

Graphical abstract

Keywords: comparative demography, PSMC, parasitism, pollination, ecology, evolution, population, bottleneck, history, effective population size, anthropocene
Abstract
Species interactions, such as pollination, parasitism and predation, form the basis of functioning ecosystems. The origins and resilience of such interactions therefore merit attention. However, fossils only occasionally document ancient interactions, and phylogenetic methods are blind to recent interactions. Is there some other way to track shared species experiences? “Comparative demography” examines when pairs of species jointly thrived or declined. By forging links between ecology, epidemiology, and evolutionary biology, this method sheds light on biological adaptation, species resilience, and ecosystem health. Here, we describe how this method works, discuss examples, and suggest future directions in hopes of inspiring interest, imitators, and critics.
1. Introduction
Biological interactions structure and maintain ecosystems: predation, pollination, parasitism, commensalism, infection, and other such interactions define how biological communities function and change. Are such dependencies enduring or ephemeral? Answering this question would help synthesize ecological, epidemiological, and evolutionary insights, advancing an integrated view of the development and functioning of myriad biological phenomena.
We recently introduced a method for comparative demography that estimates when, and for how long, pairs of populations have undergone parallel histories of growth and decline Fig. 1 . This method utilizes information in genomic sequences to reconstruct demographic histories over hundreds of thousands of years enabling quantitative assessment of correlated changes in effective population size. The method was used to examine a variety of population pairs known to have biological dependency in extant populations and the results were discussed considering what was known about these dependencies from independent lines of inquiry. Here, we extend that discussion, reviewing the biological questions on which it has already shed light, exploring promising new applications of this approach, and highlighting opportunities to further elaborate, refine, and extend the marriage of demography, phylogeny, population genetics, and genomics.
Fig. 1.

Comparative demography seeks to infer whether, and when, distinct species have undergone shared intervals of population growth and contraction. Here, temporal changes in the estimated effective population size of each of two hypothetical species is illustrated. Note that the abundance of each species grew and contracted in concert over one temporal interval (to the left of the vertical dashed line; =) but at another interval, one species grew as the other contracted (to the right; ≠).
Could we know, for example, for how long the ancestors of a given parasite infected the ancestors of its current host? Which geological or climatological events most shaped the joint fates of sympatric organisms (those that occupy the same geographic areas or overlapping ranges)? Has stability or change governed the relationships between plants and their pollinators? How markedly have human beings altered the fates of wildlife, weeds, opportunists, prey, or pathogens? And how powerfully have we altered the relationship between the species we have domesticated and the organisms that serve as their natural competitors, pathogens, or symbionts? Pursuing such answers would advance a fundamental understanding of the history of life on the planet, the forces governing key biological partnerships or antagonisms, and the episodes and landscapes that most powerfully structured interrelationships among Earth’s inhabitants.
Understanding the durability of such biological interactions might also enhance our capacity to manage current and future problems. For example, the fields of emerging infectious disease, weed science, agroecology, and conservation biology seek to understand and better manage imminent threats such as the spread of Ebola virus, the sustainability of crop monocultures, the decline of crop pollinators, and the loss of biodiversity through habitat fragmentation and destruction. Each problem illustrates the harm that can be wrought from precipitous changes in the distribution of species and the nature of their interactions; yet we lack a firm basis to suggest how stable such interactions should be. How much natural dynamism should be expected in (for example) host associations, or are we correct in perceiving today’s challenges as of unnatural and recent origin? Do they represent obvious departures from historical norms? How best can we prevent future losses to biodiversity, to ecosystem health, and to the value of the services they provide to humans and other animals?
