

Graphical abstract

Keywords: c-Jun N-terminal kinase (JNK), Inhibitor, Complex structure, Structure-activity relationship, Gatekeeper
Abstract
The activation of c-Jun N-terminal kinases (JNKs) plays an important role in physiological processes including neuronal function, immune activity, and development, and thus, JNKs have been a therapeutic target for various diseases such as neurodegenerative diseases, inflammation, and cancer. Efforts to develop JNK-specific inhibitors have been ongoing for several decades. In this process, the structures of JNK in complex with various inhibitors have contributed greatly to the design of novel compounds and to the elucidation of structure-activity relationships. Almost 100 JNK structures with various compounds have been determined. Here we summarize the information gained from these structures and classify the inhibitors into several groups based on the binding mode. These groups include inhibitors in the open conformation and closed conformation of the gatekeeper residue, non-ATP site binders, peptides, covalent inhibitors, and type II kinase inhibitors. Through this work, deep insight into the interaction of inhibitors with JNKs can be gained and this will be helpful for developing novel, potent, and selective inhibitors.
1. Introduction to JNK and related diseases
JNKs belong to the mitogen-activated protein kinases (MAPKs) [1], which are activated in response to environmental stress and pro-inflammatory cytokines [2]. JNKs are serine/threonine kinases phosphorylating several substrates such as c-Jun and activating transcription factor 2. There are three closely related genes, JNK1, JNK2, and JNK3, encoding 10 splice variants of JNKs [3]. While JNK1 and JNK2 are expressed in a variety of tissues, JNK3 is almost exclusively expressed in the brain, with very low levels expressed in the heart and testes [4]. The JNK cascade has been under investigation for the past 20 years. For JNK signaling, two MAPK kinases, MKK4 and MKK7, directly activate JNKs. MKK4 and MKK7 preferentially phosphorylate JNK on tyrosine and threonine within the JNK’s activation loop, respectively [5]. Since the dual phosphorylation of JNK on tyrosine and threonine is needed for full activation, MKK4 and MKK7 may cooperate to active JNK [6]. Both MKK4 and MKK7 are activated by various MAPKK kinases, including mixed lineage protein kinases (MLKs), apoptosis signal-regulating kinases (ASKs) and dual leucine zipper kinase (DLK) [7]. The JNK pathway is implicated in a wide range of physiological processes including neuronal function, immune activity, and embryonic development, as well as in variety of pathological conditions including neurodegenerative diseases, cancer, and inflammation [8], [9].
Several in vitro and in vivo studies have been conducted to evaluate the JNKs’ role in CNS disorders. Phosphorylated JNKs were found to be increased in postmorterm brain tissue samples of patients of Alzheimer’s disease (AD) [10]. Particularly, JNK3 was significantly expressed and phosphorylated in the brain and cerebrospinal fluid of patients with AD, and the level of JNK3 phosphorylation was connected statistically to the rate of cognitive decline [11]. It has been shown that Aβ peptides, which are deposited largely in the extracellular space of the brain in AD patients, were able to activate JNK3 in in vitro experiments on the primary cortical cultures from Wistar rat as well as in SH-SY5Y neuroblastoma cells [12], [13]. Yoon et al. showed that JNK3 was responsible for the phosphorylation of the β-amyloid precursor protein, which stimulated Aβ42 production. Depletion of the Jnk3 gene from familial AD mice led to a significant drop in the Aβ42 peptide level and neuritic plaques, and consequently increased the number of neurons and improved cognition [14]. Impaired synaptic plasticity is one of the early events in AD [15]. Several studies indicated that JNK plays a key role in the regulation of synaptic plasticity. Study of Li et al. showed that Jnk1-deficient mice (Jnk1 -/-) enhanced short-term synaptic plasticity and exhibited impairment of metabotropic glutamate receptor-dependent long term depression in hippocampus [16]. Curran et al. suggested that acute over-activation of JNK was associated with the inhibition of long-term potentiation [17]. JNK activation induced by JNK-interacting protein 1 (JIP1) negatively regulated the NMDA receptor-mediated synaptic plasticity and memory [18]. These data identified JNK3 as a potential target for therapeutic intervention in AD.
JNK signaling is also implicated in the pathological progression of Parkinson’s disease (PD). It has been shown that JNK phosphorylation increased in response to the unfolded/misfolded protein which was implicated in PD [19]. Several studies demonstrated that JNKs are activated in PD mouse models [20], [21], [22]. Jnk3 knockout mice showed a significant improvement in dopaminergic neuronal survival in the substantia nigra par compacta relative to wild-type mice in the 6-OHDA-induced neurotoxicity model [23]. Wang et al. reported that dopaminergic neurons were protected from apoptosis due to the inhibition of JNK by its specific inhibitor, SP600125 [24]. These results indicate the potential role of JNK inhibition in the treatment of PD. Altogether, current evidence suggests that JNK inhibitors may offer a promising therapeutic approach for the treatment of neurodegeneration.
The JNK signaling pathway is believed to be involved in numerous inflammatory diseases such as inflammatory bowel disease and rheumatoid arthritis. JNK regulates T cell activity and production of pro-inflammatory cytokines including TNF-α, IL-1β, and IL-6. Inhibition of JNK signaling caused a decrease in the expression of these pro-inflammatory cytokines [25] as well as chemokines CCL17 and CCL22 [26]. JNK activation was increased in the inflamed intestinal tissue of a colitis mouse model induced by dextran sodium sulphate (DSS). Treatment with a JNK inhibitor, SP600125, significantly protected the DSS mouse model from the inflammatory infiltrates in the lamina propria and edema of the colon submucosa [27], [28]. Guma et al. examined the roles of JNK1 and JNK2 in antigen-induced arthritis. Mice with genetically depleted Jnk1 showed a significant decrease in inflammatory cell infiltration and joint damage compared to the disrupted Jnk2 and wild-type mice in the methylated bovine serum albumin model [29]. These data indicate that JNK-selective or isoform-selective inhibitors hold promise in treating inflammatory disease states.
Studies in various JNK knockout mouse cancer models have led to contradictory findings. While some studies support the pro-oncogenic function of JNKs, others indicate that JNKs act as tumor suppressors. Shibata et al. reported that Jnk1 knockout mice exhibited a significant decrease in gastric carcinogenesis in the N-methyl-N-nitrosourea cancer model relative to wild-type mice [30]. Han et al. showed that myeloid cell-specific Jnk1/2 deficiency significantly suppressed the development of fulminant hepatitis and hepatocellular carcinoma (HCC) in Jnk1/2 knockout mice compared to wild-type mice [31]. However, it has been shown that Jnk1/2 disruption in hepatocytes stimulated the development of HCC in the diethylnitrosamine-induced liver cancer mouse model by increasing cell proliferation and cytokine expression [32]. These findings emphasize the importance of isoform-specific JNK inhibitors to clearly define the role of each JNK isoform in cancer promotion and suppression.
JNKs also are considered to be involved in a variety of other pathological states. A number of studies have suggested the critical role of JNKs in pulmonary fibrosis and allergic airway diseases. It has been reported that JNK1, but not JNK2, plays a causal role in the epithelial to mesenchymal transition induced by TGF-β1 and in profibrotic gene expression [33]. Jnk1-disrupted mice showed a decrease in profibrotic gene expression and accumulated less subepithelial collagen in the interstitial pulmonary fibrosis mouse model induced by TGF-β1 and bleomycin [34]. The JNKs’ role in the progression from insulin resistance to diabetes is unclear. Emerging evidence suggests that while JNK1 and JNK2 promote insulin resistance and obesity development [35], [36], JNK3 appears to protect from excessive adiposity, and possibly also loss of β-cell function [37], [38], [39]. The development of isoform-specific inhibitors will be necessary to exploit the JNKs’ role in obesity and diabetes. JNKs have also generated interest as a potential target for intervention in ischemic brain and heart injury. JNK activation stimulates inflammation and ischemic cell death [40]. Permanent occlusion of the middle cerebral artery significantly enlarged the infarction in Jnk1 knockout mice, in which JNK3 showed an increased expression in the penumbra [23]. On the other hand, Kaiser et al. showed that Jnk1 knockout and Jnk2 knockout mice exhibited less injury and cellular apoptosis in a model of myocardial ischemia-reperfusion injury [41].
In summary, the JNK pathway is involved in the pathogenesis of many diseases. The variety of physiological properties of JNK isoforms and the cross-talk between JNK signaling and other pathways preclude the use of non-specific inhibitors [42]. Development of highly selective and nontoxic inhibitors of JNK isoforms is needed to identify the specific role of each JNK isoform in multiple disease states, and further, to identify potential therapeutic agents in those disease states.
2. Structure of JNKs
The overall structure of JNKs greatly resembles the structure of other closely related MAP kinases such as ERK2 and p38 [42]. It consists of a smaller N-terminal lobe composed of mostly β strands and a larger C-terminal lobe with mainly α helices that connects to the N-terminal domain through a flexible hinge-like region (Fig. 1A). The interface of N and C lobes possesses a deep cleft which contains the ATP-binding site. Currently, the majority of drug discovery efforts for JNKs have targeted the ATP-binding site or the interaction with the scaffold protein JNK interacting protein (JIP). Most reported inhibitors are type I kinase inhibitors that bind to the highly conserved ATP-binding pocket. The major problem with this is that the ATP-binding site shares a very high percentage of homology across JNK isoforms, up to 98%, hence the JNK inhibitors lack selectivity. Nevertheless, there are differences in amino acid sequence in hydrophobic region, where the inhibitors are accessible by the gatekeeping residue Met146 (numbering in JNK3, Fig. 1B). Those amino acids are Ile92 and Met115 for JNK3 and the corresponding ones for JNK2 are Val54 and Leu77, respectively. Leu144 for JNK3 corresponds to Ile106 for JNK1. The accessibility of inhibitors to this region is very important to the inhibitory activity of inhibitor in terms of not only potency but also selectivity. For ATP binding to JNK, the gatekeeper is in closed state, thus, the hydrophobic region does not contact with ATP molecule. This feature of the protein structure may facilitate selectivity against JNK isoforms, and an increasing number of studies are reporting attempts to design JNK isoform-selective inhibitors. The efforts in the development of JNK inhibitors have been reviewed by Siddiqui and Reddy [43], Graczyk [9], and Koch et al. [44]. Current review of Koch summarizes the progress in the development of reversible and irreversible JNK3 inhibitors [45]. This review will summarize the information gained from JNK-ligand structures and classify the inhibitors into several groups including inhibitors in the open conformation and closed conformation of the gatekeeper residue, non-ATP site binders, peptides, covalent inhibitors, and type II kinase inhibitors. Through this work, deep insight into the interaction of inhibitors with JNKs can be gained and this will be helpful for developing novel, potent, and selective inhibitors.
Fig. 1.

Structure of JNK3 in complex with AMP (PDB 4KKE). (A) The overall structure of JNK3 is displayed in the gray ribbon diagram, AMP is shown as a yellow stick model, and the gatekeeper residue, Met146, in its “closed” conformation, is highlighted in green. (B) Detailed view of hydrophobic pocket, which is gated by Met146. Key residues are displayed as sticks. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3. Inhibitors with the closed conformation of the gatekeeper
Inhibitors with the closed conformation of the gatekeeper Met146 showed broad spectrum of inhibition against all JNK isoforms.
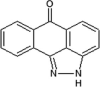
Anthrapyrazolone SP600125 was one of the early pan-JNK inhibitors reported by Celgene (compound 1 in Table 1) [46]. This inhibitor was competitive with ATP and exhibited selectivity toward a small number of kinases. SP600125 inhibited the phosphorylation of c-Jun and modulated the expression of inflammatory cytokines such as IL-2 and TNF-α. It has been used in numerous studies to define the role of JNKs in multiple pathological states such as PD, diabetes, and inflammatory diseases [27], [28], [47], [48], [49].
Table 1.
