

Answer questions and earn CME
Abbreviations
- A1AT
alpha‐1‐antitrypsin
- A1ATD
alpha‐1‐antitrypsin deficiency
- AASLD
American Association for the Study of Liver Diseases
- AAV
adenovirus‐associated vector
- ALD
alcoholic liver disease
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CFTR
cystic fibrosis transmembrane conductance regulator
- COPD
chronic obstructive pulmonary disease
- ER
endoplasmic reticulum
- ESLD
end‐stage liver disease
- GGT
gamma‐glutamyltransferase
- H&E
hematoxylin and eosin
- HCC
hepatocellular carcinoma
- INR
international normalized ratio
- iPSC
induced pluripotent stem cell
- NAFLD
nonalcoholic fatty liver disease
- NF‐κB
nuclear factor‐κB
- NSAID
nonsteroidal anti‐inflammatory drug
- PAS
periodic acid–Schiff
- siRNA
small interfering RNA
- TGF‐β
transforming growth factor‐β
- UPR
unfolded protein response
Alpha‐1‐Antitrypsin (A1AT) deficiency (A1ATD) is typically discussed in the context of lung disease as a major cause of panacinar emphysema because of impaired inhibition of neutrophil elastase. SERPINA1, the gene encoding A1AT, has an autosomal recessive inheritance with codominant expression. Large numbers of mutations in the gene are associated with lung disease and a subset with liver disease. Mutations in SERPINA1 causing liver disease do so by the formation of harmful aggregates of mutant A1AT protein within hepatocytes, which results in diverse manifestations, from devastating neonatal cholestasis to late‐onset cirrhosis and hepatocellular carcinoma (HCC) in adults. Here, we summarize advances in our understanding and management of A1ATD liver disease with particular focus on the significance of A1ATD heterozygosity and its putative role as a cofactor in common causative factors for liver disease.
Molecular Basis and Pathophysiology
A1AT is a large, 52‐kDa serum glycoprotein abundantly produced by the liver. It is a serine protease inhibitor whose primary function depends on its secretion from the liver and its physiological action in the lungs to prevent excessive tissue destruction by neutrophil elastase. A1AT is also an acute‐phase reactant with a presumed inflammatory role.1
Certain missense mutations in A1AT result in the accumulation of toxic aggregates of misfolded protein in hepatocytes and, consequently, deficiency in the circulation. These A1AT aggregates appear as periodic acid–Schiff (PAS)‐positive, diastase‐resistant inclusions, the pathological hallmark of A1ATD liver disease (Fig. 1, right). The prototype mutation is associated with the misfolded protein pGlu342Lys. It is classified as the PiZ allele based on its distinctive migration on isoelectric focusing, with the normal allele designated PiM. When both mutant PiZ alleles are inherited, the severe homozygous PiZZ form of A1ATD results, compared with the heterozygous PiMZ presentation, which is of mild‐to‐intermediate severity (Tables 1 and 2). Although hundreds of polymorphisms have been identified within the SERPINA1 gene, it is predominantly the Z mutant allele and, to a lesser extent, the S mutant allele that are attributed to clinically significant liver disease.
Figure 1.
