue | PALB2 | C>C/A | 16 | snv | missense_variant | D/Y | Gac/Tac |
| 6 | cfDNA | PALB2 | C>C/A | 16 | snv | missense_variant | D/Y | Gac/Tac |
| 6 | Paraffin embedded | PALB2 | C>C/A | 16 | snv | missense_variant | D/Y | Gac/Tac |
| 6 | Tissue | FANCA | T>T/A | 16 | snv | synonymous_variant | P | ccA/ccT |
| 6 | cfDNA | FANCA | T>T/A | 16 | snv | synonymous_variant | P | ccA/ccT |
| 6 | Paraffin embedded | FANCA | T>T/A | 16 | snv | synonymous_variant | P | ccA/ccT |
| 6 | Paraffin embedded | KNTC1 | C>C/T | 12 | snv | synonymous_variant | S | tcC/tcT |
| 7 | Tissue | CDK12 | A>A/G | 17 | snv | synonymous_variant | P | ccA/ccG |
| 7 | cfDNA | CDK12 | A>A/G | 17 | snv | synonymous_variant | P | ccA/ccG |
| 7 | Paraffin embedded | CDK12 | A>A/G | 17 | snv | synonymous_variant | P | ccA/ccG |
| 7 | Tissue | MSH2 | A>A/G | 2 | snv | missense_variant | T/A | Acc/Gcc |
| 7 | cfDNA | MSH2 | A>A/G | 2 | snv | missense_variant | T/A | Acc/Gcc |
| 7 | Paraffin embedded | MSH2 | A>A/G | 2 | snv | missense_variant | T/A | Acc/Gcc |
| 7 | Tissue | MSH3 | TG>TG/T | 5 | deletion | frameshift_variant | G/X | Gga/ga |
| 7 | cfDNA | MSH3 | TG>TG/T | 5 | deletion | frameshift_variant | G/X | Gga/ga |
| 7 | Paraffin embedded | MSH3 | TG>TG/T | 5 | deletion | frameshift_variant | G/X | Gga/ga |
| 8 | Tissue | FANCM | A>A/G | 14 | snv | synonymous_variant | ||
| 8 | cfDNA | FANCM | A>A/G | 14 | snv | synonymous_variant | ||
| 8 | Paraffin embedded | FANCM | A>A/G | 14 | snv | upstream_gene_variant | ||
| 8 | Tissue | POLD1 | C>C/T | 19 | snv | missense_variant | R/W | Cgg/Tgg |
| 8 | Paraffin embedded | POLD1 | C>C/T | 19 | snv | missense_variant | R/W | Cgg/Tgg |
| 9 | Tissue | FANCA | C>C/G | 16 | snv | splice_acceptor_variant | ||
| 9 | cfDNA | FANCA | C>C/G | 16 | snv | splice_acceptor_variant | ||
| 9 | Tissue | KNTC1 | T>T/A | 12 | snv | synonymous_variant | I | atT/atA |
| 9 | cfDNA | KNTC1 | T>T/A | 12 | snv | synonymous_variant | I | atT/atA |
| 9 | Paraffin embedded | NO sample | ||||||
| 10 | Tissue | ERCC2 | C>C/T | 19 | snv | intron_variant | ||
| 10 | cfDNA | ERCC2 | C>C/T | 19 | snv | intron_variant | ||
| 10 | Paraffin embedded | ERCC2 | C>C/T | 19 | snv | intron_variant | ||
| 10 | Tissue | KNTC1 | A>A/T | 12 | snv | missense_variant | M/L | Atg/Ttg |
| 10 | cfDNA | KNTC1 | A>A/T | 12 | snv | missense_variant | M/L | Atg/Ttg |
| 10 | Paraffin embedded | KNTC1 | A>A/T | 12 | snv | missense_variant | M/L | Atg/Ttg |
In previous studies, DNA repair gene mutations usually cause chromosomal instability (CIN). 18 Thus, the association between CIN signatures and 18 DDR gene mutations was investigated. To confirm the association of CIN signatures and 18 DDR gene mutations, CIN70 19 score was calculated in tumor samples with at least one mutation in the 18 DDR genes and wild‐type group using TCGA database. In statistics, the mutation of these genes was significantly associated with high CIN70 in 12 cancers (total 33 cancers; Supplementary figure 2). Moreover, chromosomal copy number was analysed in tissue samples from nine patients described in Figure 5a and b. Copy number abnormality was only detected in two patients. But, DDR gene mutations were detected in eight patients (Supplementary table 2), suggesting that CIN signatures are not 18 DDR genes associated factors.