Structures and in vitro characteristics of inhibitors binding to JNK with Met146 in “closed conformation”
| Compounds | In vitro characteristics | Compounds | In vitro characteristics |
|---|---|---|---|
 |
IC50 (JNK1) = 40 nM IC50 (JNK2) = 40 nM IC50 (JNK3) = 90 nM |
 |
IC50 (JNK1) = 1.6 μM IC50 (JNK2) = 23 μM IC50 (JNK3) = 2.2 μM IC50 (p38α) > 50 μM IC50 (ERK2) > 50 μM |
 |
IC50 (JNK1) = 48 nM IC50 (JNK2) = 470 nM IC50 (JNK3) = 77 nM IC50 (p38α) > 50 μM IC50 (ERK2) > 50 μM |
 |
IC50 (JNK1) = 14 nM IC50 (JNK2) = 43 nM IC50 (JNK3) = 11 nM IC50 (p38α) > 50 μM IC50 (ERK2) > 50 μM |
 |
IC50 (JNK1) = 2 nM IC50 (JNK2) = 5 nM IC50 (JNK3) = 9 nM IC50 (p38α) > 50 μM IC50 (ERK2) > 50 μM |
 |
IC50 (JNK1) = 92 nM IC50 (JNK2) = 67 nM IC50 (JNK3) = 412 nM IC50 (p38α) = 4863 nM IC50 (CDK2) = 412 nM |
 |
IC50 (JNK1) = 13 nM IC50 (JNK2) = 25 nM IC50 (JNK3) = 57 nM IC50 (p38α) = 5423 nM IC50 (CDK2) = 1517 nM |
 |
IC50 (JNK1) = 22 nM IC50 (JNK2) = 5 nM IC50 (JNK3) = 5 nM IC50 (CDK2) > 10 μM IC50 (c-Jun) = 3.8 μM |
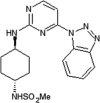 |
IC50 (JNK1) = 24 nM IC50 (JNK2) = 97 nM IC50 (JNK3) = 114 nM IC50 (p38α) > 10 μM IC50 (CDK1) = 1.52 μM IC50(CDK2) = 0.30 μM |
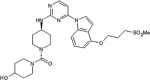 |
IC50 (JNK1) = 16 nM IC50 (JNK2) = 66 nM IC50 (c-Jun) = 1.1 μM |
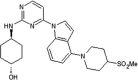 |
IC50 (JNK1) = 3 nM IC50 (JNK2) = 20 nM IC50 (c-Jun) = 0.24 μM |
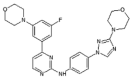 |
IC50 (JNK1) = 99 nM IC50 (JNK3) = 148 nM IC50 (p38α) > 20 μM |
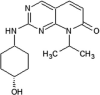 |
IC50 (JNK1) = 18 nM IC50 (JNK2) = 18 nM IC50 (JNK3) = 58 nM IC50 (p38α) > 10 μM |
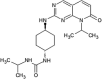 |
IC50 (JNK1) = 21 nM IC50 (JNK2) = 66 nM IC50 (JNK3) = 15 nM IC50 (p38α) > 10 μM |
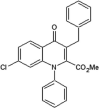 |
IC50 (JNK1) = 0.37 μM IC50 (JNK2) = 0.96 μM |
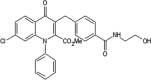 |
IC50 (JNK1) = 0.062 μM IC50 (JNK2) = 0.17 μM IC50 (p38γ) > 10 μM EC50(HK-2) = 10 μM |
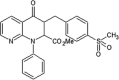 |
IC50 (JNK1) = 91 nM LYSA = < 1 μg/mL |
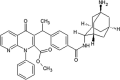 |
IC50 (JNK1) = 0.012 μM EC50(HK-2) = 4.1 μM |
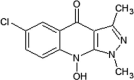 |
IC50 (JNK1) = 1 μM IC50 (p38) > 50 μM IC50 (ERK2) > 50 μM |
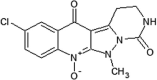 |
IC50 (JNK1) = 0.29 μM IC50 (Pc-Jun) = 0.85 μM |
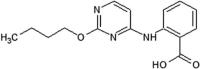 |
IC50 (JNK1) = 1.9 μM IC50 (p38) > 100 μM IC50 (ERK2) > 100 μM IC50 (c-Jun) = 22.3 μM |
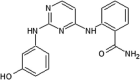 |
IC50 (JNK1) = 9 nM IC50 (p38) > 50 μM IC50 (ERK2) = 25 μM IC50 (c-Jun) = 1.22 μM |
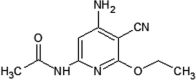 |
IC50 (JNK1) = 0.75 μM IC50 (JNK2) = 1.1 μM EC50 (Pc-Jun) = 6.8 μM |
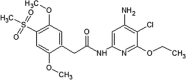 |
IC50 (JNK1) = 0.036 μM IC50 (JNK2) = 0.07 μM EC50 (Pc-Jun) = 1.75 μM |
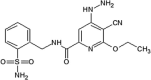 |
IC50 (JNK1) = 0.012 μM IC50 (JNK2) = 0.053 μM EC50 (Pc-Jun) = 0.11 μM |
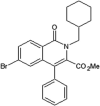 |
IC50 (JNK1) = 1.4 μM IC50 (p38) > 10 μM IC50 (ERK1) > 10 μM |
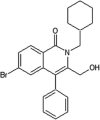 |
IC50 (JNK1) = 0.086 μM IC50 (p38) > 1.1 μM IC50 (ERK1) > 1.1 μM |
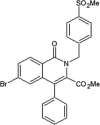 |
IC50 (JNK1) = 0.03 μM IC50 (JNK2) = 0.034 μM IC50 (p38) > 11 μMIC50 (ERK1) > 11 μM EC50 (AP-1) = 2.6 μM |
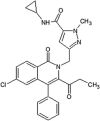 |
IC50 (JNK1) = 6 nM EC50 (AP-1) = 0.72 μM |
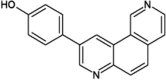 |
IC50 (JNK3) = 0.59 μM |
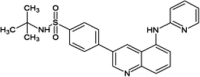 |
IC50 (JNK3) = 0.15 μM IC50 (p38) > 20 μM |
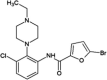 |
IC50 (JNK1) = 0.49 μM IC50 (JNK3) = 1 μM IC50 (p38) > 20 μM |
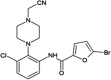 |
IC50 (JNK1) = 0.14 μM IC50 (JNK3) = 0.16 μM IC50 (p38) > 20 μM |
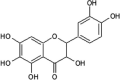 |
IC50 (JNK1) = 4.6 μM |
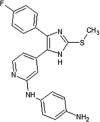 |
IC50 (JNK3) = 181 nM IC50 (p38α) = 11 nM |
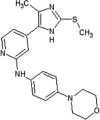 |
IC50 (JNK3) = 363 nM IC50 (p38α) > 10 μM |
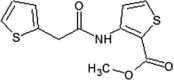
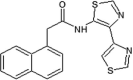
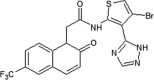
Investigators at Elan Pharmaceuticals tested thiophene- and thiazole-based JNK inhibitors for the prevention of neurodegeneration [50], [51], [52]. Compound 2 was identified as a promising hit by a high throughput screening (HTS) campaign. The X-ray crystal structure of compound 2 in complex with JNK3 (PBD 3OXI) revealed that the thiophene ring was made hydrophobic contact with the gatekeeper Met146, and the carbonyl oxygen atom formed an important hydrogen bond with the backbone amide of Met149 at the hinge region. X-ray crystal structure-guided optimization led to the identification of the improved compound 3 [50].
A second series of thiophene inhibitors originated from the hypothesis that utilizing the third substituent of tri-substituted thiophenes to explore the large hydrophobic pocket I may improve JNK3 potency and selectivity. Structure-guided structure-activity relationship (SAR) studies led to the design of compound 4, which showed JNK selectivity over p38α and ERK2 and a clean profile after screening a panel of 38 kinases. A crystal structure of JNK3 in complex with a compound in this series (PDB 3PTG) revealed that the 4-position substituent of the thiophene was close to the gatekeeper residue Met146. Guided by metabolite identification studies to improve the selectivity and pharmacokinetic properties of this series, compound 5 was obtained. The screen against a panel of 36 kinases revealed a clean profile, with only inhibition of JNK3 at the concentrations tested [51].
Alam et al. developed a series of aminopyrimidines as inhibitors for JNKs [53]. By screening the compound collection of Union Chimique Belge (UCB), compound 6 was obtained with potential JNK activity. The X-ray crystal structure of compound 7, an analog of compound 6, bound to JNK3 (PDB 2P33) revealed that the aminopyrimidine nitrogens formed two hydrogen bonds with the backbone of Met149 in the hinge region, the indole was placed in the ribose-binding site of JNK3, and the 5′-chlorine sterically interacted with the sulfur of the gatekeeper Met146. SAR analysis was employed with the hypothesis that the movement of Met146 could be utilized to induce JNK selectivity over CDK2. The optimization led to compound 8, which exhibited highly potent inhibitory activity against JNK1, JNK2, and better selectivity over CDK2. The docking model of compound 8 bound to JNK3 suggested that the phenyl group could move Met146 and access the selectivity pocket to induce JNK potency and selectivity over the closely related MAPKs.
Roche reported aminopyrimidine JNK inhibitors [54], [55]. Compound 9 exhibited selectivity for JNKs over the CDKs and p38. The cyclohexyl-amine substituents of compound 9 affected the kinase selectivity, and the 1,2,3-benzotriazole group played a role in cytochrome P450 (CYP) inhibition. Kinase profiling and structure-based drug design (SBDD) led to compound 10, with the indole and indazole scaffold replacement affording favorable selectivity and reasonable pharmacokinetic properties. The X-ray crystal structure of a compound in this series bound to JNK1β (PDB 4HYU) showed that the indazole-aminopyrimidine interacted with the backbones of amino acids Glu109, Leu110, and Met111 (numbering in JNK1β) in the hinge region via two hydrogen bonds (Fig. 2). The propoxysulfone moiety was positioned under the glycine-rich loop and made hydrogen bonds with residues Gln 37 and Lys55. In Fig. 2, it is obvious that there is no contact between the inhibitor and the hydrophobic region of JNK since the sidechain of Met108 is in the closed conformation and blocks the entry of the compound to the region. SAR-based optimization was explored and resulted in compound 11. Compound 11 showed an IC50 value of 3 nM for JNK1 and 20 nM for JNK2, which represented 40-fold greater selectivity compared to the 317 other kinases. The oral bioavailability in rat and dog was 100% and 15%, respectively, at the dose of 2 mg/kg, making it good enough for further in vivo characterization.
Fig. 2.

Structure of JNK1β in complex with aminopyrimidine derivative (PDB 4HYU). The sidechain of the gatekeeper, Met108 of JNK1β is in the closed conformation and blocks the hydrophobic region. The hydrogen bonds between the compound and JNK1β are shown with dashed red line. In the gatekeeper closed conformation, there is no interaction between the inhibitor and the hydrophobic region of kinase. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Kamenecka et al. reported a class of aminopyrimidine JNK inhibitors exhibiting the brain penetration properties [56]. Compound 12, which included 1,2,4-morpholino substituted triazoles, was selected as the most potent compound with good cell-based potency and brain penetration properties. Crystal structure of compound 12 in the complex with JNK3 (PDB 3KVX) revealed that compound 12 did not form any hydrogen bonding interaction with any residues of JNK3. The aminopyrimidine portion occupied within the adenosine binding region, and the morpholino substituent on the phenyl ring placed in the hydrophilic sugar pocket of ATP-binding site. Glycine-rich loop containing Ile70-Val78 was seen to move downwards to form a more compressed active site. The planar nature and the hydrophobic interactions appeared to contribute to the stability this compound in the ATP-binding pocket. In the biochemical assay, the IC50 values of compound 12 for the inhibition of JNK1, JNK3 and p38 were 0.099 μM, 0.148 μM, and >20 μM, respectively. Compound 12 also showed good oral exposure (AUC = 1.4 mM 3 h at 2 mg/kg dose), good oral bioavailability (45%), and reasonable half-life in rat model. Moreover, it exhibited good brain penetration with brain/plasma ratio of 0.75 in mouse. However, compound 12 possessed the high level of inhibition for some of the CYP450 enzymes.
Zheng et al. reported a novel class of 2-aminopyridopyrimidinone-based JNK inhibitors [57]. The pyridopyrimidinone 13 was identified as a potential pan-JNK inhibitor with good stability in mouse and human liver microsomes. However, its oral bioavailability was low, with F = 10% in mice. Further SAR optimization led to compounds 14 with great improvement in the oral bioavailability while maintaining the high potency in terms of JNK inhibitory activity. The crystal structure of compound 14 in complex with JNK3 (PDB 4Y5H) showed that the 2-aminopyrimidine scaffold occupied the adenine-binding site and formed two hydrogen bonding interactions with the hinge region residue Met149; the urea amide made a hydrogen bond with the side chain of residue Gln155; and the oxygen atom in the pyridopyrimidinone core formed a water-bridged hydrogen bond with the side chain amide of Lys93 (Lys55 for JNK1β). Additionally, the pyridopyrimidinone core and isopropyl group were in a network of hydrophobic interactions with nearby residues such as Val78, Ala91, Ile124, Met146, Leu148, Val196, and Leu206. The IC50 values of compound 14 for JNK1, JNK2, JNK3 were 21 nM, 66 nM, and 15 nM, respectively, in the biochemical assay, while no inhibition was observed against p38 at the tested concentration of 10 μM. Compound 14 exhibited a clean profile in terms of CYP450 inhibition, good microsomal stability, and better in vivo pharmacokinetic properties, with 87% oral bioavailability in mice.
