

Abstract
Adeno-associated virus (AAV) vector serotypes vary in their ability to transduce hepatocytes from different species. Chimeric mouse models harboring human hepatocytes have shown translational promise for liver-directed gene therapies. However, many variables that influence human hepatocyte transduction and transgene expression in such models remain poorly defined. Here, we aimed to test whether three experimental conditions influence AAV transgene expression in immunodeficient, fumaryl-acetoactetate-hydrolase-deficient (Fah−/−) chimeric mice repopulated with primary human hepatocytes. We examined the effects of the murine liver injury cycle, human donor variability, and vector doses on hepatocyte transduction with various AAV serotypes expressing a green fluorescent protein (GFP). We determined that the timing of AAV vector challenge in the liver injury cycle resulted in up to 7-fold differences in the percentage of GFP expressing human hepatocytes. The GFP+ hepatocyte frequency varied 7-fold between human donors without, however, changing the relative transduction efficiency between serotypes for an individual donor. There was also a clear relationship between AAV vector doses and human hepatocyte transduction and transgene expression. We conclude that several experimental variables substantially affect human hepatocyte transduction in the Fah−/− chimera model, attention to which may improve reproducibility between findings from different laboratories.
Keywords: humanized mice, AAV, gene therapy, liver, human hepatocytes, donor variability, hemophilia
Graphical Abstract
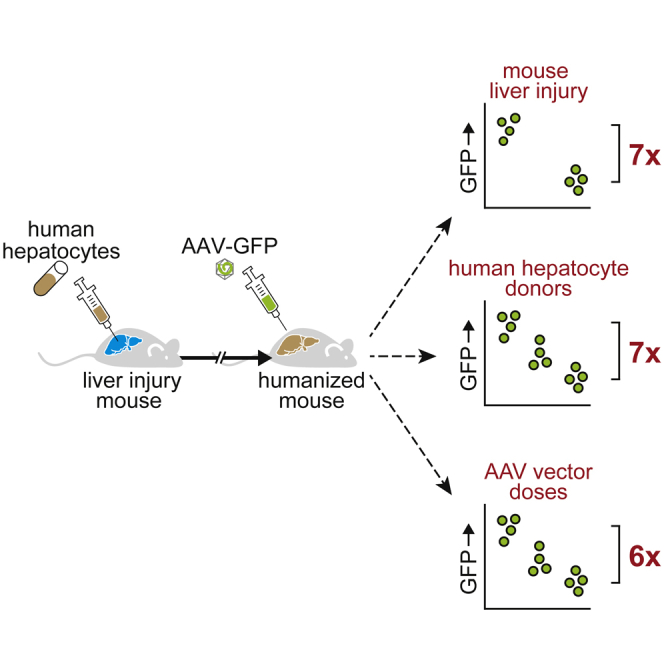
Liver chimeric mice are widely used to model human hepatocyte susceptibility to adeno-associated virus (AAV) gene vector transduction with inconsistent finding between laboratories. Zou et al. quantify to what extent three experimental conditions—namely, mouse liver injury, human donor variability, and AAV doses—affect human hepatocyte transduction in chimeric mice.
Introduction
Adeno-associated virus (AAV) gene therapy targeting hepatocytes is being pursued for many liver conditions and has progressed into clinical trials for inborn errors of metabolism and hemophilia.1 Several vectors for treatment of hemophilia by hepatic gene transfer have entered phase III evaluation.2, 3, 4 Diverse serotypes or engineered capsid variants with liver tropism are utilized in various ongoing clinical trials or are in advanced pre-clinical development. How to optimally predict performance of a specific capsid in human liver gene transfer during pre-clinical development remains a challenge. Hepatocytes from different species vary in their susceptibility to AAV vector transduction and transgene expression. This has limited the value of rodent studies in predicting AAV liver gene therapy responses in humans. Non-human primates are expected to be a better predictor but also have limitations in mimicking human gene therapy.5 While investigating AAV vector transduction of human hepatocytes holds potential to better model clinical gene therapy, in vitro studies are unlikely to predict in vivo efficacy. Hence, methods to populate mice with human hepatocytes were developed to enable studies on in vivo transduction of human hepatocytes.
Liver chimeric mice allow for the study human hepatocytes in laboratory animals. In these xenograft models, primary human hepatocytes (PHHs) are transplanted into immunodeficient mice with a form of murine liver injury.6 Exploiting the ability of healthy hepatocytes to proliferate in response to liver damage, the murine liver injury in these models drives the engraftment and proliferation of human hepatocytes. There are several forms of murine liver injury that can result in high humanization. The two most widely used models are urokinase-plasminogen activator (uPA) transgenic lines7 and fumaryl-acetoactetate-hydrolase-deficient (Fah−/−) lines.8, 9, 10 Injury in the latter model is regulated by the intermittent withdrawal of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC, or nitisinone), otherwise called NTBC cycling.11 In addition to liver injury, a second requirement shared between liver chimeric mouse models is an immunodeficiency to prevent xenorejection. While murine T cell deficiency is minimally required to accept xenografts in these models, the severely immunodeficient Fah−/− non-obese diabetic (NOD) Rag1−/− Il2rgnull (FNRG) background improves humanization over less severely immunodeficient Fah−/− mice (Y.P.d.J., unpublished data).12,13 Human chimerism in these models strongly correlates with the level of human markers in mouse serum, and, typically, human albumin (hAlb) is quantified to determine humanization levels.14, 15, 16 In our laboratory, FNRG mice typically reach peak hAlb serum levels 3–4 months after PHH transplantation and NTBC cycling.
