

Abstract
Cancer is a major burden on the healthcare system, and new therapies are needed. Recently, the development of immunotherapies, which aim to boost or use the immune system, or its constituents, as a tool to fight malignant cells, has provided a major new tool in the arsenal of clinicians and has revolutionized the treatment of many cancers.
Cellular immunotherapies are based on the administration of living cells to patients and have developed hugely, especially since 2010 when Sipuleucel-T (Provenge), a DC vaccine, was the first cellular immunotherapy to be approved by the FDA. The ensuing years have seen two further cellular immunotherapies gain FDA approval: tisagenlecleucel (Kymriah) and axicabtagene ciloleucel (Yescarta).
This review will give an overview of the principles of immunotherapies before focusing on the major forms of cellular immunotherapies individually, T cell-based, natural killer (NK) cell-based and dendritic cell (DC)-based, as well as detailing some of the clinical trials relevant to each therapy.
Keywords: Cancer, CAR-T cells, Immunotherapy, TCR-T cells
Introduction
Cancer is a disease that results from accumulation of mutations in DNA sequences that dysregulate critical cellular pathways leading to increased cell survival and proliferation, reduced cell death, and invasion and dissemination of these cells across the body [1]. Cancer is the second leading cause of death globally, and by 2040, it is predicted there will be 27.5 million new cancer diagnoses made globally each year, up from 17 million cases in 2018 [2].
The immune system has an intimate, yet conflicted, relationship with cancer. It functions akin to a double edged sword, playing complex roles in both tumour detection and elimination, as well as in tumour formation and progression [3]. Cancer cells must evade the destructive components of the immune response if they are to survive and proliferate.
There are many ways that malignant cells can avoid identification and erasure by the host defence system; these include selection of tumour variants that have a non-immunogenic phenotype, which occurs via a process known as immunoediting. This can manifest as downregulation of tumour antigen expression, creation of alternative versions of an antigen via mutations or alternative splicing, or masking of tumour antigens [4]. In addition to this, the creation of a hostile and immunosuppressive tumour microenvironment (TME) is a crucial tool for transformed cells to escape the immune response. This is characterized by hypoxia, low levels of nutrients, elevated lactate and low pH which repress the normal immune response. Recruitment of immunosuppressive cells including regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) contribute to impaired effector cell function. Cancer cells can induce immune exhaustion through several mechanisms such as decreased expression of co-stimulatory molecules, secretion of immunosuppressive factors, and expression of inhibitory ligands such as PD-L1 or CTLA-4 [5].
Cancer immunotherapy uses, or involves, components of the immune system to circumvent and reverse these evasion strategies employed by tumours and to boost the immune response against cancer cells [6].
Cancer immunotherapy can be classified into active and passive immunotherapies based on stimulation of the host immune system. Active immunotherapies boost the host defences and can be categorized as specific and non-specific. Specific immunotherapies, such as therapeutic or prophylactic vaccines, induce host immunity towards a particular antigen(s), while non-specific immunotherapy is made up of cytokines, checkpoint inhibitors and immune adjuvants which augment the host immune response on a global level. Conversely, passive immunotherapies do not boost the host immune system and involve passive transfer of tumour-targeting monoclonal antibodies, or small molecules, as well as adoptive cell transfer [7–9].
The term cellular immunotherapy refers to the administration of living cells to a patient; this type of immunotherapy can be active, such as a dendritic cell (DC) vaccine, in that the cells can stimulate an anti-tumour response in the patient, or the therapy can be passive, whereby the cells have intrinsic anti-tumour activity; this is known as adoptive cell transfer (ACT) and includes the use of autologous or allogeneic lymphocytes that may, or may not, be modified [10].
Adoptive cellular therapy involves the transfer of cells that have been primed to respond to a tumour antigen into the host; these cells are most often autologous, but progress is being made towards usage of allogeneic cells as ‘off-the-shelf’ products. Priming the cells ex vivo has the advantage of bypassing the need to activate the host immune response as this is often weak and ineffective. To achieve a successful anti-tumour effect, these cells must recognise an appropriately chosen tumour antigen, be of adequate numbers to achieve efficacy, be capable of trafficking to and infiltrating the primary tumour as well as metastatic sites, and finally, have cytotoxic functions capable of killing malignant cells [11].
In contrast to ACT, where the effector cells are ‘pre-primed’ (either by in vivo or ex vivo exposure to antigens), vaccines themselves ‘prime’ the adaptive immune system to induce a response against tumours [12].
This review will focus on cellular immunotherapies involving the use of T cells and natural killer (NK) cells in adoptive cell transfer, as well as briefly focussing on DC vaccines.
Principles of cellular immunotherapies
The last decade has seen huge strides made in terms of taking cellular immunotherapies from the ‘bench to beside’: in 2010; Sipuleucel-T (Provenge), a DC vaccine, was the first cellular immunotherapy to be approved by the FDA [13]. This marked a major milestone in the field of cancer immunotherapy, building on decades of work that was begun by William Coley in 1893, who was the first person to use immunotherapy as an anti-cancer treatment. Coley gave intratumoural injections of inactivated bacteria (Streptococcus pyogenes and Serratia marcescens) to patients with sarcomas resulting in an anti-tumour immune response (the mechanism of which was not fully understood at the time), culminating in several cases of complete tumour remission [14].
The intervening years have made further progress in this field including the approval of ‘breakthrough’ CAR-T cell (chimeric antigen receptor) therapies, tisagenlecleucel (Kymriah) and axicabtagene ciloleucel (Yescarta) [15, 16]. In parallel, the approvals of the immune checkpoint inhibitors have led to cancer immunotherapy, both active and passive, being termed the 5th pillar of cancer care by the American Association for Cancer Research (AACR) [17].
Using living cells as a ‘drug’ represents a seismic shift from the traditional view of a ‘drug’ as a small molecule or antibody that serves a single function; in contrast, cells are dynamic, living agents that can integrate a range of inputs, respond to changes in the environment, interact with other cells, and through complex signalling pathways give rise to a spectrum of responses that a conventional drug cannot [8]. In addition to this, cellular drugs can persist in the body for months to years, whereas most conventional drugs are metabolised and excreted soon after administration, adding another layer to the complexity of the patient-drug interaction [18].
The capacity of the immune system to distinguish between self- and tumour antigens underlies the attractiveness of using it as a tool to fight disease. Traditional therapies such as chemotherapy and radiotherapy lack this capacity to distinguish between normal and cancerous tissue, causing substantial toxicity to healthy tissues, which can be a major limiting factor in their use. Other treatments such as targeted agents have much better systemic toxicity profiles than chemotherapy or radiotherapy, but are limited by the tumour’s ability to evolve mechanisms of resistance [19]. The immune system has an almost limitless array of receptor specificities; this gives it the potential to adapt and adjust to the attempts of the tumour to evade destruction [20].
The immune response has other qualities that further enhance its appeal as an anti-cancer weapon. These include the ability of the immune system to form an immunological memory which can give long-term protection against disease relapse. Additionally, the immune system is thought to have the potential to kill any type of tumour cell under the right conditions, making it a potential ‘universal’ approach to cancer, although, as will become clear, many challenges exist if this is to become a reality [20].
These principles underpin the basis for adoptive cell therapy using T cells and NK cells, as well as DC-based vaccines.
T cell-based therapies
T cells are key orchestrators and effectors of the immune response and recognise peptide antigens presented on major histocompatibility complex (MHC) molecules by antigen-presenting cells (APCs) using the T cell receptor (TCR). The TCR-peptide-MHC complex forms an immunological synapse between a naïve T cell and an APC; this interaction recruits coreceptors such as CD28 and leads to initiation of a signalling cascade adding to T cell activation [21].
The outcome of the TCR interaction with the peptide-MHC complex (pMHC) is, in part, determined by co-stimulatory and co-inhibitory receptors. Molecules such as CD28, 4-IBB and CD134 cooperate with TCR signalling to enhance T cell proliferation, as well as enhance the durability of the immune response. In contrast to this, co-inhibitory receptors such as PD-1 and CTLA-4 function as negative regulators of the immune response and act as a ‘checkpoint’ to immune activation [22].
Once activated by an APC in a lymph node, a T cell begins to proliferate, differentiates into a mature effector cell and migrates to the site of infection or inflammation [23]. When a mature CD8+ T cell encounters a cell presenting the antigen complementary to its TCR, the T cell releases cytotoxic mediators such as granzyme B and perforin which leads to killing of the malignant (or virally infected) cell. The cytotoxic T lymphocyte (CTL) can also indirectly lead to tumour cell death via secretion of cytokines such as tumour necrosis factor (TNF) [24].
There are three major types of T cell-based therapies: tumour-infiltrating lymphocytes (TILs), TCR-T cells and CAR-T cells. The latter two technologies consist of genetically altered T cells where tumour specificity is induced ex vivo by inserting a chimeric antigen receptor (CAR) in the case of a CAR-T cell or alternatively, transfecting a TCR specific to a particular tumour antigen into a T cell creating a TCR-T cell. In contrast, TILs are preferentially tumour-specific prior to culture as they are extracted from tumour material and as such are primed in vivo [25]. Figure 1 details this concept visually [26]. Also, innate T cell subsets such as the γδ T cells and natural killer T (NKT) cells are emerging as areas of focus for cancer immunotherapy.
Fig. 1.
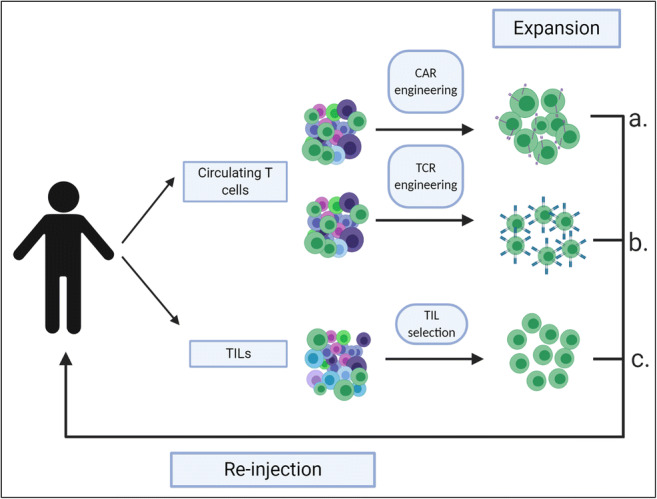
Adoptive T cell transfer therapies. Circulating T cells or tumour-infiltrating lymphocytes (TILs) are collected from a patient’s blood or tumour, respectively. Circulating T cells can be engineered to express chimeric antigen receptors (CARs) (a.) or T cell receptors (TCRs) (b.) which allow targeting of specific tumour-associated/specific antigens. These modified T cells can be expanded ex vivo and subsequently re-injected into the same patient as an allogeneic therapy. CAR-T cells and TCR-T cells can also be derived from allogeneic sources (not shown), meaning that the donor and the recipient of the cells are different people. Following tumour resection or biopsy, TILs (c.) can be extracted from the tumour material and those with specificity against tumour antigens can be selectively extracted, expanded and re-infused into the patient
TILs
ACT of tumour-infiltrating lymphocytes (TILs) was first used as an anti-cancer treatment by Rosenberg and colleagues, firstly in a mouse model and subsequently in human metastatic melanoma [27, 28]. TILs can be extracted and isolated from resected tumour and expanded ex vivo using IL-2.