Validation of the DNA repair gene panel in our cohort by cell‐free DNA in plasma
In a subsequent experiment, 64 patients with various cancer types were collected to validate the gene panel, and the patients' characteristics are summarised in Supplementary table 3. Cell‐free DNAs (cfDNAs) in the blood of 64 patients were analysed. cfDNAs in the blood of 40 patients had more than one SNVs (single‐nucleotide variants). Only 29.7% (19/64) had risk variants, showing deleterious effect (SIFT prediction). All the patients underwent pretreatment liquid biopsy and ICI treatment (Figure 5c). Nineteen (29.7%) of 64 patients had a mutated gene panel, and 11 (57.9%) of 19 patients were responsive to ICI. Conversely, 13 (28.9%) of 45 patients with a wild‐type gene panel were responsive to ICI (Figure 5d).
Discussion
In the current study, we developed a novel gene panel comprising 18 genes from the most frequent DDR gene alterations and validated this panel in two independently reported cohorts. Moreover, we confirmed that this gene panel could predict TMB and MSI status, which are important predictive biomarkers in most cancer types, in patients treated with ICIs. Finally, we validated this panel in the present cohort with patients with various cancer subtypes undergoing ICI treatment. The mutated gene panel rate was approximately 30% in different cohorts, and 60% response rates were achieved in patients with a mutated gene panel. Therefore, this gene panel is a novel and reliable tool that can provide additional information in clinical practice to predict the clinical benefit in cancer patients undergoing ICI treatment.
Parikh et al. 13 analysed 17 486 tubular gastrointestinal carcinomas to identify the correlations between DDR defects and MSI‐H or TMB‐high. A total of 17% of tumors had DDR alterations, and MSI‐H and TMB‐high were observed in 19% and 21%, respectively, of DDR‐defective tumors, indicating that DDR alterations might sensitise cancer cells to ICIs. However, this report did not demonstrate that DDR‐altered tumors were more responsive to ICIs than DDR‐intact tumors. Additionally, only two of 10 genes, CDK12 and PALB2, were included in our current gene panel; the selection of DDR genes in the gene panel is critical to maximise the sensitivity and specificity in predicting benefit of ICIs.
In our study, this gene panel was validated in patients with various cancer subtypes and exhibited distinct sensitivities and specificities in patients with different cancers undergoing treatment with ICIs. In the subtypes with more than five patients in the current study, the accuracy of the DDR gene panel was highest for urothelial carcinoma (100%) followed by cholangiocarcinoma (88.9%), esophageal cancer (62.5%), hepatocellular carcinoma (53%), and head and neck cancer (40%). The positive rate of DDR gene panel mutation was consistently approximately 30% in various cancer subtypes in the current cohort, indicating that the DDR gene panel could be a better predictive marker than MSI status or TMB, which had lower prevalence than the DDR gene panel.
This study has some limitations. No concomitant MSI and TMB status was evaluated in the patients treated with ICIs in the current study to compare the validity. However, we aimed to use cfDNA to detect DDR gene panel alterations and liquid biopsy for MSI and TMB that were developing, but had not yet matured. 20 , 21 , 22
In conclusion, a novel DDR gene panel was established and validated in different cohorts. The altered gene panel accounted for approximately 30% of various cancers and had a 60% predicted response rate in patients with a mutated gene panel undergoing treatment with ICIs. Therefore, this gene panel using liquid biopsy could be one of the best predictive tool currently available, and larger scale prospective studies are warranted to validate these findings.