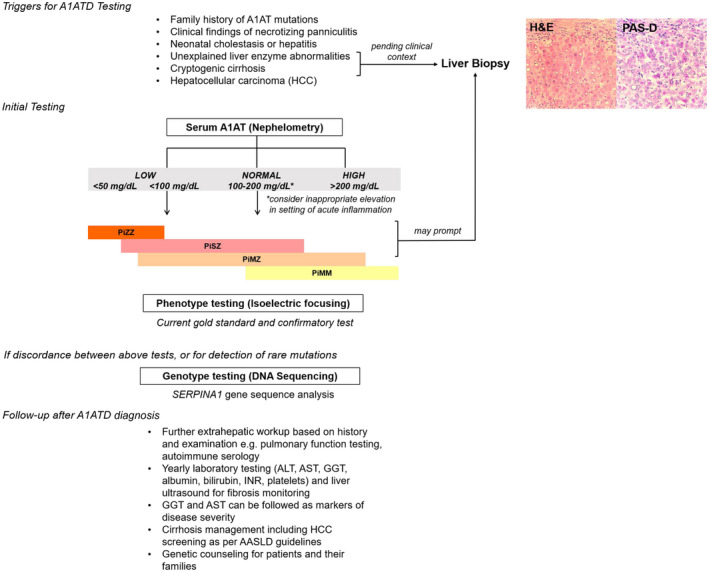
Algorithm for diagnosis of suspected A1ATD in liver disease. Serum testing should be limited to screening because it carries low sensitivity and specificity. Because of its role as an acute‐phase reactant, A1AT can be elevated up to 4‐fold in settings of inflammation, and thus normal levels cannot exclude PiMZ or PiMS heterozygous carriers. Very low levels carry high sensitivity and specificity for deficiency.2, 3 Serum level ranges for different phenotypes are graphically depicted. Phenotype testing is done by isoelectric focusing migration and is the current diagnostic gold standard and should be performed concomitantly with serum testing for confirmation. Increasingly, SERPINA1 genotype testing is performed on dried blood spots, whole blood, or saliva, and can be a useful adjunct to phenotyping results, especially when they may be discordant with serum concentrations or for detection of the rare deficiency alleles. Liver biopsy is not required for diagnosis, although often part of the work‐up for cryptogenic cirrhosis helps to reveal pathognomonic PAS‐positive, diastase‐resistant inclusions. Extrahepatic testing may be warranted and guided by thorough clinical and family history. For follow‐up, once A1ATD is identified, yearly blood work and FibroScan are recommended to screen for fibrosis. In patients with cirrhosis caused by A1ATD, routine laboratory assessment and imaging for HCC should be done according to AASLD guidelines. (Left) Adapted with permission from American Journal of Clinical Pathology.8 Copyright 2012, American Society for Clinical Pathology. (Right) Courtesy Xuchen Zhang, M.D., Ph.D., Department of Pathology, Yale School of Medicine, New Haven, CT.
Table 1.
Genetic Variants of A1ATD Involved in Liver Disease
| Alleles | Mutation | |
|---|---|---|
| Normal | M | |
| Most common mutations | S | Glu264Val |
| Z | Glu342Lys |
Table 2.
Genetic Variants of A1ATD Involved in Liver Disease
| Phenotype/Genotype | Serum A1AT Level | Phenotype Range | Phenotype Modifiers | |
|---|---|---|---|---|
| Normal | PiMM | 100% | ||
| Heterozygote | PiMZ | 60% | Asymptomatic | Age >50 years |
| Cryptogenic cirrhosis | Male sex | |||
| Decompensated liver disease/HCC | Diabetes | |||
| Severe deficiency | PiSZ | 35% | Neonatal | Steatosis (ALD, NAFLD) |
| PiZZ | <15% | Cholestasis | Viral hepatitis | |
| Fulminant hepatitis | Cystic fibrosis (CFTR) | |||
| Adult | COPD | |||
| Cryptogenic cirrhosis | NSAIDs | |||
| Decompensated liver disease/HCC | Febrile episodes | |||
| Infections |
PiZZ A1ATD “loss of function” results in pulmonary manifestations because aggregated mutant A1AT protein cannot be secreted from the endoplasmic reticulum (ER) of the hepatocyte. Because misfolded A1AT is trapped in the hepatocytes, serum levels become low, predisposing to lung disease; this aspect of phenotype can be aggravated by exposures such as smoking. Meanwhile, a “gain‐of‐function” phenotype is exhibited when the rate of accumulation of polymerized mutant A1AT protein overwhelms the hepatocyte’s protective mechanisms of recognizing, degrading, and exporting excess or misfolded proteins. Over time, this dysregulation of ER homeostasis spurs a reactive cascade that can progress clinically to the development of fibrosis, cirrhosis, and HCC4, 5 (Fig. 2).
Figure 2.