Owing to the fact that these cells were primed and activated to respond to tumour antigens in vivo, prior knowledge of which particular antigens they target is not necessary; this is further advantageous in that there is limited risk of cross-reactivity with antigens expressed on normal tissues. Additionally, rather than (as will be described later for CAR-T and TCR-T cells) having specificity towards one or two antigens, TILs are a heterogenous population of lymphocytes containing many subgroups of different antigen specificities; this leads to diverse targeting of multiple tumour antigens and a more efficient immune response [29].
Following extraction and expansion, TILs can be reintroduced into the patient as an autologous infusion following lymphodepletion using chemotherapy such as cyclophosphamide or total body irradiation [30]. The preconditioning regimen allows the TILs to exert their anti-tumour effects more efficiently by disrupting immunosuppressive cells, such as regulatory T cells, and decreasing endogenous lymphocyte competition for homeostatic regulatory cytokines, creating a ‘space’ for the TILs to expand and function [31].
Although the response rates for this therapy in melanoma refractory to previous therapies were impressive at 50 to 70% [31], there are a number of limitations to this process that have curtailed the widespread use of TILs in the clinic.
The isolation of TILs is a time-consuming laborious process and often ineffective as many tumours have limited numbers of TILs available. The access of TILs to tumours is largely thought to be influenced by tumour characteristics such as size, location and immunogenicity [32]. Furthermore, although TILs extracted from tumours are preferentially tumour-specific, a significant proportion can have suppressive rather than anti-tumour function [33]. Culturing the cells with IL-2 expands these regulatory cells which can downregulate the immune response [34]. For these reasons, the use of TILs failed to achieve widespread usage; however, they did serve as a harbinger to the genetically redirected T cells such as the TCR-T cell and CAR-T cell therapies of recent times.
TCR-T cells
T cells can be engineered to express TCRs with tumour antigen specificity; this overcomes the problems of finding a suitable subgroup of TILs with cytotoxic activity among the heterogenous population of tumour-derived immune cells. These engineered TCR-T cells can be expanded ex vivo and administered in adequate numbers to drive a successful anti-tumour response against malignant cells [35, 36].
Genetic modification of T cells can be performed using a variety of methods. Viral vectors, such as lentivirus or retrovirus, are often used due to their high transduction efficiency; however, these systems carry the risk of activating oncogenes leading to clonal expansion [37, 38]. Other methods which can be employed are transposons such as Sleeping Beauty or PiggyBac, electroporation, and gene-editing platforms such as CRISPR/Cas9, TALENs or Zinc-Finger Nucleases [39–42] (see Box 1).
Due to the fact that intracellular proteins are displayed on MHC molecules, TCR-T cells can target almost any tumour-specific or tumour-associated intracellular protein that is processed by this pathway which constitutes a major advantage of this cellular immunotherapy [8]. To avoid interactions of living drugs with normal cells, the choice of antigen specificity for the TCR is highly important. This is a common theme across all forms of cellular immunotherapies, with the exception of TILs.
The ideal antigen target is specific to tumour cells and is not expressed on normal cells. Identification of such antigens is difficult as most tumour antigens are not exclusive to cancer cells and often tend simply to be antigens that are overexpressed in comparison to normal cells; this leads to the possibility of on-target/off-tumour toxicity, where immune responses are directed at healthy cells due to expression of a poorly chosen target antigen [19, 42].
The use of neoantigens, i.e., those that are tumour-specific and result due to mutations or aberrant splicing of normal, conserved proteins, is generally recommended due to their high immunogenicity as well as lack of expression in normal tissues [43]. Identification of these neoantigens can be a challenge as truly specific antigens tend to not only be cancer-specific but patient-specific and may require sequencing of patients’ tumours which is impractical in rapidly progressing diseases [44].
Several clinical trials have been carried out which have validated the effectiveness of TCR-T cells as a therapeutic intervention. Johnson et al. generated TCR-T cells that recognized either MART-1 or gp100, both of which are melanoma-melanocyte antigens. They observed objective cancer regressions in 30% and 19% of patients who received the anti-MART-1 or anti-gp100 TCR-T cells, respectively [36]. Rapoport and colleagues showed in 2015 that TCR-T cells targeting NY-ESO-1 induced a response in 16 of 20 patients (80%) with advanced multiple myeloma [45]. This echoed a similar report showing durable responses in patients with metastatic melanoma and synovial cell sarcoma when treated with NY-ESO-1 targeting TCR-T cells [46, 47]. Similarly, recent data in relapsed, refractory, or high-risk multiple myeloma reported median overall survival of 35.1 months when treated with TCR-T cells against NY-ESO-1 peptide, with 2 patients displaying disease progression-free after almost 5 years [48]. As will be discussed later, the most recent developments in the TCR-T cell arena came this year when Stadtmauer et al. showed that gene-edited NY-ESO-1 TCR-T cells were both safe and feasible in metastatic melanoma and sarcoma patients [49].
Limitations of TCR-T cell therapies
TCR-T cell technology is not without limitations, these take two main forms: toxicities and disease progression.
As illustrated above, on-target/off-tumour toxicity ensues when the antigen targeted by the engineered cell is not exclusively expressed on the tumour. In the study described above by Johnson et al., the presence of melanocytes in the skin, eyes, and ears led to severe toxicity for some patients which had to be managed with local steroid administration to treat uveitis and hearing loss [36].
A similar pattern emerged in the case of Parkhust et al., who used a high-affinity TCR-T cell against carcinoembryonic antigen (CEA) in the setting of metastatic colorectal carcinoma. Although significant decreases in serum CEA levels were seen in all patients and one patient experienced an objective reduction in metastases, the trial was halted when all 3 patients experienced life-threatening colitis and colonic haemorrhage [50].
Two years later in 2013, this salient point was reinforced following the death of 2 patients who received anti-MAGE-A3 TCR-T cells. MAGE-A3 was chosen as a target as it was highly expressed on the 9 patient tumours (comprising metastatic melanoma, synovial sarcoma and oesophageal carcinoma) and was not known to be expressed in normal tissue. The therapy resulted in 5 partial responses; however, in several patients, the TCR-T cells homed to the brain due to the expression of MAGE-A12, a similar but related peptide to MAGE-A3; this led to severe damage to brain grey matter resulting in 2 treatment-related mortalities [51].
Owing to the fact that TCR-T cell targets are MHC-restricted, there is a risk of graft vs host disease (GVHD) due to mismatch between exogenous and endogenous chains of the TCR which could lead to deleterious recognitions of self-antigens [19]. This also poses a problem in that TCR-T cells must be derived from an autologous source which, given the time of manufacture, represents a challenge for patients with rapidly progressing disease [52].
CAR-T cells
CAR-T cell therapy is similar in principle to other forms of ACTs especially TCR-T cell therapy; however, instead of introducing TCR genes specific to tumour antigens, a chimeric antigen receptor (CAR) is designed and transfected into harvested lymphocytes [53]. The basic design of a first-generation CAR involves an extracellular single-chain variable fragment (scFv) (composed of variable heavy and variable light chains linked by a peptide) of a monoclonal antibody which has specificity for a tumour cell surface antigen; this confers the possibility of MHC-unrestricted recognition of tumour markers on the cell surface, expanding the specificity and customisability of the response. The scFv region is linked to a hinge region, which in turn is connected to a transmembrane domain and an intracellular tail composed of the CD3ζ subunit from the CD3 protein which facilitates intracellular signalling and T cell activation [54].
Evolution of the CAR design by the addition of co-stimulatory domains was important in countering the rapid anergy observed in the first-generation CAR-T cells, which led to a lack of cell persistence [55]. Incorporation of a single (second generation) or a pair of (third generation) co-stimulatory domains, the most common of which are CD28, 4-1BB or ICOS (inducible T cell co-stimulator/CD278), increased the ability of the receptors to promote cytokine secretion, T cell expansion and proliferation facilitating persistence [56]. These are illustrated visually below in Fig. 2.
Fig. 2.

Structure of CARs. All chimeric antigen receptors (CARs) have the same extracellular structure consisting of single-chain variable fragment (ScFV) part of an antibody which is specific for a target tumour antigen. First-generation CARs contain only one intracellular signalling component: CD3ζ. Addition of a co-stimulatory domain, such as CD28 or 4-1BB, to the CD3ζ created second-generation CARs. Third-generation CARs contain two co-stimulatory domains in addition to CD3ζ. Fourth-generation CARs can activate downstream transcription factors, such as NFAT, following CAR antigen recognition resulting in cytokine production
Further enhancements and developments to the CAR design have been described in the literature. Fourth-generation CARs co-express cytokine genes such as IL-12, IL-15 and IL-17 which aid in the clonal expansion of cells carrying such receptors [57]. It has been shown that secretion of such factors can modulate the immunosuppressive tumour microenvironment and enhance the host response against antigen-negative tumour cells [58, 59].
Clinical impact
CAR-T cells are designed as for use as a clinical therapeutic; therefore, they should be primarily judged on their performance in the clinical arena. In this respect, the results of CAR-T cell therapies can be summarised into two main categories; clinical trials of CAR-T cells in haematological malignancies have shown unprecedented response rates, especially in B cell malignancies, whereas the use of CAR-T cells in solid tumours has been less successful [60, 61].
Targeting of the CD19 antigen using CD19-specific CAR-T cells has been described as a ‘gamechanger’ for B cell acute lymphoblastic leukaemia (B-ALL) with complete remission rates of 70–94% seen in some clinical trials [18, 62, 63]. Such results led to the approval of tisagenlecleucel (Kymriah) by the FDA for treatment of relapsed or refractory B-ALL in patients < 25 years old [15]. However, in 30–50% of cases, the response was not sustained and relapse occurred most often within a year of treatment [18, 62].