The stability of specific relationships may be of practical interest. Non-human primates have been frequent sources of zoonotic infection, presumably owing to the “low barrier to entry” for microbes adapted to physiologically similar environs (Wolfe et al., 2007). Livestock have been another important source, presumably owing to their population densities and physical proximity to people. Why might bats serve as frequent sources of zoonotic infections, such as that of SARS and the COVID-19 coronavirus? Has this long been the case, or do such instances ensue from novel habitat incursions? Farming, especially of crop monocultures, invites specialist crop pests, weeds, and pathogens; can we learn something about the typical sources of such uninvited guests? Can we better harness nature's bounty using approaches modeled on more resilient natural systems? The “parasite release” hypothesis holds that invasive species thrive when rid of infections that held them in check in their places of origin (Keane and Crawley, 2002, Shea and Chesson, 2002, Torchin et al., 2003, Torchin et al., 2001, Torchin and Lafferty, 2009). Might this mean that long-term ecosystem dynamics are bound by longstanding antagonisms from competitors and pathogens? Or do such outcomes instead ensue from a revolving cast of pathogens? When we introduce natural enemies in hopes of reducing the harms wrought by invasive species, for how long can we hope such biocontrol to endure?
Assessing the longevity of biological associations traditionally required direct physical evidence (i.e. fossilized parasites (Morris, 1981, Peñalver et al., 2017, Poinar, 2003, Poinar and Boucot, 2006)) or indirect evidence derived from comparative phylogeny (Galbreath et al., 2009, Hafner et al., 1995, Hafner et al., 1994, Hafner and Nadler, 1988, Hoberg and Brooks, 2015). Before reviewing our new method, we will briefly describe the promise and limitations of the preceding two approaches.
Direct physical evidence of a longstanding association comes in the form of fossils that attest to ancient pairings. Such evidence illuminates, for example, for how long particular insect groups served as pollinators for particular plant groups (Poinar, 2003). Parasitic mites, preserved in amber, have been documented on feathered dinosaurs (Peñalver et al., 2017). DNA originating from Borrelia spirochetes was isolated from ticks in museum collections, preserved a century before the recognition of Lyme Disease in North America (Hubbard et al., 1998, Marshall et al., 1994, Persing et al., 1990). Such findings excite the imagination by showing us how the ancient world resembled the present. Moreover, subsequent evolutionary radiations affirm that, once established, new biological relationships (i.e. insect pollination of land plants; the establishment of photosynthetic symbioses in coral and in lichens) changed the subsequent course of life on the planet (Pellmyr, 1992). Grazers require grasslands; limpets require seas. It is hard to imagine anything more persuasive than direct physical evidence to establish the minimum age of any form of biological interaction. However, most of life's partnerships and pathogens have not been fossilized. What can be said about their origins and persistence?
In the absence of physical specimens, phylogenies reconstructed from extant species help determine whether interdependent organisms have diversified in parallel Harmon et al. (2019). Sympatric organisms experiencing environmental change (i.e. the formation of glaciers, volcanic mountain ranges, seas) sometimes bear the evidence of their shared history. The notable overlap in phylogenetic branching order among groups of parasites and their hosts (for example, the chewing lice of pocket gophers (Hafner et al., 1995, Hafner and Nadler, 1988) requires explanation; the simplest explanation would seem to be a causal one (the lice became subdivided owing to allopatric speciation of their solitary hosts). Stated more generally, the stability or transience of biological interactions has been profitably explored through the joint consideration of phylogenies, despite debates as to the fairest interpretation of concordant (or discordant) patterns of evolutionary diversification. Strict co-speciation may have been too-often accepted, when a looser process of “ecological fitting” (accommodating frequent switching among often related host groups) suffices (Brooks and Hoberg, 2007, Hoberg and Brooks, 2015, Hoberg and Brooks, 2008).
Recently, we introduced a third means to evaluate evidence for shared histories, premised on the simple idea that given pairs of species may have undergone parallel episodes of population growth and decline (Hecht et al., 2018) (Fig. 1). In the short term, antagonisms between host and parasite or predator and prey might engender inversely correlated population sizes; indeed, the classical Lotka-Volterra equations modeling predator/prey dynamics assume that growth in a predator population suppresses prey populations, cyclically limiting the growth of each (Wilson and Bossert, 1971). Over longer intervals, we suggest that biological dependencies engender concerted population growth or contraction, whether driven by climate, resource availability, or other factors. That is, climates favorable to expanding the savannah would favor growth of its various inhabitants and their pathogens, commensals, and parasites.
