

Abstract
Introduction
We developed an automated liquid chromatography‐tandem mass spectrometry high performance liquid chromatography tandem mass spectrometry (HPLC‐MS/MS) method for multiplex quantification of wild‐type (wt) amyloid β (Aβ) peptides 1‐40 (Aβ40) and 1‐42 (Aβ42) and detection of variant Aβ peptides in cerebrospinal fluid.
Methods
The multiplex Aβ HPLC‐MS/MS assay was validated in a clinically accredited laboratory following regulatory guidelines, with Aβ42 calibration assigned to the ERM/IFCC certified reference material; sequence variants were additionally multiplexed into the method.
Results
Sample preparation was fully automated on a liquid handler. The assay quantified wt‐Aβ42 and wt‐Aβ40 and detected sequence variants, when present, within the Aβ42 sequence.
Discussion
Extension of the HPLC‐MS/MS approach for quantification of wt‐Aβ42 and wt‐Aβ40 to include known sequence variants increases analytical accuracy of the mass spectrometric approach and enables identification of cases of autosomal dominant Alzheimer's disease. Development of an automated workflow and selection of appropriate instrumentation enabled deployment of this method in routine clinical testing.
Keywords: Alzheimer's disease, amyloid‐β peptides, automation, autosomal dominant Alzheimer's disease, cerebrospinal fluid, certified reference material, mass spectrometry, variant
1. INTRODUCTION
As a biomarker of amyloid pathology, the concentration of amyloid β (Aβ) peptides in cerebrospinal fluid (CSF) is used in the ante‐mortem diagnosis of Alzheimer's disease (AD). In CSF, a reduction in Aβ concentration—specifically the 1‐42 residue isoform Aβ42—is highly correlated with AD, and associated with the sequestration of Aβ42 in extracellular aggregates in the brain. 1 Confirmatory diagnosis of AD can only be made by identification of Aβ aggregates in brain tissue (most commonly by post‐mortem analysis), with the exception of autosomal dominant forms of the disease, which can be identified by DNA sequencing. Aβ peptides arise from cleavage of the trans‐membrane amyloid‐precursor protein (APP) by endogenous proteases, resulting in peptides of various lengths, including residues 1‐40 and 1‐42, referred to herein as Aβ40 and Aβ42, respectively (Figure 1). Genetic forms of AD (ie, autosomal dominant AD) include highly penetrant pathogenic sequence variants within the APP gene and two presenilin genes (PSEN1 and PSEN2). 2 The term “mutation” was previously used to describe such sequence variants, but has been replaced with “variant” further annotated with its pathogenicity or lack thereof. 3
FIGURE 1.
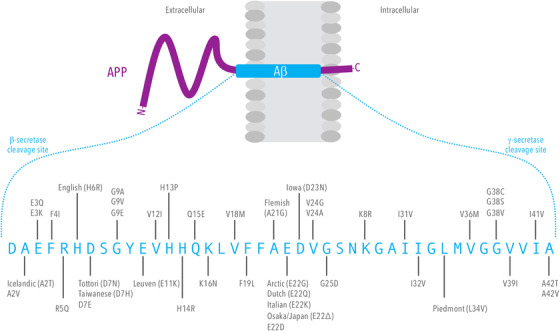
The transmembrane amyloid precursor protein (APP) is cleaved by β‐ and γ‐secretase to form the 1‐40 and 1‐42 residue isoforms of amyloid beta (Aβ). In addition to proteolytic isoforms, there are known amino acid variants, both pathogenic and benign, within the Aβ42 sequence
As the ante‐mortem diagnosis of AD moves from one identifying clinical syndromes to a biochemical definition reflective of the pathological accumulation of amyloid plaques and neurofibrillary tangles, 4 methods for accurate, precise, and selective quantification of these biomarkers gains greater importance. Historically, Aβ in CSF has been measured predominantly via immunoassay; however, immunoassays for Aβ have demonstrated high intra‐and inter‐laboratory variability, 5 which spurred the development of alternate approaches, namely automated immunoassays 6 , 7 and high performance liquid chromatography tandem mass spectrometry (HPLC‐MS/MS). 5 , 8 , 9 , 10 With HPLC‐MS/MS methods, intra‐ and inter‐laboratory precision were improved; 11 however, variability associated with multiple operators, which would occur in a clinical laboratory setting, remained unaddressed.