CAR-T cell therapy has also been used to treat refractory large B cell lymphoma with outstanding success. Use of Axicabtagene ciloleucel (Yescarta) in this cancer type brought about an objective response rate of 82%, and a complete response rate of 54%; this response was sustained in a subset of patients with 40% continuing to have a complete response after 15.4 months of follow-up [64]. This therapy was approved by the FDA for usage in this indication in October 2017 [16].
Treatment of B cell malignancies using CAR-T cells results in a near ablation of the B cell repertoire; this is a consequence that CD19 is expressed on normal B cells and represents an instance of an on-target/off-tumour effect. In clinical practice, this toxicity is managed using intravenous immunoglobulin administration; however, a lack of strategies to manage depletion of other haematopoietic lineages may curtail the use of CAR-T cells to discrete cell lineages [42].
Solid tumours
Difficulty in choosing an appropriate target antigen is one of the critical factors that frustrate the advancement of CAR-T cells in solid tumours. It has been difficult to identify solid tumour surface antigens that are tumour specific as most solid tumour antigens are merely overexpressed leading to the risk of on-target/off-tumour effects [8, 42]. A further confounding factor in selecting a target antigen is the concept that solid tumours are highly heterogenous, meaning that there is irregularity in the expression levels of an antigen across tumour cells. This allows the tumour to remove the most immunogenic antigens via the process of immunoediting leading to antigen-negative tumour escape [60, 65].
Given that irregular antigen expression hinders CAR-T cell progress, it would appear to be helpful to know essential levels of antigen expression to induce tumour regression. However, as explored in a review by Newick et al., there is a lack of consensus on this issue; this is due to the phenomenon of epitope spreading, whereby killing of malignant cells by the CAR-T cell releases novel, immunogenic antigens which stimulate an endogenous adaptive host immune response against the tumour, leading to a ‘chain reaction’ of tumour cell killing that is difficult to predict in advance [60].
It has been proposed that in order to overcome low and variable expression of tumour-specific antigens, multiple antigens could be targeted simultaneously using dual CAR-T cells that express two CARs on their surface [66], two CAR-T cell lines both with different antigen specificity, or using more novel CAR designs such as tandem CAR-T cells, combinatorial CARs, or synNotch receptors. [67, 68]. A tandem CAR contains two different scFvs in a single CAR molecule and can be arranged in series as a stack or in an interrupted looped structure as shown in Fig. 3 [67]. Combinatorial CARs are similar to dual CARs except that one CAR expresses the CD3ζ signalling domain and its partner incorporates the co-stimulatory signalling domain [67]. Synthetic Notch (synNotch) receptors engender the transcription of a second CAR after antigen recognition of their corresponding antigen [67].
Fig. 3.

Nuances on CAR-T cell design. (a.) Co-expression of two discrete CARs in one cell is known as a dual CAR. (b. and c.) Tandem CARs express two different scFvs in a single CAR molecule and can be arranged in series as a stack (b.) or in an interrupted looped structure (c). In the case of dual and tandem CAR-T cells, identification of either antigen A or antigen B is sufficient for activation. (d.) Combinatorial CARs are composed of two partners; one incorporates the CD3ζ chain while the other includes the co-stimulatory signalling domain. (e.) Synthetic Notch (synNotch) receptors cause the transcription of a second CAR after antigen identification of their corresponding antigen. Combinatorial and synNotch CARs require both antigens to be recognised for full T cell activation. (f.) ON-Switch CARs require the presence of specific activating molecules to promote fully functional receptor assembly. (g.) Inhibitory CARs (iCARs) include inhibitory signalling motifs such as PD-1 or CTLA-4 that block T cell activation following antigen recognition
Poor trafficking of CAR-T cells to solid tumours once injected is a barrier that must be overcome for successful outcomes. This process is dependent on the correct matching of the chemokines secreted by the tumour and the chemokine receptor on the CAR-T cell; as might be expected, there is often a mismatch which leads to unproductive infiltration of the tumour [60]. This quandary can be resolved by using CAR-T cells with better-partnered chemokine receptors [69, 70].
Following successful homing to the tumour, CAR-T cells face an array of challenges that restrict their ability to engage transformed cells successfully. Physical barriers such as fibrous caps or thick stromal layers deter successful entry into the tumour. Once within the tumour milieu, the CAR-T cells encounter a hostile and inhibitory environment shaped by hypoxia, elevated lactate levels, low nutrient levels, high levels of adenosine and immunosuppressive cytokines such as TGFβ, as well as immunosuppressive cells such as MDSCs and Tregs. These factors serve to limit T cell proliferation, activation, motility and effector function [42, 60]. In order to circumvent some of the immunomodulatory effects of the TME, CAR-T cells expressing a dominant negative TGFβ receptor (which is truncated and therefore, absence of the necessary intracellular signalling domains precludes downstream pathway activation) have been generated which renders them resistant to TGFβ suppression [71].
Toxicities
As with TCR-T cells, CAR-T cells can react against self-antigens inducing on-target/off-tumour toxicity. Morgan et al. described a case where CAR-T cells with an scFv region homologous to the monoclonal antibody trastuzumab (anti-ERBB2) were used in a patient with metastatic colon cancer who succumbed due to low levels of ERBB2 expression on lung epithelial cells which caused diffuse alveolar damage (DAD) leading to the clinical presentation of acute respiratory distress syndrome (ARDS) [72]. This again highlights the ever-present challenge, in both CAR-T cell and TCR-T cell therapy, of knowing which antigens to target and which antigens to avoid because of mimicry leading to autoreactivity.
Other major adverse effects of CAR-T cells include cytokine release syndrome (CRS) and neurotoxicity (sometimes referred to as ICANS, immune effector cell-associated neurotoxicity syndrome). CRS occurs due to production and secretion of large amounts of cytokines and inflammatory molecules from CAR-T cells which can precipitate multiorgan failure [73]. The incidence of CRS was estimated by Wang et al. to range from 30 to 94% of patients [74]. Factors in the severity of CRS include the dose of T cells administered, the degree of cell activation and the extent of T cell expansion. Risk factors for severe CRS include patients with large tumour burdens, those with existing comorbidities and those who develop symptoms within 3 days of initiation of therapy [73].
Neurotoxicity, presenting most commonly as encephalopathy, headache, or delirium, has been reported to occur in 20–64% of patients receiving CAR-T cell therapy [74]. In the most severe of cases, cerebral oedema has been reported, which proved fatal during a Juno Therapeutics clinical trial for anti-CD19 CAR-T in patients with ALL (NCT02535364) [75]. The pathophysiology of neurotoxicity in ACT is currently being elucidated; however, it has been postulated that excess systemic cytokines and endothelial dysfunction in the blood-brain barrier may be at fault [76]. There is some evidence that CD19 may be expressed on brain tissue; however, Titov et al. noted that evidence in favour of absence outweighs its presence [77]. Another potential factor may be the co-stimulatory domain included in the CAR-T cell: CD28-containing cells have been associated with higher rates of neurotoxicity compared to 4-1BB CAR-T cells [78].
The management of both CRS and neurotoxicity events depend highly on prompt identification, grading of severity levels and monitoring of patients with such adverse events. Aggressive supportive therapy, such as fluid resuscitation, vasopressors, anti-epileptic drugs, anti-arrhythmic drugs and intubation should be prescribed as necessary [73]. Tocilizumab is an anti-IL-6 receptor monoclonal antibody that is FDA-approved for use in CAR-T cell CRS [79]; it is often used alongside corticosteroids [80].
Nuances on CAR-T cell design
Encoding CAR-T cells (or indeed TCR-T cells) with genetically encoded suicide switches has been used as a mechanism for controlling potentially dangerous and auto-reactive T cells which can be activated in the case of adverse events. The most commonly used switches are those based on herpes simplex virus thymidine kinase, which allows T cells to be ablated via ganciclovir administration, or using inducible caspase-9 (iCasp9), which upon administration of a small; otherwise, biologically inert molecule, AP1903, causes dimerization and activation of iCasp9 leading to selective CAR-T cell death [81].
This concept of small molecule manipulation of CAR-T cell responses is also evident in the ‘On-switch CAR-T cells’; these receptors are non-functional until specific activating molecules are present, which aids formation of an active receptor [82].
As alluded to above, split-signal CARs such as dual, combinatorial, tandem, or synNotch CARs can be used to increase sensitivity of CAR-T cells for tumours with variability in antigen expression. Similar mechanisms can be employed to reduce the risk of off-target toxicity. Inhibitory CARs (iCARs) can be co-expressed with a conventional activating CAR on a T cell; the iCAR has specificity for antigens expressed on normal tissues and generates a dominant negative signal, often via PD-1 or CTLA-4 signalling pathways, which inhibits the response of the cell, counteracting signals received from the activating receptor. In this way, normal tissues that also express the tumour antigen can be spared selectively via the iCAR [83].
In 2016, the development of a synthetic Notch (synNotch) receptor allowed for huge flexibility in designing CAR-T cells with programmable functions; this receptor consists of a standard antigen binding scFv region coupled to a transcription factor of the users’ choice [84]. An application of this new technology is to engineer T cells with dual-receptor circuits, whereby a synNotch receptor for one antigen induces the expression of a CAR for a second antigen increasing the specificity of the response [85]. The nuances on this technology are virtually limitless given the modularity of the synNotch platform; among the proposals are the use of programmable T cells for controlled cytokine secretion, delivery of therapeutics such as antibodies, or even instructing cells to form complex multi-cellular structures [8, 86].
Gene editing of T cells has been used to decrease the risk of GVHD by depleting the endogenous TCR complex by disrupting the genes TRAC or TRBC (which encode components of the TCR alpha and beta chains, respectively) using a variety of tools including CRISPR/Cas9, TALENs or Zinc-Finger [42, 87]. Other genes which may be advantageous to edit include PDCD1 which encodes for PD-1, a key immune checkpoint regulator that is often targeted by malignant cells by upregulating its ligand, PD-L1. In a preclinical model, CRISPR/Cas9-genearted PD-1-deficient CAR-T cells were resistant to PD-1-mediated immunosuppression [88]. A recent clinical trial exploring both hypotheses (NCT03399448) investigated the safety, feasibility and efficacy of NY-ESO-1 targeted autologous TCR-T cells with CRISPR-edited endogenous TCR and PD-1 in multiple myeloma and sarcoma [45]. These altered cells had enhanced persistence in circulation with stable levels observed for 9 months; this compares favourably to previously reported half-lives for NY-ESO-1–targeted T cells of approximately 1 week. While the results pertaining to efficacy were perhaps a little disappointing (residual tumour was found on biopsies in two of three patients, with a decrease in tumour antigen suggesting antigen-negative escape), the absence of clinical toxicities is highly encouraging. Given that this was a first-in-human trial of multiplex CRISPR/Cas9-edited T cells, future work will undoubtedly build on this early success.