Such intuition was used to explain evidently concordant expansions and contractions in the populations of leaf-eating monkeys and pandas, two unrelated consumers of similar plant resources (Zhou et al., 2014). The spread and retreat of glaciers, the genesis of a floodplain, or the desertification of a grassland similarly impose shared opportunities and limitations for a broad suite of organisms, irrespective of the nature or strength of their biological interdependencies. Bearing in mind such caveats, and cognizant that no correlation suffices to establish causation, comparing demographic histories is a valuable and, until recently, entirely unexplored avenue to study the temporal durability of biological dependencies. Concerted growth and contraction in species pairs (or groups) should be viewed within a broader consideration of their historical and/or extant interconnections, and can serve to generate hypotheses and better delimit the plausible historical depth of their interaction. By contrast, extant dependent species pairs (or groups) lacking a parallel pattern of population growth and decline through time might be suspected as only recently establishing ecological or epidemiological dependency. Our prior work suggests exactly that for potatoes and the oomycete agent of potato blight (Phytophthera infestans): it appears they did not commence growing in tandem until potatoes were domesticated.
2. Reconstructing demographic history
It is difficult enough to estimate the extant size of a population; how can past population size be estimated? We direct the interested reader to other available resources that provide detailed methodological guidance (Ho and Shapiro, 2011, Spence et al., 2018). Here we simply broadly overview this family of inferential tools.
Larger and older populations should generally harbor more neutral genetic diversity than smaller and younger populations. This idea serves as the foundation for modern population genetics writ large, and the neutral theory of molecular evolution, in particular (Kimura, 1983). The study of island biogeography well illustrates the idea, establishing empirically that small founding populations (at the extreme, a single clonal individual or a single breeding pair) bequeath to descendent populations a small fraction of variation then circulating in source populations (i.e. those on the mainland (Johnson et al., 2000, MacArthur and Wilson, 2001). Diversity among the descendants of such populations will remain low for extended intervals (because new mutations are slow to accrue) unless infused by new variants imported by additional immigrants.
Building on this intuition, a system of inferential tools has been constructed to express the expected rate at which new neutral alleles are birthed, their probabilities of survival, and their probabilities of ultimate fixation. Viewed in reverse, these models describe the coalescence of extant alleles into their shared common ancestors. These probabilities are well-characterized for theoretical populations of constant size, and data may be evaluated against expectations from other demographic scenarios (Rosenberg and Nordborg, 2002, Eriksson et al., 2010) (Fig. 2 ). As in the case of island populations founded by small numbers of individuals, any population experiencing a rapid population decline will limit ensuing variability for estimable intervals (Galtier et al., 2000). Long-lived population bottlenecks most profoundly reduce diversity long after a population's census size has rebounded. Each allelic lineage surviving such a bottleneck may foster its own clusters of descendant lineages. By contrast, rapid population expansion produces “star phylogenies” wherein many extant variants differ only minimally from one another, and without deeper subdivisions among them (Kuhner et al., 1998, Slatkin and Hudson, 1991).
Fig. 2.

Changes in population size influence when alleles coalesce. A. Here, the expected temporal distribution of branching in a population of constant size (above, orange) is contrasted with that expected for a population that has undergone a bottleneck (below, blue) (after Eriksson et al., 2010). In the constant-sized population, fewer coalescences are ascribed to the interval represented by the grey shading. B. The age of alleles diminishes exponentially in populations of constant size, but bottlenecks accelerate allelic loss, temporally concentrating coalescence events. C. Therefore, intervals of notably frequent coalescence events may demarcate periods of past population decline. Similar reasoning underlies procedures used to identify periods of population growth. Other factors (selection, population subdivision) may similarly influence such temporal patterns.
Such patterns are most readily apparent in non-recombining loci (such as most mitochondrial haplotypes), as recombination ultimately obscures the history of any single descent history (by generating myriad, ephemeral genotypes each incorporating innovations arising from all reproductively successful progenitors). Coherent blocks of linked variants of common descent cannot long persist in sexual populations (excepting where recombination is locally or globally suppressed), meaning that their occurrence and distribution provides additional means to draw inferences about a population's past size. Models of recombination provide well-founded expectations for how genetic variation should be distributed throughout the genome of individuals sampled from sexual populations of constant size (Hudson and Kaplan, 1988). Departures from these expectations provide a basis to recognizing population inconstancy (Donnelly and Tavare, 1995, Schraiber and Akey, 2015).