Roche reported a series of 4-quinolone JNK inhibitors for the indication of asthma [58]. Lead compound 15 was derived from the HTS of a compound library combined with the results on similar JNK inhibitors published by Takeda. The goal was to reduce the lipophilicity while improving the JNK potency of this series. Optimization at the 4-position of the benzyl ring and the 7-position of the quinolone resulted in compound 16, with a good balance of potency and properties. The X-ray structure of an inhibitor from this series bound to JNK1 (PDB 4G1W) suggested that the quinolone carbonyl oxygen moiety interacted with the hinge region residue Met111, while the 4-position substituent of the benzyl group was exposed to the solvent. Compound 16 was profiled against a panel of 317 kinases and showed excellent JNK potency versus other kinases. Later, the 4-quinolone series was evolved into the 8-azaquinolone series for intravenous administration in acute kidney treatment [59]. Compound 17 served as an initial point to profile substituents at the para position of the benzyl ring for the improvement of solubility and cellular potency. Compound 18, which had a trans-adamantyl-OH substituent, showed a good balance between cellular potency, solubility, and microsomal stability. Compound 18 also possessed an excellent selectivity profile in a screen versus a 451-kinase panel, where only JNKs were inhibited by more than 50% at the concentration of 10 μM. The X-ray structure of a compound in the azaquinolone series with JNK1β1-JIP (PDB 4E73) revealed that the carbonyl group of the azaquinolone made a hydrogen bond with the hinge residue Met111, the phenyl was positioned in the space between the side chain of Ile32 and the Asp112/Ala113 plane, the aryl chain pointed out of the ATP pocket, and the amide-linked trans-adamantyl moiety sat in the shallow pocket outside of the N-terminal portion of the kinase active site, which is present only in JNKs. The 8-azaquinolone compound 18 showed better in vitro and cellular potency compared to the 4-quinolone, with four times higher JNK1 IC50 values and 2.5 times higher cellular potency.
Abbott reported a class of 1,9-dihydro-9-hydroxypyrazolo[3,4-b]quinolin-4-ones as JNK inhibitors [60]. Compound 19 was identified by HTS of the Abbott compound collection with an IC50 value of approximately 1 μM for JNK1 and higher than 50 μM for p38 and ERK2. Replacement of the chlorine subtituent of the phenyl ring by a hydrogen atom or an aryl group led to inactive compounds; in contrast, bromine substitution showed only a slight decrease of inhibitory activity. The removal of the N-hydroxyl group resulted in the loss of JNK activity, while the cellular potency improved in the absence of the acidic hydroxyl proton. The X-ray crystal structure of compound 19 bound to JNK1 (PDB 2G01) was obtained. It revealed that the chlorine substituent of the phenyl ring was oriented towards the small specificity pocket, while the oxygen at N9 position formed a hydrogen bond with the backbone amide of the hinge region residue Met111. Compound 20 was obtained by chance with an IC50 value around 0.29 μM for JNK1 and submicromolar cellular potency.
Abbott reported a class of 4-anilinopyrimidines as potent JNK inhibitors [61]. Pyrimidine compound 21 was identified from HTS and was selective for JNK1 over p38 and ERK2. Guided by the NMR-assisted JNK1 active site model, it was considered that the ether portion of compound 21 could interact with Met111, therefore the ether linkage was replaced by a mono-substituted amino substituent in order to donate a hydrogen bond to the backbone carbonyl of Met111. To increase the cell penetrating capacity, a primary amide was used to replace the acid group at the 2-position of the 4-anilino group. Compound 22 was found to exhibit a significant improvement in JNK1 potency in both enzymatic and cell-based assays, and displayed greater selectivity over other kinases. An X-ray crystal structure of compound 22 bound to JNK1 (PDB 2NO3) revealed that the pyrimidine N1 and the 2-anilino group formed 2 crucial hydrogen bonds with the hinge region Met111. The pyrimidine ring and the 4-amide group were oriented towards hydrophobic pocket by an intramolecular hydrogen bond occurring between the 4-amino group and the carbonyl group.
Abbott discovered a series of aminopyridine-based JNK inhibitors [62], [63]. HTS identified acetamide compound 23, with a JNK1 IC50 = 750 nM as well as competitive and reversible binding to the ATP-binding site. An X-ray crystal structure of a compound in this series bound to JNK1 (PDB 2GMX) revealed that the pyridine ring lies within the adenosine binding region, the C-4 amide and the amide carbonyl oxygen formed two weak out-of-plane hydrogen bonds with the hinge region (Glu109 and Met111), the nitril–CN made a weak hydrogen bond with Lys55, and the ethyl ether adapted an orientation towards the ribose binding region. SAR analysis guided by the X-ray crystal structure resulted in a more potent JNK1 inhibitor, compound 24. Reverse amides provided inhibitors with improved metabolic and oral bioavailability, resulting in identification of compound 25 with better enzymatic potency and cellular activity [63].
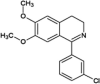
Scientists at Takeda reported a series of isoquinolone JNK inhibitors as novel agents for heart failure [64], [65]. HTS identified isoquinolone derivative 26 as a JNK1 inhibitor. Compound 27 was selected after the initial SAR analysis of the hydroxymethyl group at the 3-position and the cyclohexylmethyl group at the 2-position. Docking studies of compound 27 to JNK1 based on crystallographic data for JNK3 revealed that the inhibitor occupied the adenine-binding site by forming a hydrogen bond between the carbonyl oxygen of the isoquinolone and the backbone NH group of Met111 in the hinge region of JNK1. Further SAR optimization led to compound 28. It showed similarly potent inhibition against all JNK isotypes and showed extremely high selectivity for JNK over other serine-threonine kinases tested. Compound 28 exhibited significant suppression of cardiac hypertrophy at a dose of 10 mg/kg in rat pressure-overload models without affecting blood pressure. The crystal structure of JNK3 and an isoquinolone derivative (PDB 2ZDU) showed that Met149 formed a hydrogen bond with the 1-position carbonyl oxygen, there was an electrostatic interaction between the 4-carboxyl group of the compound and Lys68, and a hydrophobic space around the 6-bromine atom. Further modifications to increase the JNK inhibitory potency in a cell-based assay led to compound 29. Compound 29 exhibited moderate metabolic stability in human and rat with highly potent inhibition in an H9c2 cell-based assay.
Jiang et al. reported a class of 3,5-disubstituted quinolines as JNK inhibitors [66]. Firstly, a phenanthroline scaffold was discovered by screening of a Merck compound collection which had JNK3 IC50 = 590 nM and no inhibition activity observed for p38 (compound 30). By SBDD, 3,5-disubstituted quinoline compound 31 was developed as a new scaffold for JNK inhibitors. The crystal structure of JNK3 and compound 31 (PDB 2R9S) showed an unusual binding mode that did not utilize the space of the ATP binding site. Two nitrogen atoms of the aminopyridinyl group formed two hydrogen bonds with hinge region residue Met149. This positioned the rest of the molecule to surround the hinge region and stretch out into the solvent.
Shin et al. reported a series of piperazine amides as inhibitors of JNK [67]. HTS identified piperazine amide compound 32 with JNK1 IC50 = 0.49 μM, JNK3 IC50 = 1.0 μM, and no selectivity against p38. SAR analysis at the 3-position of the phenyl ring showed that larger substituents caused loss of inhibitory activity. Modification of the bromo-furan group did not lead to an increase in JNK potency. Further SAR analysis of compound 32 led to a small boost in potency in compound 33 with JNK3 IC50 = 0.16 μM and JNK1 IC50 = 0.14 μM. The X-ray crystal structure of compound 33 bound to JNK3 (PDB 3FV8) revealed that the bromofuran ring was positioned deep within the active site, with the furan oriented towards the gatekeeper residue Met146. Piperazine compound 33 made van der Waals contact with Met146, Ile70, Val78, Val196, and Leu206. The amide oxygen interacted with the backbone amide of Met 149 and formed an electrostatic interaction with the backbone carbonyl of Glu147. The sum of many weak interactions provided profound benefit to the binding mode of this series to JNK.
Baek et al. described the binding mode of flavonoid quercetagetin (compound 34) on JNK1 [68]. The crystal structure of quercetagetin bound to JNK1 (PDB 3V3V) was determined. It revealed that upon the binding of quercetagetin to the ATP-binding pocket, the N-terminal loop forming the active site was repositioned downwards. The 4-hydroxy group of the catechol moiety formed a hydrogen bond network with Lys55 and Glu73, which may be facilitated by the movement of the N-terminal lobe. Hydrogen bond interactions of the hinge region with the benzopyran were observed. The glycine-rich loop shifted down to cover the binding site. This rearrangement appeared to improve the binding of ligands to the substrate binding region. Quercetagetin attenuated JNK1 activity with an IC50 value of 4.6 μM, which is even more potent than SP600125 (IC50 = 5.2 μM), in the immobilized metal ion affinity-based fluorescence polarization assay. In the cellular assay, quercetagetin suppressed the phosphorylation of c-Jun and reduced cell transformation. In a mouse model of skin carcinogenesis, quercetagetin delayed the development of the tumors over 20 weeks of treatment. This suggested a promising future for quercetagetin in the prevention and therapy of cancer or other chronic diseases.
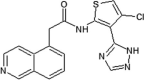
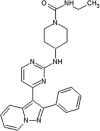
Ansideri et al. discovered a series of JNK3 inhibitors starting from a pyridinylimidazole scaffold of known p38α inhibitors [69]. Compound 35 was a balanced dual JNK3/p38α MAPK inhibitor with JNK3 IC50 = 181 nM and p38 IC50 = 11 nM. Optimization of compound 35 by modification of the five-membered heterocylic core, the aryl moiety at the imidazole-C4 position, and the pyridine-C2 amino function resulted in compound 36. Inhibitor 36 displayed substantial selectivity for JNK3 over p38α. In the screening against a panel of 45 kinases, 10 kinases were inhibited more than 50% at a concentration of 10 μM of compound 36. The crystal structure of JNK3 in complex with compound 36 (PDB 6EKD) was obtained to elucidate the binding mode. Compound 36 interacted with Met149 in the hinge region of JNK3 via two hydrogen bonds. A hydrophobic interaction network was observed with proximal residues comprising Ile70, Val78, Met146, Val196, and Leu206. These interactions were favorable for JNK3 because they were unable to occur in the larger binding site of p38α. The methyl substituent was not able to occupy the hydrophobic pocket of p38α, but pointed toward the gatekeeper Met146 in the optimal length. Hence, this methyl group facilitated the JNK3 selectivity against p38α. The binding of compound 36 repositioned the flexible Gly-rich loop downward, leading to the compression of the binding pocket.
4. Inhibitors with the open conformation of the gatekeeper
4.1. Pan-JNK inhibitors
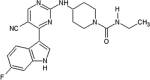
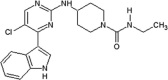
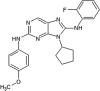
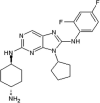
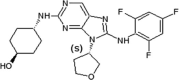
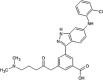
Investigators at Celgene Corporation reported a series of aminopurine-based JNK inhibitors for the prevention of ischemia reperfusion [70], [71]. Compound 37 (Table 2) was identified as an ATP-competitive inhibitor of JNK during a HTS campaign. The crystal structure of compound 37 bound to JNK3 (PDB 3TTJ) revealed that the amino pyrimidine moiety of the aminopurine scaffold formed a bidentate binding motif to Met149 at the hinge region of the binding pocket. The C8 anilinyl moiety occupied an induced-fit pocket due to the movement of the gatekeeper residue Met146. SAR-guided optimization led to compound 38 with a ~10-fold higher JNK2/JNK3 inhibition selectivity compared to JNK1 and p38a. Inhibitor 38 was selected as a potential clinical candidate for the prevention of ischemia reperfusion injury [70]. Further optimization of compound 38 for oral administration led to compound 39. The crystal structure of compound 39 and JNK1 (PBD 3TTI) revealed that the oxygen of the THF substituent has a favorable electrostatic interaction with the side chain NH2 of Asn152. Compound 39 exhibited selectivity against a panel of 240 kinases. The efficacy of compound 39 showed potential in the LPS-induced TNFα rat model and the bleomycin-induced pulmonary fibrosis mouse model [71]. Compound 39 underwent two phase II clinical trials in January 2011 in idiopathic pulmonary fibrosis patients [72]. These trials were terminated due to an inappropriate benefit/risk profile.