Given the time-consuming nature of the manufacture of autologous CAR-T cells, the disruption of endogenous TCR complex opens the possibility of allogeneic, or ‘off-the-shelf’ forms of CAR-T cell therapy, which has huge advantages in terms of cost of production, speed of production, and availability for patients’ with rapidly progressive disease [89].
The SUPRA CAR is also an important development in the progress towards a ‘universal’ CAR-T therapy (Fig. 4). There are two elements to the split, universal and programmable (SUPRA) CAR: a zipCAR, which is composed of an extracellular leucine zipper linked to intracellular T cell signalling domains, and a separate zipFv, which comprises a matching leucine zipper paired to an antigen-specific scFv. The pairing of the two leucine zippers transmits the extracellular signal to the intracellular domains leading to T cell activation [90]. This design allows for the scFv in the zipFv to be altered as needed without the requirement to re-engineer cells as the new zipFv will also have binding capability to the zipCAR T cells of the original batch. This feature has the potential to introduce revolutionary flexibility in antigen targeting that may lead to increased therapeutic effectiveness, as well as decreased antigen escape and disease recurrence underpinned by the ability to target multiple antigens simultaneously and sequentially [91].
Fig. 4.
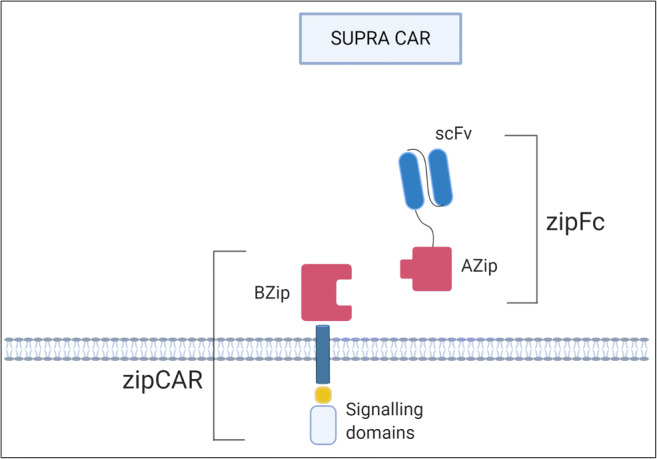
SUPRA CAR. SUPRA CARs consist of a zipCAR and zipFv. The extracellular portion of the CAR is known as a zipCAR which consists of a leucine zipper linked to intracellular signalling domains. The zipFv contains a scFv joined to a corresponding leucine zipper that can bind to the leucine zipper of the zipCAR.
Innate T cells
γδ T cells
Gamma-delta (γδ) T cells are a subset of T cells with innate immune system characteristics, such as enhanced speed of response, and ability to recognise antigens in an MHC-unrestricted manner. They are defined by the presence of the γδ TCR and have a plethora of roles in both health and disease states, including immune surveillance, cytokine production and antigen presentation [92].
Just as αβ T cells vastly outnumber γδ T cells in terms of peripheral blood composition [93], research involving T cell-based immunotherapies have mainly focussed on the more prevalent and better understood αβ T cell subset. However, γδ T cells have been shown to have extremely potent anti-tumour actions in both in vitro and in vivo settings. This has implications beyond the bench and at the bedside; Gentles and colleagues investigated the correlations between the composition of the immune tumour microenvironment and prognosis and found that the presence of infiltrating γδ T cells was the most reliable predictor of a positive outcomes across a diverse range of cancer types [94].
A naturally occurring corollary from this principle is to use engineered γδ T cells much in the same way conventional αβ T cells are manipulated to produce a targeted anti-tumour response. Similar to TCR-T cells described above, Harrer et al. showed that transferring an αβTCR into γδ T cells can allow the γδ T cell to target a specific tumour-associated peptide [95]; these engineered cells, when compared to CD8+ T cells, demonstrated an equivalent/comparable cytolytic capacity as well as a preserving their ‘innate’ anti-tumour ability and potential to lyse MHC-deficient cells. However, it was noted by the authors that this came at a price of decreased cytokine production; this is an aspect of the technology that could be improved to enhance its anti-tumour functionality.
The lure of using this innate T cell subset stems from the fact that the endogenous γδ TCRs do not recognize MHC molecules which precludes the development of graft-versus-host disease following adoptive cell therapy of such cells into allogeneic hosts. Similarly, transfecting αβ T cells with a new αβ TCR can lead to mispairing of host chains with the engineered receptor leading to alloreactivity; in the context of γδ T cells, this is unlikely to arise owing to the fact that the endogenous γδ TCR does not pair with engineered α- or β-chains [95]. This potentially provides a platform for the elusive ‘universal’ or ‘off-the-shelf’ therapy as the γδ T cells could in theory be derived from numerous sources, even encompassing immunocompetent and healthy patients [95].
Another proposed mechanism of directing γδ T cells towards transformed cells is by using chimeric antigen receptors (CARs) [96]. Capsomidis and colleagues developed a γδ CAR-T cell (second generation) targeting GD2 and tested the in vitro efficacy against neuroblastoma cell lines [97]: they found the cytotoxicity levels of their γδ CAR-T cells were comparable to conventional αβ CAR-T cells. Also noted was the retained ability of the transduced cells to act as antigen-presenting cells and cross-present tumour antigens to αβ T cells; this is an exciting feature of this anti-cancer modality as such features may lead to the phenomenon of ‘epitope spreading’ as mentioned for conventional CAR-T cells.
Despite the fanfare around the use of engineered γδ T cells as a new strategy in the fight against cancer, the transition of γδ T cells into clinical trials and towards a clinically useful product has been muted. Most of the currently recruiting or running clinical trials are focussed on un-engineered γδ T cells, i.e., extraction, ex vivo amplification and reinfusion of γδ T cells. Some of the cancer types under investigation include acute myeloid leukaemia (NCT04008381 and NCT03790072), hepatocellular liver cancer (NCT02425735) and lung cancer (NCT03183232) with the latter two trials having been completed in 2016 and 2019, respectively, although both sets of results have yet to be published.
One interesting case report published recently by Alnaggar et al. described the use of allogeneic γδ T cell treatment in a patient with stage IV cholangiocarcinoma. They provided proof of principle that this therapy may be clinically useful as they showed that the γδ T cell treatments depleted the size of the patient’s lymph node metastasis in addition to being well-tolerated and safe, with no adverse effects reported [98]. As this is a case report, further research and studies are needed in this area to expand the scope and power of this immunotherapy into an approved therapeutic.
NKT cells
Natural killer T (NKT) cells are another specialised subset of T cells that have an emerging role in the arena of cellular immunotherapies. While NKTs share common features with conventional T cells such as development in the thymus and expression of a TCR, these cells differ from αβ T cells in that their TCR repertoire is much less diverse and their TCRs recognise lipid antigens presented by CD1d (a conserved and non-polymorphic MHC class I-like molecule) [99]. Furthermore, following antigen recognition, there is a rapid effector response consisting of production of large amounts of cytokines and chemokines (especially IL-4 and IFNγ) which can influence both innate and adaptive immune components [100]. These features place the NKT cell at the interface between the innate and adaptive immune system as they fuse the precision antigen specificity of the adaptive immune system with the speed of the innate immune system [99].
There are two types of NKT cells; type I or ‘invariant’ cells contain an invariant TCRα chain partnered with a heterogenous TCRβ chain; in contrast, type II or ‘variant’ NKT cells express a more varied range of α and β TCR chains and lack an invariant chain [100]. Whereas type II NKT cells are implicated in suppression of anti-tumour responses [101], type I NKT cells are proposed to have an anti-tumour role. Reduced quantity and quality of NKT cells in peripheral blood is observed in many different cancer patients which hints at their important role in eliminating cancer cells [102]. Additionally, a greater frequency of peripheral blood type I NKT cells in cancer patients is a favourable prognostic indicator [103].
Type I NKT cells have similar tools to conventional T cells for ensuring destruction of CD1d-expressing malignant cells; these include mediators such as perforin, granzyme B, Fas ligand (FasL) or TNFα-mediated cytotoxic pathways in addition to release of cytokines which recruit, prime and activate cells of both the innate and adaptive immune system, including DCs, NK cells, B cells, and T cells [99]. This latter point has pertinence as it allows NKT cells to contemporaneously target both MHC-positive and MHC-negative tumour cells via activation of both antigen-specific CD8+ T cells (directly and indirectly via DC activation) and NK cells [99].
In terms of harnessing the potential of the NKT cell for cancer immunotherapy, several approaches exist, although no therapy has been approved as of yet. Early attempts at modulating NKT cells in vivo involved administration of soluble α-galactosylceramide (α-GalCer), which is the prototypical antigen for type I NKT cells owing to the presence of a common invariant α chain; while this was well-tolerated, it failed to show a clinical response, mostly attributed to low baseline levels of NKT cells [104]. Building on this, several groups attempted to activate NKT cells using ex vivo-generated DCs pulsed with α-GalCer. This approach was successful in expanding type I NKT cells in vivo as well as increased their capacity to secrete IFNγ; the translation of laboratory values into clinical responses was less successful although some patients exhibited partial responses or stable disease [105–107].
Another strategy that can be employed is the adoptive transfer of autologous ex vivo-expanded NKT cells. Exley et al. used this technique in a phase I clinical trial of patients with advanced melanoma; the expanded invariant NKT cells produced IFNγ, which were broadly well-tolerated, and although no formal assessment of efficacy can be drawn from such a study, 3 of 9 patients had not progressed after a median follow-up of 63 months [108]. Further clinical trials are currently investigating the safety, feasibility and efficacy of this approach in non-small cell lung cancer (NCT03198923), hepatocellular carcinoma (NCT04011033) and melanoma (NCT00631072).
Engineering of NKT cells is at a very primitive stage in contrast to other cellular modalities; however, one group is currently recruiting patients with neuroblastoma for treatment with expanded NKT cells that have been engrafted with a second-generation chimeric antigen receptor (CAR) specific for GD2, which is expressed almost ubiquitously on neuroblastoma cells (NCT03294954).
Future directions
Despite both γδ T cells and NKT cells often being classified as ‘innate’ T cells, cell activation is predicated upon recognition of a specific antigen. As manipulation and engineering of these cell types advances and they become equipped with novel receptors allowing for a more expanded scope of tumour antigen targeting, caution must be exercised in the choice of an appropriate tumour antigen to avoid similar on-target/off-tumour toxicities that have led to morbidity and mortality in several of the TCR-T cell and CAR-T cell trials discussed above.