Several tools have been developed to mine extant genetic and genomic information to reconstruct a population's demographic past (Heled and Drummond, 2008, Li and Durbin, 2011, Pybus et al., 2000, Schiffels and Durbin, 2014, Terhorst et al., 2017). Each infers a history of demographic stability, growth, or contraction from information regarding the frequency distribution and/or coalescence history of extant alleles. By incorporating estimates of per-generation mutation rate, generation time, and recombination rates, these tools enable estimates of approximately when ancestral populations experienced stasis or change. These methods for demographic history reconstruction cover more recent timescales than most comparative phylogenetic approaches, typically ranging from thousands to hundreds of thousands of years for most mammals (Box). The absolute timescale depends on the generation time and mutation rate of the species in question, as well as its levels of genetic diversity (Liu and Hansen, 2017).
Box 1: Comparison of programs for demographic reconstruction.
Since the development of theory for how varying population size would be reflected in a coalescent tree (Donnelly and Tavare 1995), numerous programs have been written to assess the likelihood of complex demographic histories based on the inferred genealogy of a population sample. Comparing two or more demographies focuses on the “shape” of those histories - the order and relative timing of population growth and decline. The absolute size of either population, at any given time, seems less important to know. Several families of method satisfy this requirement (Table 1). Each method performs at its own optimal range of timescales, primarily depending on its investment in sample size or genome coverage.
Fundamental progress on model-agnostic demographic reconstruction was made by Pybus, Rambaut and Harvey (2000) with the introduction of the “skyline plot.” The skyline plot has seen several iterations, leading to the “extended Bayesian skyline plot” (EBSP; Heled and Drummond 2008), widely used for inferring relatively recent demographic histories from multilocus population samples. The most recent past is illuminated by sampling a few loci from numerous individuals.
The development of the pairwise sequentially Markovian coalescent (PSMC) model by Li and Durbin (2011) marked the beginning of a new approach to demographic history reconstruction. It exploits the fact that the genome of a single diploid individual is a mosaic of genes from numerous ancestors, assembled through recombination (Fig. 3). The approach infers past population size by evaluating the extent and genomic distribution of heterozygous positions, comparing whole genome data to a coalescent model incorporating recombination (Fig. 3). Whole-genome methods are most effective in probing a population's demographic history on the order of 102 to 105 generations in the past.
As the cost of whole genome resequencing has fallen, more recent methods have sought to combine the advantages of PSMC and the EBSP. For example, MSMC and SMC++ utilize whole genomes of multiple individuals to resolve population sizes over a wider timescale than either PSMC or the EBSP. For example, SMC++ utilizes the allele frequency spectrum (AFS) of a population sample, while MSMC uses haplotype phase data. Such additional information reduces uncertainty about the recent past, which can be difficult to discern based on heterozygosity alone because substantial differentiation among alleles requires time. One limitation of most existing whole-genome methods (the SMC family) is that confidence in their demographic reconstructions is only expressed in terms of bootstrap variability. The EBSP more formally allows estimation of posterior probabilities.
In principle, any method producing a credible estimate of demographic history could be used as a basis to compare past population dynamics in two or more species. Table 1 summarizes some of those now available, and describes their individual strengths, requirements, and precision. Among these is a method that mines information from the range of coalescent histories represented by maternal and paternal alleles at the many loci in the genome of a single diploid individual (Fig. 3, Fig. 4 ). The Pairwise Sequentially Markovian Coalescent (PSMC) model (Li and Durbin, 2011), has been shown to accurately reconstruct demographic histories from simulated data. When applied to human genomes, the method demonstrated an impressive ability to discern “out of Africa” and other salient features of human demography supported by a strong foundation in physical anthropology, linguistics, and population genetics. Consequently, this method has found broad application to gather insight in a range of biological taxa (Liu et al., 2016, Miller et al., 2012, Nadachowska-Brzyska et al., 2016, Zhou et al., 2018). The sufficiency of but a single diploid genome makes this method attractive for studying non-model organisms, providing a convenient starting point for investigating demographic history for any diploid taxon of interest.