Methods
Human samples
Specimens from 10 patients for Figure 5a were retrieved from Linkou Chang Gung Memorial Hospital, and the study was approved by the Institutional Review Board of Linkou Chang‐Gung Memorial Hospital (201601745B0 and 201701922B0). Specimens from a total of 64 patients for Figure 5c were retrieved from Linkou Chang Gung Memorial Hospital (36 patients) and Taipei Veterans General Hospital (28 patients). The study was approved by the Institutional Review Board of Linkou Chang‐Gung Memorial Hospital (201601461B0 and 201701922B0) and Taipei Veterans General Hospital (2015‐07‐002BC).
Target gene sequencing
We pooled all of the top 20 mutated DDR genes of individual cancer type from the COSMIC databank, and a total of 37 mutated DDR genes were included in this study. 14 Eighteen DDR genes were selected and validated in three cohorts. 9 , 10 , 16
We used high‐throughput genome sequencer, the Illumina NGS system, to comprehensively explore the DNA sequence of all exons of the 18 well‐selected genes (BLM, CDK12, ERCC2, EXO1, FANCA, FANCM, KNTC1, MDC1, MLH3, MSH2, MSH3, PALB2, POLD1, POLE, PRKDC, RAD50, SHPRH and TOPBP1). Amount of 250 ng DNA from each cancer tissue and 16 μL cfDNA extracted from 4 mL plasma were used to construct the sample library using QIAseq Ultralow Input Library Kit (Qiagen 180495, QIAGEN, Inc., Hilden, Germany). Target DNA of DNA repair‐related genes was enriched using the probe‐based methods. The probes were synthesised by Integrated DNA Technologies, Inc. (IDT; Coralville, IA, USA) according to our previously designed probe sequences, and the capture procedure was performed following the IDT guidelines. After probe‐based enrichment, libraries of each pool were amplified with 14 cycles. The amplified libraries were quantified using a quantitative polymerase chain reaction (qPCR) system, were transferred into new 1.5 mL tubes and produced a 10 nm pooled DNA libraries. The final pool was used for sequencing (Illumina MiSeq/MiniSeq/NextSeq sequencer, 2 × 150 bp). The raw output of each individual library was > 300 Mb, and the average depth of target regions was > 1000×. Sequences of each read were trimmed based on the quality score (Q30), and lengths of each read < 45 bp were discarded in the following analysis. Reads were aligned to the human hg19 reference genome using Burrows–Wheeler aligner‐maximum exact matches (BWA‐MEM; http://bio‐bwa.sourceforge.net/), and the GATK Unified Genotyper (GATKLite version 2.3–9) was used for calling variants. After variant calling, we used the Variant Effect Predictor (http://grch37.ensembl.org/Homo_sapiens/Tools/VEP) to annotate the identified variants for the following statistical analysis.
Association between mutation status in DDR gene panel and TMB in various malignancies
To investigate the association between the identified DDR gene panel and TMB, we used somatic mutation data obtained from The Cancer Genome Atlas (TCGA) multi‐centre mutation calling in multiple cancers (MC3) project, 23 which includes samples from 33 different cancer types. After retrieving the somatic mutation data, we first screened for samples of primary solid tumors or primary blood‐derived cancer. On one hand, the sample with at least one nonsynonymous mutation in one of the 18 identified DDR genes listed in Table 1 was classified as the mutated group. On the other hand, if there was no nonsynonymous mutation detected in the 18 identified DDR genes, the sample was classified as the wild‐type group. The nonsynonymous mutations were defined by the ‘Variant_Classification’ information present in the somatic mutation data, and the categories considered to be nonsynonymous mutations ‘Missense_Mutation’ ‘Nonsense_Mutation’ ‘Nonstop_Mutation’ ‘Splice_Site’ ‘Translation_Start_Site’ ‘Frame_Shift_Del’ ‘Frame_Shift_Ins’ ‘In_Frame_Del’ and ‘In_Frame_Ins’. Because the TCGA MC3 project used whole exome sequencing and seven variant calling methods to identify mutations, in this study, TMB was defined as the total number of mutations in a sample. For each cancer type, based on the two groups (wild‐type group and mutated group) and their corresponding TMB for each sample, the Wilcoxon rank‐sum test was used to test the association between the mutation status in the DDR gene panel and TMB by comparing the TMB in two groups. A P‐value < 0.05 was considered to be significant, indicating that the mutation status in the DDR gene panel was significantly associated with TMB.