In recent years, Fah−/− chimera models have been used to study AAV biology in human hepatocytes. Their uses include the creation of bioengineered capsids with superior human hepatocyte transduction characteristics,5,17,18 assessment of human hepatocyte transduction comparing different serotypes,19, 20, 21, 22 and the correction of human gene defects.19 All these studies tested a number of different AAV serotypes expressing a green fluorescent protein (GFP); and while several studies used similar AAV serotypes, results substantially diverged not only as to the percentage of transduced human hepatocytes but also with regard to serotype hierarchies. This may be due to the fact that each study used different PHH donors and that several other experimental parameters also varied or were not clearly specified. Not knowing to what extent experimental parameters affect human hepatocyte AAV transduction limits the generalizability of results from Fah−/− chimera studies. We, therefore, set out to quantify the effects of three experimental conditions on human hepatocyte transduction with a limited number of AAV serotypes expressing GFP. We tested how NTBC cycling affects human hepatocyte GFP expression, quantified the variability between PHH donors, and studied vector dose responses.
Results
Murine Liver Injury Affects AAV Transgene Expression in Humanized FNRG Mice
Maintenance of the human hepatocyte graft in the FNRG model requires ongoing NTBC cycling. Here, we sought to investigate whether NTBC cycling affects AAV-GFP transduction of the human graft. We compared the timing of vector challenge in relationship to how humanized FNRG (huFNRG) mice were cycled. A schematic illustrates the workflow of these experiments in Figure 1A. PHH donor 1 (PHH1) was transplanted into FNRG mice that underwent NTBC cycling for approximately 3 months to reach plateau hAlb serum values. huFNRG mice were then transduced with 1 × 1011 vector genomes (VGs) of AAV-GFP at various times of the NTBC cycle (Figure 1B). Thirteen days later, hepatocytes were harvested from chimeric livers, and GFP expression in human hepatocytes was assessed by flow cytometry.21 Hepatocytes were enriched by density centrifugation and stained for human leukocyte antigen I (HLA-I) and mouse CD81 (mCD81). After exclusion of doublets and mCD81+ hepatocytes (Figure S1A), the percentage of GFP+ human hepatocytes was quantified (Figure 1C). Comparing three transduction protocols, a similar pattern of GFP+ fractions was observed across several AAV serotypes previously used in hemophilia gene therapy clinical studies (Figure 1D).5,21,23 AAV transduction of huFNRG mice that were off NTBC (termed “poor” protocol) resulted in a very low percentage of GFP-expressing human hepatocytes. When mice were transduced 1 day after NTBC re-introduction (“medium” protocol), the percentage of GFP-expressing hepatocytes substantially increased, with a further slight enhancement when vector administration was preceded by a longer time off NTBC (“efficient” protocol). When different AAV serotypes were pooled by transduction protocol as a percentage of the “efficient” protocol for each serotype, the “medium” protocol resulted in 23% fewer GFP-expressing human hepatocytes, while the “poor” protocol diminished the portion of GFP+ hepatocytes by 86% (Figure 1E). These data show that, in this Fah−/− chimera model, the timing of AAV vector administration in relationship to the NTBC cycle severely affects the percentage of GFP-expressing human hepatocytes.
Figure 1.

The Fah−/− Liver Injury Cycle Affects Human Hepatocyte AAV-GFP Expression
(A) Timeline outlining the creation of human liver chimeric mice, challenge with AAV-GFP, and flow-cytometric analysis of hepatocytes isolated from chimeric livers. PHH, primary human hepatocyte; FNRG, Fah−/− NOD Rag1−/−Il2rgnull; NTBC, nitisinone; huFNRG, humanized FNRG; mCD81, mouse CD81. (B) Schematic of three AAV-GFP challenge and hepatocyte harvest protocols in huFNRG mice. AAV vectors were injected while mice were 1 day on NTBC after 2 weeks off (“efficient”), on NTBC 1 day after a short 2-day interval off (“medium”), or off the protective drug NTBC for 3 days (“poor”). (C) Flow cytometry examples of GFP expression on HLA-I+ hepatocytes from mice challenged with various AAV-GFP vectors. Mice are from independent experiments. (D) GFP+ human hepatocyte percentages by AAV serotype comparing the three challenge protocols. Symbols represent individual mice; black bar represents the mean of group. (E) Mice challenged with various AAV serotypes were graphed as percentage GFP+ hepatocytes of the mean of the “efficient” protocol for each serotype. Symbols represent individual mice; black bar represents the mean of group. The p values were calculated by Mann-Whitney test.
Variable Transduction between Human Hepatocyte Donors
Clinical trials with liver-directed gene therapies show a range of transgene product plasma levels within study populations. To test whether hepatocyte susceptibility to AAV transduction differs between individuals, we transplanted FNRG mice with eight different PHH donors. FNRG livers were humanized to varying degrees, as determined by mean plateau hAlb levels in mouse sera (Table 1). After peak humanization was reached, huFNRG mice were transduced with 1 × 1011 VGs AAV-GFP using the “efficient” protocol (Figure 1B), and GFP expression in human hepatocytes was quantified by flow cytometry. For AAVLK03, the mean human hepatocyte GFP+ fraction ranged from 22% for PHH1 to 3% for PHH4 (7.2-fold difference) (Figure 2A). Within experiments, the GFP expression patterns varied between animals transplanted with the same donor (Figure S1B). We then tested whether liver humanization levels correlated with the fraction of GFP+ hepatocytes using hAlb serum levels as a surrogate for liver humanization.14, 15, 16 For these eight PHH donors, only a weak negative correlation between the percentage of GFP+ human hepatocytes and peak serum hAlb levels was observed that did not reach statistical significance (Figure 2B). There was also no relationship between PHH gender and GFP expression, with an average 13.6% human GFP+ cells with five male donors and 12.4% for three female donors (p = 0.85). Next, a subset of five PHH donors was used to test donor variability with another AAV serotype. Mean percentages of GFP+ human hepatocytes in mice transduced with AAVSpark100, an AAV serotype with established human hepatocyte tropism,23 varied from 3.8% (PHH5) to 24.1% (PHH7), a 6.2-fold difference (Figure 2C). For some donors, striking variability was again observed between mice in individual experiments (Figure S1C). Individual donors tended to respond similarly to AAVSpark100 and AAVLK03 administration. Indeed, a strong correlation between AAVSpark100 and AAVLK03 mean transduction efficiency was observed within the same PHH donor (Figure 2D). Similarly to AAVLK03, only a very weak negative correlation was found between hAlb serum levels and mean GFP+ fractions for AAVSpark100 (r = −0.25, p = 0.69). These data show that PHH donors vary up to 7-fold in their susceptibility to AAV vectors independent of liver humanization, and individual PHH donors are similarly susceptible to AAVSpark100- and AAVLK03-delivered GFP expression.