As both γδ T cell and NKT cell immunotherapies are relatively in their infancy, compared to more established T cell-based therapies, this is an exciting area that is sure to develop and progress rapidly over the coming years.
NK cell-based therapies
Cellular immunotherapies based on conventional T cells target tumour-associated antigens (TAAs) using antigen receptors, TCRs in the case of TILs and TCR-T cells, and CARs in the setting of CAR-T cell therapy. This strategy has the disadvantage that the cells must be sensitised to produce cytotoxic activity against cells bearing such antigens. In contrast to cells of the adaptive immune system, innate immune effector cells, such as NK cells, demonstrate considerable cytotoxicity against cancer and virus-infected cells without prior sensitisation. NK-mediated cytotoxicity occurs in a TAA-independent MHC-unrestricted fashion, thus bypassing the difficulty in identifying defined tumour-specific antigens while retaining accurate discrimination between normal and malignant cells [109, 110].
Regulation of NK cells has been shown to depend on an equilibrium between positive and negative signals. Ligation of inhibitory receptors such as inhibitory killer Ig-like receptors (iKIRs) by MHC class I molecules prevents cytotoxicity. In the context of malignancy, where transformed cells downregulate MHC class I molecules to avoid detection by the adaptive immune system, the inhibitory receptors are not engaged, and the NK cell recognises a ‘missing self’ leading to killing of the tumour cell. In concert with this, DNA damage and cellular stress can result in the expression of ‘stress ligands’ which can ligate positive receptors on the NK cell leading to target cell killing [111]. NK-mediated cytotoxicity can occur via direct release of cytoplasmic granules containing perforin and granzyme, as well as induction of apoptosis in the target cell via TNF-related apoptosis-inducing ligand (TRAIL) or FasL [112].
The effector cells used in TAA-independent ACT are, with the exception of NK cells, indiscrete heterogenous cell populations of expanded PBMCs that have proliferated and acquired killing activity on ex vivo culture; these populations are considered NK cell developmental intermediates that result from incubation of PBMCs in cytokine culture, and suboptimal pure NK cell expansion. They are not true NK cells per se, but rather mimic their killing functions. Cells of this classification include lymphokine-activated killer (LAK) cells, cytokine-induced killer (CIK) cells and cytokine-induced natural killer (CINK) cells [109].
LAK cells
Rosenberg et al. demonstrated that incubation of lymphocytes with high-dose IL-2 led to such cells acquiring the capacity to lyse tumour cells normally not sensitive to NK cell lysis; they used the term lymphokine-activated killers (LAK) cells to distinguish them from classic CTLs [113]. Further analysis of the cells revealed they were composed of a melange of NK cells, NK T cells (NKTs) and T cells [114]. However, the clinical utility of LAKs did not deliver on the promise suggested by the preclinical work. Initially, it seemed that ACT of LAK cells in concert with high-dose IL-2 administration had a benefit in patients with renal cancer and melanoma, and the effect was in fact attributable solely to the high-dose IL-2 [115]. This result, along with the development of more potent killer cells such as CIK cells, consigned LAK cells to a place in the annals of immunology rather than in clinical practice [109].
CIK cells
Addition of interferon-γ (IFNγ) and anti-CD3 stimulation to high-dose IL-2 led to the culture of a more cytotoxic set of cells termed cytokine-induced killer (CIK) cells [116]. Similar to LAKs, CIKs are heterogenous and are composed of NK cells, NKT cells and T cells. Although some cells express the TCR, their cytotoxicity is non-MHC-restricted and non-TCR-dependent [117]. CIKs have shown success in clinical trials, especially in the setting of patients with hepatocellular carcinoma and glioblastoma [118–120].
Cytokine-induced natural killer (CINK) cells are a variation on CIK cells whereby the proportion of NK cells in CIK preparations is increased leading to higher cytotoxic capacities per cell; however, CINK cells have yet to gain traction in the clinical arena [109].
CAR-NK cells
Genetic editing of CAR-T cells to disrupt the TCR and thus minimise the risk of GVHD in ACT of T cells has been proposed and discussed above; however, another strategy to overcome this risk is to use innate immune cells such as NK cells as donor cells. NK cells are predicted not to cause GVHD following experiments using murine models in addition to evidence arising from clinical trials in patients with hematologic or solid malignancies using haploidentical and cord blood-derived NK cell infusions [121]. This prediction has been shown to hold true in human trials of ACT using NK cells [122] which has increased interest in using CAR-NK as an ‘off-the-shelf’ cellular therapy for cancer [123].
Another potential benefit of using CAR-NK cells rather than CAR-T cells is a decreased risk of toxicities such as CRS; this is predicted due to limited in vivo persistence and a lack of clonal expansion of adoptively transferred NK cells [110]. Furthermore, CAR-NK cells preserve their intrinsic set of innate immune receptors which enable them to carry out their native functions; this may make downregulation of the CAR antigen a less successful ploy for tumour escape in the case of CAR-NK cells, compared to CAR-T cells [110].
Several groups have shown the efficacy of using CAR-NK cells as cytotoxic mediators against colorectal cancer and acute myeloid leukaemia (AML) cell lines in preclinical in vitro studies [124, 125]. There are several clinical trials currently recruiting patients to assess CAR-NK safety, feasibility and efficacy in B lymphoid malignancies (NCT03056339) and pancreatic cancer (NCT03941457) as well as trials recruiting patients with mix of solid tumours types (NCT03415100 and NCT03940820). The results of these studies have not been reported to date.
Resistance to ACT
Disease progression and/or relapse is perhaps the major issue that curbs the efficacy of ACT of lymphocytes; as mentioned above, the initial remission rates of 70–94% seen in some trials of tisagenlecleucel (Kymriah) were not long-lasting with 30–50% of patients relapsing, commonly within a year of treatment [18, 62].
Antigen-positive relapse occurs due to inadequate cell persistence in the body, allowing the tumour to simply regrow and recover. Persistence of transferred cells is governed by intrinsic factors of the cell such as the quality, age, and presence of co-stimulatory domains in the setting of CAR-T/NK cells [4].
Strategies to prevent antigen-positive relapse have been mentioned previously and include incorporation of co-stimulatory domains such as CD28 or 4-1BB in CAR design, lymphodepletion or gene editing of co-inhibitory pathways such as PD-1 [31, 57, 88]. Also proposed is the use of combination of immune checkpoint inhibitors with cells as well as reinfusion of cells; however, mixed evidence for efficacy of this latter strategy exists [126].
Antigen-negative relapse occurs due to the phenomenon of antigen escape whereby selective pressure drives formation of less immunogenic tumour cell phenotypes. Mechanisms of antigen escape include alterations to the antigen structure occurring via mutations or alternative splicing of transcripts. Cells may decrease expression of the target antigen or undergo a process known as lineage switching whereby, for example, cells from a B cell lineage may convert to a myeloid lineage after initiation of a cellular immunotherapy [127–129]. Antigen-negative relapse may be prevented by using cells with dual antigen specificity such as dual or tandem CARs as discussed above.
DC-based therapies
Dendritic cells (DCs) are professional antigen-presenting cells whose role in the immune response consists of capturing, processing and presenting antigenic material on MHC class I and II molecules to T lymphocytes, thus activating the adaptive immune response [130].
Under resting conditions, DCs are inactive and immature, functioning as sentinels; once exposed to stimuli such as inflammatory cytokines, molecular patterns associated with pathogens, or alarmin stimuli, DCs undergo a maturation process. This process is characterised by acquisition of chemokine receptors, T cell co-stimulatory receptors, upregulation of MHC class I and II, and the ability to secrete cytokines; these factors allow effective activation of the immune system [130]. The rationale for a DC-based therapeutic is that exposure of ex vivo DCs to tumour antigens would induce a host anti-tumour response when re-injected into a patient, as shown in Fig. 5.
Fig. 5.

Dendritic cell (DC) vaccines. (1) Gather peripheral blood mononuclear cells. (2) Produce immature DCs using a cytokine cocktail. (3) Loading tumour-derived antigens onto DC. (4) Inject vaccine comprising activated antigen-presenting DCs into the patient. (5) Vaccine triggers anti-tumour immune effector cell responses leading to T cell- and NK cell-mediated tumour killing
The starting point for the revolution of cellular immunotherapies over the last decade was the approval of the DC vaccine Sipuleucel-T (Provenge) by the FDA for use in castration-resistant metastatic prostate cancer [13]. Sipuleucel-T was shown to prolong overall survival by stimulating an immune response against the antigen PAP/PA2024. [131] Kumar et al. showed a median survival benefit of 189 days (356 days for treated group vs 147 days for the untreated group) for autologous dendritic cell-based vaccine for management of refractory solid malignancies, including prostate, ovarian, non-small cell lung and colorectal cancers [132].
DC-based vaccines can be classified according to how the antigens are loaded [133]. Pulsing DCs with peptide/protein fragments has been shown to preserve tumour selectivity while minimising the risk of autoreactivity; this approach, however, is subject to the same limitations that constrain CAR/TCR antigen specificity in that the selected peptides must not be expressed highly on normal tissues [133]. A phase I/II study investigating the efficacy of a DC vaccine pulsed with a fragment of a tumour-associated mucin 1 glycoprotein in patients following resection of pancreatic and biliary cancers showed that the vaccine was well-tolerated and that 4 of 12 patients survived for 4 years post-surgery, on a background median survival of 5 months [134].
Another method of loading antigen onto DCs for vaccine development is transfection of DCs with genes encoding for tumour antigens; this can be achieved using viral vectors, or direct transfection of DNA or RNA into DCs [133]. Miyazawa et al. showed that DCs expressing mesothelin (MSLN) induced CTL responses towards pancreatic cancer cells endogenously expressing MSLN [135].
DCs can be pulsed with tumour cells or tumour cell lysates to create vaccines that have the advantage of delivering a wide range of specific tumour antigens without the need for prior knowledge of the target [46]. A study conducted by Nakamura et al. concluded that immunotherapy with DC vaccines prepared from whole cells may prolong the lifespan of patients with refractory pancreatic cancer [136].
Finally, tumour cells can be fused with DCs to induce anti-tumour immune responses [137]. However, some controversy exists in the literature about the effectiveness of this strategy with conflicting reports presented by Ziske, in favour of the technique, and Chen, opposed [138, 139].