An advantage of HPLC‐MS/MS is exquisite analyte selectivity derived from the use of multiple reaction monitoring (MRM). HPLC is used to separate analytes in time, and MS/MS enables further selectivity by the analyte's mass‐to‐charge (m/z) ratio and subsequent confirmation of the analyte's primary structure (ie, amino acid sequence) by fragmentation. The use of MRM is akin to performing a sequencing experiment in which the peptide (ie, precursor ion) is reproducibly fragmented into a series of overlapping peptide sequences, and the fragment m/z ratios are then detected to confirm the identity of the target analyte. This level of selectivity, far exceeding that of immunoassays, is the reason the laboratory medicine community has turned to mass spectrometry for the development of clinical reference methods and for routine quantification of clinically relevant peptide and proteins, 12 , 13 including a candidate reference method for Aβ. 11 , 14
RESEARCH IN CONTEXT
Systematic review: Development of an automated and multiplex wild‐type/variant amyloid β (Aβ)40 and Aβ42 high performance liquid chromatography tandem mass spectrometry (HPLC‐MS/MS) assay that supports diagnostic workup of sporadic AD and can identify autosomal dominant AD via detection of amyloid‐precursor protein (APP) pathogenic variants within the Aβ sequence.
Interpretation: The method enables ease of operation in a clinical setting via automation and use of equipment commonly found in clinical laboratories.
Future directions: With the availability of a highly selective tool that detects Aβ variants, such cases can be investigated with greater resolution (eg, temporal changes in peptide isoform concentrations) to further our understanding of disease progression and pathology.
The selectivity of HPLC‐MS/MS, however, is a double‐edged sword in that MRM methods are so selective they ignore all peptides except those satisfying the MRM criteria, that is, an analyte (1) eluting at a specific time from the analytical column, and (2) having the predefined MRM transitions. Early/late eluting peptides are ignored by the method, as are those not meeting the m/z transition criteria—as would occur if there were an alteration to the Aβ sequence. Currently published mass spectrometric methods, including those using in vitro proteolytic digestion, do not include MRMs of known Aβ sequence variants 5 , 8 , 9 , 10 , 14 , 15 , 16 and due to the high analytical selectivity of the MRM approach, Aβ variants are not “seen” by the method. Immunoassays, for their part, cannot differentiate between Aβ variants that occur outside of the assay epitope regions, or if the amino acid change does occur within the epitope regions it may abrogate binding/detection. The presence of such sequence variants, unaccounted for in the design of an analytical method, has led to erroneous immunoassay results and misdiagnosis in various clinical contexts. 17 , 18 , 19 , 20 Therefore, a method that identifies Aβ variants within the 1‐42 sequence would prevent inaccurate reporting of the total Aβ concentration, with the added advantage of also identifying cases of autosomal dominant AD.
An additional challenge for deployment of Aβ methods (both immunometric and mass spectrometric) in routine clinical practice includes workflow and instrument compatibility with a clinical laboratory environment. A method intended for immediate clinical application should use instrumentation commonly found in clinical laboratories and consider compatibility of the assay workflow with standard laboratory practices and operation by shift‐working technicians.
Herein we describe a fully automated, clinically validated HPLC‐MS/MS method for quantification of wt‐Aβ peptides (Aβ40 and Aβ42) and identification of variant (var‐) Aβ peptides. With workflow optimization and full automation, this HPLC‐MS/MS methodology has been deployed in routine care.
2. METHODS
2.1. Samples
With research ethics board approval, human CSF was collected from patients at the University of British Columbia's Clinic for Alzheimer's Disease and Related Disorders and at St. Paul's Hospital, in Vancouver, Canada. Specimens were collected by a standardized protocol, including collection directly into polypropylene tubes. 5 Diagnostic classification was based on thorough clinical assessment by a neurologist with expertise in neurodegenerative disorders.
2.2. Sample preparation and HPLC‐MS/MS analysis
A detailed description of the HPLC‐MS/MS method (Tables S1‐S3 in supporting information), including material sources, can be found in the supporting information. In brief, 200 μL of each sample (ie, CSF specimens, quality controls [QC], and calibrators) was treated with guanidine hydrochloride (GdnHCl) containing 15N‐Aβ40 and 15N‐Aβ42 internal standards (ISs), subjected to reversed phase solid‐phase extraction (SPE), and eluted into a 96‐well plate. These steps were performed either manually or using a liquid handling robot. Analysis was performed using a C18 analytical column coupled to a triple quadrupole mass spectrometer.
2.3. Validation
Method validation was performed following the Clinical and Laboratory Standards Institute (CLSI) guidelines including C62, EP‐5A, and EP‐6A. 21 , 22 , 23 Validation experiments included assessments of: (1) recovery and linearity, (2) ion suppression and enhancement, (3) precision, and (4) quality measures—including determination of the analytical measuring interval (AMI), clinical measuring interval (CMI), lower limit of the measuring interval (LLMI), and method comparisons.