Table 1.
PHH Donor Characteristics
| Name | Vendor | Lot ID | Gender | Age (Years) | Mean Peak hAlb (μg/mL) |
|---|---|---|---|---|---|
| PHH1 | Celsis | AWG | male | 6 | 1,498 |
| PHH2 | TRL (Lonza) | 4129 | male | 1 | 3,963 |
| PHH3 | TRL (Lonza) | 4188 | female | 1 | 11,669 |
| PHH4 | TRL (Lonza) | 4131 | female | 48 | 5,836 |
| PHH5 | TRL (Lonza) | 4145 | male | 2 | 4,341 |
| PHH6 | TRL (Lonza) | 4069 | male | 30 | 3,321 |
| PHH7 | Becton Dickinson | HFCP940 | female | 2 | 629 |
| PHH8 | BioIVT | GEB | male | 48 | 797 |
Figure 2.

Hepatocyte AAV-GFP Expression Varies between Human Donors
(A) FNRG mice humanized with 8 different PHH donors were challenged with AAVLK03-GFP, and hepatocyte GFP expression was quantified by flow cytometry. Symbols represent individual mice; black bar represents the mean of the group. The p values were calculated by Mann-Whitney test and depicted when <0.05. (B) Peak mean hAlb serum levels of groups of FNRG mice transplanted with various PHH donors was plotted against mean percentage of GFP+ human hepatocytes. Symbols represent means of groups; correlation was calculated by Pearson test; colors correspond to those in (A). (C) FNRG mice humanized with 5 PHH donors were challenged with AAVSpark100-GFP, and hepatocyte GFP expression was quantified by flow cytometry. Symbols represent individual mice. (D) Human hepatocyte GFP+ percentages were compared between AAVLK03-GFP and AAVSpark100-GFP challenged mice humanized with 5 PHH donors. Symbols represent mean percentages of groups; correlation was calculated by Pearson test; colors correspond to those in (A) and (C).
AAV Dose Response in huFNRG Mice
A wide range of AAV vector doses have been used across animal and clinical studies, including in human liver chimeric Fah−/− mice from various laboratories. Here, we sought to quantify dose responses in huFNRG mice using two different readouts on human hepatocytes isolated from chimeric livers; namely, the amount of vector genomes per human hepatocyte (VGs) and the fraction of GFP-expressing human hepatocytes. To quantify VGs focusing primarily on the human hepatocytes, we needed populations highly enriched for human cells. Therefore, we generated three groups of retrorsine-preconditioned huFNRG mice with PHH3,24 which reliably humanizes the majority of the chimeric livers as determined by hAlb serum values greater than 10 mg/mL (Figure 3A). Humanization was confirmed by liver histology. Low-magnification H&E histology showed few small areas of mouse hepatocytes, which can be identified by increased hematoxylin staining compared to human areas (Figure 3B). Humanization was further confirmed by human-specific nuclear staining for NuMA1 (Figure 3B).14 These highly chimeric huFNRG mice were challenged with an AAVLK03 vector, which minimally transduces mouse hepatocytes.5 Using the “efficient” protocol (Figure 1A), mice were given three different doses: 2 × 1010, 1 × 1011, and 5 × 1011 VGs (indicated as low, medium, and high in the graphs in Figure 3B). Thirteen days later, hepatocytes were isolated from chimeric livers, and the percentages of human and mouse hepatocytes were quantified by flow cytometry (Figure 3C). Although mice that received higher vector doses showed slightly lower human hepatocyte purity after isolation from chimeric livers (Figure 3D), the large majority of isolated hepatocytes in all three groups were human. Total VGs in 1 million hepatocytes was quantified by qPCR, and the VG number per human hepatocyte was calculated based on flow cytometry purity. Increasing AAV vector doses resulted in higher VG copy numbers per human hepatocyte. On average, 5, 16, and 73 VGs per human hepatocyte were observed using the low, medium, and high AAV vector doses, respectively (Figure 3E). In a second set of experiments, the effect of AAV vector doses on transgene expression was quantified. To this end, PHH3 huFNRG mice were challenged with AAV-GFP at two different doses—namely, 1.5 × 1010 VG (“low”) and 1 × 1011 VG (“medium”)—using the “efficient” protocol. The mean percentages of GFP-expressing human hepatocytes increased from 3.2% to 9.7% (3-fold) for AAVSpark100 and from 1.4% to 8.3% (5.9-fold) for AAVLK03 (Figure 3F). These data illustrate that increasing doses of AAV result in higher VG numbers and more transgene expression in human hepatocytes isolated from huFNRG mice.
Figure 3.