Conclusion
In conclusion, the field of cellular immunotherapies is full of exciting developments especially in the area of genetically engineered lymphocytes. While it is unlikely that cellular immunotherapy will achieve that status of a ‘universal therapy’, it provides another treatment option for patients, especially those with refractory disease. Key in the progress of all technologies is the selection of appropriate tumour-specific antigens to maximise selective killing of tumour cells. Further work is needed on creating off-the-shelf products to ensure more widespread availability of these living drugs and to ensure that patients with rapidly progressive disease can access them.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.WHO. Cancer. World Health Organisation,. https://www.who.int/news-room/fact-sheets/detail/cancer (accessed 07 Nov 2019
- 2.Cancer Research UK. Worldwide cancer incidence statistics. https://www.cancerresearchuk.org/health-professional/cancer-statistics/worldwide-cancer/incidence#heading-Zero (accessed 07 November 2019)
- 3.de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6(1):24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- 4.Song MK, Park BB, Uhm JE. Resistance Mechanisms to CAR T-Cell Therapy and Overcoming Strategy in B-Cell Hematologic Malignancies. (in eng) Int J Mol Sci. 2019;20(20):5010. doi: 10.3390/ijms20205010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vinay DS, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35:S185–S198. doi: 10.1016/j.semcancer.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Lin Y, Okada H. Cellular immunotherapy for malignant gliomas. Expert Opin Biol Ther. 2016;16(10):1265–1275. doi: 10.1080/14712598.2016.1214266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D’Souza MJ, Gala RP, Ubale RV, D’Souza B, Vo TP, Parenky AC, Mulla NS, D’Sa S, D’Souza M, Braz-Gomes K, D’Souza N, Chiriva-Internati M, Zughaier S. Chapter 5 - trends in nonparenteral delivery of biologics, vaccines and cancer therapies. In: Singh M, Salnikova M, editors. Novel approaches and strategies for biologics, vaccines and cancer therapies. San Diego: Academic Press; 2015. pp. 89–122. [Google Scholar]
- 8.Paucek RD, Baltimore D, Li G. The cellular immunotherapy revolution: arming the immune system for precision therapy. Trends Immunol. 2019;40(4):292–309. doi: 10.1016/j.it.2019.02.002. [DOI] [PubMed] [Google Scholar]
- 9.Galluzzi L, et al. Classification of current anticancer immunotherapies (in eng) Oncotarget. 2014;5(24):12472–12508. doi: 10.18632/oncotarget.2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Armstrong AC, Eaton D, Ewing JC (2001) Cellular immunotherapy for cancer. BMJ 323(7324):1289–1293. https://doi.org/10.1136/bmj.323.7324.1289%J [DOI] [PMC free article] [PubMed]
- 11.Liebelt BD, Finocchiaro G, Heimberger AB (2016) Chapter 10 - principles of immunotherapy. In: Berger MS, Weller M (eds) Handbook of Clinical Neurology, Elsevier, 134:163–181. 10.1016/B978-0-12-802997-8.00010-4 [DOI] [PubMed]
- 12.Melief CJM, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. (in eng) J Clin Invest. 2015;125(9):3401–3412. doi: 10.1172/JCI80009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.FDA. "PROVENGE (sipuleucel-T)." https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/provenge-sipuleucel-t. Accessed 10 Nov 2019
- 14.Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin Orthop Relat Res. 1893;262:3–11. [PubMed] [Google Scholar]
- 15.FDA. "KYMRIAH (tisagenlecleucel)." https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/kymriah-tisagenlecleucel (accessed 09 November 2019
- 16.Bouchkouj N, et al. FDA Approval Summary: axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma. Clin Cancer Res. 2018;2018:2743. doi: 10.1158/1078-0432.CCR-18-2743. [DOI] [PubMed] [Google Scholar]
- 17.Baselga J, et al. AACR Cancer Progress Report. J Clin Cancer Res. 2015;21(19 Supplement):S1–S128. doi: 10.1158/1078-0432.CCR-15-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maude SL, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao L, Cao YJ (2019) “Engineered T cell therapy for cancer in the clinic,” (in English). Review 10(2250). 10.3389/fimmu.2019.02250 [DOI] [PMC free article] [PubMed]
- 20.Cancer Research Institute. "What is immunotherapy?" https://www.cancerresearch.org/immunotherapy/what-is-immunotherapy (accessed 08 Nov 2019
- 21.Gaud G, Lesourne R, Love PE. Regulatory mechanisms in T cell receptor signalling. Nat Rev Immunol. 2018;18(8):485–497. doi: 10.1038/s41577-018-0020-8. [DOI] [PubMed] [Google Scholar]
- 22.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13(4):227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol. 2013;13(5):309–320. doi: 10.1038/nri3442. [DOI] [PubMed] [Google Scholar]
- 24.Durgeau A, Virk Y, Corgnac S, Mami-Chouaib F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Review. 2018;9:14. doi: 10.3389/fimmu.2018.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perica K, Varela JC, Oelke M, Schneck J. “Adoptive T cell immunotherapy for cancer,” (in eng), Rambam Maimonides. Med J. 2015;6(1):e0004–e0004. doi: 10.5041/RMMJ.10179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barrett DM, Grupp SA, June CH. Chimeric antigen receptor– and TCR-modified T cells enter main street and wall street. J Immunol. 2015;195(3):755–761. doi: 10.4049/jimmunol.1500751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science (New York, NY) 1986;233(4770):1318–1321. doi: 10.1126/science.3489291. [DOI] [PubMed] [Google Scholar]
- 28.Rosenberg SA, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319(25):1676–1680. doi: 10.1056/nejm198812223192527. [DOI] [PubMed] [Google Scholar]
- 29.Khong HT, Wang QJ, Rosenberg SA. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression. J Iimmunother (Hagerstown, Md : 1997) 2004;27(3):184–190. doi: 10.1097/00002371-200405000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (New York, NY) 2015;348(6230):62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dudley ME, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26(32):5233–5239. doi: 10.1200/jco.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balch CM, et al. Patterns of Human Tumor-Infiltrating Lymphocytes in 120 Human Cancers. J Arch Surg. 1990;125(2):200–205. doi: 10.1001/archsurg.1990.01410140078012. [DOI] [PubMed] [Google Scholar]
- 33.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W (2004) “Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival,” (in eng). Nat Med 10(9):942–949. 10.1038/nm1093 [DOI] [PubMed]
- 34.Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. “Increase of regulatory T cells in the peripheral blood of cancer patients,” (in eng) Clin Cancer Res. 2003;9(2):606–612. [PubMed] [Google Scholar]
- 35.Morgan RA, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (New York, NY) 2006;314(5796):126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson LA, et al. “Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen,” (in eng) Blood. 2009;114(3):535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baum C, Fehse B (2003) Mutagenesis by retroviral transgene insertion: risk assessment and potential alternatives. Curr Opin Mol Ther Rev 5(5):458–462 [Online]. Available: https://www.scopus.com/inward/record.uri?eid=2-s2.0-0142185323&partnerID=40&md5=b390345046134988271e55e4c2419980. Accessed 8 Nov 2019 [PubMed]
- 38.Wang X, Rivière I (2016) Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther - Oncolytics 3. 10.1038/mto.2016.15 [DOI] [PMC free article] [PubMed]
- 39.Li D, et al. Genetically engineered T cells for cancer immunotherapy. Signal Transduct Target Ther. 2019;4(1):35. doi: 10.1038/s41392-019-0070-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magnani CF, et al. “Immunotherapy of acute leukemia by chimeric antigen receptor-modified lymphocytes using an improved Sleeping Beauty transposon platform,” (in eng) Oncotarget. 2016;7(32):51581–51597. doi: 10.18632/oncotarget.9955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gorabi AM, Hajighasemi S, Sathyapalan T, Sahebkar A (2019) “Cell transfer-based immunotherapies in cancer: a review,” (in eng). IUBMB Life. 10.1002/iub.2180 [DOI] [PubMed]
- 42.D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis. 2018;9(3):282. doi: 10.1038/s41419-018-0278-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bethune MT, Joglekar AV. Personalized T cell-mediated cancer immunotherapy: progress and challenges. Curr Opin Biotechnol. 2017;48:142–152. doi: 10.1016/j.copbio.2017.03.024. [DOI] [PubMed] [Google Scholar]
- 44.Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest. 2015;125(9):3413–3421. doi: 10.1172/JCI80008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rapoport AP, et al. NY-ESO-1–specific TCR–engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. 2015;21:914. doi: 10.1038/nm.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robbins PF, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robbins PF et al (2015) A pilot trial using lymphocytes genetically engineered with an NY-ESO-1–reactive T-cell receptor: long-term follow-up and correlates with response. J Clin Cancer Res 21(5):1019–1027. https://doi.org/10.1158/1078-0432.CCR-14-2708% [DOI] [PMC free article] [PubMed]
- 48.Stadtmauer EA, Faitg TH, Lowther DE, Badros AZ, Chagin K, Dengel K, Iyengar M, Melchiori L, Navenot JM, Norry E, Trivedi T, Wang R, Binder GK, Amado R, Rapoport AP. Long-term safety and activity of NY-ESO-1 SPEAR T cells after autologous stem cell transplant for myeloma. Blood Adv. 2019;3(13):2022–2034. doi: 10.1182/bloodadvances.2019000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stadtmauer EA, et al. CRISPR-engineered T cells in patients with refractory cancer. Science (New York, NY) 2020;367(6481):eaba7365. doi: 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DAN, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, Hughes MS, Kammula US, Phan GQ, Lim RM, Wank SA, Restifo NP, Robbins PF, Laurencot CM, Rosenberg SA. “T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis,” (in eng) Mol Ther. 2011;19(3):620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, Phan GQ, Hughes MS, Kammula US, Miller AD, Hessman CJ, Stewart AA, Restifo NP, Quezado MM, Alimchandani M, Rosenberg AZ, Nath A, Wang T, Bielekova B, Wuest SC, Akula N, McMahon FJ, Wilde S, Mosetter B, Schendel DJ, Laurencot CM, Rosenberg SA. “Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy,” (in eng) J Immunother (Hagerstown, Md : 1997) 2013;36(2):133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saudemont A, Jespers L, Clay T. “Current status of gene engineering cell therapeutics,” (in eng) Front Immunol. 2018;9:153–153. doi: 10.3389/fimmu.2018.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gross G, Waks T, Eshhar Z. “Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity,” (in eng) Proc Natl Acad Sci U S A. 1989;86(24):10024–10028. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stambrook PJ, Maher J, Farzaneh F. “Cancer immunotherapy: whence and whither,” (in eng) Mol Cancer Res. 2017;15(6):635–650. doi: 10.1158/1541-7786.Mcr-16-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brocker T. “Chimeric Fv-zeta or Fv-epsilon receptors are not sufficient to induce activation or cytokine production in peripheral T cells,” (in eng) Blood. 2000;96(5):1999–2001. doi: 10.1182/blood.V96.5.1999. [DOI] [PubMed] [Google Scholar]