2.3.1. Recovery and linearity
Linearity was assessed via a mixing study, following CLSI EP6‐A, 23 using a “low” concentration human CSF pool (Aβ40 = 1067 ng/L; Aβ42 = 220 ng/L) and a “high” pool (Aβ40 = 24067 ng/L; Aβ42 = 3387 ng/L), which was made by supplementing a human CSF pool with synthetic Aβ peptides.
2.3.2. Ion suppression and enhancement
Ion suppression and enhancement can result from other compounds in a complex sample matrix altering the ionization efficiency of the ions of interest. Ion suppression and enhancement was assessed by post‐column continuous infusion; 22 an extracted CSF pool (without the addition of IS) was injected into the LC stream and a solution containing 15N‐Aβ40 and 15N‐Aβ42 was directly infused into the flow at the source via at‐union. Additionally, phospholipid MRMs were monitored in extracted CSF samples in the validation phase.
2.3.3. Precision
Precision experiments were performed using a modification of the CLSI EP‐5A protocol 21 : specifically, quintuplicate measurements over 5 days using human CSF pools. Three CSF pools at low, medium, and high concentrations were assessed. The mean concentration in the medium CSF pool (composed solely of human CSF) was 3900 ng/L of Aβ40 and 522 ng/L of Aβ42. The low pool (Aβ40 = 2020 ng/L; Aβ42 = 273 ng/L) was made by diluting the medium pool with the artificial CSF and the high pool (Aβ40 = 10100 ng/L; Aβ42 = 1288 ng/L) was made by supplementing the medium pool with synthetic Aβ40 and Aβ42.
2.3.4. Accuracy and method comparison
Wt‐Aβ42 calibrators were assigned to the ERM/IFCC certified reference material (CRM), 24 and wt‐Aβ40 was calibrated against peptide‐manufacturer reported mass, adjusted for HPLC purity. For wt‐Aβ42 calibrator assignment, three CRMs—ERM‐DA480/IFCC, ERM‐DA481/IFCC, ERM‐DA482/IFCC—were run in duplicate (within run) and in three batches (between run) with the average concentrations set to the concentrations from the certificates of analyses. The calibrators were run as unknowns in the three batches and the mean of the observed concentrations used for assignment. A regression analysis was performed by weighted Deming regression between the CRM‐assigned calibrator concentrations and the pre‐CRM‐assigned concentrations (ie, manufacturer stated amino acid analysis and HPLC purity).
A method comparison was performed between the HPLC‐MS/MS and the INNOTEST enzyme‐linked immunosorbent assay (ELISA) β‐Amyloid[1‐42] (n = 155 specimens).
2.3.5. LLMI, AMI, and CMI
Acceptable LLMI criteria was defined by an imprecision <20% and a signal‐to‐noise (S/N) >10. The AMI is the range of analyte values that a method can directly measure without modification (eg, dilution). The CMI is defined as the range of analyte values a method can measure allowing for specimen dilution, concentration, or other pre‐treatment used to extend the AMI.
2.3.6. Manual versus automated sample preparation
To assess the precision of the manual and automated sample preparation methods for HPLC‐MS/MS analysis, human CSF samples (n = 40 individuals) with concentrations ranging between 839–10023 ng/L for Aβ40 and 113–1266 ng/L for Aβ42 were analyzed using each method.
2.3.7. Variants within the Aβ42 sequence
A list of known amino acid variants within the Aβ42 sequence was developed by searching existing databases including: Alzforum (http://www.alzforum.org/mutations), Single Nucleotide Polymorphism Database (dbSNP, NCBI), and Exome Aggregation Consortium (ExAC). A variant within the Aβ42 sequence was included in the MRM method if it satisfied the following criteria: (1) contained a variant within the Aβ42 sequence, and (2) was either pathogenic (average frequency not considered in this case), or, if non‐pathogenic or of unknown significance, had an average frequency in the global population of ≥4.0E‐05.
To test the wt/var‐Aβ HPLC‐MS/MS assay, CSF representative of the complex scenario in which an individual has one wild‐type and one variant APP allele was assessed. To create a series of CSF samples characteristic of this heterozygote state, synthetic Aβ40 variants were spiked into a human CSF pool (containing only wt‐Aβ) at equimolar concentration to the endogenous wt‐Aβ40 (0.157 nM).
2.4. Diagnostic performance
Diagnostic performance was assessed using CSF from 93 individuals presenting with cognitive complaints to a memory clinic. All individuals were evaluated by a standardized protocol with diagnosis made by clinical assessment: probable AD (n = 39) and cognitive complaints/impairment due to a non‐AD cause (n = 54). In addition to use of the wt/var‐Aβ HPLC‐MS/MS assay, total tau was quantified by ELISA (INNOTEST hTau Ag) and data analyzed using receiver operating characteristic (ROC) curves.
2.5. Data analysis
Instrument data were viewed and analyzed using Analyst software (SCIEX v.1.6), Excel (Microsoft), and R (v.3.4.0).