Hepatocyte Transduction with Varying AAV Vector Doses
(A) Serum hAlb levels of groups of huFNRG mice prior to challenge with increasing doses of AAVLK03 vector. Symbols represent individual mice; black bar represents the mean of group, The p values were calculated by Mann-Whitney test. (B) Histology on liver tissue from a mouse with serum hAlb level of 20 mg/mL illustrates the high human chimerism. Arrows indicate small darker stained areas that contained predominantly mouse hepatocytes (top panel, H&E staining; scale bar, 2 mm). The higher magnification left inset shows small human hepatocytes with pale cytoplasm on the right and large mouse hepatocytes with darker cytoplasm on the left (H&E; scale bar, 50 μm). The right inset shows a human-specific nuclear staining, which confirmed the small pale hepatocytes to be human while larger mouse hepatocytes stained negative (anti-human NuMA1; scale bar, 50 μm). (C) Illustrative flow cytometry plot of hepatocytes isolated from highly humanized mice that were stained for human HLA-I and mouse CD81 (mCD81). (D) The percentages of human and mouse hepatocytes isolated from huFNRG mice challenged with varying doses of AAVLK03 and determined by flow cytometry. Symbols indicate human and mouse hepatocyte percentages from individual mice; black bar represents the mean of group. The p values were calculated by Mann-Whitney test. (E) AAVLK03 vector genomes were quantified by qPCR in 1 million hepatocytes isolated from individual mice, and copy number per human hepatocyte was calculated based on purity determined by flow cytometry. Symbols represent individual mice; black bar represents the mean of group. The p values were calculated by Mann-Whitney test. VG, vector genomes. (F) Hepatocytes isolated from huFNRG mice challenged with two doses of AAVLK03-GFP or AAVSpark100-GFP were analyzed by flow cytometry. Symbols represent individual mice; black bar represents the mean of group. The p values were calculated by Mann-Whitney test.
Discussion
Chimeric mouse models are important tools to study AAV transduction differences between species but have been poorly standardized. In the Fah−/− chimera model literature, human hepatocyte transduction percentages vary wildly.5,20, 21, 22 Furthermore, studies that use similar serotypes, e.g., for AAV320, 21, 22 or AAV8,5,22 show inconsistent transduction hierarchies. We have now started to define parameters that cause variability and have not been appreciated in the past. Specifically to the widely used Fah−/− chimera model, NTBC cycling substantially affects transduction rate. This may, in part, explain differences in efficiency of gene transfer to human hepatocytes between published studies using the same serotype. After peak hAlb levels have been achieved, huFNRG mice in our facility continued to require intermittent NTBC for survival, which differs from immunodeficient Fah−/− mice in other facilities that can be maintained off NTBC.25 Conversely, we have observed the gradual loss of hAlb serum levels when huFNRG mice were maintained on continuous NTBC for months. This suggests that the human grafts in maximally huFNRG mice remain somewhat dynamic, expanding during the times off NTBC and contracting while on the drug. In immunodeficient Rag1−/− mice challenged with an AAV-hFIX, cycling of mouse hepatocytes after partial hepatectomy did not affect transduction but resulted in lower hFIX levels.26 Whether human hepatocyte proliferation in the Fah−/− chimera model contributes to the substantial variability in GFP expression observed in this study, and whether this is affected by dilution of episomal AAV vectors, will require further investigation. Such studies could become relevant to AAV vector therapies for pediatric patients and for liver conditions with active hepatocyte turnover and regeneration.
Second, we observed striking variability between PHH donors in this study, with up to 7-fold differences in the percentage of GFP-expressing hepatocytes. This finding contrasts with our previous conclusion that donor variability appeared minimal, which was based on only two donors21 or data by others with few donors.5 PHH donors vary in their ability to humanize liver chimeric models, with poor donors reaching less than 1 mg/mL hAlb or 10% human chimerism. The possible weak negative correlation that we observed between hAlb serum levels and GFP+ human hepatocytes suggests that some of the donor variability may be confounded by a dose effect as proposed by others.22 Particularly for serotypes that inefficiently transduce mouse hepatocytes, AAV vector doses per human hepatocyte may starkly differ between lowly and highly humanized mice. This explanation was hypothesized to account for discrepant AAV transduction efficiencies in chimeric Fah−/− mice humanized to vastly different levels with two donors in separate laboratories.17 In regular mice, gender differences in hepatocyte AAV vector expression have been long known,27 generally showing higher transgene levels in males than in females. Given the small numbers of PHH donors in this study, we cannot draw any conclusions regarding gender differences, particularly since PHHs from both genders were transplanted into female FNRG animals. Therefore, we now conclude that transduction efficiency between donors can vary substantially, but choice of donor does not appear to alter relative performance of one capsid versus another. Identifying these variables may help improve the consistency of clinical AAV gene therapies.
Liver-directed gene therapy trials use various AAV serotypes over wide dose ranges. Transduction efficiencies are deducted from surrogate markers in plasma, and no liver tissues have been analyzed to quantify transduction efficiencies in human liver. Experiments using Fah−/− chimeras have identified natural and engineered AAV serotypes that better transduce human hepatocytes.5,17,19, 20, 21, 22 These have led to an appreciation of how starkly many serotypes differ in their ability to transduce human and animal hepatocytes. Using a serotype that preferentially transduces human over mouse hepatocytes, we quantified a dose response based on the average number of AAV vector genomes per human hepatocyte. The percentage of GFP-expressing human hepatocytes also increased with higher AAV-GFP doses, albeit with substantial spread between huFNRG mice and more than that reported by others.20,22 With susceptible PHH donors, there was striking variability (Figures 2B, 2D, and S1C) with similar patterns. For other donors, the GFP expression pattern varied with individual experiments (Figure S1B) and between serotypes (Figure 1C). Particularly for serotypes that efficiently transduce mouse hepatocytes, e.g., AAV8, lowly GFP-expressing human hepatocytes may be falsely counted positive. This could be due to flow cytometry gating limitations or other poorly defined mechanisms such as passive transfer from mouse-expressed GFP to the human graft. To better understand the number of vector genomes required for detectable transgene expression and define the heterogeneity between individual hepatocytes, further analyses at the single-cell level are required. Even then, the translational value of findings from liver chimeras will remain largely speculative until liver tissue is analyzed from individuals undergoing AAV gene therapies.