Table 2.
Structures and in vitro characteristics of inhibitors binding to JNK with Met146 in an “open conformation”
| compounds | In vitro characteristics | compounds | In vitro characteristics |
|---|---|---|---|
 |
IC50 (JNK1) = 1.1 μM IC50 (JNK2) = 0.12 μM IC50 (JNK3) = 0.21 μM IC50 (p38α) > 30 μM |
 |
IC50 (JNK1) = 0.27 μM IC50 (JNK2) = 0.025 μM IC50 (JNK3) = 0.024 μM IC50 (p38α) = 0.22 μM |
 |
IC50 (JNK1) = 0.061 μM IC50 (JNK2) = 0.007 μM IC50 (JNK3) = 0.006 μM IC50 (p38α) = 3.4 μM |
 |
JNK3 96% inhibition @ 10 μM c-Raf 65% inhibition @ 10 μM |
 |
IC50 (JNK1) = 1.0 μM IC50 (JNK2) = 0.069 μM IC50 (JNK3) = 0.024 μM IC50 (p38α) > 50 μM IC50 (ERK2) > 50 μM |
 |
IC50 (JNK1) = 0.11 μM IC50 (JNK2) = 0.15 μM IC50 (JNK3) = 0.014 μM IC50 (p38α) na IC50 (ERK2) na |
 |
Kia (JNK3) = 2.7 μM Ki (p38α) > 100 μM Ki (ERK2) > 30 μM |
 |
Ki (JNK1) = 0.45 μM Ki (JNK2) = 0.07 μM Ki (JNK3) = 0.09 μM Ki (p38α) > 20 μM Ki (ERK2) > 50 μM |
 |
Ki (JNK3) = 0.74 μM Ki (p38α) = 0.23 μM |
 |
IC50 (JNK1) = 1.6 uM IC50 (JNK3) = 0.1 uM |
 |
pIC50b (JNK1) = 5.8 | 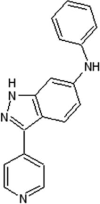 |
IC50 (JNK1) > 10 μM IC50 (JNK3) = 48 nM IC50 (p38α) = 30 nM |
 |
IC50 (JNK1) = 101 nM IC50 (JNK3) = 3 nM IC50 (p38α) = 903 nM |
 |
pIC50 (JNK3) = 5.0 |
 |
pIC50 (JNK1) = 4.0 pIC50 (JNK2) = 6.1 pIC50 (JNK3) = 7.3 pIC50 (p38α) = 5.1 pIC50 (ERK2) < 4.0 |
 |
pIC50 (JNK1) < 4.0 pIC50 (JNK2) = 5.9 pIC50 (JNK3) = 6.9 pIC50 (p38α) = 5.1 pIC50 (ERK2) < 4.0 |
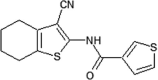 |
pIC50 (JNK3) = 6.2 pIC50 (p38α) < 4.8 |
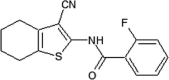 |
pIC50 (JNK1) < 5.0 pIC50 (JNK3) = 6.4 pIC50 (p38α) < 4.8 |
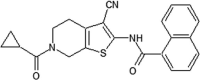 |
pIC50 (JNK1) < 5.0 pIC50 (JNK2) = 6.5 pIC50 (JNK3) = 6.6 pIC50 (p38α) < 4.8 pIC50 (ERK2) < 5.0 |
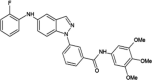 |
IC50 (JNK1) = 0.073 μM IC50 (JNK3) = 0.012 μM IC50 (p38α) = 0.003 μM |
 |
IC50 (JNK1) = 0.17 μM IC50 (JNK3) = 0.007 μM IC50 (p38α) > 20 μM |
 |
IC50 (JNK1) = 0.4 μM IC50 (JNK3) = 0.025 μM IC50 (p38α) = 3.7 μM |
 |
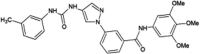
IC50 (JNK1) > 10 μM IC50 (JNK2) = 0.311 μM IC50 (JNK3) = 0.206 μM IC50 (p38α) > 10 μM |
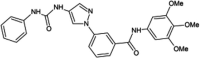
 |
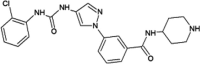
IC50 (JNK1) = 2.74 μM IC50 (JNK2) = 0.486 μM IC50 (JNK3) = 0.115 μM IC50 (JNK3L144I) > 10 μM IC50 (p38α) > 10 μM |
Since some compounds such as compounds 47 and 50 are primary hits and un-optimized ones, there were no isoform-selective data. aKi: inhibitory constant, bpIC50: -log IC50. All values are adopted from the original publications.
4.2. Isoform-selective JNK inhibitors
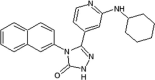
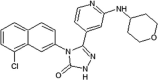
Elan pharmaceuticals also reported the triazolone class of selective JNK2 and JNK3 inhibitors [73]. Identification of compound 40 from the screening campaign and further optimization afforded compound 41 with a 10-fold higher JNK2 and JNK3 inhibitory selectivity over JNK1. This compound revealed a clean profile after screening against a panel of 38 kinases. Compound 42 was obtained by chlorine substitution on the naphthalene ring. It exhibited around 10-fold JNK3 selectivity over JNK1 and JNK2, and no measurable IC50 value against ERK2 or p38α at 10 μM. An X-ray crystal structure of a compound in this series bound to JNK3 (PDB 3OY1) revealed that the naphthalene ring penetrated deep into the hydrophobic region in Fig. 1, and sulfur-π stacking interaction was present between Met146 and the bridgehead carbons of the naphthalene ring.
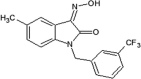
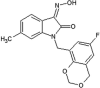
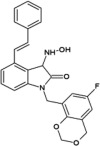
Vertex investigators reported a class of N-benzyl isatin oximes as JNK3 inhibitors [74]. Isatin oxime compound 43, an ATP-competitive JNK inhibitor, was identified by the HTS campaign as exhibiting moderate JNK3 potency (Ki = 2.7 μM) and selectivity against MAPK family members ERK2 (>30 μM) and p38 (>100 μM). SAR-guided optimization identified compound 44, with the N-substituent being 6-fluoro-4H-benzo[d][1,3]dioxin-8-ylmethyl, as the most potent inhibitor in this class, exhibiting good selectivity for JNKs over other kinases. The crystal structure of compound in this series bound to JNK3 (PDB 3G90, cyan in Fig. 3) revealed that the isatin carbonyl oxygen and the oxime hydroxyl formed two hydrogen bonds with the backbone amide of Met149 and the backbone carbonyl of Glu147 at the hinge region. In this structure, the gatekeeper, Met146 is in the closed conformation. Interestingly, it appeared that introduction of a large substituent at the 4-position of the isatin oxime induced the open conformation of Met146. Several substitutions at the 4-position of isatin oxime were obtained. The crystal structure of JNK3 in complex with compound 45 possessing the 4-styrenyl substituent (PDB 3G9L, yellow in Fig. 3) showed that the gatekeeper Met146 shifted out to accommodate the styrenyl group in the induced-fit hydrophobic pocket. However, the JNK potency of compound 45 was 5-fold lower than that of the smaller 4-vinyl substituent. It was speculated that opening and filling the pocket by a rigid side chain led to a slight loss in JNK inhibitory activity.
Fig. 3.

Overlaid structures of JNK3 in complex with the compound 44 (cyan stick, PDB 3G90) and 45 (yellow stick, PDB 3G9L). The JNK3 active site is shown in gray ribbon. Hydrogen bonds between the inhibitors and the hinge region of JNK3 are formed in both structures and displayed as red dashed lines. In the JNK3 structure in complex with the compound 45, the gatekeeper, Met146, is in the open conformation allowing the insertion of the sterenyl group to the hydrophobic region, while the gatekeeper in the structure with of the compound 44 in the closed conformation. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
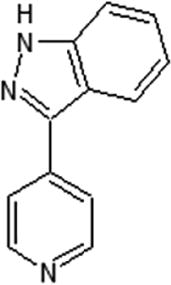
Scientists from AstraZeneca reported a series of 6-anilinoindazoles as JNK3 inhibitors [75]. From the HTS campaign, compounds 46 and 47 were obtained with JNK3 and JNK1 inhibition activity, respectively [76]. An X-ray crystal structure of compound 46 bound to JNK3 revealed that the p-fluorophenyl group occupied the selectivity pocket of JNK3. Further optimization by introducing a 6-anilino group on the 3-arylindazol scaffold of compound 47 resulted in compound 48. An X-ray crystal structure of compound 48 in the ATP pocket of JNK3 showed that the methylthioethyl chain of the gatekeeper residue Met146 moved to accommodate the aniline part of the ligand. There were hydrogen bonds formed between the indazole nitrogens and the backbone amide of Met149, and the backbone carbonyl of Glu147. Compound 48 possessed an excellent selectivity for JNK3 against JNK1. Further optimization identified compound 49 that exhibited excellent JNK3 selectivity relative to both JNK1 and p38. An X-ray crystal structure of compound 49 in complex with JNK3 (PDB 2B1P) revealed a binding mode similar to that of compound 48. Additionally, the carboxylic acid group made a hydrogen bond with the amide of Asn152, which corresponds to the acidic residue Asp112 in p38.
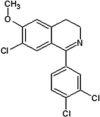
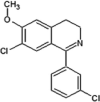
Investigators at GSK reported a series of 1-aryl-3,4-dihydroisoquinoline inhibitors of JNK3 [77]. Screening for new JNK3 inhibitors identified a weakly potent dihydroisoquinoline compound 50 with pIC50 = 5.8. Further SAR exploration resulted in compounds 51 and 52, in which the methoxy group at the 7-position of the dihydroisoquinoline was replaced by a chlorine. The crystal structure of the bromo-analog of compound 52 in complex with JNK3 (PDB 2WAJ) was obtained. It revealed that the chlorine at the 7-position of the dihydroisoquinoline made an unusual hydrogen bond with the backbone NH of Met149, and a halogen bond with the backbone carbonyl of Glu147, while the 3-chlorophenyl substituent occupied the hydrophobic selectivity pocket, which was exposed by rearrangement of the gatekeeper Met146 and was formed mainly by the sidechains of Met146, Ala91, Leu144, Ile124, Leu206, and Lys93. The bromine atom on the phenyl substituent was in contact with Leu144 (corresponding to Ile106 in JNK1) and Ala91. Compounds 51 and 52 were highly potent inhibitors, with 10-fold and 1000-fold selectivity for JNK3 over JNK2 and JNK1, respectively.
GSK reported a series of N-(3-cyano-4,5,6,7-tetrahydro-1-benzothien-2-yl)amides for JNK2 and JNK3 inhibitors [78]. Compound 53, N-(3-cyano-4,5,6,7-tetrahydro-1-benzothien-2-yl)amide, was identified as a potent hit by a screening exercise. Optimization based on the SAR resulted in compound 54, with high selectivity for JNK2/3 over JNK1. An X-ray crystal structure of compound 54 in complex with JNK3 (PDB 2O2U) revealed that the amide of Met149 donated a hydrogen bond to the 3-cyano substituent of 54, the sulfur of Met146 formed an unusual hydrogen bond with the amide nitrogen, and the Met146 sidechain moved away to allow the 2-(2-fluoro)benzamine substituent to take up the ‘induced-fit’ hydrophobic pocket, while the amide oxygen of compound 54 made a water-bridged hydrogen bond to Lys93. Guided by the X-ray structure of compound 54, compound 55 was obtained. Compound 55 was inactive against a panel of 30 kinases. The crystal structure of the JNK3-compound 55 complex (PDB 2O0U) showed that the naphthyl substituent occupied the hydrophobic selectivity pocket, with the piperidine exposed to the solvent.