3. RESULTS
3.1. Figures of merit
3.1.1. Recovery and linearity
The method was linear from at least 100–20,000 ng/L for Aβ40 and 100–3000 ng/L for Aβ42 (Figure S1 and Table S4‐S5 in supporting information).
3.1.2. Ion suppression and enhancement
There was no observable ion suppression or enhancement occurring at the retention time of the Aβ40 and Aβ42 peptides, or due to phospholipids (data not shown).
3.1.3. Precision and automated versus manual sample preparation
Intra‐ and inter‐run precision data for manual and automated sample preparation protocols can be found in Table S6 in supporting information. By regression analysis for Aβ42: Yautomated = 1.01 × Xmanual + 17.63, R2 = 0.9693, confidence interval (CI) slope: [0.942, 1.074], CI intercept: [–70.843, 36.119] (Figure 2A). By regression analysis for Aβ40: Yautomated = 0.95 × Xmanual + 0.06, R2 = 0.9728, CI slope: [0.895, 1], CI intercept: [−0.38,0.297] (Figure 2C). Difference plots for the automated versus manual workflow revealed a mean bias of –1.64%, 95% CI: [–20.94, 17.66] for Aβ42 and –6.00%, 95% CI: [–22.54, 10.55] for Aβ40 (Figure 2B and D).
FIGURE 2.

Manual and automated sample preparation prior to high performance liquid chromatography tandem mass spectrometry analysis demonstrated comparable performance for both (A and B) amyloid β 1‐42 (Aβ42) and (C and D) amyloid β 1‐40 (Aβ40). A and C, Shaded region represents the 95% confidence interval (CI) of the slope of the linear regression. B and D, Black dashed lines represent the 95% CI of the mean difference (solid line)
3.1.4. Accuracy and method comparison for Aβ42
The CRM‐assigned calibrators yielded the following regression equation to the calibrators’ pre‐CRM assignment: YCRM = 0.89 × Xpre‐CRM + 9.31, R2 = 0.9904, CI slope: [0.769, 1.04], CI intercept: [–10.676, 91.708] (Figure 3). The method comparison between the HPLC‐MS/MS assay and the predicate ELISA method revealed the following by linear regression analysis: YHPLC‐MS/MS = 2.64 × XELISA –247.4, R2 = 0.63, CI slope: [2.38,2.99], CI intercept: [–398.75, –123.67] and a mean bias of 71.5%, 95% CI: [123.6, 19.4], noting that the ELISA is not calibrated to the ERM/IFCC CRM (Figure S2 in supporting information).
FIGURE 3.

Amyloid β peptide 1‐42 (Aβ42) certified reference material (CRM)‐assigned calibrators versus calibrators pre‐CRM assignment
3.1.5. LMI, AMI, and CMI
For the calibrator with a concentration of 100 ng/L for both Aβ40 and Aβ42, the average back‐calculated concentration for Aβ40 was 106 ng/L with an average S/N of 47 and for Aβ42 was 105 ng/L with an average S/N of 23. Given the acceptable S/N ratio for both Aβ40 and Aβ42 at 100 ng/L, the reportable LLMI for both peptides was set to 100 ng/L (Figure S3 in supporting information). Due to the broad analytical range of the method no extra dilution procedures were required and thus the AMI was equivalent to the CMI at 100–20000 ng/L for Aβ40 and 100–3000 ng/L for Aβ42.
3.2. Variants within the Aβ42 sequence
From the database search (Table S7 in supporting information), a total of 20 Aβ variants were found to meet the inclusion criteria. This included 13 pathogenic variants, 1 non‐pathogenic variant, and 6 variants of unknown significance (Table S8 in supporting information). As proof of concept, nine of these variants were synthesized as var‐Aβ40 peptides (given that the sequence variants were captured within residues 1‐40), and used to create the “heterozygous” CSF samples (Table 1).
TABLE 1.