- 56.Zhang H et al (2007) 4-1BB is superior to CD28 costimulation for generating CD8+ cytotoxic lymphocytes for adoptive immunotherapy. J Immunol 179(7):4910–4918. https://doi.org/10.4049/jimmunol.179.7.4910% [DOI] [PMC free article] [PubMed]
- 57.Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15(8):1145–1154. doi: 10.1517/14712598.2015.1046430. [DOI] [PubMed] [Google Scholar]
- 58.Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, Rosenberg SA, Morgan RA. “Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment,” (in eng) Mol Ther. 2011;19(4):751–759. doi: 10.1038/mt.2010.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chmielewski M, Kopecky C, Hombach AA, Abken H. “IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression,” (in eng) Cancer Res. 2011;71(17):5697–5706. doi: 10.1158/0008-5472.Can-11-0103. [DOI] [PubMed] [Google Scholar]
- 60.Newick K, O'Brien S, Moon E, Albelda SM. “CAR T cell therapy for solid tumors,” (in eng) Annu Rev Med. 2017;68:139–152. doi: 10.1146/annurev-med-062315-120245. [DOI] [PubMed] [Google Scholar]
- 61.Martinez M, Moon EK (2019) “CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment,” (in English). Review 10(128). 10.3389/fimmu.2019.00128 [DOI] [PMC free article] [PubMed]
- 62.Park JH, et al. “Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia,” (in eng) N Engl J Med. 2018;378(5):449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Almasbak H, Aarvak T, Vemuri MC. “CAR T cell therapy: a game changer in cancer treatment,” (in eng) J Immunol Res. 2016;2016:5474602. doi: 10.1155/2016/5474602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Neelapu SS, et al. “Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma,” (in eng) N Engl J Med. 2017;377(26):2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16(3):151–167. doi: 10.1038/s41571-018-0142-8. [DOI] [PubMed] [Google Scholar]
- 66.Wilkie S, van Schalkwyk MCI, Hobbs S, Davies DM, van der Stegen SJC, Pereira ACP, Burbridge SE, Box C, Eccles SA, Maher J. “Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling,” (in eng) J Clin Immunol. 2012;32(5):1059–1070. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- 67.Guedan S, Calderon H, Posey AD, Jr, Maus MV. “Engineering and design of chimeric antigen receptors,” (in eng) Mol Ther Methods Clin Dev. 2019;12:145–156. doi: 10.1016/j.omtm.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hegde M, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Invest. 2016;126(8):3036–3052. doi: 10.1172/JCI83416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moon EK, et al. “Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor,” (in eng) Clin Cancer Res. 2011;17(14):4719–4730. doi: 10.1158/1078-0432.Ccr-11-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kershaw MH, et al. “Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2,” (in eng) Hum Gene Ther. 2002;13(16):1971–1980. doi: 10.1089/10430340260355374. [DOI] [PubMed] [Google Scholar]
- 71.Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, Maus MV, Fraietta JA, Zhao Y, June CH. “Dominant-negative TGF-beta receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication,” (in eng) Mol Ther. 2018;26(7):1855–1866. doi: 10.1016/j.ymthe.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. “Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2,” (in eng) Mol Ther. 2010;18(4):843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.S. S. Neelapu et al., "Chimeric antigen receptor T-cell therapy — assessment and management of toxicities," Nat Rev Clin Oncol, Review Article vol. 15, p. 47, 09/19/online 2017, doi: 10.1038/nrclinonc.2017.148 https://www.nature.com/articles/nrclinonc.2017.148#supplementary-information [DOI] [PMC free article] [PubMed]
- 74.Wang Z, Han W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomark Res. 2018;6(1):4. doi: 10.1186/s40364-018-0116-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.American Association for Cancer Research, JCAR015 in ALL: a root-cause investigation, Cancer discovery, Jan 2018, 8(1):4–5, 10.1158/2159-8290.Cd-nb2017-169
- 76.Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, Yeung C, Liles WC, Wurfel M, Lopez JA, Chen J, Chung D, Harju-Baker S, Özpolat T, Fink KR, Riddell SR, Maloney DG, Turtle CJ. “Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells,” (in eng) Cancer Discov. 2017;7(12):1404–1419. doi: 10.1158/2159-8290.Cd-17-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Titov A, et al. The biological basis and clinical symptoms of CAR-T therapy-associated toxicites. Cell Death Dis. 2018;9(9):897. doi: 10.1038/s41419-018-0918-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ying Z, He T, Wang X, Zheng W, Lin N, Tu M, Xie Y, Ping L, Zhang C, Liu W, Deng L, Qi F, Ding Y, Lu XA, Song Y, Zhu J. Parallel comparison of 4-1BB or CD28 co-stimulated CD19-targeted CAR-T cells for B cell non-Hodgkins lymphoma. Mol Ther - Oncolytics. 2019;15:60–68. doi: 10.1016/j.omto.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.FDA. "FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome." https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tisagenlecleucel-b-cell-all-and-tocilizumab-cytokine-release-syndrome?platform=hootsuite (accessed 9 Nov 2019
- 80.Davila ML et al (2014) Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. J Sci Transl Med 6(224):224ra25-224ra25. https://doi.org/10.1126/scitranslmed.3008226% [DOI] [PMC free article] [PubMed]
- 81.Zhou X, Dotti G, Krance RA, Martinez CA, Naik S, Kamble RT, Durett AG, Dakhova O, Savoldo B, di Stasi A, Spencer DM, Lin YF, Liu H, Grilley BJ, Gee AP, Rooney CM, Heslop HE, Brenner MK. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood. 2015;125(26):4103–4113. doi: 10.1182/blood-2015-02-628354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu C-Y, Roybal KT, Puchner EM, Onuffer J, Lim WA (2015) Remote control of therapeutic T cells through a small molecule–gated chimeric receptor. J Sci 350(6258):aab4077. https://doi.org/10.1126/science.aab4077% [DOI] [PMC free article] [PubMed]
- 83.Fedorov VD, Themeli M, Sadelain M (2013) PD-1– and CTLA-4–based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. J Sci Transl Med 5(215):215ra172-215ra172. https://doi.org/10.1126/scitranslmed.3006597% [DOI] [PMC free article] [PubMed]
- 84.Morsut L, et al. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell. 2016;164(4):780–791. doi: 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roybal KT, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell. 2016;164(4):770–779. doi: 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Toda S, Blauch LR, Tang SKY, Morsut L, Lim WA. “Programming self-organizing multicellular structures with synthetic cell-cell signaling,” (in eng) Science (New York, NY) 2018;361(6398):156–162. doi: 10.1126/science.aat0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bunse M, et al. RNAi-mediated TCR knockdown prevents autoimmunity in mice caused by mixed TCR dimers following TCR gene transfer. Mol Ther. 2014;22(11):1983–1991. doi: 10.1038/mt.2014.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rupp LJ, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7(1):737. doi: 10.1038/s41598-017-00462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Poirot L, et al. “Multiplex genome-edited T-cell manufacturing platform for “Off-the-Shelf” adoptive T-cell immunotherapies,” (in eng) Cancer Res. 2015;75(18):3853–3864. doi: 10.1158/0008-5472.Can-14-3321. [DOI] [PubMed] [Google Scholar]
- 90.Liu D, Zhao J, Song Y. Engineering switchable and programmable universal CARs for CAR T therapy. J Hematol Oncol. 2019;12(1):69. doi: 10.1186/s13045-019-0763-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen YY. “Increasing T cell versatility with SUPRA CARs,” (in eng) Cell. 2018;173(6):1316–1317. doi: 10.1016/j.cell.2018.05.030. [DOI] [PubMed] [Google Scholar]
- 92.Lawand M, Déchanet-Merville J, Dieu-Nosjean M-C. “Key features of Gamma-Delta T-cell subsets in human diseases and their immunotherapeutic implications,” (in eng) Front Immunol. 2017;8:761–761. doi: 10.3389/fimmu.2017.00761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nielsen MM, Witherden DA, Havran WL. γδ T cells in homeostasis and host defence of epithelial barrier tissues. Nat Rev Immunol. 2017;17(12):733–745. doi: 10.1038/nri.2017.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gentles AJ, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–945. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Harrer DC, et al. RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: a safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer. 2017;17(1):551. doi: 10.1186/s12885-017-3539-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fisher J, Anderson J. “Engineering approaches in human Gamma Delta T cells for cancer immunotherapy,” (in eng) Front Immunol. 2018;9:1409. doi: 10.3389/fimmu.2018.01409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Capsomidis A, Benthall G, van Acker HH, Fisher J, Kramer AM, Abeln Z, Majani Y, Gileadi T, Wallace R, Gustafsson K, Flutter B, Anderson J. Chimeric antigen receptor-engineered human Gamma Delta T cells: enhanced cytotoxicity with retention of cross presentation. Mol Ther. 2018;26(2):354–365. doi: 10.1016/j.ymthe.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Alnaggar M, et al. Allogenic Vγ9Vδ2 T cell as new potential immunotherapy drug for solid tumor: a case study for cholangiocarcinoma. J Immunol Ther Cancer. 2019;7(1):36. doi: 10.1186/s40425-019-0501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nair S, Dhodapkar MV (2017) “Natural killer T cells in cancer immunotherapy,” (in English). Review 8(1178). 10.3389/fimmu.2017.01178 [DOI] [PMC free article] [PubMed]