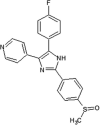
Kamenecka et al. developed a class of aminopyrazoles for JNK3 inhibitors [79]. First, a series of indazole inhibitors was developed from parent compound 56 to maintain the JNK3 potency while gaining selectivity for JNK3 over p38. However, there was no increase in JNK3 selectivity over p38 observed after SAR optimization. Second, a class of aminopyrazole urea-based inhibitors, which was comparable to the indazole structure, was investigated. SAR analysis led to compound 57, which had 2800-fold and 20-fold greater JNK3 selectivity over p38 and JNK1, respectively, in an enzymatic assay. Compound 57 showed an IC50 value of 1.3 μM for c-Jun phosphorylation inhibition in cell-based activity assays. The crystal structures of JNK3 in complex with indazole 56 (PDB 3FI3) and aminopyrazole 58 (PDB 3FI2), an analog possessing JNK3 potency similar to 57, were obtained. They showed that both inhibitors bound to the ATP-binding site in a similar fashion. The same hydrogen bonds were formed between the indazole N2 of 56 and the pyrazole N2 of 58 and the backbone amide of Met149 in JNK3. The amide nitrogen atom of compound 56 or 58 made a hydrogen bond with the backbone carbonyl of Met149. The trimethoxyphenyl ring was exposed to the solvent. The gatekeeper residue Met146 adopted a conformational change upon the binding of both inhibitors, hence the 2-fluoro-anilino group of 56 or the urea-phenyl ring of 58 occupied hydrophobic pocket. The structural features of these compounds giving rise to the selectivity in the aminopyrazole class included the highly planar nature of the pyrazole and the N-linked phenyl structures, which better occupy the smaller active site of JNK3 compared to the larger active site of p38. Further optimization of the trimethoxyphenyl ring led to compound 59, with more than 50-fold selectivity for JNK3 over JNK1 [80].
Park et al. discovered the molecular basis for JNK2/3 isoform selectivity over JNK1 of this aminopurine class [81]. Using sequence alignment between JNK3α1 and JNK1α1 in parallel with comparisons of the crystal structures of JNK3 and JNK1 binding to AMP-PNP, Leu144 in JNK3 and Ile106 in JNK1 were identified as the only distinct residues among 23 residues forming the ATP-binding pocket. A series of five aminopyrazole inhibitors (analogs of compound 57) showed a decrease >20-fold in the inhibitory activity for JNK3-L144I in comparison to WT JNK3 in an enzymatic assay. The crystal structure of aminopyrazole 60 bound to WT JNK3 (PDB 4W4V) showed that the aminopyrazole was placed in the ATP-binding pocket and formed a hydrogen bond network with JNK3, in which the phenyl ring occupied the selectivity pocket due to the rearrangement of the Met146 sidechain. This observation suggested that the hydrophobic interaction between the phenyl group and the amino acids in the hydrophobic pocket, in which Leu144 played a critical role, was greatly beneficial for JNK3 selectivity over JNK1.
5. Non-ATP site inhibitors
5.1. Small molecules
Abbott Laboratories reported the discovery of JNK1 kinase non-ATP site inhibitors [82]. An affinity-based screen was performed against the unphosphorylated JNK1 protein using affinity selection followed by mass spectroscopy. A library of ~500,000 small molecules was screened, from which 68 candidate ligands were identified, and 41 were confirmed as ligands for the protein by NMR binding studies. The majority of confirmed ligands (34 of 41 tested) bound to the ATP-binding site. Two compounds (compounds 61 and 62) (Table 3) bound exclusively to a different site on the inactive form of the protein. Crystallographic studies of JNK1 in the presence of compound 62 were performed (PDB 3O2M) (Fig. 4). The dimethyl pyrimidine ring of compound 62 occupied a “hydrophobic bend” provided by Ile231 and Tyr230, and its butyl moiety was inserted into a hydrophobic pocket partially provided by the MAP kinase insert containing Trp234 at the bottom. The biaryl tetrazole moiety was positioned in a broad space provided by the activation loop and the second helix from the MAP kinase insert. Given that no specific interactions were observed between the tetrazole group and JNK1, compound 63 was obtained by reducing the size while maintaining critical interactions to improve the cellular potency. Compounds 61, 62, and 63 were active in the MKK7/JNK1 activation cascade assay, but not in the JNK1 enzymatic activity assay. This suggested that these inhibitors inhibited JNK1 activity through a mechanism distinct from inhibition of the catalytic activity of JNK1.
Table 3.
Structures and in vitro characteristics of non-ATP site inhibitors.
| compounds | In vitro characteristics | compounds | In vitro characteristics |
|---|---|---|---|
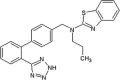 |
Kd (JNK1-u) = 11 μM Kd (JNK1-a) > 50 μM Kd (p38-u) = 18 μM Kd (ERK2-u) = 18 μM IC50 (JNK1) > 100 μM IC50 (MKK7-JNK1) = 7.8 μM EC50 (P-cJun) > 30 μM |
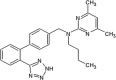 |
Kd (JNK1-u) = 16 μM Kd (JNK1-a) > 50 μM Kd (p38-u) = 17 μM Kd (ERK2-u) = 32 μM IC50 (JNK1) = 6.2 μM IC50 (MKK7-JNK1) = 7.7 μM EC50 (P-cJun) > 30 μM |
 |
IC50 (JNK1) > 100 μM IC50 (MKK7-JNK1) = 3.8 μM EC50 (P-cJun) = 4.0 μM |
Fig. 4.

Structures of compound 62 (yellow stick) bound to JNK1 (PDB 3O2M, gray ribbon). (A) The compound 62 binding site is different from the ATP-binding hinge region (colored in red). Activation loop is shown in green ribbon. MAPK insert is shown in cyan ribbon. (B) Detailed view of amino acids involved in the non-ATP binding site. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
5.2. Peptides
Kaoud et al. reported two JIP-derived peptide inhibitors with highly potent JNK2 inhibition [83]. The backbone of JIP10-△-TATi peptide comprised a 10-mer JIP144-153 peptide (PKRPTTLNLF) linked to the N-terminus of an inverted HIV-TAT49-58 sequence through a 6-aminohexanoyl group. JIP10-△-R9 peptide was similar to the aforementioned peptide except that the TAT sequence was replaced by a different nine-residue sequence. Both of these peptides significantly attenuated JNK2 activity, with IC50 values of 92 nM and 89 nM, which were 12-fold and 150-fold lower than their IC50 values against JNK1/3 and p38 or ERK2, respectively. In a cell-based assay, both peptides strongly suppressed the activation/phosphorylation of JNKs. In vivo, these JIP-based inhibitors inhibited cell migration in a breast cancer cell line.
Pan et al. engineered a JNK3 peptide inhibitor as a potential therapeutic reagent to protect dopaminergic neurons in PD [84]. A 21-residue peptide, termed JNK3-N-TAT, consisted of a 10-amino acid sequence corresponding to the C9-18 segment of the N-terminus of JNK3 connected to an HIV-TAT sequence. This peptide inhibited the binding of scaffolding protein β-arrestin 2 to the N-terminus of JNK3 and strongly attenuated the activation of JNK3 induced by MPP/MPTP in both SH-SY5Y cellular assays and PD mouse models. In an MTT assay, pretreatment of SH-SY5Y cells or primary cultured cortical neurons with 20 μM of peptide resulted in a significant decrease in cell death induced by MPP. In vivo, this inhibitor exhibited protective effects against MPTP-induced dopaminergic neuronal injury.
Chambers et al. designed a peptide of outer mitochondrial membrane protein Sab as an inhibitor of JNK mitochondrial translocation [85]. This retro-inverso peptide contained 11 amino acids corresponding to the phosphorylated JNK binding site of Sab (AVVRPGSLDLR), attached with an HIV-Tat motif to enhance cellular penetrance. In anisomycin-stressed HeLa cells, Tat-SabKIM1 blocks the mitochondrial JNK signaling, but not nuclear JNK signaling pathway, and prevents Bcl-2 phosphorylation, cell death, loss of mitochondrial membrane potential, and superoxide generation. Furthermore, this peptide protected against 6-OHDA-induced oxidative stress, mitochondrial dysfunction, and neurotoxicity in vitro and in vivo by inhibiting the JNK mitochondrial translocation [86]. Win et al. reported that Tat-SabKIM1 peptide suppressed the effect of endoplasmic reticulum stress on mitochondrial respiration by inhibiting the interaction of JNK and Sab [87].
The crystal structure of unligand JNK3 bound to JIP1-derived peptide (pepJIP1: RPKRPTTLNLF) (PDB 4H39) and Sab-derived peptide (pepSAB: VVRPGSLDLP) (PDB: 4H3B) were reported by Laughlin et al. [88] These peptides were found to bind to the C-terminal lobe of JNK3. For the binding mode of pepJIP1, Arg160 interacted to residue Glu367 of JNK3 through a salt bridge 2.7 Å, and formed hydrogen bonding interactions with Tyr168, the backbone of Trp362 of JNK3. The backbone and side chains of Thr162 and Thr163 formed extensive hydrogen bond networks with Arg165 of JNK3. PepJIP contacted with JNK3 through a van der Waals network with Tyr171, Asp364, Val197 and Val156 of JNK3. In the pepSAB to JNK3, Arg343 formed an electrostatic interaction with JNK3 Glu367. Gly345 and Ser346 showed a subset of hydrogen bonds to Arg165 of JNK3. Upon the binding of pepSAB, the activation loop of JNK3 coiled into a helix, and docked into the ATP binding pocket. The G-rich loop possessed a considerable shift to accommodate the docking of the helix. By utilizing the structure class analysis of 26 JNK3 structures with or without peptide binding, the author suggested that the binding of peptides induced large interlobe hinge and rotation motion of JNK3.
5.3. MAPK inserts
Scientists from Roche suggested that the MAPK insert played a crucial role in regulating the activation of JNK2 [89]. MAPKs share a common sequence insertion in the C-terminal kinase lobe called the “MAP kinase insert.” This segment includes two α-helices, α1L14 and α2L14, which are connected by a short loop. The crystal structure of JNK2 (PDB 3E7O) presented two protein chains, A and B, which adopted two distinct conformations of JNK2’s activation loop and the MAPK insert. In chain A, the side chains of Met181 and Met182 were buried in the pocket partially formed by the MAPK insert. The side chain of activation loop residue Tyr185, which is involved in phosphorylation by the upstream kinase MKK4, was buried inside the protein. This conformation, hence, prevented JNK2 from being activated by MKK4. In chain B, the activation loop was not resolved and the MAPK insert moved out. This observation suggested that the MAPK insert regulates JNK2 activity. Further studies should be carried out to understand the reasons behind this conformational change, which likely modulates JNK2 activity.
6. Type II JNK inhibitors
Scientists from Roche discovered for the first time the crystal structure of a JNK isoform in the “DFG-out” (type II) conformation. In this conformation, the Phe residue of the DFG (Asp, Phe, Gly) triplet on the activation loop points towards the outside of the ATP binding site, which prevents ATP from binding to the active site [90]. The X-ray structure of a complex between JNK2 and the p38α inhibitor BIRB796 (PDB 3NPC, Fig. 5A) showed that the DFG motif is swung out, allowing the inhibitor to entirely occupy the active site of JNK2 and extend into the pocket formed by helix C, the DFG triplet, and the Gly-rich loop. The Thr183 side chain, which is one of the key residues involved in JNK2 phosphorylation by the upstream kinase MKK7, pointed into the ATP binding pocket, and hence stabilized the JNK2 activation loop in the phosphorylation-incompatible conformation (Fig. 5B).
Fig. 5.