Multiple reaction monitoring transitions and retention times of wt‐Aβ40 and ‐Aβ40 variants analyzed in the APP heterozygosity experiments
| Variant | Name | Mass (Da) | Retention time (min) | Precursor ion (m/z) | Product ions (m/z) |
|---|---|---|---|---|---|
| Wild‐type | 4329.9 | 4.06 | 1083.0 | 1054.1, 1029.1,1000.8 | |
| L34V | Piedmont | 4315.8 | 3.93 | 1079.9 | 1050.5, 1011.5, 997.4 |
| A21G | Flemish | 4315.8 | 3.97 | 1079.9 | 1050.5, 1011.5, 997.4 |
| H6R | English | 4348.9 | 4.12 | 1088.2 | 1058.9, 1033.9, 1005.4 |
| D7N | Tottori | 4328.9 | 4.14 | 1083.0 | 1054.1, 1029.1, 1000.8 |
| E22G | Arctic | 4257.8 | 4.18 | 1065.5 | 1036.2, 1011.5, 997.1 |
| E22∆ | Osaka | 4200.8 | 4.22 | 1051.2 | 943.9, 914.8, 882.7 |
| E22Q | Dutch | 4328.9 | 4.24 | 1083.0 | 1054.1, 1029.1, 1000.8 |
| D23N | Iowa | 4328.9 | 4.32 | 1083.0 | 1054.1, 1029.1, 1000.8 |
| E22K | Italian | 4328.9 | 4.42 | 1083.0 | 1054.1, 1029.1, 1000.8 |
Abbreviations: Aβ, amyloid beta; APP, amyloid‐precursor protein; multiple reaction monitoring; m/z, mass‐to‐charge ratio; wt, wild type
By HPLC‐MS/MS analysis, all Aβ variants tested were resolved chromatographically from wt‐Aβ (Figure 4A‐J). This included variants that shared similar transitions to wt‐Aβ40: E22Q, D23N, D7N, and E22K. The E22G, D23N, and E22Q variants co‐eluted, with E22G readily identifiable by its unique MRMs. E22Q and D23N shared precursor masses and transitions (as currently selected) within the prescribed m/z tolerances, and therefore these two variants could not be distinguished from one another using the chromatographic conditions described (Figure 4C and F).
FIGURE 4.

The multiplex amyloid beta (Aβ) high performance liquid chromatography tandem mass spectrometry assay enabled identification of cases (A) homozygous for wild‐type amyloid‐precursor protein (wt‐APP) alleles and (B‐J) heterozygous for an Alzheimer's disease autosomal dominant APP variant, as demonstrated by the multiple reaction monitoring chromatograms. A, In the homozygous sample, a single wt‐Aβ40 peak (blue) is observed. B‐J, In the heterozygous samples, both the wt‐Aβ40 (blue) and var‐Aβ40 (magenta) peaks are observed, the latter corresponding to the following pathogenic variants: (B) Arctic (E22G), (C) Dutch (E22Q), (D) English (H6R), (E) Flemish (A21G), (F) Iowa (D23N), (G) Italian (E22K), (H) Osaka (E22∆), (I) Piedmont (L34V), and (J) Tottori (D7N)
3.3. Diagnostic performance
ROC curve analysis yielded the following area under the curve (AUC) for tau/Aβ42 (0.9137), Aβ42/Aβ40 (0.8305), Aβ42 (0.8436), and Aβ40 (0.5857; Figure 5). In this cohort, the biomarker cut‐points at the Youden indices were as follows: 950 ng/L for Aβ42, 450 ng/L for total tau, 0.28 for the total tau/Aβ42 ratio, and 0.12 for the Aβ42/Aβ40 ratio.
FIGURE 5.

ROC curves using the multiplex amyloid β (Aβ) high performance liquid chromatography tandem mass spectrometry assay for: tau/Aβ42 (dark blue), Aβ42 (green), Aβ42/Aβ40 (light blue), and Aβ40 (orange) with total tau measured by enzyme‐linked immunosorbent assay
4. DISCUSSION
There were several barriers to the deployment of an Aβ peptide assay for routine care in our health‐care setting including: (1) limited selection of Aβ42 assays licensed by our national regulatory agency, and (2) no licensed Aβ40 assays. For Aβ42, only an ELISA was available, which is an undesirable format for clinical laboratories. Based on previously reported mass spectrometric Aβ assays at the time of our assay development, challenges for clinical implementation included:
-
1)
Use of instrumentation not commonly found in clinical laboratories;
-
2)
Manual sample preparation protocols;
-
3)
Assays typically operated by a single experienced operator (and subsequently all figures of merit reported dependent on this single operator);
-
4)
Lack of calibration to the certified reference material;
-
5)
No accounting for the presence of sequence variants.
For deployment in routine clinical testing, we developed a fully automated HPLC‐MS/MS method for multiplex quantification of wt‐Aβ40 and wt‐Aβ42 and detection of var‐Aβ peptides in CSF. We utilized a liquid robotic handler, chromatography system, and mass spectrometer class commonly found in hospital laboratories. For the analytical equipment, we used HPLC (mL/min flow range) coupled to triple quadrupole MS. These systems are widely used in laboratory medicine based on their longstanding use in toxicology. 12 Lower flow liquid chromatography systems (ie, micro‐ and nano‐flow) and high resolution/accurate‐mass mass spectrometers while common in research proteomics labs are rarely used by hospital laboratories and, thus, assays on these platforms have limited uptake. As with our small molecule clinical HPLC‐MS/MS assays, we automated the sample preparation on our robotic liquid handler. The resultant precision and accuracy of the automated method was comparable to that of a single experienced operator. This allowed for implementation in a hospital laboratory where staffing varies, along with operator expertise, and where an assay cannot be assigned to a single technician.