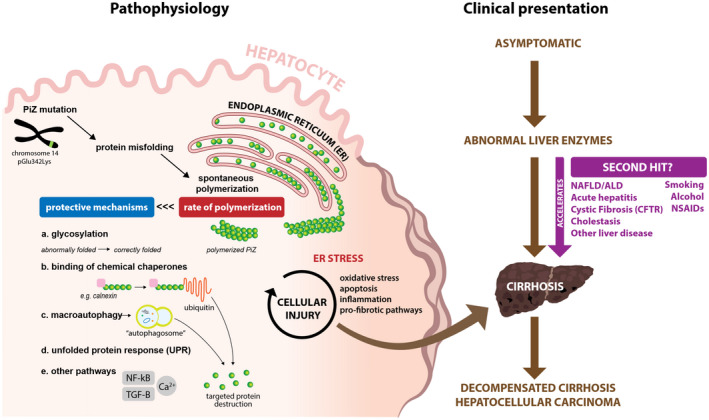
Pathophysiology with correlating clinical presentation of A1ATD in liver disease. Formation of PiZ A1AT starts with the base pair mutation that prompts protein misfolding and polymerization within the ER of the hepatocyte. Protective mechanisms for detecting and destroying mutant protein, such as PiZ A1AT, within the hepatocyte including the following: (a) glycosylation, the normal process of protein modification, which allows time for misfolded proteins to be identified and properly folded; (b) ER‐associated destruction (ERAD), such as binding to chemical chaperones like calnexin, which can target for destruction via ubiquitin‐mediated pathways; (c) formation of lysosome‐fused phagosome that can also target for destruction of mutant protein; (d) UPR, typically another defense mechanism of the cell, but often not triggered in A1ATD because PiZ polymers are not recognized as abnormal1; and (e) many other pathways, including those that are calcium mediated, such as NF‐κB and TGF‐β, also exist. A1ATD is characterized by a rate of polymerization that exceeds these protective mechanisms and leads to unregulated formation, ultimately triggering a cycle of inflammation. Clinical manifestations are variable, and ESLD may be accelerated by the presence of other risk factors.
Clinical Manifestations
The highest prevalence of homozygous PiZZ A1ATD is seen in Northern Europe, where 1/2000 individuals are affected. Because heterozygous PiMZ carriers can also experience development of disease, the at‐risk population is much greater, with an estimated 4 million individuals worldwide carrying any disease‐causing allele.6 Males appear to be affected more than females, and murine studies suggest that hormonal effects contribute to increased A1AT expression and liver injury in male PiZ carriers.7
The natural history of A1ATD liver disease deserves further research; however, like many single‐gene disorders, A1ATD displays marked clinical variability from infancy to adulthood.8 In children, acute presentation with cholestatic liver disease progressing to end‐stage liver disease is recognized. Modifiers that lead a minority of children to follow this rapidly progressive course have not been delineated.9, 10 This contrasts with adults, for whom it is more common to have a silently progressive liver disease for several decades until signs of portal hypertension develop or there is hepatic decompensation including HCC.5 Interestingly, adults may have minimal liver enzyme elevations disproportionate to their severity of liver injury, perhaps reflecting that the mode of liver cell injury in PiZZ A1ATD liver disease is predominantly driven by apoptosis.1
It is well observed that patients with A1ATD with liver disease, especially ESLD, typically do not have emphysema. Certainly, PiZZ A1ATD can lead to both obstructive lung disease and liver disease; however, because the pathogenesis differs for each organ disease, it becomes the patient’s risk factors and disease modifiers that trigger one over the other.11 A1ATD patients with emphysema and a significant smoking history may experience mortality before experiencing signs of liver disease. Regardless, patients with A1ATD liver disease are strictly counseled on smoking cessation, especially as part of candidacy for transplantation.
Diagnosis
A1ATD remains a disappointedly underreported, underdiagnosed condition often with a diagnostic delay of 5 to 10 years, by which time patients already have ESLD. Studies suggest that more than 80% to 90% of individuals with A1ATD are unaware of their condition because of lack of clinical symptoms or misdiagnosis with other cirrhosis causes, such as alcoholic liver disease (ALD) or nonalcoholic fatty liver disease (NAFLD).6
Figure 1 summarizes the current accepted testing algorithm for A1ATD liver disease.12, 13
Following diagnosis, patients found to have A1ATD are largely managed supportively with close monitoring for progression of liver disease (Fig. 1). In comparison with A1ATD lung disease for which intravenous enzyme replacement therapy is available, treatments to correct misfolding in liver disease are currently the subject of several clinical trials (Tables 1 and 2), but they are not yet commercially available or shown to improve outcomes.
Routine screening for fibrosis, as well as HCC, is recommended, although it remains unclear how much the risk for HCC development differs in A1ATD from other etiologies of liver disease. In the original Swedish cohort studies by Eriksson et al.,14 the relative risk was 5, with 28% of autopsy livers revealing HCC, much more than would be expected for cirrhosis alone. Further studies have presented conflicting data reporting both increased and decreased risk for HCC.8, 14 As a general observation, however, inherited metabolic diseases do carry an increased risk for HCC,15 which overall supports the need for surveillance.