The binding mode of type II inhibitor in the JNK2 binding pocket. (A) X-ray structure of a complex between JNK2 and the p38α inhibitor BIRB796 (PBD 3NPC). (B) Overlay of X-ray crystal structure of the activation loop in the DFG-out conformation (PDB 3NPC, cyan ribbon) and the DFG-in conformation (PDB 3E7O, purple ribbon) of JNK2. Type I inhibitor is displayed as green stick. Type II inhibitor is displayed as yellow stick. In the DFG-out conformation, Phe170 (cyan stick) pointed out; hence, the type II inhibitor BIRB796 (yellow stick) extended into the pocket provided by the Gly-rich loop, helix C and DFG triplet. Phe170 (purple stick) pointed in the ATP-binding pocket in the DFG-in conformation. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
7. Covalent inhibitors
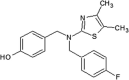
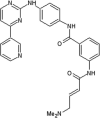
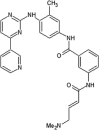
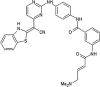
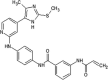
Zhang et al. reported covalent inhibitors that targeted a conserved cysteine in the ATP binding site of JNK family members [91]. Utilizing the phenylamino pyrimidine scaffold, a known c-kit type II inhibitor, to exploit a cysteine residue preceding the DFG triplet of JNKs led to compound 64 (Table 4). It exhibited a high degree of binding for JNK1/2/3 in the Z’-LYTE assay. To confirm the binding mode of this inhibitor to JNK, the crystal structure of an analog of 64 bound to JNK3 was obtained (PDB 3V6R, Fig. 6). It revealed that the inhibitor bound to JNK3 in the type I binding mode. The pyridine substituent was positioned in the adenine binding site, while the two nitrogen atoms of the aminopyrimidine adopted two hydrogen bonding interactions with Met149 of the hinge region. The amide NH formed one more hydrogen bond to the side chain amide of Asn152, which may help in orienting the acrylamide moiety towards the Cys154 to form a covalent bond. Replacing the acrylamide by an isosteric propyl amide substituent resulted in a great loss in the potency for JNKs, which demonstrated that the covalent bond played a crucial role in the JNK potency of this class. The results from biochemical assays and cell-based kinase assays of a series of 12 compounds in this class showed that inhibitors possessing a 1,4-disposition of the dianiline moiety and a 1,3-disposition of the terminal aminobenzoic acid moiety were highly potent for JNK inhibition. For example, compound 65 exhibited highly potent JNK inhibitory activity in the nanomolar range in the enzymatic and cellular assays.
Table 4.
Structures and in vitro characteristics of covalent JNK inhibitors.
| compounds | In vitro characteristics | compounds | In vitro characteristics |
|---|---|---|---|
 |
IC50 (JNK1) = 7.78 μM IC50 (JNK2) = 4.23 μM IC50 (JNK3) = 7.75 μM |
 |
IC50 (JNK1) = 1.54 nM IC50 (JNK2) = 1.99 nM IC50 (JNK3) = 0.75 nM EC50 (Hela) (P-cJun) = 130 nM EC50 (A375) (P-cJun) = 244 nM |
 |
IC50 (JNK1) = 4.67 nM IC50 (JNK2) = 18.7 nM IC50 (JNK3) = 0.98 nM EC50 (Hela) (P-cJun) = 486 nM EC50 (A375) (P-cJun) = 338 nM |
 |
IC50 (JNK1) = 13 nM IC50 (JNK2) = 11.3 nM IC50 (JNK3) = 11 nM EC50 (Hela) (P-cJun) = 605 nM EC50 (A375) (P-cJun) = 134 nM |
 |
IC50 (JNK3) = 0.3 nM IC50 (p38) = 36 nM |
 |
IC50 (JNK3) = 2 nM IC50 (p38) = 1952 nM |
The incubation time for biochemical assay (IC50): 60 min, cellular assay: 90 min (Hela cell line), 180 min (A375 cell line).
Fig. 6.

X-ray crystal structure of an analog of 64 (PDB 3V6R) binding to ATP-binding pocket of JNK3 (gray ribbon). Compound formed a covalent bond with residue Cys154. The hydrogen bonds between the compound and JNK3 are shown with dashed red line. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The series of 12 compounds was profiled against a panel of 200 kinases. Compound 66 and 67, which were prepared by replacing the pyridine moiety by a larger substituent in order to fully occupy hydrophobic pocket, possessed high selectivity for JNKs, while other analogs still strongly inhibited other kinases. Compound 66 exhibited IC50 values of 4.7, 18.7, and 1 nM for JNK1, JNK2, and JNK3, respectively, and IC50 values of more than 230 nM for other kinases. Apart from JNKs, compound 67 inhibited IRAK1, HIPK4, AKT2 with IC50 values <90 nM. Thus, compounds 66 and 67 could provide a promising platform to develop the irreversible intracellular inhibitors for the JNK family by targeting the conserved cysteine residue in the ATP binding pocket.
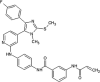
Muth et al. reported a class of pyridinylimidazoles as cysteine-directed covalent inhibitors of JNK3 [92]. Pyridinylimidazole 35, a potent scaffold for JNK3 and p38 inhibition as reported in previous work, was chosen for further optimization by SAR analysis to elaborate the covalent binding mode [93]. Docking compound 35 into JNK3 suggested that the fluorophenyl ring was placed in the selectivity pocket, while two hydrogen bonds were made with Met149. Encouraged by the binding motif of covalent inhibitor 65 to JNK3 [91], the linker connecting the scaffold and the warhead of the compound was optimized. Compound 68 showed great potency, with IC50 values of 0.3 nM and 36 nM for JNK3 and p38, respectively. This significant improvement in the JNK3 inhibitory activity suggested that compound 68 may orient towards and form a covalent bond with Cys154. Further modification to get higher selectivity for JNK3 over p38 resulted in compound 69 by replacing the 4-fluorophenyl ring with a methyl substituent. Compound 69 exhibited ~1000-fold JNK3 selectivity over p38. The cysteine-directed covalent bond formation was confirmed by mass spectrometry. Compounds 68 and 69 were screened against a panel of 410 kinases at the testing concentration of 1 μM to profile their selectivity within the kinome. Inhibitor 68 inhibited 15 kinases, while compound 69 inhibited only 5 kinases including three JNK isoforms.
8. Bivalent halogen/chalcogen bond
Lange et al. exploited the sulfur atom of the gatekeeper residue Met146 for attractive hydrogen bonding interaction in the design of JNK3 aminopyrimidine-based inhibitors [94]. Halogen/hydrogen analogs of aminopyrimidine, in which chlorine was considered to interact with Met146 [53], were generated. In this ligand series, all halogenated ligands exhibited significantly higher affinity in binding to JNK3 compared to an unsubstituted ligand. Neither the bromine nor the iodine analog was able to attain a significant advance in JNK3 binding relative to the chlorine analog in the fluorescence polarization assay and isothermal titration calorimetry. To define the role of the halogen bond in the kinase selectivity, Met146 was mutated to alanine, leucine, or threonine. Mutation to the smaller alanine showed a strong drop in the binding affinity, suggesting the importance of the gatekeeper interactions in the binding affinity of all ligands. Mutation to the threonine showed a significant loss in ligand binding affinity, which was consistent with the results with p38. This indicated that no alterative halogen bond was able to be formed with the threonine oxygen. In contrast, leucine was able to compensate for the loss in methionine interaction by making the hydrophobic/dispersive interactions with halogen atoms, and in this case, chlorine was greatly preferred over bromine or iodine. Therefore, although the choice of halogen was unable to change the ligand binding affinity to JNK3, it had an important effect on kinase selectivity. The X-ray crystal structure of the iodine derivative bound to JNK3 (PDB 4X21) revealed that the ligand was tightly placed in the adenine binding site with a network of hydrogen bonding interactions made by Met149, Glu147, and Lys93. Interestingly, Met146 was found to be engaged in not only a bivalent halogen bond with the ligand, but also a chalcogen bond with the proximal Met115 in the C-helix of hydrophobic pocket. This dual interaction strongly stabilized the Met146 flexibility. The probability of having two methionine residues at these two positions was only 9.3% in the human kinome. Among JNKs, while JNK3 and JNK1 share this feature, JNK2 does not possess this methionine combination. Therefore, this observation may be utilized in the design of selective kinase inhibitors.
9. Summary
There are more than one hundred structures of JNKs in complex with various inhibitors. In terms of the potency of JNK inhibitors, some kinds of interactions, such as the formation of hydrogen bonds between the inhibitor and the hinge region of the kinase in the ATP binding site, are generally common to the complexes, even though there are rare examples in which the inhibitor does not utilize such interactions. However, to increase the potency and to introduce selectivity of the inhibitors, additional interactions are necessary. To get further insight into the development of JNK inhibitors, we scrutinized the details of these structural complexes, including hydrophobic interactions. Especially in JNKs, there is a wide and deep hydrophobic region whose accessibility is controlled by the gatekeeping amino acid methionine. Here we have reviewed the characteristics of JNK inhibitors with the gatekeeper in ‘open’ and ‘closed’ conformations. In ‘open’ conformation of the gatekeeper, the hydrophobic region was accessible by the inhibitors where varieties of interaction including hydrophobic interaction contribute to potency and selectivity of the bound compounds. From this review, it is obvious that there are more non-specific inhibitors in the category of the ‘closed’ conformation structures and that the number of isoform selective JNK inhibitors is higher in the category of the ‘open’ conformation structure. Usually in the ‘open’ conformation structure of JNK in complex with inhibitors, spacious structural motifs occupy the hydrophobic region and provides the selectivity. From this work, we hope that investigators attain a deeper understanding of the interactions between JNK inhibitors and the kinase and find clues to design and synthesize more potent and selective inhibitors.
CRediT authorship contribution statement
Men Thi Hoai Duong: Writing - original draft. Joon-Hwa Lee: Conceptualization, Writing - review & editing. Hee-Chul Ahn: Conceptualization, Writing - original draft, Writing - review & editing.
Acknowledgements
This work was supported by the National Research Foundation of Korea [2017R1A2B2A001832 and 2020R1A2C1006909 to J.-H.L. and 2012R1A1A1014651 and 2018R1D1A1B0705097 to H.-C.A.], the Samsung Science and Technology Foundation [SSRF-BA1701-10 to J.-H.L.], and a KBSI grant [D39700]. We thank M. Stauffer, of Scientific Editing Solutions, for editing the manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.06.013.
Contributor Information
Joon-Hwa Lee, Email: joonhwa@gnu.ac.kr.