With the availability of the wt‐Aβ42 CRM, the assay was calibrated to this standard. This calibration supports efforts to standardized reporting of Aβ42 mass spectrometric assays and supports harmonization of Aβ42 assays independent of the analytical platform used. Such efforts facilitate comparisons of data (and cut‐points) across different methods, instruments, and laboratories. Based on known variability in amino acid analysis and HPLC‐based purity assessments 25 and our historical lot‐to‐lot calibrator cross‐over data, CRM assignment should be established for each new lot of peptide stocks. Aβ40 was calibrated to the peptide‐manufacturer stated product mass and purity, and with future availability of a CRM, we can apply the procedure described herein to the Aβ40 calibrators as well. Comparison of the Aβ42 assay with the INNOTEST ELISA (not calibrated to the CRM), demonstrated a regression profile consistent with that previously noted for other HPLC‐MS/MS assays with comparisons to the Luminex xMAP and INNOTEST ELISA. 26 , 27
Corroborating previous findings, tau/Aβ42 demonstrated the highest AUC in the ROC curve analysis, with phosphorylated tau not included due to the lack of an assay approved for use in patient care in Canada (at present). In this cohort, the Aβ42/40 ratio did not improve the AUC relative of Aβ42 alone. This is compatible with previous studies that have shown similar‐to‐improved diagnostic performance for Aβ42/40 versus Aβ42, compared to amyloid positron imaging tomography scans. 14 , 28 A diagnostic accuracy study, requiring autopsy confirmation, was not undertaken, nor were amyloid positron imaging tomography scans (due to a lack of clinical availability)—diagnostic studies comparing CSF Aβ to autopsy and/or amyloid imaging tracers has previously been extensively studied for these analytes. 29
While the use of mass spectrometry for quantification of peptides presents numerous analytical advantages, 13 there is a rarely acknowledged short‐coming relating to its greatest strength: analytical selectivity. The MRM approach is so selective that it precludes observation of any modifications to the wild‐type peptide structure which would alter the mass of the peptide and/or retention to the chromatographic column. In general, lack of consideration of variants during peptide or protein biomarker assay development, both for immunoassays and mass spectrometric assays, can lead to erroneous results; misdiagnosis; and, subsequently, inappropriate medical treatment. 17 , 18 , 19 , 20 , 30 In routine care for neurogenerative disorders, an underlying genetic cause can be obscured as family histories may be unknown, uncertain, or miscommunicated to the clinician. 31 , 32 Moreover, genetic analysis may not be broadly performed due to patient/family wishes and/or test availability/cost, and further complicated by the recognition of de novo (non‐Mendelian) pathogenic variants. 33 , 34 Analytical methods for Aβ peptides that do not detect or discriminate between Aβ sequence variants may be suitable in a research setting in which the genotype is known or clinical decisions are not being made based on the results; however, in clinical care such considerations rise in importance. Thus, we developed a mass spectrometric database of known Aβ variants occurring within residues 1‐42.
As proof of concept, we tested the ability of the method to detect the most challenging scenario—APP heterozygosity resulting in two different Aβ peptide sequences in circulation—and the method successfully identified the presence of a var‐Aβ peptide sequence in addition to the wt‐Aβ peptide sequence. Quantification of variants was not performed as variant identification was deemed sufficient in most cases to guide care. For example, identification of a penetrant pathogenic variant is sufficient to identify autosomal dominant AD, and DNA sequencing confirmation would then be recommended. In the case of detection of a non‐pathogenic variant, this would prevent reporting of a falsely low Aβ concentration (ie, a concentration based on just the wild‐type isoform), and inform appropriate testing (eg, using the location of the sequence variant to select a method likely to detect both isoforms). A limitation of this approach is the identification of new variants, requiring updating of the database and testing as new mutations are identified. For the purposes of demonstrating applicability to a wide range of variants, we included variants based on pathogenicity and a frequency threshold. Given the ease of multiplexing with HPLC‐MS/MS, variants monitored can and should be tailored to the laboratory's clinical population. Thus, this multiplex Aβ HPLC‐MS/MS assay serves not only as means to assess the presence of amyloid pathology via quantitation of Aβ peptides, but is also part of the diagnostic workflow for autosomal dominant AD.