Human liver chimeric mouse models are increasingly applied to address divergent findings between pre-clinical animal models and clinical studies. Despite their obvious utility for studying human hepatocyte biology in vivo, they come with a number of limitations. The murine immunodeficiency required to prevent xenorejection precludes their use in studying lymphocyte responses against AAV vectors. To overcome this limitation, mice dually humanized with hepatocytes and a human immune system have been created.14,28, 29, 30 However, the human immune cells in these models display limited functionality,31 and their utility in modeling immune responses against AAV remains to be shown. Another limitation of these models is that only hepatocytes are humanized to high chimerism, while non-parenchymal (e.g., sinusoidal endothelial, Kupffer) cells remain largely murine. This complicates studying interactions between hepatocytes and other liver cells, given the unknown number of species incompatibilities, e.g., receptor/co-receptor expression, signal transduction, and other intracellular pathways that may affect viral trafficking. Also, we do not know whether the high degree of immune deficiency alters cellular processes that impact hepatocyte transduction. In addition to these biological limitations, various technical hurdles exist to studying AAV vector biology in these models. The mixture of human and murine hepatocytes in the mouse liver poses challenges to quantify the level of AAV transgene product produced by either species. Therefore, these models have to rely on a combination of markers to identify transgene production in human hepatocytes. Commonly used readouts are immunofluorescence staining of liver tissues and flow cytometry. Although these techniques vary in the absolute percentages, both generally correlate well with regard to the hierarchy across several AAV serotypes within individual laboratories.20, 21, 22 However, detection of low-level fluorophore expression using both techniques is limited by hepatocyte autofluorescence. We have observed subtle variability in autofluorescence between human hepatocyte populations isolated from “identical” huFNRG mice. This is particularly striking in the yellow-green spectrum (illustrated by gates in Figure 1C), limiting the detection of hepatocytes expressing low levels of GFP. Despite these limitations, liver chimera mice are the only practical laboratory animals to study human hepatocyte biology. Because in vitro studies poorly predict animal or clinical data for many AAV serotypes, liver chimera models likely provide value in bridging findings from human cell culture studies toward clinical trials.
In conclusion we here have quantified several variables that influence AAV vector expression in chimeric Fah−/− mice. In order to further improve reproducibility, additional experimental conditions that were previously shown to affect outcomes in other animal models remain to be tested in chimeric Fah−/− mice. These include AAV vector design, including hepatocyte-specific or non-specific promoters, methodology of AAV vector production,32 vector injection route,33,34 transgene expression over time, and environmental variables such as rodent chow and microbiome compositions. Evaluating how these factors affect AAV vector expression in the human graft will improve reproducibility and may enhance the translational value of liver chimeric models.
Materials and Methods
Human Subjects and Animal Usage
All protocols involving human tissue were reviewed and exempted under Category 4 by the Rockefeller University Institutional Review Board (New York, NY, USA). All procedures involving mice were in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and approved by the Rockefeller University Institutional Animal Care and Use Committees (protocol 18063)
AAV Vectors
Recombinant AAV (rAAV) vectors were produced by transfection of human embryonic kidney 293 (HEK293) cells using liposomes. Three (or two) plasmid DNAs were combined to produce each rAAV serotype.35 These plasmids included (1) pAAV-CB-scGFP, containing the expression cassette (enhanced GFP under transcriptional control of cytomegalovirus-enhanced chicken beta-actin promoter) flanked by AAV inverted terminal repeats (ITRs), with one ITR modified to produce self-complementary vector genomes; (2) a helper plasmid DNA containing rep2/cap3 or rep2/cap9; and (3) pHelper plasmid (Invitrogen) containing adenoviral helper genes. For rAAV8 packaging, rep/cap and adenoviral helper genes were combined in a single construct (pXYZ5 and pDG8, respectively). HEK293 cells were expanded in 15-cm plates. Ninety micrograms of total DNA (recombinant and helpers, in equimolar amounts) per plate were applied. Cells were harvested 72 h after transfection, and the virus was purified using the iodixanol-density protocol.36 The titer of each preparation was determined by dot blot hybridization.21
Generation of Humanized Mice and AAV Gene Transfer
PHHs were obtained from vendors as shown in Table 1. Mice were transplanted with cryopreserved PHHs except for PHH3, PHH7, and PHH8, which were passaged through mice and adoptively transplanted into new animals that were preconditioned with retrorsine.24 Under anesthesia with isoflurane, human liver cell suspensions were injected intrasplenically (0.5 × 106 to 1 × 106 cells per mouse) into FNRG mice that were generated by 13-generation backcross of the Fah−/− allele11 to NOD Rag1−/− Il2rgnull animals, provided by M. Grompe (Oregon Health & Science University) and obtained from Jackson Laboratories, respectively.12 Zero to 5 days prior to transplantation, mice were cycled off the liver protective drug NTBC (Yecuris), as described elsewhere.8 The hAlb levels in mouse sera were measured by ELISA (Bethyl Laboratories). AAV vectors were injected through the tail vein at 1 × 1011 vector genomes per mouse, unless specified otherwise.