Hee-Chul Ahn, Email: hcahn@dongguk.edu.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Johnson G.L., Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 2.Ip Y.T., Davis R.J. Signal transduction by the c-Jun N-terminal kinase (JNK)–from inflammation to development. Curr Opin Cell Biol. 1998;10:205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- 3.Chang L., Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 4.Mohit A.A., Martin J.H., Miller C.A. p493F12 kinase: a novel MAP kinase expressed in a subset of neurons in the human nervous system. Neuron. 1995;14:67–78. doi: 10.1016/0896-6273(95)90241-4. [DOI] [PubMed] [Google Scholar]
- 5.Lawler S., Fleming Y., Goedert M., Cohen P. Synergistic activation of SAPK1/JNK1 by two MAP kinase kinases in vitro. Current Biol CB. 1998;8:1387–1390. doi: 10.1016/s0960-9822(98)00019-0. [DOI] [PubMed] [Google Scholar]
- 6.Dérijard B., Hibi M., Wu I.H., Barrett T., Su B., Deng T. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 7.Cuevas B., Abell A., Johnson G.L. Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene. 2007;26:3159–3171. doi: 10.1038/sj.onc.1210409. [DOI] [PubMed] [Google Scholar]
- 8.Zeke A., Misheva M., Remenyi A., Bogoyevitch M.A. JNK signaling: regulation and functions based on complex protein-protein partnerships. Microbiol Mol Biol Rev. 2016;80:793–835. doi: 10.1128/MMBR.00043-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graczyk P.P. JNK inhibitors as anti-inflammatory and neuroprotective agents. Future Med Chem. 2013;5:539–551. doi: 10.4155/fmc.13.34. [DOI] [PubMed] [Google Scholar]
- 10.Zhu X., Raina A.K., Rottkamp C.A., Aliev G., Perry G., Boux H. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. J Neurochem. 2001;76:435–441. doi: 10.1046/j.1471-4159.2001.00046.x. [DOI] [PubMed] [Google Scholar]
- 11.Gourmaud S., Paquet C., Dumurgier J., Pace C., Bouras C., Gray F. Increased levels of cerebrospinal fluid JNK3 associated with amyloid pathology: links to cognitive decline. J Psychiatry Neurosci. 2015;40:151–161. doi: 10.1503/jpn.140062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suwanna N., Thangnipon W., Soi-Ampornkul R. Neuroprotective effects of diarylpropionitrile against beta-amyloid peptide-induced neurotoxicity in rat cultured cortical neurons. Neurosci Lett. 2014;578:44–49. doi: 10.1016/j.neulet.2014.06.029. [DOI] [PubMed] [Google Scholar]
- 13.Xu N., Xiao Z., Zou T., Huang Z. Induction of GADD34 regulates the neurotoxicity of amyloid beta. Am J Alzheimers Dis Other Demen. 2015;30:313–319. doi: 10.1177/1533317514545616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoon S.O., Park D.J., Ryu J.C., Ozer H.G., Tep C., Shin Y.J. JNK3 perpetuates metabolic stress induced by Abeta peptides. Neuron. 2012;75:824–837. doi: 10.1016/j.neuron.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benarroch E.E. Glutamatergic synaptic plasticity and dysfunction in Alzheimer disease. Emerg Mech. 2018;91:125–132. doi: 10.1212/WNL.0000000000005807. [DOI] [PubMed] [Google Scholar]
- 16.Li X.M., Li C.C., Yu S.S., Chen J.T., Sabapathy K., Ruan D.Y. JNK1 contributes to metabotropic glutamate receptor-dependent long-term depression and short-term synaptic plasticity in the mice area hippocampal CA1. Eur J Neurosci. 2007;25:391–396. doi: 10.1111/j.1460-9568.2006.05300.x. [DOI] [PubMed] [Google Scholar]
- 17.Curran B.P., Murray H.J., O'Connor J.J. A role for c-Jun N-terminal kinase in the inhibition of long-term potentiation by interleukin-1beta and long-term depression in the rat dentate gyrus in vitro. Neuroscience. 2003;118:347–357. doi: 10.1016/s0306-4522(02)00941-7. [DOI] [PubMed] [Google Scholar]
- 18.Morel C., Sherrin T., Kennedy N.J., Forest K.H., Avcioglu Barutcu S., Robles M. JIP1-Mediated JNK activation negatively regulates synaptic plasticity and spatial memory. J Neurosci Off J Soc Neurosci. 2018;38:3708–3728. doi: 10.1523/JNEUROSCI.1913-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown M., Strudwick N., Suwara M., Sutcliffe L.K., Mihai A.D., Ali A.A. An initial phase of JNK activation inhibits cell death early in the endoplasmic reticulum stress response. J Cell Sci. 2016;129:2317–2328. doi: 10.1242/jcs.179127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan J., Qian J., Zhang Y., Ma J., Wang G., Xiao Q. Small peptide inhibitor of JNKs protects against MPTP-induced nigral dopaminergic injury via inhibiting the JNK-signaling pathway. Lab Invest. 2010;90:156–167. doi: 10.1038/labinvest.2009.124. [DOI] [PubMed] [Google Scholar]
- 21.Badshah H., Ali T., Shafiq-ur R., Faiz-ul A., Ullah F., Kim T.H. Protective effect of lupeol against lipopolysaccharide-induced neuroinflammation via the p38/c-Jun N-terminal kinase pathway in the adult mouse brain. J Neuroimmune Pharmacol. 2016;11:48–60. doi: 10.1007/s11481-015-9623-z. [DOI] [PubMed] [Google Scholar]
- 22.Zhang S., Gui X.H., Huang L.P., Deng M.Z., Fang R.M., Ke X.H. Neuroprotective effects of beta-asarone against 6-hydroxy dopamine-induced parkinsonism via JNK/Bcl-2/Beclin-1 pathway. Mol Neurobiol. 2016;53:83–94. doi: 10.1007/s12035-014-8950-z. [DOI] [PubMed] [Google Scholar]
- 23.Brecht S., Kirchhof R., Chromik A., Willesen M., Nicolaus T., Raivich G. Specific pathophysiological functions of JNK isoforms in the brain. Eur J Neurosci. 2005;21:363–377. doi: 10.1111/j.1460-9568.2005.03857.x. [DOI] [PubMed] [Google Scholar]
- 24.Wang W., Ma C., Mao Z., Li M. JNK inhibition as a potential strategy in treating Parkinson's disease. Drug News Perspect. 2004;17:646–654. doi: 10.1358/dnp.2004.17.10.873916. [DOI] [PubMed] [Google Scholar]
- 25.Han M.S., Jung D.Y., Morel C., Lakhani S.A., Kim J.K., Flavell R.A. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science. 2013;339:218–222. doi: 10.1126/science.1227568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hefetz-Sela S., Stein I., Klieger Y., Porat R., Sade-Feldman M., Zreik F. Acquisition of an immunosuppressive protumorigenic macrophage phenotype depending on c-Jun phosphorylation. Proc Natl Acad Sci USA. 2014;111:17582–17587. doi: 10.1073/pnas.1409700111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Assi K., Pillai R., Gomez-Munoz A., Owen D., Salh B. The specific JNK inhibitor SP600125 targets tumour necrosis factor-alpha production and epithelial cell apoptosis in acute murine colitis. Immunology. 2006;118:112–121. doi: 10.1111/j.1365-2567.2006.02349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitsuyama K., Suzuki A., Tomiyasu N., Tsuruta O., Kitazaki S., Takeda T. Pro-inflammatory signaling by Jun-N-terminal kinase in inflammatory bowel disease. Int J Mol Med. 2006;17:449–455. [PubMed] [Google Scholar]
- 29.Guma M., Ronacher L.M., Firestein G.S., Karin M., Corr M. JNK-1 deficiency limits macrophage-mediated antigen-induced arthritis. Arthritis Rheum. 2011;63:1603–1612. doi: 10.1002/art.30271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shibata W., Maeda S., Hikiba Y., Yanai A., Sakamoto K., Nakagawa H. c-Jun NH2-terminal kinase 1 is a critical regulator for the development of gastric cancer in mice. Cancer Res. 2008;68:5031–5039. doi: 10.1158/0008-5472.CAN-07-6332. [DOI] [PubMed] [Google Scholar]
- 31.Han M.S., Barrett T., Brehm M.A., Davis R.J. Inflammation mediated by JNK in myeloid cells promotes the development of hepatitis and hepatocellular carcinoma. Cell Rep. 2016;15:19–26. doi: 10.1016/j.celrep.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das M., Garlick D.S., Greiner D.L., Davis R.J. The role of JNK in the development of hepatocellular carcinoma. Genes Dev. 2011;25:634–645. doi: 10.1101/gad.1989311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alcorn J.F., Guala A.S., van der Velden J., McElhinney B., Irvin C.G., Davis R.J. Jun N-terminal kinase 1 regulates epithelial-to-mesenchymal transition induced by TGF-beta1. J Cell Sci. 2008;121:1036–1045. doi: 10.1242/jcs.019455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alcorn J.F., van der Velden J., Brown A.L., McElhinney B., Irvin C.G., Janssen-Heininger Y.M. c-Jun N-terminal kinase 1 is required for the development of pulmonary fibrosis. Am J Respir Cell Mol Biol. 2009;40:422–432. doi: 10.1165/rcmb.2008-0174OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirosumi J., Tuncman G., Chang L., Gorgun C.Z., Uysal K.T., Maeda K. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 36.Tuncman G., Hirosumi J., Solinas G., Chang L., Karin M., Hotamisligil G.S. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc Natl Acad Sci USA. 2006;103:10741–10746. doi: 10.1073/pnas.0603509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abdelli S., Puyal J., Bielmann C., Buchillier V., Abderrahmani A., Clarke P.G. JNK3 is abundant in insulin-secreting cells and protects against cytokine-induced apoptosis. Diabetologia. 2009;52:1871–1880. doi: 10.1007/s00125-009-1431-7. [DOI] [PubMed] [Google Scholar]
- 38.Abdelli S., Bonny C. JNK3 maintains expression of the insulin receptor substrate 2 (IRS2) in insulin-secreting cells: functional consequences for insulin signaling. PLoS One. 2012;7 doi: 10.1371/journal.pone.0035997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ezanno H., Pawlowski V., Abdelli S., Boutry R., Gmyr V., Kerr-Conte J. JNK3 is required for the cytoprotective effect of exendin 4. J Diabetes Res. 2014;2014 doi: 10.1155/2014/814854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davis R.J. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 41.Kaiser R.A., Liang Q., Bueno O., Huang Y., Lackey T., Klevitsky R. Genetic inhibition or activation of JNK1/2 protects the myocardium from ischemia-reperfusion-induced cell death in vivo. J Biol Chem. 2005;280:32602–32608. doi: 10.1074/jbc.M500684200. [DOI] [PubMed] [Google Scholar]
- 42.Xie X., Gu Y., Fox T., Coll J.T., Fleming M.A., Markland W. Crystal structure of JNK3: a kinase implicated in neuronal apoptosis. Structure (London, England 1993) 1998;6:983–991. doi: 10.1016/s0969-2126(98)00100-2. [DOI] [PubMed] [Google Scholar]
- 43.Siddiqui M.A., Reddy P.A. Small Molecule JNK (c-Jun N-Terminal Kinase) Inhibitors. J Med Chem. 2010;53:3005–3012. doi: 10.1021/jm9003279. [DOI] [PubMed] [Google Scholar]
- 44.Koch P., Gehringer M., Laufer S.A. Inhibitors of c-Jun N-terminal kinases: an update. J Med Chem. 2015;58:72–95. doi: 10.1021/jm501212r. [DOI] [PubMed] [Google Scholar]
- 45.Koch P. Topics in Medicinal Chemistry. Springer; Berlin, Heidelberg: 2020. Inhibitors of c-Jun N-terminal Kinase 3. [Google Scholar]
- 46.Bennett B.L., Sasaki D.T., Murray B.W., O'Leary E.C., Sakata S.T., Xu W. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han Z., Boyle D.L., Chang L., Bennett B., Karin M., Yang L. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J Clin Invest. 2001;108:73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bogoyevitch M.A., Arthur P.G. Inhibitors of c-Jun N-terminal kinases: JuNK no more? Biochim Biophys Acta. 2008;1784:76–93. doi: 10.1016/j.bbapap.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lou L., Hu D., Chen S., Wang S., Xu Y., Huang Y. Protective role of JNK inhibitor SP600125 in sepsis-induced acute lung injury. Int J Clin Exp Pathol. 2019;12:528–538. [PMC free article] [PubMed] [Google Scholar]
- 50.Hom R.K., Bowers S., Sealy J.M., Truong A.P., Probst G.D., Neitzel M.L. Design and synthesis of disubstituted thiophene and thiazole based inhibitors of JNK. Bioorg Med Chem Lett. 2010;20:7303–7307. doi: 10.1016/j.bmcl.2010.10.066. [DOI] [PubMed] [Google Scholar]
- 51.Bowers S., Truong A.P., Jeffrey Neitz R., Hom R.K., Sealy J.M., Probst G.D. Design and synthesis of brain penetrant selective JNK inhibitors with improved pharmacokinetic properties for the prevention of neurodegeneration. Bioorg Med Chem Lett. 2011;21:5521–5527. doi: 10.1016/j.bmcl.2011.06.100. [DOI] [PubMed] [Google Scholar]
- 52.Bowers S., Truong A.P., Neitz R.J., Neitzel M., Probst G.D., Hom R.K. Design and synthesis of a novel, orally active, brain penetrant, tri-substituted thiophene based JNK inhibitor. Bioorg Med Chem Lett. 2011;21:1838–1843. doi: 10.1016/j.bmcl.2011.01.046. [DOI] [PubMed] [Google Scholar]