On the one hand, Aβ sequence variants only affect accurate quantification of Aβ in a small fraction of individuals (based on reported APP variant frequencies); on the other hand, every case analyzed in clinical care is of equal importance irrespective of variant prevalence. As imprecision and accuracy of Aβ methods have been shown to be important considerations, 11 so is misreporting an Aβ concentration by 50% to 100%, as may occur in cases of APP variant heterozygosity and homozygosity, respectively. Also, there may be yet new roles for variant identification in patient care as genetic research continues to identify new variants and associations with disease, and as mutational databases expand and include more diverse populations. 35 With the availability of a highly selective tool that detects and discriminates Aβ sequence variants, this method can be applied to further our understanding of disease progression and pathology (eg, by studying temporal changes in isoform‐specific peptide concentrations).
This is the first method to identify both sporadic AD (by wt‐Aβ42 and wt‐Aβ40 concentration) and autosomal dominant AD (by identification of pathogenic APP variants) in one method, enabling application of the HPLC‐MS/MS method without a priori knowledge of the genetic makeup of an individual. Moreover, Aβ42 has been calibrated to the CRM and the method and the workflow automated on common clinical laboratory equipment. These assay characteristics enabled implementation of the method in routine clinical care.
Supporting information
Supporting Information
DeMarco ML, Nguyen Q, Fok A, Hsiung G‐YR, van der Gugten JG. An automated clinical mass spectrometric method for identification and quantification of variant and wild‐type amyloid‐β 1‐40 and 1‐42 peptides in CSF. Alzheimer's Dement. 2020;12:e12036 10.1002/dad2.12036
REFERENCES
- 1. Formichi P, Battisti C, Radi E, Federico A. Cerebrospinal fluid tau, a beta, and phosphorylated tau protein for the diagnosis of Alzheimer's disease. J Cell Physiol. 2006;208:39‐46. [DOI] [PubMed] [Google Scholar]
- 2. Bateman RJ, Aisen PS, De Strooper B, et al. Autosomal‐dominant Alzheimer's disease: a review and proposal for the prevention of Alzheimer's disease. Alzheimers Res Ther. 2011;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jack JCR, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimer's Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mattsson N, Andreasson U, Persson S, et al. CSF biomarker variability in the Alzheimer's Association quality control program. Alzheimer's Dement. 2013;9:251‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bittner T, Zetterberg H, Teunissen CE, et al. Technical performance of a novel, fully automatedm electrochemiluminescence immunoassay for the quantitation of b‐amyloid (1‐42) in human cerebrospinal fluid. Alzheimer's Dement. 2016;12:517‐526. [DOI] [PubMed] [Google Scholar]
- 7. Janelidze S, Pannee J, Mikulskis A, et al. Concordance between different amyloid immunoassays and visual amyloid positron emission tomographic assessment. JAMA Neurol. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lame ME, Chambers EE, Blatnik M. Quantitation of amyloid beta peptides Ab1‐38, Ab1‐40, and Ab1‐42 in human cerebrospinal fluid by ultra‐performance liquid chromatography–tandem mass spectrometry. Anall Biochem. 2011;419:133‐139. [DOI] [PubMed] [Google Scholar]
- 9. Shaw LM, Vanderstichele H, Knapik‐Czajka M, et al. Qualification of the analytical and clinical performance of CSF biomarker analyses in ADNI. Acta Neuropathol. 2011;121:597‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Korecka M, Waligorska T, Figurski M, et al. Qualification of a surrogate matrix‐based absolute quantification method for Amyloid‐β42 in human cerebrospinal fluid using 2d UPLC‐tandem mass spectrometry. J Alzheimer's Dis. 2014;41:441‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leinenbach A, Pannee J, Dulffer T, et al. Mass spectrometry‐based candidate reference measurement procedure for quantification of amyloid‐beta in cerebrospinal fluid. Clin Chem. 2014;60:987‐994. [DOI] [PubMed] [Google Scholar]
- 12. Holmes DT, Romney M, Angel P, DeMarco ML. Proteomics applications in laboratory medicine and pathology: present state and future prospects. Clin Biochem. 2020. in press. [DOI] [PubMed] [Google Scholar]
- 13. Hoofnagle AN, Wener MH. The fundamental flaws of immunoassays and potential solutions using tandem mass spectrometry. J Immunol Methods. 2009;347:3‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Korecka M, Figurski MJ, Landau SM, et al. Analytical and clinical performance of amyloid‐beta peptides measurements in CSF of ADNIGO/2 participants by an LC‐MS/MS reference method. Clin Chem. 2020;66:587‐597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin PP, Chen W, Yuan F, et al. An UHPLC–MS/MS method for simultaneous quantification of human amyloid beta peptides Aβ1‐38, Aβ1‐40 and Aβ1‐42 in cerebrospinal fluid using micro‐elution solid phase extraction. J Chromatogr B Analyst Technol Biomed Life Sci 2017;1070:82‐91. [DOI] [PubMed] [Google Scholar]