Hepatocyte Isolation and Flow Cytometry
Mice were anesthetized by ketamine/xylazine injection, after which the inferior vena cava was cannulated with an angiocath (BD Biosciences) for in situ liver perfusion. After perfusion with heparin, the large liver lobe was tied off in order to prevent its digestion, removed, and fixed in 10% formalin for immunohistochemistry. The remainder of the liver was then perfused with PBS with 0.5 mM EDTA, followed by PBS with 0.05% collagenase. The collagenase-perfused liver lobes were resected and put over a 100-μm cell strainer before Percoll purification and fixation in 4% buffered formalin. After permeabilization with BD Cytofix/Cytoperm (BD Biosciences), cells were stained with anti-human HLA-I-APC/Cy7 (BioLegend) and anti-mCD81-PE (BD PharMingen). Data were acquired on an LSR II Flow Cytometer (BD Biosciences) and analyzed using FlowJo software. Sequential gating strategy was as follows: inclusion of hepatocytes by forward/side scatter, exclusion of doublets, exclusion of mCD81+ cells, and the inclusion of HLA-I+. The GFP+ fraction was determined as percent positive of the HLA-I+ population.
Quantification of Vector Genomes in Isolated Hepatocytes
After isolation from chimeric livers and density purification as described earlier, total hepatocytes were counted. Cell pellets containing 1 million hepatocytes were frozen at −20°C until further processing. Vector copy number per genome was assessed using a qualified quantitative PCR (qPCR) assay. Briefly, DNA was extracted using the ZR Genomic DNA Tissue Midi Prep Kit (Zymo Research) as per the manufacturer’s protocol. DNA was stored at −80°C. Samples were diluted to 200 ng/μL in nuclease-free water and further diluted to 100 ng/μL with equal volumes of 2× Diluent S (0.002% Pluronic F-68, 4 ng/μL salmon sperm DNA in nuclease-free water). Ten microliters or 1 μg DNA was used in the PCR reaction. Quantitative real-time PCR was performed using primers and probes targeting the transgene (5′-TGAGGAGGCTGAAGACTATGA-3′; reverse primer, 5′- CCACAGACCTGATCTGAATGAA-3′; and a probe, 5′-56-FAM-TGGATGTGG/ZEN/TGAGGTTTGATGATGACA-3IABkFQ-3′). ZEN Double-Quenched Probes containing a 5′ fluorophore, 3′ Iowa Black FQ quencher, and internal ZEN quencher were ordered through Integrated DNA Technologies. A linearized plasmid bearing the target-specific sequence was used for the preparation of a standard curve.
Immunohistochemistry
Immunohistochemistry and H&E staining was performed on formalin-fixed, paraffin-embedded murine liver tissues using polyclonal rabbit NuMA (Abcam) staining performed as described previously.14 In brief, sections were deparaffinized in xylene (3 changes), rehydrated through graded alcohols (3 changes 100% ethanol, 3 changes 95% ethanol), and rinsed in distilled water. Antibody incubation and detection were carried out on a Discovery XT instrument (Ventana Medical Systems Tucson, AZ, USA) using Ventana’s Reagent Buffer and Detection Kit. Heat-induced epitope retrieval was performed in a 1,200-W microwave oven at 100% power in 10 mM sodium citrate buffer (pH 6.0) for 20 min. Sections were allowed to cool for 30 min and then rinsed in distilled water. Endogenous peroxidase activity was blocked with hydrogen peroxide. Rabbit anti-human NuMA was diluted in Tris-BSA (25 mM Tris, 15 mM NaCL, 1% BSA [pH 7.2]) and incubated overnight at room temperature. Primary antibody was detected with goat anti-rabbit horseradish peroxidase (HRP) conjugated multimer for 8 min. The complex was visualized with 3,3 diaminobenzidine and enhanced with copper sulfate. Matched immunoglobulin isotype, at equivalent concentration and diluted in PBS, was used as negative control. Upon completion of staining, all slides were washed in distilled water, counterstained with hematoxylin, dehydrated, and mounted with permanent media. Stained slides were scanned at 40× magnification using the Leica Microsystems SCN 400F Whole Slide Scanner. Images were viewed and captured using SlidePath’s Digital Image Hub (Leica Microsystems, Buffalo Grove, IL, USA).
Statistical Analyses
Groups of animals were compared by unpaired t test, and correlations were calculated by Pearson’s r, using Prism 8 software (GraphPad).
Author Contributions
C.Z., K.O.A.V., E.M., M.K., I.Z., C.Q., and M.Y. conceived and performed experiments. X.M.A. and K.A.H. conceived experiments and provided expertise and feedback. K.O.A.V., R.W.H., and Y.P.d.J. wrote the manuscript. R.W.H. and Y.P.d.J. secured funding.
Conflicts of Interest
Y.P.d.J. received funding from Spark Therapeutics. M.Y., X.M.A., and K.A.H. are employees of Spark Therapeutics. The remaining authors report no competing interests.
Acknowledgments
This work was supported by NIH grants R01HL131093 (to R.W.H. and Y.P.d.J.), K08DK090576 (to Y.P.d.J.), and F32DK107164 (to E.M.). Further support was provided by Spark Therapeutics (to Y.P.d.J.). K.O.A.V. was supported by a fellowship of the Belgian American Educational Foundation. The project was co-sponsored by the Center for Basic and Translational Research on Disorders of the Digestive System through the generosity of the Leona M. and Harry B. Helmsley Charitable Trust (to E.M.). We thank the Rockefeller University Flow Cytometry Resource Center for help with flow design and analyses.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.05.033.