- 53.Alam M., Beevers R.E., Ceska T., Davenport R.J., Dickson K.M., Fortunato M. Synthesis and SAR of aminopyrimidines as novel c-Jun N-terminal kinase (JNK) inhibitors. Bioorg Med Chem Lett. 2007;17:3463–3467. doi: 10.1016/j.bmcl.2007.03.078. [DOI] [PubMed] [Google Scholar]
- 54.Palmer W.S., Alam M., Arzeno H.B., Chang K.C., Dunn J.P., Goldstein D.M. Development of amino-pyrimidine inhibitors of c-Jun N-terminal kinase (JNK): kinase profiling guided optimization of a 1,2,3-benzotriazole lead. Bioorg Med Chem Lett. 2013;23:1486–1492. doi: 10.1016/j.bmcl.2012.12.047. [DOI] [PubMed] [Google Scholar]
- 55.Gong L., Han X., Silva T., Tan Y.C., Goyal B., Tivitmahaisoon P. Development of indole/indazole-aminopyrimidines as inhibitors of c-Jun N-terminal kinase (JNK): optimization for JNK potency and physicochemical properties. Bioorg Med Chem Lett. 2013;23:3565–3569. doi: 10.1016/j.bmcl.2013.04.029. [DOI] [PubMed] [Google Scholar]
- 56.Kamenecka T., Jiang R., Song X., Duckett D., Chen W., Ling Y.Y. Synthesis, biological evaluation, X-ray structure, and pharmacokinetics of aminopyrimidine c-jun-N-terminal kinase (JNK) inhibitors. J Med Chem. 2010;53:419–431. doi: 10.1021/jm901351f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng K., Park C.M., Iqbal S., Hernandez P., Park H., LoGrasso P.V. Pyridopyrimidinone derivatives as potent and selective c-Jun N-terminal kinase (JNK) inhibitors. ACS Med Chem Lett. 2015;6:413–418. doi: 10.1021/ml500474d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gong L., Tan Y.C., Boice G., Abbot S., McCaleb K., Iyer P. Discovery of a novel series of 4-quinolone JNK inhibitors. Bioorg Med Chem Lett. 2012;22:7381–7387. doi: 10.1016/j.bmcl.2012.10.066. [DOI] [PubMed] [Google Scholar]
- 59.Haynes N.E., Scott N.R., Chen L.C., Janson C.A., Li J.K., Lukacs C.M. Identification of an Adamantyl Azaquinolone JNK Selective Inhibitor. ACS Med Chem Lett. 2012;3:764–768. doi: 10.1021/ml300175c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu M., Xin Z., Clampit J.E., Wang S., Gum R.J., Haasch D.L. Synthesis and SAR of 1,9-dihydro-9-hydroxypyrazolo[3,4-b]quinolin-4-ones as novel, selective c-Jun N-terminal kinase inhibitors. Bioorg Med Chem Lett. 2006;16:2590–2594. doi: 10.1016/j.bmcl.2006.02.046. [DOI] [PubMed] [Google Scholar]
- 61.Liu M., Wang S., Clampit J.E., Gum R.J., Haasch D.L., Rondinone C.M. Discovery of a new class of 4-anilinopyrimidines as potent c-Jun N-terminal kinase inhibitors: synthesis and SAR studies. Bioorg Med Chem Lett. 2007;17:668–672. doi: 10.1016/j.bmcl.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 62.Szczepankiewicz B.G., Kosogof C., Nelson L.T., Liu G., Liu B., Zhao H. Aminopyridine-based c-Jun N-terminal kinase inhibitors with cellular activity and minimal cross-kinase activity. J Med Chem. 2006;49:3563–3580. doi: 10.1021/jm060199b. [DOI] [PubMed] [Google Scholar]
- 63.Zhao H., Serby M.D., Xin Z., Szczepankiewicz B.G., Liu M., Kosogof C. Discovery of potent, highly selective, and orally bioavailable pyridine carboxamide c-Jun NH2-terminal kinase inhibitors. J Med Chem. 2006;49:4455–4458. doi: 10.1021/jm060465l. [DOI] [PubMed] [Google Scholar]
- 64.Asano Y., Kitamura S., Ohra T., Aso K., Igata H., Tamura T. Discovery, synthesis and biological evaluation of isoquinolones as novel and highly selective JNK inhibitors (1) Bioorg Med Chem. 2008;16:4715–4732. doi: 10.1016/j.bmc.2008.02.027. [DOI] [PubMed] [Google Scholar]
- 65.Asano Y., Kitamura S., Ohra T., Itoh F., Kajino M., Tamura T. Discovery, synthesis and biological evaluation of isoquinolones as novel and highly selective JNK inhibitors (2) Bioorg Med Chem. 2008;16:4699–4714. doi: 10.1016/j.bmc.2008.02.028. [DOI] [PubMed] [Google Scholar]
- 66.Jiang R., Duckett D., Chen W., Habel J., Ling Y.Y., LoGrasso P. 3,5-Disubstituted quinolines as novel c-Jun N-terminal kinase inhibitors. Bioorg Med Chem Lett. 2007;17:6378–6382. doi: 10.1016/j.bmcl.2007.08.054. [DOI] [PubMed] [Google Scholar]
- 67.Shin Y., Chen W., Habel J., Duckett D., Ling Y.Y., Koenig M. Synthesis and SAR of piperazine amides as novel c-jun N-terminal kinase (JNK) inhibitors. Bioorg Med Chem Lett. 2009;19:3344–3347. doi: 10.1016/j.bmcl.2009.03.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baek S., Kang N.J., Popowicz G.M., Arciniega M., Jung S.K., Byun S. Structural and functional analysis of the natural JNK1 inhibitor quercetagetin. J Mol Biol. 2013;425:411–423. doi: 10.1016/j.jmb.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ansideri F., Macedo J.T., Eitel M., El-Gokha A., Zinad D.S., Scarpellini C. Structural Optimization of a Pyridinylimidazole Scaffold: Shifting the Selectivity from p38alpha Mitogen-Activated Protein Kinase to c-Jun N-Terminal Kinase 3. ACS Omega. 2018;3:7809–7831. doi: 10.1021/acsomega.8b00668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krenitsky V.P., Delgado M., Nadolny L., Sahasrabudhe K., Ayala L., Clareen S.S. Aminopurine based JNK inhibitors for the prevention of ischemia reperfusion injury. Bioorg Med Chem Lett. 2012;22:1427–1432. doi: 10.1016/j.bmcl.2011.12.028. [DOI] [PubMed] [Google Scholar]
- 71.Plantevin Krenitsky V., Nadolny L., Delgado M., Ayala L., Clareen S.S., Hilgraf R. Discovery of CC-930, an orally active anti-fibrotic JNK inhibitor. Bioorg Med Chem Lett. 2012;22:1433–1438. doi: 10.1016/j.bmcl.2011.12.027. [DOI] [PubMed] [Google Scholar]
- 72.van der Velden J.L., Ye Y., Nolin J.D., Hoffman S.M., Chapman D.G., Lahue K.G. JNK inhibition reduces lung remodeling and pulmonary fibrotic systemic markers. Clin Transl Med. 2016;5:36. doi: 10.1186/s40169-016-0117-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Probst G.D., Bowers S., Sealy J.M., Truong A.P., Hom R.K., Galemmo R.A., Jr. Highly selective c-Jun N-terminal kinase (JNK) 2 and 3 inhibitors with in vitro CNS-like pharmacokinetic properties prevent neurodegeneration. Bioorg Med Chem Lett. 2011;21:315–319. doi: 10.1016/j.bmcl.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 74.Cao J., Gao H., Bemis G., Salituro F., Ledeboer M., Harrington E. Structure-based design and parallel synthesis of N-benzyl isatin oximes as JNK3 MAP kinase inhibitors. Bioorg Med Chem Lett. 2009;19:2891–2895. doi: 10.1016/j.bmcl.2009.03.043. [DOI] [PubMed] [Google Scholar]
- 75.Swahn B.M., Huerta F., Kallin E., Malmstrom J., Weigelt T., Viklund J. Design and synthesis of 6-anilinoindazoles as selective inhibitors of c-Jun N-terminal kinase-3. Bioorg Med Chem Lett. 2005;15:5095–5099. doi: 10.1016/j.bmcl.2005.06.083. [DOI] [PubMed] [Google Scholar]
- 76.Stocks M.J., Barber S., Ford R., Leroux F., St-Gallay S., Teague S. Structure-driven HtL: design and synthesis of novel aminoindazole inhibitors of c-Jun N-terminal kinase activity. Bioorg Med Chem Lett. 2005;15:3459–3462. doi: 10.1016/j.bmcl.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 77.Christopher J.A., Atkinson F.L., Bax B.D., Brown M.J., Champigny A.C., Chuang T.T. 1-Aryl-3,4-dihydroisoquinoline inhibitors of JNK3. Bioorg Med Chem Lett. 2009;19:2230–2234. doi: 10.1016/j.bmcl.2009.02.098. [DOI] [PubMed] [Google Scholar]
- 78.Angell R.M., Atkinson F.L., Brown M.J., Chuang T.T., Christopher J.A., Cichy-Knight M. N-(3-Cyano-4,5,6,7-tetrahydro-1-benzothien-2-yl)amides as potent, selective, inhibitors of JNK2 and JNK3. Bioorg Med Chem Lett. 2007;17:1296–1301. doi: 10.1016/j.bmcl.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 79.Kamenecka T., Habel J., Duckett D., Chen W., Ling Y.Y., Frackowiak B. Structure-activity relationships and X-ray structures describing the selectivity of aminopyrazole inhibitors for c-Jun N-terminal kinase 3 (JNK3) over p38. J Biol Chem. 2009;284:12853–12861. doi: 10.1074/jbc.M809430200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zheng K., Iqbal S., Hernandez P., Park H., LoGrasso P.V., Feng Y. Design and synthesis of highly potent and isoform selective JNK3 inhibitors: SAR studies on aminopyrazole derivatives. J Med Chem. 2014;57:10013–10030. doi: 10.1021/jm501256y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Park H., Iqbal S., Hernandez P., Mora R., Zheng K., Feng Y. Structural basis and biological consequences for JNK2/3 isoform selective aminopyrazoles. Sci Rep. 2015;5:8047. doi: 10.1038/srep08047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Comess K.M., Sun C., Abad-Zapatero C., Goedken E.R., Gum R.J., Borhani D.W. Discovery and characterization of non-ATP site inhibitors of the mitogen activated protein (MAP) kinases. ACS Chem Biol. 2011;6:234–244. doi: 10.1021/cb1002619. [DOI] [PubMed] [Google Scholar]
- 83.Kaoud T.S., Mitra S., Lee S., Taliaferro J., Cantrell M., Linse K.D. Development of JNK2-selective peptide inhibitors that inhibit breast cancer cell migration. ACS Chem Biol. 2011;6:658–666. doi: 10.1021/cb200017n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pan J., Li H., Zhang B., Xiong R., Zhang Y., Kang W.Y. Small peptide inhibitor of JNK3 protects dopaminergic neurons from MPTP induced injury via inhibiting the ASK1-JNK3 signaling pathway. PLoS ONE. 2015;10 doi: 10.1371/journal.pone.0119204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chambers J.W., Cherry L., Laughlin J.D., Figuera-Losada M., Lograsso P.V. Selective inhibition of mitochondrial JNK signaling achieved using peptide mimicry of the Sab kinase interacting motif-1 (KIM1) ACS Chem Biol. 2011;6:808–818. doi: 10.1021/cb200062a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chambers J.W., Howard S., LoGrasso P.V. Blocking c-Jun N-terminal kinase (JNK) translocation to the mitochondria prevents 6-hydroxydopamine-induced toxicity in vitro and in vivo. J Biol Chem. 2013;288:1079–1087. doi: 10.1074/jbc.M112.421354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Win S., Than T.A., Fernandez-Checa J.C., Kaplowitz N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis. 2014;5:e989. doi: 10.1038/cddis.2013.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Laughlin J.D., Nwachukwu J.C., Figuera-Losada M., Cherry L., Nettles K.W., LoGrasso P.V. Structural mechanisms of allostery and autoinhibition in JNK family kinases. Structure (London, England: 1993) 2012;20:2174–2184. doi: 10.1016/j.str.2012.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shaw D., Wang S.M., Villasenor A.G., Tsing S., Walter D., Browner M.F. The crystal structure of JNK2 reveals conformational flexibility in the MAP kinase insert and indicates its involvement in the regulation of catalytic activity. J Mol Biol. 2008;383:885–893. doi: 10.1016/j.jmb.2008.08.086. [DOI] [PubMed] [Google Scholar]
- 90.Kuglstatter A., Ghate M., Tsing S., Villasenor A.G., Shaw D., Barnett J.W. X-ray crystal structure of JNK2 complexed with the p38alpha inhibitor BIRB796: insights into the rational design of DFG-out binding MAP kinase inhibitors. Bioorg Med Chem Lett. 2010;20:5217–5220. doi: 10.1016/j.bmcl.2010.06.157. [DOI] [PubMed] [Google Scholar]
- 91.Zhang T., Inesta-Vaquera F., Niepel M., Zhang J., Ficarro S.B., Machleidt T. Discovery of potent and selective covalent inhibitors of JNK. Chem Biol. 2012;19:140–154. doi: 10.1016/j.chembiol.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Muth F., El-Gokha A., Ansideri F., Eitel M., Doring E., Sievers-Engler A. Tri- and tetrasubstituted pyridinylimidazoles as covalent inhibitors of c-Jun N-terminal kinase 3. J Med Chem. 2017;60:594–607. doi: 10.1021/acs.jmedchem.6b01180. [DOI] [PubMed] [Google Scholar]
- 93.Ansideri F., Lange A., El-Gokha A., Boeckler F.M., Koch P. Fluorescence polarization-based assays for detecting compounds binding to inactive c-Jun N-terminal kinase 3 and p38alpha mitogen-activated protein kinase. Anal Biochem. 2016;503:28–40. doi: 10.1016/j.ab.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 94.Lange A., Gunther M., Buttner F.M., Zimmermann M.O., Heidrich J., Hennig S. Targeting the gatekeeper MET146 of C-Jun N-terminal kinase 3 induces a bivalent halogen/chalcogen bond. J Am Chem Soc. 2015;137:14640–14652. doi: 10.1021/jacs.5b07090. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.