- 16. Weber DM, Tran D, Goldman SM, et al. High‐throughput mass spectrometry assay for quantifying β‐Amyloid 40 and 42 in cerebrospinal fluid. Clin Chem. 2020;65:1572‐1580. [DOI] [PubMed] [Google Scholar]
- 17. Drees JC, Stone JA, Reamer CR, et al. Falsely undetectable TSH in a cohort of South Asian euthyroid patients. J Clin Endocrinol Metab. 2014;99:1171‐1179. [DOI] [PubMed] [Google Scholar]
- 18. Servant‐Delmas A, Mercier‐Darty M, Ly TD, et al. Variable capacity of 13 hepatitis B virus surface antigen assays for the detection of HBsAg mutants in blood samples. J Clin Virol. 2012;53:338‐345. [DOI] [PubMed] [Google Scholar]
- 19. Hendrickson B, Kamili S, Timmons T, Iwen PC, Pedati C, Safranek T. Notes from the field: false‐negative hepatitis B surface antigen test results in a hemodialysis patient—Nebraska, 2017. MMWR Morb Mortal Wkly Rep. 2018;67:311‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Plantier JC, Djemai M, Lemée V, et al. Census and analysis of persistent false‐negative results in serological diagnosis of human immunodeficiency virus type 1 group O infections. J Clin Microbiol. 2009;47:2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. CLSI . Evaluation of Precision of Quantitative Measurement Procedures; Approved Guideline. CLSI document EP‐5A. 3rd ed Wayne, PA: Clinical and Laboratory Standards Institute; 2014. [Google Scholar]
- 22. CLSI . Liquid Chromatography‐Mass Spectrometry Methods; Approved Guideline. CLSI document C62‐A. Wayne, PA: Clinical and Laboratory Standards Institute; 2014. [Google Scholar]
- 23. CLSI . Evaluation of the Linearity of Quantitative Measurement Procedures: A Statistical Approach; Approved Guideline. CLSI document EPo6‐A. Wayne, PA: Clinical and Laboratory Standards Institute; 2003. [Google Scholar]
- 24. Kuhlmann J, Boulo S, Andreasson U, et al. Certification report: the certification of Amyloid β1‐42 in CSF in ERM®‐DA480/IFCC, ERM®‐DA481/IFCC and ERM®‐DA482/IFCC Luxembourg2017.
- 25. Hoofnagle AN, Whiteaker JR, Carr SA, et al. Recommendations for the generation, quantification, storage, and handling of peptides used for mass spectrometry–based assays. Clin Chem. 2016;62:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Korecka M, Waligorska T, Figurski M, et al. Qualification of a surrogate matrix‐based absolute quantification method for amyloid‐beta(42) in human cerebrospinal fluid using 2D UPLC‐tandem mass spectrometry. J Alzheimers Dis. 2014;41:441‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shaw LM, Hansson O, Manuilova E, et al. Method comparison study of the Elecsys® β‐Amyloid (1‐42) CSF assay versus comparator assays and LC‐MS/MS. Clin Biochem. 2019;72:7‐14. [DOI] [PubMed] [Google Scholar]
- 28. Janelidze S, Pannee J, Mikulskis A, et al. Concordance between different amyloid immunoassays and visual amyloid positron emission tomographic assessment. JAMA Neurol. 2017;74:1492‐1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jack Jr CR, Bennett DA, Blennow K, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimer's Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ketha H, Singh RJ. Clinical assays for quantitation of insulin‐like‐growth‐factor‐1 (IGF1). Methods. 2015;81:93‐98. [DOI] [PubMed] [Google Scholar]
- 31. Goldman JS, Hahn SE, Catania JW, et al. Genetic counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet Med. 2011;13:597‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alexander EL, Butler RK, Guimond C, Butler B, Sadovnick AD. Accuracy of reported family history and effectiveness of medical record requests in genetic counseling for Alzheimer disease. J Genet Couns. 2011;20:129‐135. [DOI] [PubMed] [Google Scholar]
- 33. Rovelet‐Lecrux A, Charbonnier C, Wallon D, et al. De novo deleterious genetic variations target a biological network centered on Aβ peptide in early‐onset Alzheimer disease. Mol Psychiatry. 2015;20:1046‐1056. [DOI] [PubMed] [Google Scholar]
- 34. Butler R, Dwosh E, Beattie BL, et al. Genetic counseling for early‐onset familial Alzheimer disease in large aboriginal kindred from a remote community in British Columbia: unique challenges and possible solutions. J Genet Couns. 2011;20:136‐142. [DOI] [PubMed] [Google Scholar]
- 35. Jia L, Fu Y, Shen L, et al. PSEN1, PSEN2, and APP mutations in 404 Chinese pedigrees with familial Alzheimer's disease. Alzheimer's Dement. 2020;16:178‐191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information