

Abstract
Protein glycosylation, resulting from glycosyl transferase reactions under complex control in the secretory pathway, consists of a distribution of related glycoforms at each glycosylation site. Because the biosynthetic substrate concentration and transport rates depend on architecture and other aspects of cellular phenotypes, site-specific glycosylation cannot be predicted accurately from genomic, transcriptomic, or proteomic information. Rather, it is necessary to quantify glycosylation at each protein site and how this changes among a sample cohort to provide information about disease mechanisms. At present, mature mass spectrometry-based methods allow for qualitative assignment of the glycan composition and glycosylation site of singly glycosylated proteolytic peptides. To make such quantitative comparisons, it is necessary to sample the glycosylation distribution with sufficient coverage and accuracy for confident assessment of the glycosylation changes that occur in the biological cohort. In this Perspective, we discuss the unmet needs for mass spectrometry acquisition methods and bioinformatics for the confident comparison of protein site-specific glycosylation among sample cohorts.
Graphical Abstract
Approximately 30% of the vertebrate transcriptome passes through the secretory pathway, the majority of which becomes glycosylated.1 Glycoproteins located on the plasma membrane, in the pericellular matrix and extracellular matrix, define the cellular antigenicity and the microenvironments in which vertebrate biology occurs. Numerous protein–glycan binding interactions govern multicellular phenotypes and host–host and host–pathogen interactions.2 Micro- and macroheterogeneous protein and lipid glycosylation enables long-lived multicellular species to survive in the presence of rapidly evolving pathogens.3 In addition, glycoproteins display accelerated evolution during vertebrate radiation, driven by the need to elaborate the protein interaction properties of plasma membranes and extracellular matrices.4 Therefore, to understand the roles of glycoproteins in the numerous disease mechanisms that depend on glycan–protein binding, it is necessary to quantify the distribution of glycosylation that resides at each glycosite. Beyond this, it is necessary to define the molecular populations that tune the functional properties of glycoproteins that exist in a given biological context. Viewed simply, the theoretical numbers of such glycosylated proteoforms are multiples of the number of glycans that exist at each protein site. However, for proteins with several sites of glycosylation, this results in theoretical numbers of glycoprotein structural variants that are too large to exist biologically.5 It is therefore important to define the subset of such glycosylated proteoforms that exist among biological cohorts.
We address the challenge of comparing how glycoprotein proteoform populations differ among sample cohorts in this Perspective. Mass spectrometry-based methods can now identify the glycan composition and glycosylation site for singly glycosylated peptides. Methods for acquiring MS data to quantify site-specific glycosylation with sufficient confidence for identification of changes that occur during disease mechanisms are now emerging.
SUMMARY OF LC-TANDEM MS METHODS FOR ASSIGNMENT OF SITE-SPECIFIC GLYCOSYLATION
Mass spectrometry-based omics studies rely on statistical analysis to identify the most confident data assignment. This requires careful consideration of rates of true and false identifications and sources of experimental error. The glycoproteome is orders of magnitude more complex than the proteome, owing to the non-template-driven glycan biosynthetic pathways. Given this complexity, what is the limit of sample complexity that can reasonably be analyzed with the present technology? We summarize the state of the glycoproteomics field with this question in mind.
Discovery-based bottom-up proteomics often uses data-dependent acquisition (DDA) whereby ions are selected automatically during data acquisition based on abundance for subsequent tandem MS acquisition. In such experiments, a finite instrument acquisition speed limits quantitative reproducibility due to the stochastic nature of the ion selection. Proteomics database searching can be used to identify small post-translational modifications, such as those that occur most often in relation to intracellular processes, including phosphorylation, acetylation, methylation, and acylation.6 It is not well suited, however, to analysis of glycosylated peptides for a few reasons. The glycan undergoes preferential collisional dissociation, resulting in the need for specialized MS acquisition methods, including the use of collision energy ranges higher than those for unmodified peptides of a similar amino acid composition. For complex glycans, the glycan size is often >1000 Da and it is heterogeneous. Thus, glycosylation heterogeneity divides the total ion signal, necessitating the enrichment of the glycosylated molecules. Even with enrichment, the high number of glycoforms at a given site (often >30) that elute within a small retention time window during an LC-MS experiment makes it impossible to select all precursors for tandem MS quantification of the complete glycopeptide distribution. Such heterogeneity also limits the number of precursor ions that can be quantified using tandem MS inclusion lists. Therefore, some of the glycopeptide glycoforms cannot be identified unambiguously in the absence of a tandem MS scan.
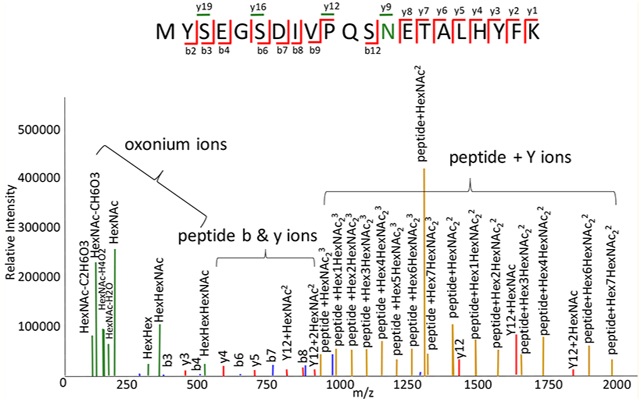
During a tandem MS event, glycopeptides dissociate to form low-molecular weight oxonium ions, high-molecular weight ions from loss of saccharide units from the precursor ions (here termed peptide + Y ions), and peptide backbone product ions (Figure 1).
Figure 1.