| PSMC | SMC++ | MSMC | EBSP | |
|---|---|---|---|---|
| Approach | Coalescent | Coalescent + AFS | Coalescent | Coalescent |
| Number of samples | One | Tens to hundreds | One to eight | Tens to hundreds |
| Number of loci | Whole genome | Whole genome | Whole genome | At least one |
| Extra information | None | None | Phasing | Time calibration |
| Timescale (generations) | ~102–105 | ~101–105 | ~102–105 | ~101–103 |
| Uncertainty | Bootstraps | Bootstraps | Bootstraps | MCMC posterior probability distribution |
| Reference | Li and Durbin., 2011 | Terhorst et al., 2017 | Schiffels and Durbin, 2014 | Heled and Drummond, 2008 |
Fig. 3.

A single diploid genome can be harnessed to estimate the age distribution of maternal and paternal alleles. Blocks of the genome are characterized by their heterozygote density, which is assumed to reflect for how long they have been accumulating differences.
Fig. 4.

Inferring historical demography from the genomic distribution of heterozygosity. A. In a genomic walk, loci are categorized according to the density of heterozygous positions (for illustration purposes, depicted according to the accompanying heatmap where the darkest loci harbor the greatest density of heterozygous positions). B. A frequency distribution summarizes the proportion maternal and paternal alleles differing by few or many heterozygous positions. Loci distinguished by few heterozygotes are assumed to have diverged more recently than loci distinguished by many heterozygotes. C. Population decline favors allelic loss, whereas population growth favors allelic persistence. Adjusted for statistical expectations (which assume that most alleles in any given population are young), past population size estimates (red line) are inversely related to the age distribution of alleles in the genome (black histogram).
The methods we developed to compare demographic histories inferred from the PSMC (Hecht et al 2018 and summarized below) could in principle be applied, with little modification, to demographic histories reconstructed using any of several approaches (summarized in Table 1). The elaborations and improvements we envision ought likewise to be applied to other members of this family of methods.
3. Introducing comparative demographic history
To the extent that the survival and reproduction of individuals of one species depend on the bodies or ecosystem services of another species, their numbers should rise and fall in tandem. A classic example of this is found in models of predator–prey dynamics, where a predator's reproductive rate depends on the abundance of its prey (Kendall et al., 1998). Shared success should occasion species that thrive under shared conditions; conversely, adverse events (such as glaciation) threaten many species simultaneously. Temporal correlations have been noted between specific environmental variables and the abundance of unrelated species that depend on shared habitats and food resources (Zhou et al., 2014). Comparative demography examines ecological dependency when extrapolated over hundreds of generations, using patterns of similarity or difference to discern for how long the ecological dependency has held.
Our initial approach to comparative demographic history did not concern itself with absolute estimates of population size or the magnitude of population change because we reasoned that even species locked in a tight mutualistic relationship might differ in their reproductive rates, and that other factors would also influence their respective population sizes (Wells et al., 2017). For example, a plant might be limited by nutrient availability even if its pollinators are abundant (Mattila and Kuitunen, 2000), and a parasite's population size may be limited by the availability of both primary and secondary hosts, or by the density of its host population in addition to its absolute size. We therefore limited our focus to directional change in estimated population size, identifying intervals of growth, stasis, or decline. By this (intentionally simple) formulation, we endeavored to understand when pairs of species grew or contracted in concert.