- 100.Wu L, Van Kaer L. “Natural killer T cells in health and disease,” (in eng) Front Biosci (Schol Ed) 2011;3:236–251. doi: 10.2741/s148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dhodapkar MV, Kumar V. “Type II NKT cells and their emerging role in health and disease,” (in eng) J Immunol (Baltimore, Md : 1950) 2017;198(3):1015–1021. doi: 10.4049/jimmunol.1601399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Molling JW, et al. “Peripheral blood IFN-gamma-secreting Valpha24+Vbeta11+ NKT cell numbers are decreased in cancer patients independent of tumor type or tumor load,” (in eng) Int J Cancer. 2005;116(1):87–93. doi: 10.1002/ijc.20998. [DOI] [PubMed] [Google Scholar]
- 103.Schneiders FL et al. (2012) Circulating invariant natural killer T-cell numbers predict outcome in head and neck squamous cell carcinoma: updated analysis with 10-year follow-up. 30(5), pp. 567–570, doi: 10.1200/jco.2011.38.8819 [DOI] [PubMed]
- 104.Giaccone G, et al. “A phase I study of the natural killer T-cell ligand alpha-galactosylceramide (KRN7000) in patients with solid tumors,”(in eng) Clin Cancer Res. 2002;8(12):3702–3709. [PubMed] [Google Scholar]
- 105.Nicol AJ, Tazbirkova A, Nieda M (2011) Comparison of clinical and immunological effects of intravenous and intradermal administration of α-galactosylceramide (KRN7000)-pulsed dendritic cells. J Clin Cancer Res 17(15):5140–5151. https://doi.org/10.1158/1078-0432.CCR-10-3105% [DOI] [PubMed]
- 106.Nagato K, et al. Accumulation of activated invariant natural killer T cells in the tumor microenvironment after α-galactosylceramide-pulsed antigen presenting cells. J Clin Immunol. 2012;32(5):1071–1081. doi: 10.1007/s10875-012-9697-9. [DOI] [PubMed] [Google Scholar]
- 107.Uchida T, Horiguchi S, Tanaka Y, Yamamoto H, Kunii N, Motohashi S, Taniguchi M, Nakayama T, Okamoto Y. “Phase I study of alpha-galactosylceramide-pulsed antigen presenting cells administration to the nasal submucosa in unresectable or recurrent head and neck cancer,” (in eng) Cancer Immunol Immunother. 2008;57(3):337–345. doi: 10.1007/s00262-007-0373-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Exley MA, et al. “Adoptive transfer of invariant NKT cells as immunotherapy for advanced melanoma: a phase I clinical trial,” (in eng) Clin Cancer Res. 2017;23(14):3510–3519. doi: 10.1158/1078-0432.Ccr-16-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Torabi-Rahvar M, Aghayan H-R, Ahmadbeigi N (2019) Antigen-independent killer cells prepared for adoptive immunotherapy: one source, divergent protocols, diverse nomenclature. J Immunol Methods:112690. 10.1016/j.jim.2019.112690 [DOI] [PubMed]
- 110.Rezvani K, Rouce R, Liu E, Shpall E. Engineering natural killer cells for cancer immunotherapy. Mol Ther. 2017;25(8):1769–1781. doi: 10.1016/j.ymthe.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Campbell KS, Hasegawa J. Natural killer cell biology: an update and future directions. J Allergy Clin Immunol. 2013;132(3):536–544. doi: 10.1016/j.jaci.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Topham NJ, Hewitt EW. “Natural killer cell cytotoxicity: how do they pull the trigger?,” (in eng) Immunology. 2009;128(1):7–15. doi: 10.1111/j.1365-2567.2009.03123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Grimm EA, Mazumder A, Zhang HZ, Rosenberg SA. “Lymphokine-activated killer cell phenomenon. Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes,” (in eng) J Exp Med. 1982;155(6):1823–1841. doi: 10.1084/jem.155.6.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Herberman RB, Hiserodt J, Vujanovic N, Balch C, Lotzova E, Bolhuis R, Golub S, Lanier LL, Phillips JH, Riccardi C, Ritz J, Santoni A, Schmidt RE, Uchida A. “Lymphokine-activated killer cell activity characteristics of effector cells and their progenitors in blood and spleen,” (in eng) Immunol Today. 1987;8(6):178–181. doi: 10.1016/0167-5699(87)90035-1. [DOI] [PubMed] [Google Scholar]
- 115.Law TM, et al. “Phase III randomized trial of interleukin-2 with or without lymphokine-activated killer cells in the treatment of patients with advanced renal cell carcinoma,” (in eng) Cancer. 1995;76(5):824–832. doi: 10.1002/1097-0142(19950901)76:5<824::aid-cncr2820760517>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 116.Lu PH, Negrin RS. “A novel population of expanded human CD3+CD56+ cells derived from T cells with potent in vivo antitumor activity in mice with severe combined immunodeficiency,” (in eng) J Immunol (Baltimore, Md : 1950) 1994;153(4):1687–1696. [PubMed] [Google Scholar]
- 117.Schmidt-Wolf IG, et al. “Phenotypic characterization and identification of effector cells involved in tumor cell recognition of cytokine-induced killer cells,” (in eng) Exp Hematol. 1993;21(13):1673–1679. [PubMed] [Google Scholar]
- 118.Lee JH, et al. “Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma,” (in eng) Gastroenterology. 2015;148(7):1383–91.e6. doi: 10.1053/j.gastro.2015.02.055. [DOI] [PubMed] [Google Scholar]
- 119.Yoon JS, Song BG, Lee JH, Lee HY, Kim SW, Chang Y, Lee YB, Cho EJ, Yu SJ, Sinn DH, Kim YJ, Lee JH, Yoon JH. “Adjuvant cytokine-induced killer cell immunotherapy for hepatocellular carcinoma: a propensity score-matched analysis of real-world data,” (in eng) BMC Cancer. 2019;19(1):523–523. doi: 10.1186/s12885-019-5740-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kong D-S, et al. “Phase III randomized trial of autologous cytokine-induced killer cell immunotherapy for newly diagnosed glioblastoma in Korea,” (in eng) Oncotarget. 2017;8(4):7003–7013. doi: 10.18632/oncotarget.12273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mehta RS, Rezvani K. “Chimeric antigen receptor expressing natural killer cells for the immunotherapy of cancer,” (in eng) Front Immunol. 2018;9:283–283. doi: 10.3389/fimmu.2018.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Curti A, Ruggeri L, D'Addio A, Bontadini A, Dan E, Motta MR, Trabanelli S, Giudice V, Urbani E, Martinelli G, Paolini S, Fruet F, Isidori A, Parisi S, Bandini G, Baccarani M, Velardi A, Lemoli RM. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood. 2011;118(12):3273–3279. doi: 10.1182/blood-2011-01-329508. [DOI] [PubMed] [Google Scholar]
- 123.Rezvani K, Rouce RH (2015) “The application of natural killer cell immunotherapy for the treatment of cancer,” (in English). Review 6(578). 10.3389/fimmu.2015.00578 [DOI] [PMC free article] [PubMed]
- 124.Zhang Q, Zhang H, Ding J, Liu H, Li H, Li H, Lu M, Miao Y, Li L, Zheng J. “Combination therapy with EpCAM-CAR-NK-92 cells and regorafenib against human colorectal cancer models,” (in eng) J Immunol Res. 2018;2018:4263520. doi: 10.1155/2018/4263520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tettamanti S, Marin V, Pizzitola I, Magnani CF, Giordano Attianese GMP, Cribioli E, Maltese F, Galimberti S, Lopez AF, Biondi A, Bonnet D, Biagi E. “Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor,” (in eng) Br J Haematol. 2013;161(3):389–401. doi: 10.1111/bjh.12282. [DOI] [PubMed] [Google Scholar]
- 126.DiNofia AM, Maude SL (2019) Chimeric antigen receptor T-cell therapy clinical results in pediatric and young adult B-ALL. 3(4), p. e279, doi: 10.1097/hs9.0000000000000279 [DOI] [PMC free article] [PubMed]
- 127.Evans AG, Rothberg PG, Burack WR, Huntington SF, Porter DL, Friedberg JW, Liesveld JL. “Evolution to plasmablastic lymphoma evades CD19-directed chimeric antigen receptor T cells,” (in eng) Br J Haematol. 2015;171(2):205–209. doi: 10.1111/bjh.13562. [DOI] [PubMed] [Google Scholar]
- 128.Gardner R, et al. “Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy,” (in eng) Blood. 2016;127(20):2406–2410. doi: 10.1182/blood-2015-08-665547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jacoby E, et al. “CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity,” (in eng) Nat Commun. 2016;7:12320. doi: 10.1038/ncomms12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.T. Worbs, S. I. Hammerschmidt, and R. Förster, "Dendritic cell migration in health and disease," Nat Rev Immunol, Review Article vol. 17, p. 30, 11/28/online 2016, doi: 10.1038/nri.2016.116 [DOI] [PubMed]
- 131.Kantoff PW, et al. “Sipuleucel-T immunotherapy for castration-resistant prostate cancer,” (in eng) N Engl J Med. 2010;363(5):411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 132.Kumar C, Kohli S, Chiliveru S, Bapsy PP, Jain M, Suresh Attili VS, Mohan J, Vaid AK, Sharan B. “A retrospective analysis comparing APCEDEN((R)) dendritic cell immunotherapy with best supportive care in refractory cancer,” (in eng) Immunotherapy. 2017;9(11):889–897. doi: 10.2217/imt-2017-0064. [DOI] [PubMed] [Google Scholar]
- 133.Yang J, Shangguan J, Eresen A, Li Y, Wang J, Zhang Z (2019) Dendritic cells in pancreatic cancer immunotherapy: vaccines and combination immunotherapies. Pathol Res Pract:152691. 10.1016/j.prp.2019.152691 [DOI] [PubMed]
- 134.Lepisto AJ, et al. “A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors,” (in eng) Cancer Ther. 2008;6(B):955–964. [PMC free article] [PubMed] [Google Scholar]
- 135.Miyazawa M, et al. Dendritic cells adenovirally-transduced with full-length mesothelin cDNA elicit mesothelin-specific cytotoxicity against pancreatic cancer cell lines in vitro. Cancer Lett. 2011;305(1):32–39. doi: 10.1016/j.canlet.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 136.Nakamura M, Wada J, Suzuki H, Tanaka M, Katano M, Morisaki T. “Long-term outcome of immunotherapy for patients with refractory pancreatic cancer,” (in eng) Anticancer Res. 2009;29(3):831–836. [PubMed] [Google Scholar]
- 137.Markov O, Oshchepkova A, Mironova N (2019) “Immunotherapy based on dendritic cell-targeted/−derived extracellular vesicles—a novel strategy for enhancement of the anti-tumor immune response,” (in English). Review 10(1152). 10.3389/fphar.2019.01152 [DOI] [PMC free article] [PubMed]
- 138.Ziske C, Etzrodt PE, Eliu AS, Gorschlüter M, Strehl J, Flieger D, Messmer D, Schmitz V, Gonzalez-Carmona MA, Sievers E, Brossart P, Sauerbruch T, Schmidt-Wolf IGH. “Increase of in vivo antitumoral activity by CD40L (CD154) gene transfer into pancreatic tumor cell-dendritic cell hybrids,” (in eng) Pancreas. 2009;38(7):758–765. doi: 10.1097/MPA.0b013e3181ae5e1a. [DOI] [PubMed] [Google Scholar]
- 139.Chen J, Guo X-Z, Li H-Y, Wang D, Shao X-D. Comparison of cytotoxic T lymphocyte responses against pancreatic cancer induced by dendritic cells transfected with total tumor RNA and fusion hybrided with tumor cell. Exp Biol Med. 2015;240(10):1310–1318. doi: 10.1177/1535370215571884. [DOI] [PMC free article] [PubMed] [Google Scholar]