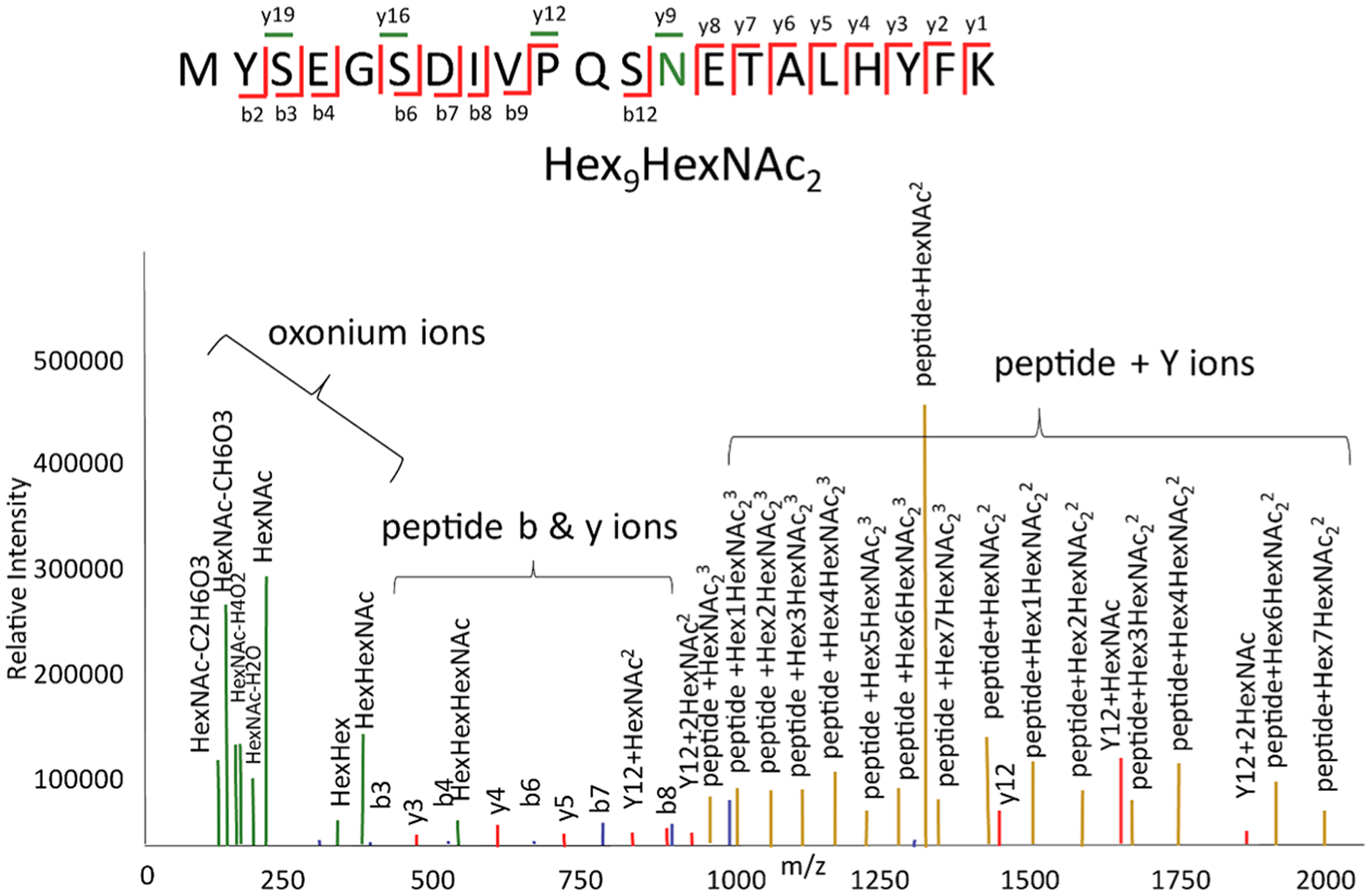
Types of glycopeptide product ions produced by collisional dissociation. Oxonium ions are colored green, peptide bn ions blue, yn ions red, and peptide + Y ions goldenrod. In the fragmentation diagram, the glycosylated peptide residue is colored green. Peptide yn ions that are glycosylated are colored green.
The relative abundances of these product ions depend on the analyzer and dissociation method. For example, with beam-type collisional dissociation, including so-called higher-energy dissociation (HCD), peptide backbone product ions are observed in relatively low abundances provided that sufficient collision energy is used.7–9 When resonant collisional excitation is used, for example with ion traps, product ions formed from the precursor are cooled rapidly and peptide backbone dissociation product ions may not be observed. For electron transfer dissociation-based methods, it is typically necessary to use supplemental vibrational excitation to mitigate nondissociative electron transfer, whereby product ions are held together by noncovalent forces in the gas phase. Examples are electron transfer higher-energy collisional dissociation (EThcD)10–12 and activated ion electron transfer dissociation (AI-ETD).13,14 Thus, although ETD cleaves primarily the peptide backbone, there is a degree of dissociation of the glycan from vibrational excitation when using it for analysis of glycopeptides.
The presence of oxonium ions is diagnostic for a glycosylated peptide. The abundances of peptide + Y ions depend on the collision energy. If the collision energy is relatively low, then ions resulting from loss of one or a few saccharide units will be abundant. If the collision energy is relatively high, then peptide + Y ions corresponding to peptide plus mono- to trisaccharides may be abundant.15,16 The tandem mass spectra that result in the most confident glycopeptide identifications include all three types of product ions, being able to accurately sequence the peptide and characterize the glycan composition. To produce such product ion patterns for the highest percentage of precursor ions, it is necessary to use a stepped collision energy.7,9,17,18 The obvious disadvantage to this approach is that it reduces the number of precursor ions that can be analyzed in a given time window.
For glycopeptides, the most confident assignments come from tandem mass spectra with product ions with resolved isotopic clusters due to the propensity for product ions to be multiply charged. Such high-resolution LC-tandem mass spectral data can be acquired using QTOF or Orbitrap analyzers. Note that many proteomics data sets acquired using ion trap–Orbitrap hybrid instruments employ the low-resolution ion trap for product ion mass spectra. Such low-resolution tandem mass spectra yield low-confidence glycopeptide assignments because the product ion compositions cannot be assigned from a list of theoretical structures with high accuracy.15,16,19–21 The advantage of high-resolution glycopeptide LC-tandem MS analysis is that the same chromatography columns, mobile phases, and collisional tandem MS instruments that were used widely in proteomics laboratories can be used for glycopeptides.
Activated electron dissociation results in preferential dissociation of the peptide backbone with minimal dissociation to glycopeptide glycans.22 Electron transfer dissociation has undergone several cycles of engineering improvements on commercial mass spectrometry systems and has been applied to biological glycoscience studies.10,11,23,24 The advantage to ETD is that it is capable of producing peptide backbone dissociation for larger peptides than for collisional dissociation. Some commercial instruments are capable of both collisional and electron transfer dissociation in the same LC-MS run. Electron transfer dissociation requires considerably lower scan frequencies than does collisional dissociation, with the result that ETD should be used in LC-MS workflows, for which efficient use of analyzer time is paramount, only for precursor ions that do not dissociate effectively using HCD. Newer instruments can select among activation methods and parameters based upon precursor mass and charge state, which can be used to perform ETD only on sufficiently large and charged precursors for a subset of samples with multiply glycosylated peptides.25 Not all multiply glycosylated peptides are substantially larger than the singly glycosylated peptides in a sample, particularly when considering O-glycopeptides, which can make it difficult to determine which precursors to dissociate with ETD on-the-fly for such samples.