Association between mutation status of DDR genes and MSI status as well as TMB
Microsatellite instability status (MSI‐H or MSS) for each sample in the TCGA MC3 dataset was predicted by the MSIseq tool. 17 In order to verify the prediction accuracy of MSIseq, we first tested it on the five cancer types with MSI status in the clinical data from TCGA, that is colon adenocarcinoma (COAD), rectum adenocarcinoma (READ), stomach adenocarcinoma (STAD), uterine corpus endometrial carcinoma (UCEC) and uterine carcinosarcoma (UCS). The results indicated that MSIseq can assess the MSI status with high accuracy (98.8% in COAD, 98.7% in READ, 99.3% in STAD, 95.1% in UCEC, and 96.5% in UCS). Therefore, the samples without MSI status in their clinical data can also be predicted using MSIseq with high confidence. For each sample, it can be classified as MSI‐H or MSS by MSIseq. Moreover, they could be classified as mutated or wild‐type based on the mutation status of the 18 genes in the DDR gene panel. In this situation, the association between mutation status in DDR gene panel and MSI status can be determined by Fisher's exact test for each cancer type. Further, the association between mutation status in DDR gene panel and TMB was also investigated in MSS samples using the Wilcoxon rank‐sum test. In both cases, a P‐value < 0.05 was considered to be significant.
Survival analysis for patients with mutated and wild‐type DDR genes in the validation cohorts
For the independent validation cohorts with both somatic mutation data and treatment response information, the Kaplan–Meier survival curves were plotted for survival analysis comparing patients with mutated and wild‐type 18 DDR genes. The log‐rank test was used to determine the significance. A P‐value < 0.05 was considered to be significant. The supplementary materials for chromosomal instability are described in Supplementary appendix 1.
Conflict of interest
The authors declare no conflict of interest.
Author contribution
Yi‐Ru Pan: Data curation; Investigation; Project administration; Writing‐review & editing. Chiao‐En Wu: Data curation; Investigation; Project administration; Writing‐review & editing. Yu‐Chao Wang: Data curation; Formal analysis; Software; Supervision; Writing‐original draft. Yi‐Chen Yeh: Funding acquisition; Resources; Visualization. Meng‐Lun Lu: Data curation; Formal analysis. Yi‐Ping Hung:
Formal analysis; Resources. Yee Chao: Funding acquisition; Resources. Da‐Wei Yeh: Data curation; Formal analysis. Chien‐Hsing Lin: Formal analysis; Investigation; Software; Validation. Jason Chia‐Hsun Hsieh: Resources. Ming‐Huang Chen: Conceptualization; Investigation; Project administration; Supervision. Chun‐Nan Yeh: Conceptualization; Investigation; Project administration; Supervision; Writing‐original draft.
Supporting information
Acknowledgments
We thank the Tissue Bank, Chang Gung Memorial Hospital, Linkou for technical support. This work was supported by grants from Linkou Chang‐Gung Memorial Hospital (CRRPG3F0031~3, CMRPG3I0231, CMRPG3I0241, CORPG3J0251, NMRPG3F6021~2 and NMRPG3H6211~2 to CNY and CMRPG3I0451, CMRPG3J0971 and NMRPG3J0011 to CEW); the Ministry of Science and Technology (105‐2314‐B‐182A‐041‐MY2 and 107‐2314‐B‐182A‐134‐MY3 to CNY, 108‐2314‐B‐182A‐007 to CEW, 106‐2221‐E‐010‐019‐MY3 to YCW and 104‐2314‐B‐075‐064‐MY2 to MHC); and Taipei Veterans General Hospital (V109C‐028 to MHC)
Contributor Information
Ming‐Huang Chen, Email: mhchen9@vghtpe.gov.tw.