Liver transplantation remains the sole curative option for A1ATD liver disease, with the healthy donor liver both eliminating the mutant phenotype and restoring normal circulating levels of A1AT. Reassuringly, patients transplanted for A1ATD, who represent 1% of all liver transplants performed, have excellent 5‐year graft and patient survival rates.1
Because of the lack of medical treatment options, there is an emphasis on efforts to prevent progression of A1ATD‐related liver injury by reduction of modifiable risk factors. These include weight loss, cessation of tobacco and alcohol use, avoidance of nonsteroidal anti‐inflammatory drugs (NSAIDs; because their properties may increase synthesis of mutant A1AT protein in the liver), prompt treatment of febrile illness, and up‐to‐date hepatitis vaccinations.16
Emerging Treatments
The advances in delineation of disease mechanisms have revealed promising molecular therapeutic targets. Disease models for A1ATD include human PiZZ transgenic mice and Caenorhabditis elegans expressing Z allele, which have been informative for understanding disease mechanisms and screening molecules as potential therapies. Although most of these potential treatments are in early stages of clinical development, some are using established US Food and Drug Administration–approved medications that may be repurposed for use in A1ATD and could, therefore, bypass the laborious regulatory process for new drugs. One such example is carbamazepine, which appears to enhance autophagy and clearance of mutant Z protein aggregates. It is currently in clinical trial. Another promising approach is RNA interference therapy to reduce production of mutant A1AT.17
Table 3 summarizes the major target pathways for treatment, therapies under study, and their potential limitations.
Table 3.
Summary of A1ATD Treatment Targets Under Investigation
| Target Pathway | Treatment | Studies | Current Status | Limitations |
|---|---|---|---|---|
| Prevent polymerization | siRNA to downregulate endogenous Z mutant A1AT* | Cruz et al. 200723 | Clinical trials phases 1 and 2 (Alnylam Pharmaceuticals and Arrowhead Research Corporation) | Will suppress all A1AT production and require lifelong supplementation or augmentation therapy unless concurrent gene therapy is done to transfect patients with normal A1AT |
| Guo et al. 201424 | ||||
| AAV‐gene vector transfer of normal A1AT to muscle cells* | Multiple studies | Clinical trials phases 1 and 2 | ||
| Autologous grafting of iPSC from Pi*ZZ individuals reprogrammed using CRISPR technology to correct Z mutation | Rashid et al. 201025 | In vitro studies using iPSCs | ||
| Yusa et al. 201126 | ||||
| Tafaleng et al. 201527 | ||||
| Wilson et al. 201528 | ||||
| Detect and improve protein misfolding | Histone deacetylase 7 inhibitor suberoylanilide hydroxamic acid can restore secretion of Z‐type A1AT from epithelial cells | Bouchecareilh et al. 201229 | In vitro study | Difficulties creating medicine molecules for use in animal models |
| Use of chemical chaperones stabilizes native A1AT and combats misfolded proteins | 4‐Phenylbutyric acid* | Burrows et al. 200030 | Clinical trials phase 2 | |
| Teckman et al. 200431 | ||||
| Trimethylamine N‐oxide | Devlin et al. 200132 | In vitro study | ||
| Increase degradation via autophagy | Carbamazepine* | Hidvegi et al. 201033 | Clinical trials phase 2, PiZZ‐associated cirrhosis | Lowest effect doses needed to impact A1ATD exceed safety profiles for humans |
| Bile acid 24‐norursodeoxycholic acid | Tang et al. 201634 | In vivo study in PiZ mice | ||
| Sirolimus | Kaushal et al. 201035 | In vivo study in PiZ mice; appears to be more effective when given in weekly doses rather than daily | ||
| Ezetimibe | Yamamura et al. 201436 | In vitro study | ||
| Other agents: lithium, valproic acid, pimozide, fluphenazine, fluspirilene, cantharidin, tamoxifen, glucosamine, N‐acetylglucosamine | Gosai et al. 201037 | In vivo study in C. elegans | ||
| Tat‐beclin 1 peptide | Mallya et al. 200738 | |||
| Gene transfer of transcription factor TFEB using adenovirus‐mediated approaches | Pastore et al. 201339 | In vivo study in PiZ mice | ||
| Increase clearance of accumulated protein | Small‐molecule therapy using bile duct ligation | Khan et al. 201740 | In vivo study in PiZ mice | |
| Glucosidase inhibitors: castanospermine | ||||
| Mannosidase inhibitors | ||||
| Reducing hepatic injury/inflammation | Cyclosporin A reduces mitochondrial injury | Teckman et al. 200441 | In vitro study with HeLa cells | |
| In vivo study in PiZ mice | ||||
| Immunosuppressants (cyclophosphamide, cyclosporine) with variable results | Multiple studies |
Treatments undergoing clinical trial.