Collisional dissociation typically dissociates the glycan and therefore does not distinguish sites of glycosylation on peptides with multiple glycosylation sites. ETD methods are more suited for this purpose. One disadvantage to ETD is that positive charge states are diminished by the addition of an electron, with the result that small glycopeptides may not carry sufficient charge for efficient dissociation. Another problem is that ETD products may be observed as gas phase pairs with m/z values equivalent to the loss of charge from the precursor, a phenomenon known as ETnoD. Dissociating such ions requires a supplemental dissociation step using collisional dissociation, known as electron transfer high-energy dissociation (EThcD), a method that is available on a commercial instrument. Another approach requires supplemental excitation with an infrared laser, which requires a customized instrument.13
QUANTIFICATION OF SITE-SPECIFIC GLYCOSYLATION
Multiple-reaction monitoring (MRM), the most sensitive method for quantification of compounds using mass spectrometry, requires use of stable isotope-labeled internal standards for absolute quantification. For best practices in proteomics, three tiers of targeted quantitative assays have been defined.26 For exploratory studies (tier 3), no reference standards are used and the precision is similar to that of label free discovery proteomics. For research use quantification (tier 2), limited reference standards are used and a moderately high precision is achieved with a 20–35% coefficient of variation. For clinical bioanalysis (tier 1), reference standards are used for every peptide quantified and a high precision is achieved (<20–25% CV). For glycopeptides, there are few such standards available. Thus, MRM methods generally produce relative quantification that is useful for following biological changes to glycosylation.27 Mechref et al. introduced the approach of using oxonium ion MRM transitions for glycopeptides, simplifying the task of identifying transitions at the possible expense of specificity.28 Goldman et al. used MRM to quantify plasma haptoglobin glycopeptides related to liver disease.29,30 These authors purified the haptoglobin to alleviate concerns about the specificity of the MRM transitions in a complex glycoprotein background. They also used neuraminidases to limit the glycan heterogeneity. Lebrilla et al. have used MRM to quantify 64 immunoglobulin glycopeptides from serum without enrichment.31
Parallel reaction monitoring (PRM) has the advantage that a full-range tandem mass spectrum is acquired for every isolated precursor ion, allowing users to optimize the specificity of the assay. Larsson et al. quantified an amyloid β-glycopeptide modified with a core 1 (Hex-HexNAc) disaccharide using a glycopeptide standard synthesized for this purpose.32 Yoo et al. used a glycopeptide PRM method to determine extent of fucosylation of α-fetoprotein immunoprecipitated from human serum as a biomarker for hepatocellular carcinoma.33 These authors extended this approach to analyze glycopeptides from several glycoproteins enriched using multiplexed immunoprecipitations.34
While data-independent acquisition (DIA) allows tandem MS of precursor ions without the need for ion isolation, spectral libraries are used widely for interpretation of overlapping patterns of precursor and product ions in DIA data sets.35 Spectral clustering36 and spectral library searching use experimentally collected mass spectra identified by a separate mass spectrometry experiment.37 This method works well for peptides, and for small molecules where there are no canonical fragmentation pathways that can be generalized to all molecules.38 It has also been used for glycopeptides in GPQuest39 and SweetNET.40 Spectral libraries lack portability, as the library spectra are specific to the collision energies used during acquisition. Thus, this approach becomes less sensitive as the acquisition conditions differ from those from the original library construction. This is particularly important for glycopeptides, for which the relative abundances of oxonium ions, peptide + glycan ions, and peptide backbone dissociation ions vary considerably as collision energy is increased. Fortunately, the use of stepped collision energies helps mitigate this variability.9 Spectral libraries have seen extensions for clustering both identified and unidentified spectra41 and for MRM identification and quantification with SWATH.42
Tandem MS results in similar monosaccharide residue losses and oxonium ions for the series glycoforms present for a given glycopeptide. Given this, the extent to which DIA will generate acceptable specificity for quantification of such series of glycopeptide glycoforms is not clear. DIA methods have been used for analysis of immunoglobulin G glycoforms, for which there is very limited glycan heterogeneity due to steric constraints.43 Ye et al. also showed DIA with in silico-augmented libraries may be tractable for sufficiently small glycans with limited heterogeneity, as found for common smaller O-GalNAc glycans, though requiring additional metrics to calibrate identification confidence.44 Molloy et al. compared PRM and DIA for analysis of targeted glycopeptides from blood plasma.45 They reported that PRM was effective for quantification but limited by duty cycle. They analyzed DIA results using a theoretical targeted glycopeptide database rather than a spectral library. While this quantified more glycopeptides than with the PRM method, the specificity whereby targeted glycopeptides can be identified in the presence of complex and uncharacterized plasma glycoproteins was not defined. There is a general challenge in achieving appropriate fragmentation for molecules spanning several neutral mass ranges falling within the same wide isolation window in DIA, where a single collision energy is applied to the entire window.46,47 This incomplete fragmentation exacerbates the problem discussed earlier in acquiring high-quality assignments.
While DIA and targeted methods borrowed from proteomics may be applicable to glycoproteomics samples, they still suffer from the increased complexity of the sample, and the greater redundancy among product ions of related glycoforms. Chromatogram libraries are particularly vulnerable to corruption by co-isolation of adjacent glycoforms overlapping in elution time, and the problem is resistant to the solution for small modifications.48 However, it has been shown that relative elution time shifts are induced by differences in monosaccharide composition. Recently, tools have been developed to exploit these relationships49–52 for estimating confident identifications of lower-quality structural assignments relying on only precursor mass and apex elution time. This method shows potential for overcoming some of the limitations of DIA for glycopeptides.
In summary, DIA has the potential to improve the ability to quantify targeted peptides, but its specificity for quantification of targeted glycopeptides in complex biological samples with or without spectral libraries remains undefined.
QUANTIFICATION OF GLYCOPEPTIDES USING ISOBARIC LABELS
Lee et al. demonstrated that TMT-labeled N-linked glycopeptides produce HCD reporter ions.53 They used this approach to quantify vitronectin N-linked glycopeptides as candidate markers for hepatocellular carcinoma and identified glycoforms with significantly increased levels in diseased plasma. In further work, this group used their approach for simultaneous qualitative and quantitative comparison of site-specific N-glycopeptide glycoforms.54 Zhang et al. used TMT tagging to quantify fucosylated glycopeptides enriched using lectin affinity chromatography as part of a study of changes in glycosylation associated with aggressive prostate cancer cell lines.55 For these studies, proteins were extracted from cells, digested with trypsin, enriched using lectin chromatography, labeled with TMT, and then analyzed using LC-HCD-MS. This group also developed a workflow for simultaneous identification of glycoand phosphopeptides co-isolated from breast tumor patient-derived xenografts.56 Palmisano et al. mapped N- and O-glycoproteins from urine to discriminate prostate cancer from benign prostatic hyperplasia.57 For these studies, the total extracted proteins were digested and labeled with TMT10plex. Glycopeptides were enriched using a combination of hydro-philic interaction and titanium dioxide solid phase extraction and quantified using LC-HCD-MS.
Viner et al. demonstrated that ETD produces four distinct isobaric product ions when using the TMT-6 plex amino-specific label.58 They then demonstrated quantification of OGalNAc-modified glycopeptides from crystallin protein tryptic digests based on the ETD-produced reporter ions. Such peptides undergo dissociation of the O-GalNAc residue during HCD, making it impossible to quantify with confident identification of the glycosylation site. Ye et al. compared glycosylation among 10 lots of human chorionic gonadotropin drug products using a combination of HCD and ETD.59 The protein tryptic digests were labeled with TMT10plex using high-resolution LC-MS with alternating HCD and ETD methods. They used partial least-squares discriminant analysis to identify the glycopeptides that differed among the protein lots.
THE QUESTION OF APPROPRIATE SAMPLE COMPLEXITY IN GLYCOPROTEOMICS: HOW COMPLEX IS TOO COMPLEX?
As shown in Figure 2, assignment of glycopeptides from tandem mass spectra entails defining a search space consisting of the theoretical protein sequence, the peptides generated therefrom, and theoretical glycan compositions.15,16 The most rigorous approach is to measure the proteome and glycome of the sample and use the data to build a glycopeptide search space. A typical glycoprotein glycosite has ≥30 glycoforms, vastly increasing the complexity of the search space relative to that of a proteome with no glycosylation sites. While this effect is appreciated generally, there is no consensus regarding the use of confidence measures to ensure that glycopeptide assignments are correct in a given biological background. We demonstrated that the ability to assign glycopeptides confidently decreases as the sample complexity increases.19 We suggest that investigators spike in a well-characterized glycoprotein from a different species to complex biological samples from which glycopeptides are assigned. In doing so, they will be able to determine the extent to which assignment confidence deteriorates for the well-characterized glycopeptides in the presence of a complex background relative to the pure glycoprotein.
Figure 2.
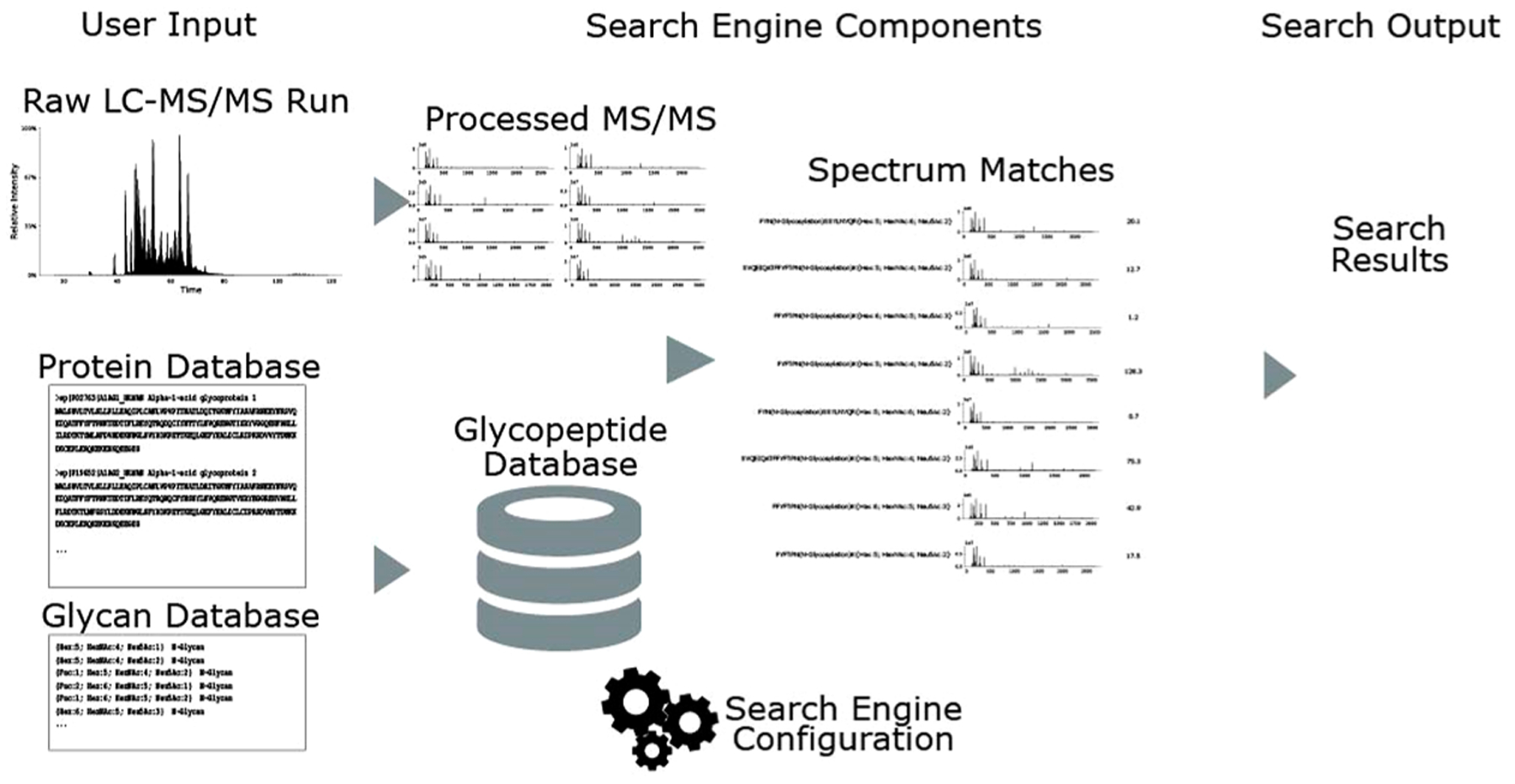
Schematic diagram of a glycoproteomics search engine, including inputs, outputs, and intermediate steps.
Software for identification of glycopeptides can be broken into the following categories: peptide-centric, glycan-centric, combined, and complete. Peptide-centric60 methods focus on identifying peptide backbone sequences. While they may use peptide + Y ions, they do not control for the FDR of the glycan component. Glycan-centric methods61,62 focus on identifying the attached glycan, without sequencing the peptide. A combined method51,63,64 uses a single score to capture both peptide and glycan components and controls the total uncertainty but does not control the uncertainty of both components separately. A complete method18,65,66 works by controlling the uncertainty of both components separately and jointly. Though one might desire to use a complete method for any experimental data, it requires that the experiment produces the appropriate fragmentation. Complete methods usually require relatively complex fragmentation reactions, in some cases spanning multiple spectra, limiting their throughput.
Some data-dependent tandem MS methods51,63,64,67 look at the oxonium ions in a tandem mass spectrum to constrain which glycans could realistically be attached to a given peptide. These tools can be used on most glycoproteomics data as oxonium ions are expected to be present in all collisional dissociation spectra, and they provide orthogonal signature ions to the large intact peptide + Y ions for assigning glycan compositions. They are vulnerable to contamination caused by co-isolation, such as when a glycan with a sialic acid is co-isolated with an unsialylated glycan. The prevalence of co-isolation is expected to be a function of sample complexity. While there is value in using these signals for the determination of the class (N, O, or GAG linker) and subclass (e.g., NeuAc-bearing, NeuGc-bearing, high-mannose N-glycans), isolation purity has yet to be addressed either experimentally or algorithmically.
BIOINFORMATICS DEVELOPMENTS NEEDED FOR QUANTITATIVE GLYCOPROTEOMICS
In an LC-MS experiment, the limited number of precursor ions that can be analyzed in a time window and the stochastic nature of precursor ion isolation result in missing values that limit the quantitative value of DDA methods.68 Likewise, there are a limited number of ions that can be quantified using a tandem MS inclusion list. Until recently,63,64,67 most software for glycopeptide identification rarely included built-in support for label free quantification (LFQ), and when they did, the features of these methods were not always complete. Byonic can integrate glycopeptide identification and quantification because of the underlying Byologic platform for LFQ of normal proteomics. The glyXtool was built on top of OpenMS,69 which includes a collection of different LFQ implementations and support tools for match between runs (MBR), feature finding and alignment, and integration, providing a bridge between glycopeptide representation within their application code and OpenMS’s glyco-agnostic algorithms. These techniques can help to diminish the effect of missing values caused by stochastic precursor selection. While these steps move toward streamlining quantification of site-specific glycosylation, we argue that unsolved problems remain.
The most popular approach to reducing missing values, MBR, requires that an analyte be fragmented and identified at least once in a study to transfer that identification to other acquisitions in that study.70 Recent work has shown that transferred identifications that were identified in only a small fraction of the acquisitions were more likely to be false positives.71 Integration of techniques for validating glycopeptide identifications by glycan-induced retention time shifts discussed earlier would be expected to reduce these false transferred identifications. When an analyte is never fragmented across any data acquisition, a larger issue for smaller studies, we may still be able to infer its identity from its mass and retention time offsets from other identified glycopeptides. While this may be done in an ad hoc manner for manual annotation, a high-throughput approach that incorporates some amount of statistical rigor has not been developed.
Quantitative targeted proteomics software for building acquisition lists and analyzing the results is designed for unmodified peptides or those that have small, well-defined, modifying groups.42 They do not at present address the problem of defining large, heterogeneous glycan post-translational modifications for peptides. As a result, it is necessary for users to construct long precursor target lists and MRM/PRM transitions using time-consuming manual effort or approximate in silico methods.44,72 There is a need to adapt existing tools and algorithms from quantitative proteomics to account for heterogeneous glycosylation as a post-translational modification. Part of the problem is that formats for defining and representing small molecule protein modifications differ from those needed to define the distribution of glycoforms that exist at a given protein site. This effort is made challenging by the lack of proteomics data exchange formats that can be applied to glycosylation (see the discussion below). Even if the existing tools were used to encode such large modifications, they would not be able to accurately account for the non-peptide-derived fragments unless the user manually entered an exhaustive list.
There is a clear, unmet, need to communicate glycoproteomics results to data repositories and knowledge bases, as well as between software. This is a challenge due to the extreme complexity of the domain, with an effectively intractable number of biosynthetically feasible glycan structures and the ways in which they may be represented.73 In proteomics, the mzIdentML data standard74–76 was developed by the Proteome Standards Initiative in collaboration with instrument and software vendors to facilitate storage and transfer of proteomics identification data. This standard provides a stable vendor-independent means for data exchange that has vastly improved the dissemination of proteomics data, and a growing ecosystem inter-operating software. Skyline can read mzIdentML files produced by compliant identification software to understand which peptides were identified when in which samples when building targets or transition lists.77 OpenMS similarly can read and write mzIdentML, mzQuantML, and mzTab files that, as previously mentioned, lets pipeline builders weave together tools from different authors, where each piece of software needs to know how to do only one thing rather than everything.78 PRIDE can automatically infer which scans were used to identify which peptides and which raw data files they were taken from by inspecting an mzIdentML file, automating the project submission.79 PRIDE can also generate annotated spectra from such submissions, obviating the requirement for authors to submit manually annotated spectra with articles.
The mzIdentML format was designed for representing peptides and a user-specified number of modifications such as those defined in the Unimod database,80 or free-text descriptions. As we have emphasized here, glycans at a given protein site exist as a set of related glycoforms, the ontologies for representing which need to be incorporated into an exchange format. In the absence of such an exchange format, each piece of glycoproteomics software must create a method for encoding glycans, making it difficult to compare or combine their results. As a result, exchange of glycoproteomics data remains a serious barrier to incorporation of glycopro teomics results into repositories and knowledge bases. Such an exchange format should use an externally hosted glycan database, such as GlyTouCan,81,82 represented in Open Biomedical Ontology (OBO) format, or another acceptable exchange format. An example where this has begun to happen is in GlyConnect, a database of glycoproteins based on manual curation from published research and high-throughput studies.83 It still relies on those high-throughput studies being able to provide enough information to connect the glycan notation used by their software to the glycan represented in GlyTouCan. However, GlyConnect is linked to UniProt84 and NeXtProt85,86 knowledge bases that are used by experts in many branches of biology, not just glycobiology. Through this connection, these Web sites now communicate to their users that the function of the protein that they are browsing is not one uniform molecule but dozens to hundreds of different structures inpotentia depending upon biological conditions.
To create an exchange format, the field must reach some consensus on the level of detail with which to represent glycans. While it might be ideal to specify explicitly all structures exactly,87 including stereoisomeric identities and linkage, current experimental and computational methods are incapable of reliably and uniformly discriminating these properties, such as separating GlcNAc and GalNAc or 2–3-and 2–6-linked NeuAc. Should it still be an option to explicitly specify these properties, does this mandate that any consumer of glycoproteomics results must be able to recognize the subsumption relationship87 between GlcNAc2Man6 and HexNAc2 Hex6? Both could be valid identifications of the same experimental evidence, and without the ability to recognize that one is just a specialization of the other. If subsumption is a feature of the standard, then the cost to implement the standard grows considerably, but leaving it off will lead to noncomparable results. A fair middle ground might be to define the standard with the freedom to be specific, but to recommend that generic names be used wherever appropriate, and for subsumption resolution to be required at the monosaccharide level but not at the aggregate level.
CONCLUSIONS
Tandem mass spectrometry methods have advanced to the point that singly glycosylated peptides can be assigned in LC-MS workflows using software available in the commercial and academic domains. The most appropriate use of these methods is for characterization of glycoproteins from enriched samples. This is because the extreme heterogeneity of glycoproteins from complex biological samples limits the ability to understand sources of error in glycopeptide assignments. Fortunately, the field is moving rapidly toward defining operating procedures and data exchange formats to allow widespread dissemination of accurate and reproducible glycoproteomics data.
Funding
Financial support was provided by National Institutes of Health Grant U01CA221234.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Varki A, Cummings R, Esko J, Stanley P, Hart G, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, and Seeberger PH (2017) Essentials of Glycobiology, 3rd ed., Cold Spring Harbor Laboratory Press, Plainview, NY. [PubMed] [Google Scholar]
- (2).Cummings RD, and Pierce JM (2014) The challenge and promise of glycomics. Chem. Biol 21, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Varki A (2006) Nothing in glycobiology makes sense, except in the light of evolution. Cell 126, 841–845. [DOI] [PubMed] [Google Scholar]
- (4).Dennis JW (2017) Genetic code asymmetry supports diversity through experimentation with posttranslational modifications. Curr. Opin. Chem. Biol 41, 1–11. [DOI] [PubMed] [Google Scholar]
- (5).Aebersold R, Agar JN, Amster IJ, Baker MS, Bertozzi CR, Boja ES, Costello CE, Cravatt BF, Fenselau C, Garcia BA, Ge Y, Gunawardena J, Hendrickson RC, Hergenrother PJ, Huber CG, Ivanov AR, Jensen ON, Jewett MC, Kelleher NL, Kiessling LL, Krogan NJ, Larsen MR, Loo JA, Ogorzalek Loo RR, Lundberg E, MacCoss MJ, Mallick P, Mootha VK, Mrksich M, Muir TW, Patrie SM, Pesavento JJ, Pitteri SJ, Rodriguez H, Saghatelian A, Sandoval W, Schluter H, Sechi S, Slavoff SA, Smith LM, Snyder MP, Thomas PM, Uhlen M, Van Eyk JE, Vidal M, Walt DR, White FM, Williams ER, Wohlschlager T, Wysocki VH, Yates NA, Young NL, and Zhang B (2018) How many human proteoforms are there? Nat. Chem. Biol 14, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Pagel O, Loroch S, Sickmann A, and Zahedi RP (2015) Current strategies and findings in clinically relevant post-translational modification-specific proteomics. Expert Rev. Proteomics 12, 235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Khatri K, Staples GO, Leymarie N, Leon DR, Turiák L, Huang Y, Yip S, Hu H, Heckendorf CF, and Zaia J (2014) Confident Assignment of Site-Specific Glycosylation in Complex Glycoproteins in a Single Step. J. Proteome Res 13, 4347–4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hinneburg H, Stavenhagen K, Schweiger-Hufnagel U, Pengelley S, Jabs W, Seeberger PH, Silva DV, Wuhrer M, and Kolarich D (2016) The Art of Destruction: Optimizing Collision Energies in Quadrupole-Time of Flight (Q-TOF) Instruments for Glycopeptide-Based Glycoproteomics. J. Am. Soc. Mass Spectrom 27, 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Liu MQ, Zeng WF, Fang P, Cao WQ, Liu C, Yan GQ, Zhang Y, Peng C, Wu JQ, Zhang XJ, Tu HJ, Chi H, Sun RX, Cao Y, Dong MQ, Jiang BY, Huang JM, Shen HL, Wong CCL, He SM, and Yang PY (2017) pGlyco 2.0 enables precision N-glycoproteomics with comprehensive quality control and one-step mass spectrometry for intact glycopeptide identification. Nat. Commun 8, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Saba J, Dutta S, Hemenway E, and Viner R (2012) Increasing the productivity of glycopeptides analysis by using higher-energy collision dissociation-accurate mass-product-dependent electron transfer dissociation. Int. J. Proteomics 2012, 560391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yu Q, Wang B, Chen Z, Urabe G, Glover MS, Shi X, Guo LW, Kent KC, and Li L (2017) Electron-Transfer/Higher-Energy Collision Dissociation (EThcD)-Enabled Intact Glycopeptide/Glycoproteome Characterization. J. Am. Soc. Mass Spectrom 28, 1751–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Khatri K, Pu Y, Klein JA, Wei J, Costello CE, Lin C, and Zaia J (2018) Comparison of Collisional and Electron-Based Dissociation Modes for Middle-Down Analysis of Multiply Glycosylated Peptides. J. Am. Soc. Mass Spectrom 29, 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Riley NM, Hebert AS, Westphall MS, and Coon JJ (2019) Capturing site-specific heterogeneity with large-scale N-glycoproteome analysis. Nat. Commun 10, 1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Riley NM, Westphall MS, and Coon JJ (2017) Activated Ion-Electron Transfer Dissociation Enables Comprehensive Top-Down Protein Fragmentation. J. Proteome Res 16, 2653–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hu H, Khatri K, and Zaia J (2017) Algorithms and design strategies towards automated glycoproteomics analysis. Mass Spectrom. Rev 36, 475–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hu H, Khatri K, Klein J, Leymarie N, and Zaia J (2016) A review of methods for interpretation of glycopeptide tandem mass spectral data. Glycoconjugate J. 33, 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bollineni RC, Koehler CJ, Gislefoss RE, Anonsen JH, and Thiede B (2018) Large-scale intact glycopeptide identification by Mascot database search. Sci. Rep 8, 2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Xiao K, and Tian Z (2019) GPSeeker Enables Quantitative Structural N-Glycoproteomics for Site- and Structure-Specific Characterization of Differentially Expressed N-Glycosylation in Hepatocellular Carcinoma. J. Proteome Res 18, 2885–2895. [DOI] [PubMed] [Google Scholar]
- (19).Khatri K, Klein JA, and Zaia J (2017) Use of an informed search space maximizes confidence of site-specific assignment of glycoprotein glycosylation. Anal. Bioanal. Chem 409, 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lee LY, Moh ES, Parker BL, Bern M, Packer NH, and Thaysen-Andersen M (2016) Toward Automated N-Glycopeptide Identification in Glycoproteomics. J. Proteome Res 15, 3904–3915. [DOI] [PubMed] [Google Scholar]
- (21).Solntsev SK, Shortreed MR, Frey BL, and Smith LM (2018) Enhanced Global Post-translational Modification Discovery with MetaMorpheus. J. Proteome Res 17, 1844–1851. [DOI] [PubMed] [Google Scholar]
- (22).Håkansson K, Cooper HJ, Emmett MR, Costello CE, Marshall AG, and Nilsson CL (2001) Electron capture dissociation and Infrared multiphoton dissociation MS/MS of an N-glycosylated tryptic peptide to yield complementary sequence information. Anal. Chem 73, 4530–4536. [DOI] [PubMed] [Google Scholar]
- (23).Mechref Y (2012) Use of CID/ETD mass spectrometry to analyze glycopeptides Current Protocols in Protein Science, Chapter 12, Unit 12.11.1–12.11.11, Wiley. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Singh C, Zampronio CG, Creese AJ, and Cooper HJ (2012) Higher energy collision dissociation (HCD) product ion-triggered electron transfer dissociation (ETD) mass spectrometry for the analysis of N-linked glycoproteins. J. Proteome Res 11, 4517–4525. [DOI] [PubMed] [Google Scholar]
- (25).Rose CM, Rush MJ, Riley NM, Merrill AE, Kwiecien NW, Holden DD, Mullen C, Westphall MS, and Coon JJ (2015) A calibration routine for efficient ETD in large-scale proteomics. J. Am. Soc. Mass Spectrom 26, 1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Carr SA, Abbatiello SE, Ackermann BL, Borchers C, Domon B, Deutsch EW, Grant RP, Hoofnagle AN, Huttenhain R, Koomen JM, Liebler DC, Liu T, Maclean B, Mani DR, Mansfield E, Neubert H, Paulovich AG, Reiter L, Vitek O, Aebersold R, Anderson L, Bethem R, Blonder J, Boja E, Botelho J, Boyne M, Bradshaw RA, Burlingame AL, Chan D, Keshishian H, Kuhn E, Kinsinger C, Lee J, Lee SW, Moritz R, Oses-Prieto J, Rifai N, Ritchie J, Rodriguez H, Srinivas PR, Townsend RR, Van Eyk J, Whiteley G, Wiita A, and Weintraub S (2014) Targeted Peptide Measurements in Biology and Medicine: Best Practices for Mass Spectrometry-based Assay Development Using a Fit-for-Purpose Approach. Mol. Cell. Proteomics 13, 907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Goldman R, and Sanda M (2015) Targeted methods for quantitative analysis of protein glycosylation. Proteomics: Clin. Appl 9, 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Song E, Pyreddy S, and Mechref Y (2012) Quantification of glycopeptides by multiple reaction monitoring liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom 26, 1941–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Sanda M, Pompach P, Brnakova Z, Wu J, Makambi K, and Goldman R (2013) Quantitative liquid chromatography-mass spectrometry-multiple reaction monitoring (LC-MS-MRM) analysis of site-specific glycoforms of haptoglobin in liver disease. Mol. Cell. Proteomics 12, 1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Darebna P, Novak P, Kucera R, Topolcan O, Sanda M, Goldman R, and Pompach P (2017) Changes in the expression of N- and O-glycopeptides in patients with colorectal cancer and hepatocellular carcinoma quantified by full-MS scan FT-ICR and multiple reaction monitoring. J. Proteomics 153, 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hong Q, Ruhaak LR, Stroble C, Parker E, Huang J, Maverakis E, and Lebrilla CB (2015) A Method for Comprehensive Glycosite-Mapping and Direct Quantitation of Serum Glycoproteins. J. Proteome Res 14, 5179–5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Nilsson J, Brinkmalm G, Ramadan S, Gilborne L, Noborn F, Blennow K, Wallin A, Svensson J, Abo-Riya MA, Huang X, and Larson G (2019) Synthetic standard aided quantification and structural characterization of amyloid-beta glycopeptides enriched from cerebrospinal fluid of Alzheimer’s disease patients. Sci. Rep 9, 5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kim KH, Lee SY, Hwang H, Lee JY, Ji ES, An HJ, Kim JY, and Yoo JS (2018) Direct Monitoring of Fucosylated Glycopeptides of Alpha-Fetoprotein in Human Serum for Early Hepatocellular Carcinoma by Liquid Chromatography-Tandem Mass Spectrometry with Immunoprecipitation. Proteomics: Clin. Appl 12, 1800062. [DOI] [PubMed] [Google Scholar]
- (34).Kim KH, Park GW, Jeong JE, Ji ES, An HJ, Kim JY, and Yoo JS (2019) Parallel reaction monitoring with multiplex immunoprecipitation of N-glycoproteins in human serum for detection of hepatocellular carcinoma. Anal. Bioanal. Chem 411, 3009–3019. [DOI] [PubMed] [Google Scholar]
- (35).Rosenberger G, Bludau I, Schmitt U, Heusel M, Hunter CL, Liu Y, MacCoss MJ, MacLean BX, Nesvizhskii AI, Pedrioli PGA, Reiter L, Rost HL, Tate S, Ting YS, Collins BC, and Aebersold R (2017) Statistical control of peptide and protein error rates in large-scale targeted data-independent acquisition analyses. Nat. Methods 14, 921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Frank AM, Bandeira N, Shen Z, Tanner S, Briggs SP, Smith RD, and Pevzner PA (2008) Clustering millions of tandem mass spectra. J. Proteome Res 7, 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Griss J (2016) Spectral library searching in proteomics. Proteomics 16, 729–740. [DOI] [PubMed] [Google Scholar]
- (38).Benton HP, Wong DM, Trauger SA, and Siuzdak G (2008) XCMS2: processing tandem mass spectrometry data for metabolite identification and structural characterization. Anal. Chem 80, 6382–6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Toghi Eshghi S, Shah P, Yang W, Li X, and Zhang H (2015) GPQuest: A Spectral Library Matching Algorithm for Site-Specific Assignment of Tandem Mass Spectra to Intact N-glycopeptides. Anal. Chem 87, 5181–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Nasir W, Toledo AG, Noborn F, Nilsson J, Wang M, Bandeira N, and Larson G (2016) SweetNET: A Bioinformatics Workflow for Glycopeptide MS/MS Spectral Analysis. J. Proteome Res 15, 2826–2840. [DOI] [PubMed] [Google Scholar]
- (41).Frank AM, Monroe ME, Shah AR, Carver JJ, Bandeira N, Moore RJ, Anderson GA, Smith RD, and Pevzner PA (2011) Spectral archives: extending spectral libraries to analyze both identified and unidentified spectra. Nat. Methods 8, 587–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Rost HL, Rosenberger G, Navarro P, Gillet L, Miladinovic SM, Schubert OT, Wolski W, Collins BC, Malmstrom J, Malmstrom L, and Aebersold R (2014) Open-SWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol 32, 219–223. [DOI] [PubMed] [Google Scholar]
- (43).Sanda M, and Goldman R (2016) Data Independent Analysis of IgG Glycoforms in Samples of Unfractionated Human Plasma. Anal. Chem 88, 10118–10125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ye Z, Mao Y, Clausen H, and Vakhrushev SY (2019) Glyco-DIA: a method for quantitative O-glycoproteomics with in silico-boosted glycopeptide libraries. Nat. Methods 16, 902–910. [DOI] [PubMed] [Google Scholar]
- (45).Lin C-H, Krisp C, Packer NH, and Molloy MP (2018) Development of a data independent acquisition mass spectrometry workflow to enable glycopeptide analysis without predefined glycan compositional knowledge. J. Proteomics 172, 68–75. [DOI] [PubMed] [Google Scholar]
- (46).Tsou C-C, Avtonomov D, Larsen B, Tucholska M, Choi H, Gingras A-C, and Nesvizhskii AI (2015) DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 12, 258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Searle BC, Pino LK, Egertson JD, Ting YS, Lawrence RT, MacLean BX, Villén J, and MacCoss MJ (2018) Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat. Commun 9, 5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Rosenberger G, Liu Y, Rost HL, Ludwig C, Buil A, Bensimon A, Soste M, Spector TD, Dermitzakis ET, Collins BC, Malmstrom L, and Aebersold R (2017) Inference and quantification of peptidoforms in large sample cohorts by SWATHMS. Nat. Biotechnol 35, 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Hu Y, Shihab T, Zhou S, Wooding K, and Mechref Y (2016) LC-MS/MS of permethylated N-glycans derived from model and human blood serum glycoproteins. Electrophoresis 37, 1498–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wang B, Tsybovsky Y, Palczewski K, and Chance MR (2014) Reliable determination of site-specific in vivo protein N-glycosylation based on collision-induced MS/MS and chromatographic retention time. J. Am. Soc. Mass Spectrom 25, 729–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Choo MS, Wan C, Rudd PM, and Nguyen-Khuong T (2019) GlycopeptideGraphMS: Improved Glycopeptide Detection and Identification by Exploiting Graph Theoretical Patterns in Mass and Retention Time. Anal. Chem 91, 7236–7244. [DOI] [PubMed] [Google Scholar]
- (52).Ang E, Neustaeter H, Spicer V, Perreault H, and Krokhin OV (2019) Retention Time Prediction for Glycopeptides in Reversed Phase Chromatography for Glycoproteomic Applications. Anal. Chem 91, 13360. [DOI] [PubMed] [Google Scholar]
- (53).Lee HJ, Cha HJ, Lim JS, Lee SH, Song SY, Kim H, Hancock WS, Yoo JS, and Paik YK (2014) Abundance-ratio-based semiquantitative analysis of site-specific N-linked glycopeptides present in the plasma of hepatocellular carcinoma patients. J. Proteome Res 13, 2328–2338. [DOI] [PubMed] [Google Scholar]
- (54).Ye H, Hill J, Gucinski AC, Boyne MT 2nd, and Buhse LF (2015) Direct site-specific glycoform identification and quantitative comparison of glycoprotein therapeutics: imiglucerase and velaglucerase alfa. AAPS J. 17, 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Zhou J, Yang W, Hu Y, Hoti N, Liu Y, Shah P, Sun S, Clark D, Thomas S, and Zhang H (2017) Site-Specific Fucosylation Analysis Identifying Glycoproteins Associated with Aggressive Prostate Cancer Cell Lines Using Tandem Affinity Enrichments of Intact Glycopeptides Followed by Mass Spectrometry. Anal. Chem 89, 7623–7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Cho KC, Chen L, Hu Y, Schnaubelt M, and Zhang H (2019) Developing Workflow for Simultaneous Analyses of Phosphopeptides and Glycopeptides. ACS Chem. Biol 14, 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Kawahara R, Ortega F, Rosa-Fernandes L, Guimarães V, Quina D, Nahas W, Schwammle V, Srougi M, Leite KRM, Thaysen-Andersen M, Larsen MR, and Palmisano G (2018) Distinct urinary glycoprotein signatures in prostate cancer patients. Oncotarget 9, 33077–33097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Viner RI, Zhang T, Second T, and Zabrouskov V (2009) Quantification of post-translationally modified peptides of bovine alpha-Crystallin using tandem mass tags and electron transfer dissociation. J. Proteomics 72, 874–885. [DOI] [PubMed] [Google Scholar]
- (59).Zhu H, Qiu C, Ruth AC, Keire DA, and Ye H (2017) A LC-MS All-in-One Workflow for Site-Specific Location, Identification and Quantification of N-/O- Glycosylation in Human Chorionic Gonadotropin Drug Products. AAPS J. 19, 846–855. [DOI] [PubMed] [Google Scholar]