Supplemental Information
References
- 1.Ginocchio V.M., Ferla R., Auricchio A., Brunetti-Pierri N. Current Status on Clinical Development of Adeno-Associated Virus-Mediated Liver-Directed Gene Therapy for Inborn Errors of Metabolism. Hum. Gene Ther. 2019;30:1204–1210. doi: 10.1089/hum.2019.151. [DOI] [PubMed] [Google Scholar]
- 2.Butterfield J.S.S., Hege K.M., Herzog R.W., Kaczmarek R. A Molecular Revolution in the Treatment of Hemophilia. Mol. Ther. 2020;28:997–1015. doi: 10.1016/j.ymthe.2019.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herzog R.W., Pierce G.F. Liver Gene Therapy: Reliable and Durable? Mol. Ther. 2019;27:1863–1864. doi: 10.1016/j.ymthe.2019.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perrin G.Q., Herzog R.W., Markusic D.M. Update on clinical gene therapy for hemophilia. Blood. 2019;133:407–414. doi: 10.1182/blood-2018-07-820720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lisowski L., Dane A.P., Chu K., Zhang Y., Cunningham S.C., Wilson E.M., Nygaard S., Grompe M., Alexander I.E., Kay M.A. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature. 2014;506:382–386. doi: 10.1038/nature12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grompe M., Strom S. Mice with human livers. Gastroenterology. 2013;145:1209–1214. doi: 10.1053/j.gastro.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 7.Mercer D.F., Schiller D.E., Elliott J.F., Douglas D.N., Hao C., Rinfret A., Addison W.R., Fischer K.P., Churchill T.A., Lakey J.R. Hepatitis C virus replication in mice with chimeric human livers. Nat. Med. 2001;7:927–933. doi: 10.1038/90968. [DOI] [PubMed] [Google Scholar]
- 8.Azuma H., Paulk N., Ranade A., Dorrell C., Al-Dhalimy M., Ellis E., Strom S., Kay M.A., Finegold M., Grompe M. Robust expansion of human hepatocytes in Fah-/-/Rag2-/-/Il2rg-/- mice. Nat. Biotechnol. 2007;25:903–910. doi: 10.1038/nbt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bissig K.D., Le T.T., Woods N.B., Verma I.M. Repopulation of adult and neonatal mice with human hepatocytes: a chimeric animal model. Proc. Natl. Acad. Sci. USA. 2007;104:20507–20511. doi: 10.1073/pnas.0710528105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grompe M., al-Dhalimy M., Finegold M., Ou C.N., Burlingame T., Kennaway N.G., Soriano P. Loss of fumarylacetoacetate hydrolase is responsible for the neonatal hepatic dysfunction phenotype of lethal albino mice. Genes Dev. 1993;7(12A):2298–2307. doi: 10.1101/gad.7.12a.2298. [DOI] [PubMed] [Google Scholar]
- 11.Grompe M., Lindstedt S., al-Dhalimy M., Kennaway N.G., Papaconstantinou J., Torres-Ramos C.A., Ou C.N., Finegold M. Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I. Nat. Genet. 1995;10:453–460. doi: 10.1038/ng0895-453. [DOI] [PubMed] [Google Scholar]
- 12.de Jong Y.P., Dorner M., Mommersteeg M.C., Xiao J.W., Balazs A.B., Robbins J.B., Winer B.Y., Gerges S., Vega K., Labitt R.N. Broadly neutralizing antibodies abrogate established hepatitis C virus infection. Sci. Transl. Med. 2014;6:254ra129. doi: 10.1126/scitranslmed.3009512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Naugler W.E., Tarlow B.D., Fedorov L.M., Taylor M., Pelz C., Li B., Darnell J., Grompe M. Fibroblast Growth Factor Signaling Controls Liver Size in Mice With Humanized Livers. Gastroenterology. 2015;149:728–740.e15. doi: 10.1053/j.gastro.2015.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Billerbeck E., Mommersteeg M.C., Shlomai A., Xiao J.W., Andrus L., Bhatta A., Vercauteren K., Michailidis E., Dorner M., Krishnan A. Humanized mice efficiently engrafted with fetal hepatoblasts and syngeneic immune cells develop human monocytes and NK cells. J. Hepatol. 2016;65:334–343. doi: 10.1016/j.jhep.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawahara T., Toso C., Douglas D.N., Nourbakhsh M., Lewis J.T., Tyrrell D.L., Lund G.A., Churchill T.A., Kneteman N.M. Factors affecting hepatocyte isolation, engraftment, and replication in an in vivo model. Liver Transpl. 2010;16:974–982. doi: 10.1002/lt.22099. [DOI] [PubMed] [Google Scholar]
- 16.Vanwolleghem T., Libbrecht L., Hansen B.E., Desombere I., Roskams T., Meuleman P., Leroux-Roels G. Factors determining successful engraftment of hepatocytes and susceptibility to hepatitis B and C virus infection in uPA-SCID mice. J. Hepatol. 2010;53:468–476. doi: 10.1016/j.jhep.2010.03.024. [DOI] [PubMed] [Google Scholar]
- 17.Paulk N.K., Pekrun K., Zhu E., Nygaard S., Li B., Xu J., Chu K., Leborgne C., Dane A.P., Haft A. Bioengineered AAV Capsids with Combined High Human Liver Transduction In Vivo and Unique Humoral Seroreactivity. Mol. Ther. 2018;26:289–303. doi: 10.1016/j.ymthe.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pekrun K., De Alencastro G., Luo Q.J., Liu J., Kim Y., Nygaard S., Galivo F., Zhang F., Song R., Tiffany M.R. Using a barcoded AAV capsid library to select for clinically relevant gene therapy vectors. JCI Insight. 2019;4:e131610. doi: 10.1172/jci.insight.131610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bissig-Choisat B., Wang L., Legras X., Saha P.K., Chen L., Bell P., Pankowicz F.P., Hill M.C., Barzi M., Leyton C.K. Development and rescue of human familial hypercholesterolaemia in a xenograft mouse model. Nat. Commun. 