To achieve this, - demographic histories must first be synchronized. Using methods briefly summarized below, we devised a method of curve-fitting constrained by reasonable estimates of the relative annual mutation rate of each member of the species pair (Fig. 5 ). Each interval was coded for each species as one of growth (1), stasis (0), or decline (-1). Subtracting the value for one species from the other in each interval identifies intervals of agreement or conflict in growth history. For example, if both species experienced growth, the difference in this index (1–1) is zero. The same would be true for intervals where each species declined, or each experienced stasis. Slight disagreement would be registered if, say, one population was stable but the other grew or declined (absolute value of the difference = 1). Conflict (one population growing while the other is contracting) yields an index difference with an absolute value of 2. When compared across all time intervals in the demographic curves, species pairs that have the smallest average slope differences deserve further consideration. (It would be notable as well if two species had average slope differences close to 2, as this would suggest that they respond in opposite ways to variation in prevailing conditions). In theory, unrelated species might have average slope differences close to 1; in our experience, species sharing no obvious ecological association have averaged differences of 0.5. We cannot say whether this apparent resemblance derives from actual commonalities among all species so far considered, or whether this reflects some bias in the reconstruction process. That is, shared environmental responses may explain non-random episodes of population growth and decline in many species. Any systematic bias in estimating population size (say, consistent exaggeration of the most distant past) would inflate the extent of correlation between pairs of demographic estimates. That said, our prior work demonstrated significantly better fits when comparing true host-parasite species pairs to arbitrary species pairs. Additional evaluation of diverse biological data, and further exploration of data simulated under defined (known) demographic histories, are needed to further clarify this question.
Fig. 5.
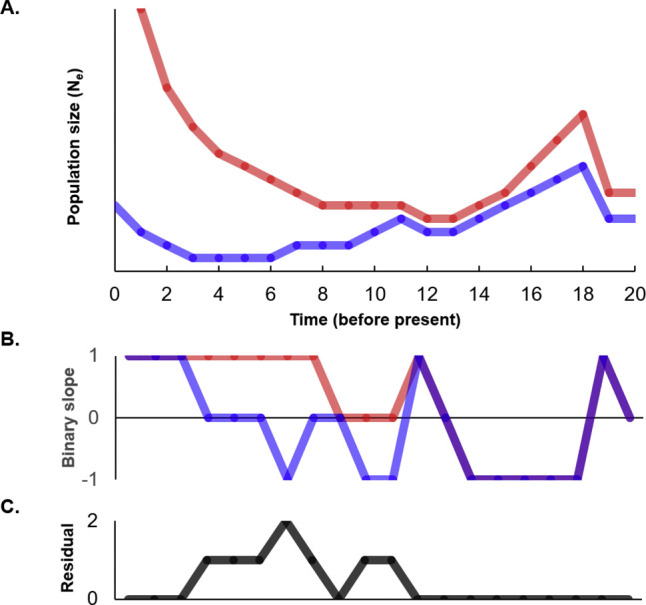
Each segment of a temporally aligned pair of demographic history curves (A) is scored based on whether the population is growing, declining, or unchanged over that period (B). The residual differences in slope direction identifies periods of consistent or inconsistent population dynamics and summarize the overall fit between the pair of demographic history curves (C).
We took up several case studies when introducing comparative PSMC (C-PSMC). These included the agent of severe human malaria (Plasmodium falciparum) in relation to its mosquito vectors and the great apes (gorillas, chimpanzees, and human beings). Here, we saw a striking correspondence between the ancient historical growth patterns of the parasite with its Anopheline vectors and with all great ape lineages in the ancient past, followed by a striking population explosion unique to the rise of human beings, alone (Fig. 6 ). That is to say, early human demographic history resembles patterns shared by non-human primates, but more recent primate declines contrast markedly with human exponential growth. It stands to reason that human-feeding mosquitoes that thrive in human-disturbed habitats would flourish in the Anthropocene, and that parasites reproducing in such mosquitoes and in us would likewise flourish: patterns of genomic variation in each appear entirely consistent with exactly this expectation.
Fig. 6.

The demographic histories of the malaria parasite (Plasmodium falciparum) and its mosquito vector (Anopheles gambiae) mirror human population growth; especially their increase in the last 10,000 years. Based on Hecht (2018) Fig. 1a. Effective population size is shown relative to each species’ average over the period.