Chun‐Nan Yeh, Email: yehchunnan@gmail.com.
References
- 1. Hodi FS, O'Day SJ, McDermott DF et al Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363: 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hodi FS, Chiarion‐Sileni V, Gonzalez R et al Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4‐year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol 2018; 19: 1480–1492. [DOI] [PubMed] [Google Scholar]
- 3. Hamid O, Robert C, Daud A et al Five‐year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE‐001. Ann Oncol 2019; 30: 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA approval summary: pembrolizumab for the treatment of microsatellite instability‐high solid tumors. Clin Cancer Res 2019; 25: 3753–3758. [DOI] [PubMed] [Google Scholar]
- 5. Overman MJ, McDermott R, Leach JL et al Nivolumab in patients with metastatic DNA mismatch repair‐deficient or microsatellite instability‐high colorectal cancer (CheckMate 142): an open‐label, multicentre, phase 2 study. Lancet Oncol 2017; 18: 1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Overman MJ, Lonardi S, Wong KYM et al Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair‐deficient/microsatellite instability‐high metastatic colorectal cancer. J Clin Oncol 2018; 36: 773–779. [DOI] [PubMed] [Google Scholar]
- 7. Tray N, Weber JS, Adams S. Predictive biomarkers for checkpoint immunotherapy: current status and challenges for clinical application. Cancer Immunol Res 2018; 6: 1122–1128. [DOI] [PubMed] [Google Scholar]
- 8. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer 2019; 19: 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Snyder A, Makarov V, Merghoub T et al Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N Engl J Med 2014; 371: 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rizvi NA, Hellmann MD, Snyder A et al Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science 2015; 348: 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hellmann MD, Callahan MK, Awad MM et al Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small‐cell lung cancer. Cancer Cell 2018; 33: 853–861.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rosenberg JE, Hoffman‐Censits J, Powles T et al Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum‐based chemotherapy: a single‐arm, multicentre, phase 2 trial. Lancet 2016; 387: 1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parikh AR, He Y, Hong TS et al Analysis of DNA damage response gene alterations and tumor mutational burden across 17,486 tubular gastrointestinal carcinomas: implications for therapy. Oncologist 2019; 24: 1340–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chae YK, Anker JF, Carneiro BA et al Genomic landscape of DNA repair genes in cancer. Oncotarget 2016; 7: 23312–23321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Van Allen EM, Miao D, Schilling B et al Genomic correlates of response to CTLA‐4 blockade in metastatic melanoma. Science 2015; 350: 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hugo W, Zaretsky JM, Sun L et al Genomic and transcriptomic features of response to anti‐PD‐1 therapy in metastatic melanoma. Cell 2016; 165: 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang MN, McPherson JR, Cutcutache I, Teh BT, Tan P, Rozen SG. MSIseq: software for assessing microsatellite instability from catalogs of somatic mutations. Sci Rep 2015; 5: 13321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instablity‐an evolving hallmark of cancer. Nat Rev Mol Cell Bio 2010; 11: 220–228. [DOI] [PubMed] [Google Scholar]
- 19. Carter SL, Eklund AC, Kohane IS et al A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet 2006; 38: 1043–1048. [DOI] [PubMed] [Google Scholar]
- 20. Georgiadis A, Durham JN, Keefer LA et al Noninvasive detection of microsatellite instability and high tumor mutation burden in cancer patients treated with PD‐1 blockade. Clin Cancer Res 2019; 25: 7024–7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Willis J, Lefterova MI, Artyomenko A et al Validation of microsatellite instability detection using a comprehensive plasma‐based genotyping panel. Clin Cancer Res 2019; 25: 7035–7045. [DOI] [PubMed] [Google Scholar]
- 22. Fenizia F, Pasquale R, Roma C, Bergantino F, Iannaccone A, Normanno N. Measuring tumor mutation burden in non‐small cell lung cancer: tissue versus liquid biopsy. Transl Lung Cancer Res 2018; 7: 668–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ellrott K, Bailey MH, Saksena G et al Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Systems 2018; 6: 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials