

Abstract
Pheochromocytomas (PCC) and paragangliomas (PGL) are rare neuroendocrine tumors. Pheochromocytomas arise from chromaffin cells in the adrenal medulla, and PGLs arise from chromaffin cells in the ganglia of the autonomic nervous system. Paragangliomas originate from sympathetic or parasympathetic ganglia in the abdomen, thorax, and pelvis. The majority of PCC and sympathetic PGL are endocrine active tumors causing clinical symptoms by secreting excess catecholamines (norepinephrine, epinephrine, dopamine) and their metabolites. The incidence of PCC and PGL ranges between 2 and 8 per million, with a prevalence between 1:2500 and 1:6500. It peaks between the 3rd and 5th decades of life, and approximately 20% of cases are pediatric patients. The prevalence among patients with hypertension in outpatient clinic ranges between 0.1-0.6% in adults and between 2-4.5% in the pediatric age group. 10-49% of these tumors is detected incidentally in imaging techniques performed for other reasons. However, 4-8% of adrenal incidentalomas are PCCs. Of these neuroendocrine tumors, 80-85% are PCCs and 15-20% are PGLs.
Up to 40% of patients with PCC and PGL has disease-specific germline mutations and the situation is hereditary. Of 60% of the remaining sporadic patients, at least 1/3 has a somatic mutation in predisposing genes. 8% of the sporadic cases, 20-75% of the hereditary cases, 5% of the bilateral, adrenal cases, and 33% of the extra-adrenal cases at first presentation are metastatic. Although PCCs and PGLs have scoring systems for histological evaluation of the primary tumor, it is not possible to diagnose whether the tumor is malignant since there is no histological system approved for the biological aggressiveness of this tumor group. Metastasis is defined as the presence of chromaffin tissue in non-chromaffin organs, such as lymph nodes, liver, lungs and bone. Although most of the PCC and PGL are benign, the metastatic disease may develop in 15-17%. Metastatic disease is reported between 2-25% in PCCs and 2.4-60% in PGLs. The TNM staging system of the American Joint Committee on Cancer (AJCC) was developed to predict the prognosis, based on the specific anatomical features of the primary tumor and the occurrence of metastasis.
Keywords: Catecholamine synthesis and metabolism, paraganglioma, pheochromocytoma
Pheochromocytoma (PCC) and paraganglioma (PGL) are rare neuroendocrine tumors. Pheochromocytomas arise from chromaffin cells in the adrenal medulla, and PGLs arise from chromaffin cells in the ganglia of the autonomic nervous system.[1, 2] Paragangliomas originate from sympathetic or parasympathetic ganglia in the abdomen, thorax, and pelvis. Paragangliomas may also originate from parasympathetic ganglia at the base of the skull and along the glossopharyngeal nerve and vagus nerve in the neck.[2] The majority of PCCs and sympathetic PGLs are endocrine active tumors, and they cause clinical symptoms by secreting excess catecholamines (norepinephrine, epinephrine, dopamine) and their metabolites. However, approximately 2/3 of PGLs of parasympathetic origin in the head and neck region are not endocrine active.[3] Paragangliomas are sometimes called extra-adrenal PCCs. These tumors are abbreviated in the literature as PCC/PGL or PPGL; in this study, PPGL abbreviation will be used.[1, 2, 4]
Epidemiology and Incidence
The incidence of PPGL ranges between 2 and 8 per million, with a prevalence between 1:2500 and 1:6500.[5, 6] The annual incidence in the USA is approximately 500-1600 cases per year, which is lower than the prevalence of PPGL (0.05-0.1%) detected in the autopsy series incidentally.[6] PPGL peaks between the 3rd and 5th decades of life, and approximately 20% of cases is pediatric patients.[5, 7] The prevalence among patients with hypertension in outpatient clinic ranges between 0.1-0.6% in adults and between 2-4.5% in the pediatric age group.[8] 10-49% of PPGLs is detected incidentally in imaging techniques performed for other reasons. However, 4-8% of adrenal incidentalomas are PCCs.[5] Of these neuroendocrine tumors, 80-85% is PCCs, and 15-20% is PGLs.[1, 2] 70-80% of PGLs arises from infradiaphragmatic sympathetic ganglia. It most commonly originates from the Zuckerkandl organ located near the origin of the inferior mesenteric artery at the aortic bifurcation. It can be less frequently found in the retroperitoneal area around the kidney and adrenal and in the bladder; however, it can be seen in all anatomical areas in the abdomen where sympathetic or parasympathetic ganglia are localized. Approximately 10% of the PGLs are in the mediastinal and pericardial localization within the thorax, and approximately 20% is localized in the head and neck region.[2, 9] Up to 40% of patients with PPGL have disease-specific germline mutations and the situation is hereditary. Of the 60% of the remaining sporadic patients, at least 1/3 has a somatic mutation in predisposing genes.[10] 8% of the sporadic cases, 20-75% of the hereditary ones, 5% of the bilateral, adrenal cases, and 33% of the extradrenal ones at first presentation are metastatic.[5]
Although the 10’s rule, which Graham defined in 1951 for PPGL, describing 10% extradrenal, 10% bilateral, and 10% malignancy, is still included in many textbooks, it has expired today.[11]
Catecholamine Synthesis and Metabolism
Synthesis of catecholamine hormones takes place in the adrenal medulla, sympathetic and central nervous system. Catecholamine synthesis begins with the conversion of L-tyrosine, which is taken from the diet or synthesized from phenylalanine in the liver, to L- dihydroxyphenylalanine (DOPA) by catalyzation of the tyrosine hydroxylase enzyme.[1] This stage is the rate-limiting step of catecholamine synthesis; hence, the source of the catecholamines depends on the presence of this enzyme.[12] Tyrosine hydroxylase enzyme is largely found in the catecholaminergic nerve endings of the brain and in the sympathetic nervous system in the periphery, in the adrenal medulla, and extraadrenal chromaffin cells.[12] DOPA is converted into dopamine with L-aromatic amino acid decarboxylase enzyme. Dopamine is transported by vesicular monoamine transporter into storage vesicles inside the cell. Dopamine is converted intravesically to norepinephrine with a dopamine-beta-hydroxylase enzyme, which is one of the components of storage vesicles (Fig. 1).[1, 12] Norepinephrine is converted to epinephrine (adrenaline) with the phenylethanolamine N-methyl transferase (PNMT) enzyme found only in the adrenal medulla and the Zuckerkandl organ, sympathetic ganglion (Fig. 1). Since PNMT is a cytosolic enzyme, epinephrine synthesis is dependent on the infiltration of norepinephrine from its synthesis site, vesicles to the cytoplasm. There are two subgroups of chromaffin cells. Adrenergic cells (epinephrine cells) contain PNMT and store mainly epinephrine, and these cells contain medium-density large granules containing epinephrine.[12] Adrenergic cells are localized close to the cortex and are exposed to high cortisol levels of the cortex. Cortisol is required for the induction of the PNMT enzyme.[13] Norepinephrine cells do not contain PNMT; they mainly store norepinephrine and contain small electron density granules containing norepinephrine.[12] Failure to secrete epinephrine in extra-adrenal tissues may also arise from the absence of cortisol for PNMT stimulation.[13] The circulating epinephrine is secreted by the adrenal medulla, is released directly from the medulla to the circulation, creating a hormonal effect on cells in other tissues and its basal value is 0.1-0.3 nmol/L. Norepinephrine acts as a neurotransmitter in some neurons in the brain and peripheral sympathetic neurons. Norepinephrine is secreted into the synaptic cleft for activation and taken back into the nerve from the synaptic cleft. As a result, a small amount of norepinephrine is released into the circulation by escaping from the sympathetic nerves. More than 90% of circulating norepinephrine originates from sympathetic nerves and approximately 7% from the adrenal medulla and innervates the mesenteric organs, kidney and skeletal muscles. The plasma level in basal conditions is 0.6-2 nmol/L. However, circulating epinephrine is released only from the adrenal medulla, and its level is 0.1-0.3 nmol/L. Dopamine, which is not converted to norepinephrine and epinephrine, is metabolized in the gastrointestinal tract.[12, 13] When the adrenal medulla is stimulated, the secretory vesicles join the cell membrane in chromaffin cells, and the entire vesicle content is released to the circulation through its rich vascular network. These vesicles contain catecholamines, as well as ATP, calcium, magnesium, chromogranins and other neuropeptides.[14] Catecholamine content may vary depending on enzyme expression in PCCs. Most PCCs produce predominantly norepinephrine. Some produce both norepinephrine and epinephrine, and rarely epinephrine. Since dopamine is predominantly converted to norepinephrine, in very rare cases, the mainly produced molecule is dopamine.[12] Since there is no PNMT in PGLs, norepinephrine is produced predominantly.
Figure 1.
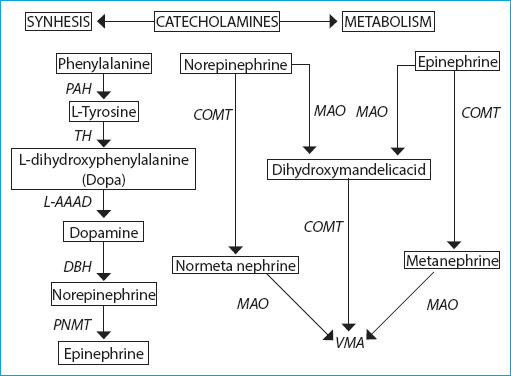
Catecholamine synthesis and metabolism.
PAH: Phenylalaninehydroxylase; TH: Tyrosine hydroxylase; L-AAAD: L- aromatic amino acid decarboxylase; DBH: Dopamine-beta-hydroxylase; PNMT: Phenyletanolamine-N-methyltransferase; MAO: Monoamine oxidase; COMT: Catechol O-methyltransferase; VMA: (vanillylmandelic acid) 3-methoxy-4-hydroxymandelic acid.
Catecholamines show their biological effects by binding to transmembrane adrenergic receptors, which are G protein-coupled receptors. There are adrenergic receptors with different affinity for catecholamines in different tissues and these are α, β1, β2 receptors. The receptor affinities for α receptors are; epinephrine > norepinephrine >> isoproterenol; for β1 receptors; isoproterenol > epinephrine = norepinephrine; and for β2 receptors; isoproterenol > epinephrine >> norepinephrine.[14] The effects of catecholamines in the organs of where receptors are present through these receptors can be summarized as follows: While α1 receptors cause vasoconstriction in the blood vessels, decreased motility in the intestine, increased sphincter tone, decreased insulin and glucagon release in the pancreas, glycogenolysis and gluconeogenesis in the liver, dilation of the pupils, uterine contraction and sweating, α2 receptors inhibit norepinephrine release in synapses and stimulate platelet aggregation. While β1 receptors cause chronotropic and inotropic effects on the heart, vasodilation in coronary vessels, lipolysis in adipose tissue, decreased motility and increased sphincter tone in the intestine, increase in insulin and glucagon release in the pancreas; β2 receptors cause vasodilation in the blood vessels, bronchodilation in the lungs and uterine relaxation.[14]
Many of the catecholamines are metabolized in the neural cells in which they are produced; catecholamines produced in the adrenal medulla are metabolized with adrenal medulla. The stimulation half-life of the hormones released to the blood is very short, about 1-2 minutes, and they are taken by the non-neural tissues. The metabolism of catecholamines given to circulation takes place primarily in the liver and kidneys. Norepinephrine and epinephrine are metabolized to normetanephrine and metanephrine with catechol-o-methyltransferase (COMT) in the adrenal medulla and non-neural tissues. These metabolites are metabolized to the end product, vanillylmandelic acid (VMA), with monoamine oxidase enzyme (MAO). Norepinephrine and epinephrine can also be metabolised to VMA using MAO and then with COMT. VMA is excreted in urine as conjugated with sulfuric and glucuronic acid, which happens in the liver and intestine. Small amounts of free dopamine, epinephrine and norepinephrine are excreted in the urine (Fig. 1).[1, 12, 13] Catecholamines are secreted from adrenal medulla intermittently and their half-life is very short. Therefore, these may reflect the sympathoadrenal activity at the time the sample was taken. Oxymethyl derivatives of catecholamines are continuously produced in tumor cells. The plasma half-life of these metabolites is much longer than norepinephrine and epinephrine. Their plasma levels are relatively more stable. Plasma metanephrine level is often independent of sympathoadrenal stimulation.[1, 13]
Pathophysiology
PPGLs release an uncontrolled excess of catecholamines, which bind to receptors in different organs, causing some physiological changes. Norepinephrine stimulates α1, α2, and β1 receptors, whereas epinephrine stimulates only β1 and β2 receptors. While normal levels of dopamine do not affect adrenergic receptors, it can stimulate both α and β receptors when dopamine increases in circulation, as in case of tumors that secrete dopamine. The α1 adrenergic receptor is primarily located in the smooth muscles of the peripheral arteries and veins (such as coronary, peripheral and renal), and their stimulation causes vasoconstriction. Presynaptic α2 adrenergic receptors inhibit norepinephrine secretion in sympathetic ganglia. Stimulation of α2 receptors in smooth muscles causes arterial vasodilation and coronary vasoconstriction. β1 receptors are primarily localized in the heart and have positive inotropic and chronotropic effects on the heart. In addition, β1 receptors increase renin secretion, resulting in an increase in mean arterial pressure. β2 receptors lead to vasodilation in skeletal muscle arteries. It increases the release of norepinephrine from sympathetic nerve endings. Dopamine stimulates D1 and D2 receptors. D1 receptors cause vasodilation in the renal arteries. Stimulation of D2 receptors inhibits the secretion of norepinephrine in the sympathetic nerve endings, and it has a slightly negative inotropic effect on the heart. In addition, high levels of dopamine stimulate α and β1 receptors, causing vasoconstriction and increased heart rate.[15, 16]
PCCs often secrete both epinephrine and norepinephrine at different rates. PGLs, on the other hand, are not able to synthesize epinephrine since they do not contain the PNMT enzyme. They only secrete norepinephrine. Different catecholamines and their ratios may cause different vasoactive effects. In some cases, clinical findings can be extremely informative.
Prolonged and recurrent norepinephrine secretion results in prolonged vasoconstriction and increased blood pressure, and venous vasoconstriction results in decreased blood volume in the circulation. If preoperative α blockade is not applied and volume support is not provided in these patients, acute hypovolemia may occur as the vasoconstriction caused by norepinephrine disappears after PPGL extraction in surgery. In tumors that predominantly secrete epinephrine, tachyarrhythmia arises in addition to hypertension.[1, 5] In addition, postural hypotension occurs due to vasodilation in skeletal muscles with epinephrine stimulation. Hypertension and palpitations may not be present in tumors producing dopamine.[2, 15] Increased plasma catecholamine level, increased glycogenolysis and inhibition of insulin secretion in pancreatic islet cells result in findings of diabetes. Increased catecholamines can cause stress-induced cardiomyopathy (Takotsubo cardiomyopathy).[5]
The name pheochromocytoma crisis is the name given to a group of symptoms. It is a rare condition resulting from uncontrolled catecholamine secretion characterized by multisystem organ failure, high fever, encephalopathy, severe hypertension or hypotension. If not recognized and not treated urgently, it may progress to metabolic acidosis and lead to death.[2, 5]
Genetic Basis and Genetic Testing
Up to 40% of patients with PPGL have disease-specific germline mutations and are one of the most common hereditary tumors. At least 1/3 of the remaining 60% sporadic cases have a somatic mutation in predisposing genes and approximately 60% of patients with PPGL have germline or somatic mutation. More than 20 gene mutations have been detected to date.[10, 17, 18]
Patients with PPGL with these mutations can be divided into three groups according to the cancer genome map. These groups are:[19]
Cluster 1 - pseudohypoxic Krebs cycle-related genes
Cluster 2 - Kinase Signaling-Related Genes
Cluster 3 - Wnt Signaling-Related Genes
Cluster 1 - Pseudohypoxic Krebs Cycle-Related Genes
This group is called the pseudohypoxic pathway because it mimics cellular hypoxia.[20]
This group can be divided into two main subgroups. Those associated with tricarboxylic acid cycle contain germline mutations in genes encoding SDHx (succinate dehydrogenase) subunits (SDHA, SDHB, SDHC, SDHD), SDHAF2 (succinate dehydrogenase complex assembly factor-2), FH (fumarate hydratase), MDH2 (malate dehydrogenase 2), IDH (isocitrate dehydrogenase 1) and the majority of patients with hereditary PGL are associated with these mutations. Due to mutations in genes encoding enzymes in this group, the phosphorylation in the Krebs cycle is disrupted, glycolysis increases and results in the accumulation of oncometabolites, such as succinate, malate and fumarate. PPGLs, especially related to mutations in SDHx subunits, are often multiple, aggressive and metastatic tumors compared to those originating from other mutations, especially cluster 2 mutations.[7]
Mutations related to the other subgroup VHL/EPAS1 genes contain germline mutations in PHD1/2 (Egl-9 prolyl hydroxylase-1 and -2), VHL (von Hippel–Lindau tumor suppressor) and HIF2A/EPAS1/2 (hypoxia-inducible factor 2α) genes. 95-100% of patients with VHL syndrome are related to the mutation in the VHL tumor suppressor gene. By reduction in hydroxylation and reduction of these HIF proteins, HIF-α stabilizes and accumulates.[20]
This results in cellular proliferation, decreased apoptosis and angiogenesis. EPAS1 related gene mutations also increase the transcription of HIF-2α genes, similar to VHL.[7] In addition, it has recently been suggested that mutations in EPAS genes have unique molecular signatures in PPGLs.[21]
There were different effects of genes related to HIF transcription, oxidative phosphorylation, and angiogenesis compared with VHL- and SDHx-related PPGLs. In the future, mutations in VHL and EPAS genes can be divided into two specific subgroups.[7, 20]
Almost all tumors with mutations in cluster 1 are tumors with a noradrenergic biochemical phenotype. These produce norepinephrine and dopamine, not epinephrine.[20]
Cluster 2 - Kinase Signaling-Related Genes
Cluster 2 kinase signaling pathway is associated with germline or somatic mutations in the RET (rearranged-during-transfection) proto-oncogene, NF 1 (neurofibromin 1) tumor suppressor, HRAS, TMEM127 (transmembrane protein 127) and MAX (Myc-associated factor X) genes. Mutations in these genes stimulate cell proliferation, cell surveillance, cancer development, and angiogenesis by activating PI3K/AKT (phosphatidylinositol-3-kinase), RAS/RAF/ERK, mTORC1 (mammalian target of rapamycin)/p70S6K (p70S6 kinase) signaling pathways.[20]
The most common hereditary syndrome in this group is MEN syndromes.[20] Many of the cluster 2 mutations have adrenergic biochemical phenotype and are associated with increased epinephrine production as a result of high PNMT expression. Norepinephrine may increase or noradrenaline may be at normal levels.[20]
Most PCCs associated with these mutations are benign but have a high rate of recurrence and multifocality.[22]
Cluster 3 - Wnt Signaling-Related Genes
CSDE1 (Cold Shock Domain Containing E1) gene and the MAML3 (mastermind-like transcriptional coactivator 3) fusion gene contain PCCs with somatic mutations that activate the Wnt and Hedgehog (Hh) signaling pathways. These tumors strongly express chromogranin A.[20] It has been suggested that this category is related to aggressive PPGLs, because some of the patients with this mutation admitted with recurrence and metastasis.[19]
The germline mutation of this category is unknown.[22] The biochemical phenotype of this group is also unknown.[20]
In general, the risk of metastatic disease from high to low is as follows: mutations associated with cluster 1 tricarboxylic acid cycle, mutations associated with group 1VHL/ EPAS1 genes, cluster 3, cluster 2.[7]
Genetic Syndromes Associated with Pheochromocytoma and Paraganglioma
Multiple Endocrine Neoplasia Type 2 (MEN 2) (Table 1)
Table 1.
Genetic Syndromes Associated with Pheochromocytoma and Paraganglioma
| Syndrome | Subtype | Mutation Gen | Biochemical Phenotype | PCC/PGL penetrance | Penetrance PCC | Penetrance pPGL | Penetrance sPGL | Frequency All PCC | Bilaterality | Proportion Malignant | Other Manifestations |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MEN | MEN2A MEN2B | RET (10q11.2) | A | 50% | High | Very low | Very low | %5 | 50-60% | <5% | MTC pHPT MTC Mucosal neuroma, colonic ganglioneuromatosis, marfanoidfacies, skeletal abnormalities. |
| VHL | Type 1 | VHL (3p25-p26) | NA | 20% | High | Very low | Low | 5-10% | 40-50% | <5% | Hemangioblastoma in the retina and central nervous system, multiple abdominal neoplasms and cysts, RCC, and PCC |
| NF1 | NF1 (17q11.2) | A | 1-5% | Low | Very low | Very low | 1% | 20% | 10% | Cafe-au-lait macules, malignglioma, neurofibromas, freckling in the axillary or inguinal region, GIST, osseous lesion, and PCC | |
| FPGL | FPGL1 | SDHD (11q23) | NA, D | unknown | Low | High multifocal | Low | 5-10% | low | <5% | RCC GIST |
| FPGL2 | SDHAF2 (11q13.1) | NA, A | unknown | Very low | High multifocal | Very low | rare | unknown | pituitary adenoma | ||
| FPGL3 | SDHC (1q21) | NA, D | unknown | Low | Low | Low | rare | unknown | |||
| FPGL4 | SDHB (1p35-p36) | NA, D | unknown | Low | Intermediate | Intermediate | 10-20% | low | 30-70% | ||
| FPGL5 | SDHA (5p15.33) | NA, D | unknown | Very low | Very low | Very low | <5% | low | 10% |
PCC: Pheochromocytoma; pPGL: Parasympathetic paraganglioma; sPGL: Sympathetic paraganglioma; MTC: Medullary thyroid cancer; pHPT: Primary hyperparathyroidism; NF1: Neurofibromatosis type 1 (von Recklinghausen’s disease); VHL: Von Hippel Lindau syndrome; MEN: Multiple Endocrine Neoplasia; MEN2A: Multiple Endocrine Neoplasia type 2A; MEN2B: Multiple Endocrine Neoplasia type 2B; RCC: Renal cell carcinoma; GIST: Gastrointestinal stromal tumor; FPGL: Familial paraganglioma syndromes.
MEN2 syndromes are autosomal dominant diseases associated with RET proto-oncogene mutation located on chromosome 10. There is a strong phenotype-genotype relationship in these syndromes. More than a hundred RET point mutations have been identified, approximately 90% have single point mutations.[23]
There are three subtypes of MEN syndrome. 75% of MEN syndromes are MEN2A, 5% are MEN2B and 20% are familial medullary thyroid cancer. The familial medullary thyroid cancer subgroup has no known components other than medullary cancer.[24]
Medullary cancer develops in almost 100% of the patients with MEN2A, PCC in 50%, primary hyperparathyroidism in 10%. Approximately 100% of the patients with MEN2B develop medullary cancer, 50% develop PCC, and 95% develop the gastrointestinal system, mucosal neuroma in the mouth and lips and Marfanoid appearance.[24] In MEN syndromes, the tumor is often localized in the adrenal medulla and PGL is very rare.[7] PCCs in this syndrome make up 5% of all PCCs.[5] Bilateral adrenal involvement occurs in approximately 50-60%.[25, 26] Bilateralism can be synchronous or metachronous.[27] Its biochemical phenotype is adrenergic.[28]
Metastatic pheochromocytoma is seen below 5%.[5, 25] In addition, very rarely, head and neck PGL is defined in MEN2 syndromes.[28]
PCC should be investigated before any intervention in MEN2 syndrome, and if any, PCC should be treated before surgery for other components of the syndrome.
Von Hippel Lindau Syndrome (Table 1)
Von Hippel Lindau Syndrome is an autosomal dominant syndrome that arises from the germline mutation in the VHL tumor suppressor gene on the 3p25-26 chromosome.[25] Several different mutations in the VHL gene may cause this syndrome and affect multiple organs. Its prevalence is one in 36000 people.[24] Cerebellar and spinal hemangioblastoma, retinal hemangioma, renal cysts, clear cell renal carcinoma, pancreatic neuroendocrine tumors, cysts and cystadenomas of the pancreas, PPGLs, cystadenomas in gonads, benign asymptomatic lung lesions, benign asymptomatic liver lesions, liver cysts, endolymphatic cystic tumors can be seen in this syndrome.[25] In 20% of the patients with VHL, PCC usually occurs and is seen at a young age. Half of the pediatric age group, which presents with PPGL, has VHL syndrome.[2]
Patients with PPGL with VHL syndrome constitute 5-10% of all PPGL patients.[5] 40-50% of PCCs in this syndrome are bilateral.[25] The biochemical phenotype is noradrenergic. Metastatic PCC is rare.[2] In VHL syndrome, PGL is rare, usually of parasympathetic origin and does not secrete a significant amount of catecholamines.[29] VHL syndrome is divided into two subgroups, as type 1 if there is no PCC or type 2 if PCC is present. Type 1 includes cerebellar and spinal hemangioblastoma, retinal hemangioma, multiple abdominal cysts and tumors, clear cell renal carcinoma, but no PCC. Type 2 is divided into three types according to the presence and aggressiveness of renal carcinoma. Cerebellar and spinal hemangioblastoma, retinal hemangioma, low-risk renal carcinoma and PCC are present in Type 2A, cerebellar and spinal hemangioblastoma, retinal hemangioma, high-risk renal carcinoma, endocrine neoplasms, pancreatic cysts and PCC are present in Type 2B and only PCC is present in Type 2C.[29]
Cases with PCC are often asymptomatic and, unlike sporadic PCCs, they do not release excess catecholamines.[29]
Neurofibromatosis 1 (Von Recklinghausen’s Disease (VRD) (Table 1)
Neurofibromatosis 1 is an autosomal dominant syndrome that arises from a mutation in the NF1 gene, and its prevalence is 1 in 3000 people. This gene is localized on the 17qll.2 chromosome and encodes the neurofibromin protein. In the absence or decreased levels of neurofibromin with mutations in the NF1 gene, cell proliferation through various pathways and inhibited apoptosis occur.[30] NF1 is characterized by multiple neuromas and is characterized by 15% peripheral nerve sheath tumors.[2] In addition, café-au-lait spots, freckles in the axilla and inguinal areas, malignant glioma, bone lesions, gastrointestinal stromal tumors, and PPGL may occur.[5, 31]
PPGL develops in 1-5% of patients with NF-1. Almost all of these tumors are PCC, and PGL occurs rarely. They constitute 1% of all patients with PCC.[5] They are 20% bilateral.[1]
Hereditary Paraganglioma Syndromes (PGL Syndromes) TYPE 1-5 (SDH COMPLEX) (Table 1)
There are four subunits of succinate dehydrogenase (SDHA, SDHB, SDHC, SDHD), which cause syndromes with the autosomal dominant inheritance that occur with germline mutations in genes on different chromosomes that encode different subunits. Type 1 is associated with SDHD mutations, type 2 with SDHAF2 mutations, type 3 with SDHC mutations, type 4 with SDHB mutations, and type 5 with SDHA mutations.[5, 31] The prevalence is estimated as 1 in 1 million people.[24] These syndromes are more often associated with PGL. Head and neck PGLs are common. PCCs are seen in all five types. Malignancy rate is higher in SDHB mutations and 30-70% malignancy has been reported. In other types, the malignancy rate is low.[2]
Apart from these, renal cell carcinoma, gastrointestinal stromal tumors, pituitary adenomas can be seen. There is a risk of papillary thyroid cancer and breast cancer in SDHB mutations.[32]
Other Mutations Associated with Pheochromocytoma/Paraganglioma
PPGL can also be seen in TMEM127 mutations, MAX mutations, KIF1B mutations, EGLN1 mutations, MDH2 mutations, FH mutations, KIF1β mutations, HIF2A, CSDE1, MAML3 Pacak-Zhuang Syndrome, Carney complex, Carney-Stratakis Syndrome, MEN1 Syndrome.[1, 20, 31]
Clinical and Biological Behavior of Pheochromocytoma/Paraganglioma
Although PPGLs have scoring systems for histological evaluation of the primary tumor, it is not possible to diagnose whether the tumor is malignant, since there is no histological system approved for the biological aggressiveness of this tumor group.[33] Malignancy in these tumors can be defined as metastasis at presentation or during subsequent follow-up. Metastasis is defined as the presence of chromaffin tissue in non-chromaffin organs, such as lymph nodes, liver, lungs and bone.[34] The proposed benign and malignant definitions for PCC in the 3rd version of the World Health Organization (WHO) published on the classification of endocrine tumors in 2004 were removed in the 4th version published in 2017.[35, 36] Because all PCCs and PGLs may have metastatic potential. Therefore, in version 4, the “metastatic” term is used instead of “malignant” for both PCCs and PGLs. This also eliminated the confusion that may occur between locally invasive and distant metastatic tumors.[9] PGLs are divided into two groups as sympathetic paragangliomas and parasympathetic paragangliomas according to their clinical and biological behavior in version 4. Parasympathetic PGLs are often localized primarily in the head and neck region, rarely localized in the thorax and pelvis. For this reason, PGLs are examined under two subtitles as sympathetic PGLs and head and neck PGLs.[36]
Metastatic Pheochromocytomas/Paragangliomas
Although most of the PPGLs are benign, the metastatic disease may develop in 15-17%. Metastatic disease is reported between 2-25% in PCCs and 2.4-60% in PGLs.[37, 38]
Head and neck PGLs constitute 20% of all PGLs, but the rate of metastatic disease is 5%.[9] Synchronous metastasis is present in 35-52% of patients with metastatic PPGL and metachronous in 48-65%.[37, 38] While at least 30% of the patients with PPGL have a hereditary predisposition, almost 50% of metastatic patients have germline mutations.[39] In metastatic disease, male gender, advanced age, synchronous metastasis, larger primary tumor, patients without primary tumor resection, patients with sympathetic PGL and tumors with increased dopamine are associated with short-term survival.[37, 38] The mean tumor diameter is larger and 8.2 cm in patients with metastatic PPGL, while 4.9 cm in non-metastatic tumors. 16% of metastatic tumors are under 5 cm in size, most of them (78.5%) are PGL, and only 21.5% of them are PCC.[37] Germline SDHB mutation is thought to be one of the most important risk factors for the development of metastases in patients with PPGL.[40] More than half of patients with metastatic PGL and 5-10% of patients with metastatic PCC have germline SDHB mutation. MAX mutation carriers have a 10-25% risk of developing metastatic disease, but MAX mutations are rare. VHL and SDHD mutation carriers have less than 5% risk of metastatic disease.[39] The course of metastatic disease is heterogeneous and the 5-year survival rate ranges between 40-85%.[33] The most common metastasis sites in metastatic PPGL are 80% local and distant lymph nodes, 72% bones, 50% liver, and 50% lungs.[39] Bone and lymph node metastasis in PGL is more than PCC, while liver metastasis is more common in PCC.[40] 91% of bone metastasis are lytic lesions, 81% are in vertebrae and 67% in the pelvis. Bone lesions were detected in CT or MRI with a rate of 81% and the detection rate in FDG-PET/CT is 95%. Survival in bone metastases is better than liver and lung metastases. Median total survival was 12 years in those with only bone metastasis, 7.5 years in liver and lung metastases, and five years in patients with bone, liver and lung metastases.[41] The TNM staging system of the American Joint Committee on Cancer (AJCC) was developed to predict the prognosis, based on the specific anatomical features of the primary tumor and the occurrence of metastasis. In the light of the data accumulated in the literature, for the first time, staging for PPGL was done in the updated version 8 of the TNM staging system (Table 2).[42] TNM staging system for PPGL was implemented on January 01, 2017. It will guide clinicians in determining the type and extension of surgery, indications for neoadjuvant and adjuvant treatments and the follow-up needed by metastatic PPGL patients. Comprehensive staging and survival data will be collected over the coming years. These data are expected to further our understanding of PPGL and facilitate the development of effective treatments.[40]
Table 2.
TNM classifications of pheochromocytoma and paraganglioma
| Primary Tumor (T) | |
| Tx | Tumor cannot be assessed |
| T1 | <5 cm in greatest diameter, no extra-adrenal invasion |
| T2 | >5 cm or sympathetic paraganglioma of any size, no extra-adrenal invasion |
| T3 | Any size with invasion into surrounding tissues (e.g., liver, pancreas, spleen, and and kidneys) |
| Nodal Involvement (N) | |
| Nx | Regional lymph nodes cannot be Assessed |
| N0 | No regional lymph node metastasis |
| N1 | Regional lymph node metastasis |
| Distant metastases (M) | |
| M0 | No distant metastasis |
| M1 | Distant metastasis |
| M1a | Metastasis to only bone |
| M1b | Metastasis to only distant lymph nodes/liver or lung |
| M1c | Metastasis to the bone and multiple |
| other sites | |
| Stage Groups | |
| Stage 1 | T1, N0, M0 |
| Stage 2 | T2, N0, M0 |
| Stage 3 | T1, N1, M0 or T2, N1, M0 or T3, |
| Any N, M0 | |
| Stage 4 | Any T, Any N, Any M1 |
Disclosures
Peer-review: Externally peer-reviewed.
Conflict of Interest: None declared.
Authorship Contributions: Concept – M.U., N.A.; Design – M.U., N.A.; Supervision – M.U.; Materials – M.U., N.A.; Data collection &/or processing – M.U., N.A.; Analysis and/or interpretation – M.U.; Literature search – M.U., N.A.; Writing – M.U., N.A.; Critical review – M.U.
References
- 1.Tevosian SG, Ghayee HK. Pheochromocytomas and Paragangliomas. Endocrinol Metab Clin North Am. 2019;48:727–50. doi: 10.1016/j.ecl.2019.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Patel D, Phay JE, Yen TWF, Dickson PV, Wang TS, Garcia R, et al. Update on Pheochromocytoma and Paraganglioma from the SSO Endocrine/Head and Neck Disease-Site Work Group. Part 1 of 2:Advances in Pathogenesis and Diagnosis of Pheochromocytoma and Paraganglioma. Ann Surg Oncol. 2020;27:1329–37. doi: 10.1245/s10434-020-08220-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schreiner F, Beuschlein F. Disease monitoring of patients with pheochromocytoma or paraganglioma by biomarkers and imaging studies. Best Pract Res Clin Endocrinol Metab. 2019 Oct 21; doi: 10.1016/j.beem.2019.101347. [Epub ahead of print], doi:10.1016/j.beem.2019.101347. [DOI] [PubMed] [Google Scholar]
- 4.Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, Naruse M, Pacak K, Young WF, Jr Endocrine Society. Pheochromocytoma and paraganglioma:an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915–42. doi: 10.1210/jc.2014-1498. [DOI] [PubMed] [Google Scholar]
- 5.Kiernan CM, Solórzano CC. Pheochromocytoma and Paraganglioma:Diagnosis, Genetics, and Treatment. Surg Oncol Clin N Am. 2016;25:119–38. doi: 10.1016/j.soc.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 6.Chen H, Sippel RS, O'Dorisio MS, Vinik AI, Lloyd RV, Pacak K North American Neuroendocrine Tumor Society (NANETS) The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors:pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39:775–83. doi: 10.1097/MPA.0b013e3181ebb4f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crona J, Taïeb D, Pacak K. New Perspectives on Pheochromocytoma and Paraganglioma:Toward a Molecular Classification. Endocr Rev. 2017;38:489–515. doi: 10.1210/er.2017-00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bholah R, Bunchman TE. Review of Pediatric Pheochromocytoma and Paraganglioma. Front Pediatr. 2017;5:155. doi: 10.3389/fped.2017.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lam AK. Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours. Endocr Pathol. 2017;28:213–27. doi: 10.1007/s12022-017-9484-5. [DOI] [PubMed] [Google Scholar]
- 10.Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma:from genetics to personalized medicine. Nat Rev Endocrinol. 2015;11:101–11. doi: 10.1038/nrendo.2014.188. [DOI] [PubMed] [Google Scholar]
- 11.Graham JB. Pheochromocytoma and hypertension;an analysis of 207 cases. Int Abstr Surg. 1951;92:105–21. [PubMed] [Google Scholar]
- 12.Eisenhofer G, Huynh TT, Hiroi M, Pacak K. Understanding catecholamine metabolism as a guide to the biochemical diagnosis of pheochromocytoma. Rev Endocr Metab Disord. 2001;2:297–311. doi: 10.1023/a:1011572617314. [DOI] [PubMed] [Google Scholar]
- 13.Cisco RM, Grondal S, Hamberger B. Adrenal physiology. In: Clark OH, Duh QY, Kebebew E, Gosnell JE, Shen WT, editors. Textbook of Endocrine Surgery. 3rd ed. Jaypee Brothers Medical Publishers; 2016. pp. 973–8. [Google Scholar]
- 14.Lal G, Clark OH. Thyroid, parathyroid and adrenal. In: Brunucardi FC, Andersen DK, Billiar TR, Kao LS, Hunter JG, Matthews JB, et al., editors. Schwartz's Principles of Surgery. 11th ed. New York: McGraw Hill; 2019. pp. 1625–704. [Google Scholar]
- 15.Zuber SM, Kantorovich V, Pacak K. Hypertension in pheochromocytoma:characteristics and treatment. Endocrinol Metab Clin North Am. 2011;40:295–311. doi: 10.1016/j.ecl.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gunawardane PTK, Grossman A. Phaeochromocytoma and Paraganglioma. Adv Exp Med Biol. 2017;956:239–59. doi: 10.1007/5584_2016_76. [DOI] [PubMed] [Google Scholar]
- 17.Jochmanova I, Pacak K. Genomic Landscape of Pheochromocytoma and Paraganglioma. Trends Cancer. 2018;4:6–9. doi: 10.1016/j.trecan.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khatami F, Mohammadamoli M, Tavangar SM. Genetic and epigenetic differences of benign and malignant pheochromocytomas and paragangliomas (PPGLs) Endocr Regul. 2018;52:41–54. doi: 10.2478/enr-2018-0006. [DOI] [PubMed] [Google Scholar]
- 19.Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell. 2017;31:181–93. doi: 10.1016/j.ccell.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nölting S, Ullrich M, Pietzsch J, Ziegler CG, Eisenhofer G, Grossman A, et al. Current Management of Pheochromocytoma/Paraganglioma:A Guide for the Practicing Clinician in the Era of Precision Medicine. Cancers (Basel) 2019;11:1505. doi: 10.3390/cancers11101505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhuang Z, Yang C, Lorenzo F, Merino M, Fojo T, Kebebew E, et al. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med. 2012;367:922–30. doi: 10.1056/NEJMoa1205119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antonio K, Valdez MMN, Mercado-Asis L, Taïeb D, Pacak K. Pheochromocytoma/paraganglioma:recent updates in genetics, biochemistry, immunohistochemistry, metabolomics, imaging and therapeutic options. Gland Surg. 2020;9:105–23. doi: 10.21037/gs.2019.10.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raue F, Frank-Raue K. Update on Multiple Endocrine Neoplasia Type 2:Focus on Medullary Thyroid Carcinoma. J Endocr Soc. 2018;2:933–43. doi: 10.1210/js.2018-00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grogan RH. The Importance of Family History in the Management of Endocrine Disease. Surg Clin North Am. 2019;99:711–20. doi: 10.1016/j.suc.2019.04.016. [DOI] [PubMed] [Google Scholar]
- 25.Iacobone M, Citton M, Viel G, Schiavone D, Torresan F. Surgical approaches in hereditary endocrine tumors. Updates Surg. 2017;69:181–91. doi: 10.1007/s13304-017-0451-y. [DOI] [PubMed] [Google Scholar]
- 26.Frunzac RW, Grant CS. Pheochromocytoma. In: Clark OH, Duh QY, Kebebew E, Gosnell JE, Shen WT, editors. Textbook of Endocrine Surgery. 3rd ed. Jaypee Brothers Medical Publishers; 2016. pp. 1047–67. [Google Scholar]
- 27.Asari R, Scheuba C, Kaczirek K, Niederle B. Estimated risk of pheochromocytoma recurrence after adrenal-sparing surgery in patients with multiple endocrine neoplasia type 2A. Arch Surg. 2006;141:1199–205. doi: 10.1001/archsurg.141.12.1199. [DOI] [PubMed] [Google Scholar]
- 28.Boedeker CC, Erlic Z, Richard S, Kontny U, Gimenez-Roqueplo AP, Cascon A, et al. Head and neck paragangliomas in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. J Clin Endocrinol Metab. 2009;94:1938–44. doi: 10.1210/jc.2009-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crespigio J, Berbel LCL, Dias MA, Berbel RF, Pereira SS, Pig¬natelli D, et al. Von Hippel-Lindau disease:A single gene, sev¬eral hereditary tumors. J Endocrinol Investig. 2018;41:21–31. doi: 10.1007/s40618-017-0683-1. [DOI] [PubMed] [Google Scholar]
- 30.Shen MH, Harper PS, Upadhyaya M. Molecular genetics of neurofibromatosis type 1 (NF1) J Med Genet. 1996;33:2–17. doi: 10.1136/jmg.33.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu P, Li M, Guan X, Yu A, Xiao Q, Wang C, et al. Clinical Syndromes and Genetic Screening Strategies of Pheochromocytoma and Paraganglioma. J Kidney Cancer VHL. 2018;5:14–22. doi: 10.15586/jkcvhl.2018.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martucci VL, Pacak K. Pheochromocytoma and paraganglioma:diagnosis, genetics, management, and treatment. Curr Probl Cancer. 2014;38:7–41. doi: 10.1016/j.currproblcancer.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corssmit EPM, Snel M, Kapiteijn E. Malignant pheochromocytoma and paraganglioma:management options. Curr Opin Oncol. 2020;32:20–6. doi: 10.1097/CCO.0000000000000589. [DOI] [PubMed] [Google Scholar]
- 34.Plouin PF, Amar L, Dekkers OM, Fassnacht M, Gimenez-Roqueplo AP, Lenders JW, et al. Guideline Working Group. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol. 2016;174:G1–10. doi: 10.1530/EJE-16-0033. [DOI] [PubMed] [Google Scholar]
- 35.DeLellis RA, Lloyd RV, Heitz PU, Eng C. 3rd ed. Lyon: IARC; 2004. WHO classification of tumours:pathology and genetics of tumours of endocrine organs. [Google Scholar]
- 36.Lloyd RV, Osamura RY, Kloppel G, Rosai J. 4th ed. Lyon: IARC; 2017. WHO classification of tumours:pathology and genetics of tumours of endocrine organs. [Google Scholar]
- 37.Ayala-Ramirez M, Feng L, Johnson MM, Ejaz S, Habra MA, Rich T, et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas:primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab. 2011;96:717–25. doi: 10.1210/jc.2010-1946. [DOI] [PubMed] [Google Scholar]
- 38.Hamidi O, Young WF, Jr, Iñiguez-Ariza NM, Kittah NE, Gruber L, Bancos C, Tamhane S, Bancos I. Malignant Pheochromocytoma and Paraganglioma:272 Patients Over 55 Years. J Clin Endocrinol Metab. 2017;102:3296–305. doi: 10.1210/jc.2017-00992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jimenez C, Rohren E, Habra MA, Rich T, Jimenez P, Ayala-Ramirez M, et al. Current and future treatments for malignant pheochromocytoma and sympathetic paraganglioma. Curr Oncol Rep. 2013;15:356–71. [Google Scholar]
- 40.Roman-Gonzalez A, Jimenez C. Malignant pheochromocytoma-paraganglioma:pathogenesis, TNM staging, and current clinical trials. Curr Opin Endocrinol Diabetes Obes. 2017;24:174–83. doi: 10.1097/MED.0000000000000330. [DOI] [PubMed] [Google Scholar]
- 41.Ayala-Ramirez M, Palmer JL, Hofmann MC, de la Cruz M, Moon BS, Waguespack SG, et al. Bone metastases and skeletal-related events in patients with malignant pheochromocytoma and sympathetic paraganglioma. J Clin Endocrinol Metab. 2013;98:1492–7. doi: 10.1210/jc.2012-4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jimenez C, Libutti SK, Landry CS, Lloyd RV, McKay RR, Rohren E, et al. Adrenal-neuroendocrine tumors. In: Amin MB, Edge S, Greene F, Byrd DR, Brookland RK, Washington MK, et al., editors. AJCC cancer staging manual. 8th ed. New York: Springer; 2017. pp. 919–27. [Google Scholar]