We also examined the demographic history of the parasite Trichinella spiralis in relation to swine in Europe and Asia, and noted with great interest a regional distinction in the growth histories of parasite that mirrors a corresponding (and previously published (Groenen et al., 2012)) difference in the population histories of Asian and European swine. The regional specificity of evidently shared population histories in the parasite and this host provides additional confidence that historical information can be extracted with these tools, and supports phylogenetic inferences indicating that Asian and European populations of the parasite have been distinct since long before the domestication of swine in each region (Thompson et al. in prep).
We determined that the long-term growth history of a photosynthetic alga mirrored that of the anemone species with which it continues to partner in a symbiotic relationship. Given that warming waters cause coral bleaching events (Berkelmans and van Oppen, 2006) we had suspected that no such pattern of long-term concerted population growth might result. The data indicated otherwise, supporting a much stronger correlation of population growth between the species involved in an extant symbiotic relationship than with an unrelated coral from a different geographic region. Thus, short-term fluctuations in symbiotic associations may be bounded, reverting to long-established affinities.
Lastly, we applied this analysis to the potatoes and the oomycete agent of potato blight. Here, comparing demographic histories yielded a provocative insight: only since the age of potato domestication has both the plant and its pathogen exploded in abundance. Prior to that time, we discern no convincing parallelism in their respective growth histories. A fair interpretation of these data suggests that the origins of this plant-pathogen coincided with the time of potato domestication (not earlier). It is tempting to speculate that domestication itself created conditions conducive to the establishment and spread of potato blight. Such a causal interpretation cannot be proven by mere temporal correlation; but this interpretation gains plausibility by examining phylogenies of the pathogen and plant species, which indicate rampant facultative opportunism in the ancestors of Phytophthora (Cooke et al., 2000, Gómez-Alpizar et al., 2007, Haas et al., 2009, Martin et al., 2014).
4. Methodological issues
4.1. Temporal scaling
Demographic histories cannot be read directly from genetic data, but instead require fitting a demographic model. Most methods for doing this are based on the principle of the coalescent, which assumes that effective population size is proportional to the average number of genetic differences between individuals in the population (Rosenberg and Nordborg, 2002). However, this number depends not only on the population size, but also the rate at which new mutations are introduced. More genetically variable samples derive from larger, older ancestral populations; but scaling this requires knowing (or estimating) the mutation rate. In PSMC, the independent histories sampled by each of a diploid individual’s two parents provides the basis for estimating the relative size and age of the population (Li and Durbin, 2011). However, to scale this history into absolute terms of effective population size and years, for comparison with other populations’ histories, an annual mutation rate must be specified.
The annual mutation rate can be derived empirically from the data when using methods that incorporate asynchronous sampling (including ancient DNA), such as the Bayesian Skyline Plot (Drummond et al., 2005, Heled and Drummond, 2008)), or assumed based on previously published estimates. If a per-generation mutation rate has been previously published, the annual mutation rate can be estimated by dividing the generational mutation rate by the population’s generation time. The generation time can, in turn, be estimated from life tables as the average age of mothers, or the age at first reproduction can be assumed as a lower bound (Staerk et al., 2019).
For rare or under-studied species, none of this information may be available. In such cases, a further option is to determine plausible ranges of mutation rate and/or generation time and determine which parameters best align a distinctive feature of the demographic history (Ne peaks/troughs) to a known historical event, such as the Last Glacial Maximum or the date an invasive species was introduced. The demographic history of an ecologically related species may also be used. Such a fitting procedure can be facilitated by the C-PSMC program.
4.2. Relationship between effective and census population sizes
Once demographic history plots have been appropriately scaled, historical effective population sizes can be compared. However, it is important to remember that effective population size estimates (Ne) are not mere estimates of census size (N). Generally, effective population size is a fraction of census population size, reflecting variance in the proportion of individuals who actually reproduce and contribute to the long-term genetic diversity of the population (Frankham, 1995). However, the ratio Ne/N may vary over time if evolutionary or environmental changes lead to different rates of reproductive success (Turner et al., 2006). To our knowledge, no study of long-term demographic history has yet attempted to account for these natural vagaries in Ne/N, though short-term studies have documented them (e.g. (Shrimpton and Heath, 2003)). Domesticating animals and plants has significantly altered how long they live and what proportion of them reproduce. Indeed, exponential growth of the human population has resulted in approximately 24-fold growth in only the last thousand years, and 7-fold growth (an extra 6 billion people) in only the last 100 years. Because effective population size is computed as a harmonic mean that is sensitive to lower values, extant human variation resembles what would be expected for a population numbering only tens of thousands; human beings will likely never accumulate genetic variability commensurate with our current abundance (as it would take more time than the Earth will likely remain habitable) (Kliman et al, 2008). Such distinctions are worth remembering when inferring ecological information from genetic evidence.