Relevance in Common Liver Diseases
A1ATD has gained recognition as a comodifier to other liver diseases. A longitudinal study by Tanash and Piitulainen8 found that risk factors for progression of A1ATD liver disease included male sex, age older than 50 years, viral hepatitis, and diabetes. Recent studies have also linked the PiMZ phenotype to a higher risk for development of cirrhosis in patients with NAFLD or chronic alcohol use,18 as well as cystic fibrosis.19 Perlmutter et al. were among the first to recognize from retrospective studies that a large proportion of patients transplanted for A1ATD were heterozygous and appeared to have a “second hit” that accelerated progression to ESLD.20 Putative modifiers of A1ATD liver disease include single‐nucleotide polymorphisms in SERPINA1 and in genes involved in the unfolded protein response (UPR) to control accumulation of mutant misfolded protein and environmental factors. In a 2018 study by Schaefer et al.20, the PiMZ genotype was found to be an independent risk factor for advanced cirrhosis, decompensation by hepatic encephalopathy, and ascites, as well as a higher risk factor for death and need for transplantation. Although the mechanisms behind these associations are not well understood, the additive effect of multiple stressors on the hepatocellular system are likely contributing factors for increased inflammation and cell death. Therefore, A1ATD is emerging as a genetic modifier that predisposes to clinically significant liver disease akin to the role of Patatin‐like phospholipase domain‐containing protein 3 (PNPLA3) polymorphism in NAFLD and alcoholic liver disease (ALD). Given the high prevalence of the PiZ carrier state, identification of individuals and appropriate genetic counseling should be prioritized and offered.
Future Directions
The precise mechanisms underlying the variability in clinical presentations of patients with A1ATD have not been delineated. To date, no studies exist on the liver transplant outcomes for recipients with A1AT heterozygosity. More robust studies are also needed to confirm the degree of risk associated with the PiMZ phenotype and faster development of cirrhosis. A better definition of the natural history of A1ATD is also needed and would be beneficial in the pursuit of new detection strategies and biomarkers for the disease. Certainly, as established treatments emerge for A1ATD, its early and accurate diagnosis will become more of a priority. Furthermore, an in‐depth understanding of the variants of A1ATD will facilitate precision medicine for individuals not only with this genetic disorder but with cirrhosis from other causative factors.
Acknowledgment
The authors thank Dr. Xuchen Zhang for providing pathology images included in Fig. 1 and Mikko Sallinen for significant contributions to Fig. 2. P.N. would also like to thank Dr. Kymberly D. Watt for serving as a longitudinal mentor, as well as a mentor for the 2018 Emerging Liver Scholars program from which stemmed the invitation for this submission.
Potential conflict of interest: Nothing to report.