2015;6:7339. doi: 10.1038/ncomms8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shao W., Pei X., Cui C., Askew C., Dobbins A., Chen X., Abajas Y.L., Gerber D.A., Samulski R.J., Nichols T.C. Superior human hepatocyte transduction with adeno-associated virus vector serotype 7. Gene Ther. 2019;26:504–514. doi: 10.1038/s41434-019-0104-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vercauteren K., Hoffman B.E., Zolotukhin I., Keeler G.D., Xiao J.W., Basner-Tschakarjan E., High K.A., Ertl H.C.J., Rice C.M., Srivastava A. Superior In Vivo Transduction of Human Hepatocytes Using Engineered AAV3 Capsid. Mol. Ther. 2016;24:1042–1049. doi: 10.1038/mt.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L., Bell P., Somanathan S., Wang Q., He Z., Yu H., McMenamin D., Goode T., Calcedo R., Wilson J.M. Comparative Study of Liver Gene Transfer With AAV Vectors Based on Natural and Engineered AAV Capsids. Mol. Ther. 2015;23:1877–1887. doi: 10.1038/mt.2015.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.George L.A., Sullivan S.K., Giermasz A., Rasko J.E.J., Samelson-Jones B.J., Ducore J., Cuker A., Sullivan L.M., Majumdar S., Teitel J. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N. Engl. J. Med. 2017;377:2215–2227. doi: 10.1056/NEJMoa1708538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michailidis E., Vercauteren K., Mancio-Silva L., Andrus L., Jahan C., Ricardo-Lax I., Zou C., Kabbani M., Park P., Quirk C. Expansion, in vivo-ex vivo cycling, and genetic manipulation of primary human hepatocytes. Proc. Natl. Acad. Sci. USA. 2020;117:1678–1688. doi: 10.1073/pnas.1919035117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bissig K.D., Wieland S.F., Tran P., Isogawa M., Le T.T., Chisari F.V., Verma I.M. Human liver chimeric mice provide a model for hepatitis B and C virus infection and treatment. J. Clin. Invest. 2010;120:924–930. doi: 10.1172/JCI40094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miao C.H., Nakai H., Thompson A.R., Storm T.A., Chiu W., Snyder R.O., Kay M.A. Nonrandom transduction of recombinant adeno-associated virus vectors in mouse hepatocytes in vivo: cell cycling does not influence hepatocyte transduction. J. Virol. 2000;74:3793–3803. doi: 10.1128/jvi.74.8.3793-3803.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davidoff A.M., Ng C.Y., Zhou J., Spence Y., Nathwani A.C. Sex significantly influences transduction of murine liver by recombinant adeno-associated viral vectors through an androgen-dependent pathway. Blood. 2003;102:480–488. doi: 10.1182/blood-2002-09-2889. [DOI] [PubMed] [Google Scholar]
- 28.Bility M.T., Cheng L., Zhang Z., Luan Y., Li F., Chi L., Zhang L., Tu Z., Gao Y., Fu Y. Hepatitis B virus infection and immunopathogenesis in a humanized mouse model: induction of human-specific liver fibrosis and M2-like macrophages. PLoS Pathog. 2014;10:e1004032. doi: 10.1371/journal.ppat.1004032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strick-Marchand H., Dusséaux M., Darche S., Huntington N.D., Legrand N., Masse-Ranson G., Corcuff E., Ahodantin J., Weijer K., Spits H. A novel mouse model for stable engraftment of a human immune system and human hepatocytes. PLoS ONE. 2015;10:e0119820. doi: 10.1371/journal.pone.0119820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson E.M., Bial J., Tarlow B., Bial G., Jensen B., Greiner D.L., Brehm M.A., Grompe M. Extensive double humanization of both liver and hematopoiesis in FRGN mice. Stem Cell Res. (Amst.) 2014;13(3 Pt A):404–412. doi: 10.1016/j.scr.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shultz L.D., Keck J., Burzenski L., Jangalwe S., Vaidya S., Greiner D.L., Brehm M.A. Humanized mouse models of immunological diseases and precision medicine. Mamm. Genome. 2019;30:123–142. doi: 10.1007/s00335-019-09796-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schnödt M., Büning H. Improving the Quality of Adeno-Associated Viral Vector Preparations: The Challenge of Product-Related Impurities. Hum. Gene Ther. Methods. 2017;28:101–108. doi: 10.1089/hgtb.2016.188. [DOI] [PubMed] [Google Scholar]
- 33.Dane A.P., Wowro S.J., Cunningham S.C., Alexander I.E. Comparison of gene transfer to the murine liver following intraperitoneal and intraportal delivery of hepatotropic AAV pseudo-serotypes. Gene Ther. 2013;20:460–464. doi: 10.1038/gt.2012.67. [DOI] [PubMed] [Google Scholar]
- 34.Mingozzi F., Schüttrumpf J., Arruda V.R., Liu Y., Liu Y.L., High K.A., Xiao W., Herzog R.W. Improved hepatic gene transfer by using an adeno-associated virus serotype 5 vector. J. Virol. 2002;76:10497–10502. doi: 10.1128/JVI.76.20.10497-10502.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsushita T., Elliger S., Elliger C., Podsakoff G., Villarreal L., Kurtzman G.J., Iwaki Y., Colosi P. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther. 1998;5:938–945. doi: 10.1038/sj.gt.3300680. [DOI] [PubMed] [Google Scholar]
- 36.Ayuso E., Mingozzi F., Montane J., Leon X., Anguela X.M., Haurigot V., Edmonson S.A., Africa L., Zhou S., High K.A. High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 2010;17:503–510. doi: 10.1038/gt.2009.157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.