- (60).Toghi Eshghi S, Yang W, Hu Y, Shah P, Sun S, Li X, and Zhang H (2016) Classification of Tandem Mass Spectra for Identification of N- and O-linked Glycopeptides. Sci. Rep 6, 37189–37189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Lakbub JC, Su X, Zhu Z, Patabandige MW, Hua D, Go EP, and Desaire H (2017) Two New Tools for Glycopeptide Analysis Researchers: A Glycopeptide Decoy Generator and a Large Data Set of Assigned CID Spectra of Glycopeptides. J. Proteome Res 16, 3002–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Chandler KB, Pompach P, Goldman R, and Edwards N (2013) Exploring Site-Specific N-Glycosylation Microheterogeneity of Haptoglobin Using Glycopeptide CID Tandem Mass Spectra and Glycan Database Search. J. Proteome Res 12, 3652–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Klein JA, Meng L, and Zaia J (2018) Deep sequencing of complex proteoglycans: a novel strategy for high coverage and site-specific identification of glycosaminoglycan-linked peptides. Mol. Cell. Proteomics 17, 1578–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Bern M, Kil YJ, and Becker C (2012) Byonic: advanced peptide and protein identification software. Curr. Protoc. Bioinf 40, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).He L, Xin L, Shan B, Lajoie GA, and Ma B (2014) GlycoMaster DB: software to assist the automated identification of N-linked glycopeptides by tandem mass spectrometry. J. Proteome Res 13, 3881–3895. [DOI] [PubMed] [Google Scholar]
- (66).Mayampurath A, Yu CY, Song E, Balan J, Mechref Y, and Tang H (2014) Computational framework for identification of intact glycopeptides in complex samples. Anal. Chem 86, 453–463. [DOI] [PubMed] [Google Scholar]
- (67).Pioch M, Hoffmann M, Pralow A, Reichl U, and Rapp E (2018) glyXtool(MS): An Open-Source Pipeline for Semiautomated Analysis of Glycopeptide Mass Spectrometry Data. Anal. Chem 90, 11908–11916. [DOI] [PubMed] [Google Scholar]
- (68).Karpievitch Y, Stanley J, Taverner T, Huang J, Adkins JN, Ansong C, Heffron F, Metz TO, Qian WJ, Yoon H, Smith RD, and Dabney AR (2009) A statistical framework for protein quantitation in bottom-up MS-based proteomics. Bioinformatics 25, 2028–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Rost HL, Schmitt U, Aebersold R, and Malmstrom L (2014) pyOpenMS: A Python-based interface to the OpenMS mass-spectrometry algorithm library. Proteomics 14, 74–77. [DOI] [PubMed] [Google Scholar]
- (70).Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, and Mann M (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Lim MY, Paulo JA, and Gygi SP (2019) Evaluating False Transfer Rates from the Match-between-Runs Algorithm with a Two-Proteome Model. J. Proteome Res 18, 4020–4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Phung TK, Zacchi LF, and Schulz BL (2019) DIALib: an automated ion library generator for data independent acquisition mass spectrometry analysis of peptides and glycopeptides. bioRxiv. [DOI] [PubMed] [Google Scholar]
- (73).Akune Y, Lin CH, Abrahams JL, Zhang J, Packer NH, Aoki-Kinoshita KF, and Campbell MP (2016) Comprehensive analysis of the N-glycan biosynthetic pathway using bioinformatics to generate UniCorn: A theoretical N-glycan structure database. Carbohydr. Res 431, 56–63. [DOI] [PubMed] [Google Scholar]
- (74).Vizcaino JA, Mayer G, Perkins S, Barsnes H, Vaudel M, Perez-Riverol Y, Ternent T, Uszkoreit J, Eisenacher M, Fischer L, Rappsilber J, Netz E, Walzer M, Kohlbacher O, Leitner A, Chalkley RJ, Ghali F, Martinez-Bartolome S, Deutsch EW, and Jones AR (2017) The mzIdentML Data Standard Version 1.2, Supporting Advances in Proteome Informatics. Mol. Cell. Proteomics 16, 1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Jones AR, Eisenacher M, Mayer G, Kohlbacher O, Siepen J, Hubbard SJ, Selley JN, Searle BC, Shofstahl J, Seymour SL, Julian R, Binz PA, Deutsch EW, Hermjakob H, Reisinger F, Griss J, Vizcaino JA, Chambers M, Pizarro A, and Creasy D (2012) The mzIdentML data standard for mass spectrometry-based proteomics results. Mol. Cell. Proteomics 11, M111.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Eisenacher M (2011) mzIdentML: an open community-built standard format for the results of proteomics spectrum identification algorithms. Methods Mol. Biol 696, 161–177. [DOI] [PubMed] [Google Scholar]
- (77).Maclean B, Tomazela DM, Abbatiello SE, Zhang S, Whiteaker JR, Paulovich AG, Carr SA, and Maccoss MJ (2010) Effect of collision energy optimization on the measurement of peptides by selected reaction monitoring (SRM) mass spectrometry. Anal. Chem 82, 10116–10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Rost HL, Sachsenberg T, Aiche S, Bielow C, Weisser H, Aicheler F, Andreotti S, Ehrlich HC, Gutenbrunner P, Kenar E, Liang X, Nahnsen S, Nilse L, Pfeuffer J, Rosenberger G, Rurik M, Schmitt U, Veit J, Walzer M, Wojnar D, Wolski WE, Schilling O, Choudhary JS, Malmstrom L, Aebersold R, Reinert K, and Kohlbacher O (2016) OpenMS: a flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 13, 741–748. [DOI] [PubMed] [Google Scholar]
- (79).Vizcaino JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, Xu QW, Wang R, and Hermjakob H (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, 11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Creasy DM, and Cottrell JS (2004) Unimod: Protein modifications for mass spectrometry. Proteomics 4, 1534–1536. [DOI] [PubMed] [Google Scholar]
- (81).Tiemeyer M, Aoki K, Paulson J, Cummings RD, York WS, Karlsson NG, Lisacek F, Packer NH, Campbell MP, Aoki NP, Fujita A, Matsubara M, Shinmachi D, Tsuchiya S, Yamada I, Pierce M, Ranzinger R, Narimatsu H, and Aoki-Kinoshita KF (2017) GlyTouCan: an accessible glycan structure repository. Glycobiology 27, 915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Aoki-Kinoshita K, Agravat S, Aoki NP, Arpinar S, Cummings RD, Fujita A, Fujita N, Hart GM, Haslam SM, Kawasaki T, Matsubara M, Moreman KW, Okuda S, Pierce M, Ranzinger R, Shikanai T, Shinmachi D, Solovieva E, Suzuki Y, Tsuchiya S, Yamada I, York WS, Zaia J, and Narimatsu H (2016) GlyTouCan 1.0 - The international glycan structure repository. Nucleic Acids Res. 44, D1237–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Alocci D, Mariethoz J, Gastaldello A, Gasteiger E, Karlsson NG, Kolarich D, Packer NH, and Lisacek F (2019) GlyConnect: Glycoproteomics Goes Visual, Interactive, and Analytical. J. Proteome Res 18, 664–677. [DOI] [PubMed] [Google Scholar]
- (84).Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, and Yeh LS (2004) UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 32, D115–D119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Gaudet P, Argoud-Puy G, Cusin I, Duek P, Evalet O, Gateau A, Gleizes A, Pereira M, Zahn-Zabal M, Zwahlen C, Bairoch A, and Lane L (2013) neXtProt: organizing protein knowledge in the context of human proteome projects. J. Proteome Res 12, 293–298. [DOI] [PubMed] [Google Scholar]
- (86).Gaudet P, Michel PA, Zahn-Zabal M, Britan A, Cusin I, Domagalski M, Duek PD, Gateau A, Gleizes A, Hinard V, Rech de Laval V, Lin J, Nikitin F, Schaeffer M, Teixeira D, Lane L, and Bairoch A (2017) The neXtProt knowledgebase on human proteins: 2017 update. Nucleic Acids Res. 45, D177–D182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Matsubara M, Aoki-Kinoshita KF, Aoki NP, Yamada I, and Narimatsu H (2017) WURCS 2.0 Update To Encapsulate Ambiguous Carbohydrate Structures. J. Chem. Inf. Model 57, 632–637. [DOI] [PubMed] [Google Scholar]