References
- 1. Greene CM, Marciniak SJ, Teckman J, et al. α1‐Antitrypsin deficiency. Nat Rev Dis Primers 2016;2:16051. [DOI] [PubMed] [Google Scholar]
- 2. Corda L, Bertella E, Pini L, et al. Diagnostic flow chart for targeted detection of alpha1‐antitrypsin deficiency. Respir Med 2006;100:463‐470. [DOI] [PubMed] [Google Scholar]
- 3. Greulich T, Averyanov A, Borsa L, et al. European screening for alpha1‐antitrypsin deficiency in subjects with lung disease. Clin Respir J 2017;11:90‐97. [DOI] [PubMed] [Google Scholar]
- 4. Fairbanks KD, Tavill AS. Liver disease in alpha 1‐antitrypsin deficiency: a review. Am J Gastroenterol 2008;103:2136‐2141; quiz 42. [DOI] [PubMed] [Google Scholar]
- 5. Chu AS, Perlmutter DH, Wang Y. Capitalizing on the autophagic response for treatment of liver disease caused by alpha‐1‐antitrypsin deficiency and other genetic diseases. Biomed Res Int 2014;2014:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Serres F, Blanco I. Role of alpha‐1 antitrypsin in human health and disease. J Intern Med 2014;276:311‐335. [DOI] [PubMed] [Google Scholar]
- 7. Mitchell EL, Khan Z. Liver disease in alpha‐1 antitrypsin deficiency: current approaches and future directions. Curr Pathobiol Rep 2017;5:243‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tanash HA, Piitulainen E. Liver disease in adults with severe alpha‐1‐antitrypsin deficiency. J Gastroenterol 2019;54:541‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sveger T. Liver disease in alpha1‐antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 1976;294:1316‐1321. [DOI] [PubMed] [Google Scholar]
- 10. Hadzic N. Therapeutic Options in Alpha‐1 Antitrypsin Deficiency: Liver Transplantation. Methods Mol Biol 2017;1639:263‐265. [DOI] [PubMed] [Google Scholar]
- 11. Tomashefski JF Jr, Crystal RG, Wiedemann HP, et al. Alpha 1‐antitrypsin deficiency registry study G. The bronchopulmonary pathology of alpha‐1 antitrypsin (AAT) deficiency: findings of the Death Review Committee of the national registry for individuals with Severe Deficiency of Alpha‐1 Antitrypsin. Hum Pathol 2004;35:1452‐1461. [DOI] [PubMed] [Google Scholar]
- 12. Donato LJ, Jenkins SM, Smith C, et al. Reference and interpretive ranges for alpha(1)‐antitrypsin quantitation by phenotype in adult and pediatric populations. Am J Clin Pathol 2012;138:398‐405. [DOI] [PubMed] [Google Scholar]
- 13. Silverman EK, Sandhaus RA. Clinical practice. Alpha1‐antitrypsin deficiency. N Engl J Med 2009;360:2749‐2757. [DOI] [PubMed] [Google Scholar]
- 14. Eriksson S. Alpha 1‐antitrypsin deficiency and liver cirrhosis in adults. An analysis of 35 Swedish autopsied cases. Acta Med Scand 1987;221:461‐467. [PubMed] [Google Scholar]
- 15. Antoury C, Lopez R, Zein N, et al. Alpha‐1 antitrypsin deficiency and the risk of hepatocellular carcinoma in end‐stage liver disease. World J Hepatol 2015;7:1427‐1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Erez A, Shchelochkov OA, Plon SE, et al. Insights into the pathogenesis and treatment of cancer from inborn errors of metabolism. Am J Hum Genet 2011;88:402‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kandregula CAB, Smilin Bell Aseervatham G, Bentley GT, et al. Alpha‐1 antitrypsin: associated diseases and therapeutic uses. Clin Chim Acta 2016;459:109‐116. [DOI] [PubMed] [Google Scholar]
- 18. Teckman JH. Emerging concepts and human trials in alpha‐1‐antitrypsin deficiency liver disease. Semin Liver Dis 2017;37:152‐158. [DOI] [PubMed] [Google Scholar]
- 19. Strnad P, Buch S, Hamesch K, et al. Heterozygous carriage of the alpha1‐antitrypsin Pi*Z variant increases the risk to develop liver cirrhosis. Gut 2019;68:1099‐1107. [DOI] [PubMed] [Google Scholar]
- 20. Bartlett JR, Friedman KJ, Ling SC, et al. Genetic modifiers of liver disease in cystic fibrosis. JAMA 2009;302:1076‐1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chu AS, Chopra KB, Perlmutter DH. Is severe progressive liver disease caused by alpha‐1‐antitrypsin deficiency more common in children or adults?. Liver Transpl 2016;22:886‐894. [DOI] [PubMed] [Google Scholar]
- 22. Schaefer B, Mandorfer M, Viveiros A, et al. Heterozygosity for the alpha‐1‐antitrypsin Z allele in cirrhosis is associated with more advanced disease. Liver Transpl 2018;24:744‐751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cruz PE, Mueller C, Cossette TL, et al. In vivo post‐transcriptional gene silencing of alpha‐1 antitrypsin by adeno‐associated virus vectors expressing siRNA. Lab Invest 2007;87:893‐902. [DOI] [PubMed] [Google Scholar]
- 24. Guo S, Booten SL, Aghajan M, et al. Antisense oligonucleotide treatment ameliorates alpha‐1 antitrypsin‐related liver disease in mice. J Clin Invest 2014;124:251‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rashid ST, Corbineau S, Hannan N, et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J Clin Invest 2010;120:3127‐3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yusa K, Rashid ST, Strick‐Marchand H, et al. Targeted gene correction of alpha1‐antitrypsin deficiency in induced pluripotent stem cells. Nature 2011;478:391‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tafaleng EN, Chakraborty S, Han B, et al. Induced pluripotent stem cells model personalized variations in liver disease resulting from alpha1‐antitrypsin deficiency. Hepatology 2015;62:147‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilson AA, Ying L, Liesa M, et al. Emergence of a stage‐dependent human liver disease signature with directed differentiation of alpha‐1 antitrypsin‐deficient iPS cells. Stem Cell Reports 2015;4:873‐885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bouchecareilh M, Hutt DM, Szajner P, et al. Histone deacetylase inhibitor (HDACi) suberoylanilide hydroxamic acid (SAHA)‐mediated correction of alpha1‐antitrypsin deficiency. J Biol Chem 2012;287:38265‐38278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant alpha 1‐antitrypsin (alpha 1‐AT) Z: a potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1‐AT deficiency. Proc Natl Acad Sci U S A 2000;97:1796‐1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Teckman JH. Lack of effect of oral 4‐phenylbutyrate on serum alpha‐1‐antitrypsin in patients with alpha‐1‐antitrypsin deficiency: a preliminary study. J Pediatr Gastroenterol Nutr 2004;39:34‐37. [DOI] [PubMed] [Google Scholar]
- 32. Devlin GL, Parfrey H, Tew DJ, et al. Prevention of polymerization of M and Z alpha1‐Antitrypsin (alpha1‐AT) with trimethylamine N‐oxide. Implications for the treatment of alpha1‐at deficiency. Am J Respir Cell Mol Biol 2001;24:727‐732. [DOI] [PubMed] [Google Scholar]
- 33. Hidvegi T, Ewing M, Hale P, et al. An autophagy‐enhancing drug promotes degradation of mutant alpha1‐antitrypsin Z and reduces hepatic fibrosis. Science 2010;329:229‐232. [DOI] [PubMed] [Google Scholar]
- 34. Tang Y, Fickert P, Trauner M, et al. Autophagy induced by exogenous bile acids is therapeutic in a model of alpha‐1‐AT deficiency liver disease. Am J Physiol Gastrointest Liver Physiol 2016;311:G156‐G165. [DOI] [PubMed] [Google Scholar]
- 35. Kaushal S, Annamali M, Blomenkamp K, et al. Rapamycin reduces intrahepatic alpha‐1‐antitrypsin mutant Z protein polymers and liver injury in a mouse model. Exp Biol Med (Maywood) 2010;235:700‐709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yamamura T, Ohsaki Y, Suzuki M, et al. Inhibition of Niemann‐Pick‐type C1‐like1 by ezetimibe activates autophagy in human hepatocytes and reduces mutant alpha1‐antitrypsin Z deposition. Hepatology 2014;59:1591‐1599. [DOI] [PubMed] [Google Scholar]
- 37. Gosai SJ, Kwak JH, Luke CJ, et al. Automated high‐content live animal drug screening using C. elegans expressing the aggregation prone serpin alpha1‐antitrypsin Z. PLoS One 2010;5:e15460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mallya M, Phillips RL, Saldanha SA, et al. Small molecules block the polymerization of Z alpha1‐antitrypsin and increase the clearance of intracellular aggregates. J Med Chem 2007;50:5357‐5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pastore N, Ballabio A, Brunetti‐Pierri N. Autophagy master regulator TFEB induces clearance of toxic SERPINA1/alpha‐1‐antitrypsin polymers. Autophagy 2013;9:1094‐1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Khan Z, Yokota S, Ono Y, et al. Bile Duct Ligation Induces ATZ Globule Clearance in a Mouse Model of alpha‐1 Antitrypsin Deficiency. Gene Expr 2017;17:115‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Teckman JH, An JK, Blomenkamp K, et al. Mitochondrial autophagy and injury in the liver in alpha 1‐antitrypsin deficiency. Am J Physiol Gastrointest Liver Physiol 2004;286:G851‐G862. [DOI] [PubMed] [Google Scholar]