Population structure is another cause of variation in Ne/N. In a metapopulation, subdivision restricts an individual to a subset of possible ancestors; this reduces Ne/N. Conversely, recent admixture can inflate the Ne/N ratio, by pooling the inheritance of long-separated demes (Baalsrud et al., 2014). In either case, the apparent change in Ne/N is relative to the researcher’s understanding of the true population structure. In demographic history reconstruction, these problems can largely be overcome by analyzing population structure before grouping samples into putative populations for coalescent analyses (Heller et al., 2013). However, population structure may also have varied over time, introducing artifacts to the reconstructed historical effective population sizes. For example, after a glacial episode, populations previously isolated in refugia may then appear to grow in effective population size due to their subsequent admixture, even if the total population has not grown (Mazet et al., 2016).
4.3. Determining direction of causation
A range of ecological processes could explain correlated demographic histories. Specialized predation, mutualism, parasitism or strong commensalism would all be expected to lead to demographic dependence in one or both species. On the other hand, a pair of species might simply be limited by a common environmental constraint (Hardie and Hutchings, 2010), as may often hold true for populations in extreme environments (e.g. very cold or dry) or during extreme events such as glacial periods or El Niño years (Trillmich and Limberger, 1985). However, shared dependency on a third factor is unlikely to account for tens of thousands of years of demographic correlation between any randomly selected pair of species in the temperate zone, judging by the variety of demographic histories which have been published.
The dependency of an obligate parasite on its host simplifies the process of determining the cause of any correlation between their demographic histories. When a parasite is entirely dependent upon a single host species, its population is all but certain to be regulated by the availability of hosts (Arneberg et al., 1998, Buck and Lutterschmidt, 2017). Of course, without strong a priori expectations about the identity of the host, attribution can still be complicated if there are multiple plausible host species which exhibit similar demographic histories.
Species with complex demographic histories (i.e. those involving multiple discrete bottleneck and expansion events) will be most amenable to this comparative approach, since models involving more discrete demographic events afford more opportunities to identify concordant or discordant periods of growth or decline, making it easier to determine if and when a pair of populations experienced specifically intertwined fates.
4.4. Complications with sequencing complete genomes
The methods we have employed require accurate assessment of heterozygosity across an organism’s genome; certain genomic features and limitations of data collection may complicate this goal. For example, haploid organisms are devoid of heterozygosity, limiting the application of PSMC. Detecting heterozygosity may also be confounded by misalignment of repetitive regions (which can confuse duplicated regions as alleles of the same locus). Finally, most available data of this sort has been generated in short reads (at most 300 bp with current next generation sequencing technology). While 50 base pairs generally suffice to locate a read in a genome, the presence of repetitive elements may cause small misalignments of reads to the reference sequence, resulting in systematic local overestimation of heterozygosity.
Haploid genomes are not uncommon among parasites. Falciparum malaria, for example, has been repeatedly sequenced in its haploid state. Nevertheless, a reasonable assessment of heterozygosity can be made from these organisms by imagining the diploid state that would be formed by joining two such haploid sporozoites in the ookinete (akin to any fertilized oocyte or ovule). There are several means to synthesize such hypothetical diploid genomes (pseudodiploids) from two haploid genome sequencing projects. The consensus sequences for two haploid individuals may be generated from a typical genome sequencing project and then aligned over the entire length of chromosomes (or contigs and scaffolds depending on the status of the genome assembly) using any number of alignment tools (Armstrong et al., 2019). However, these alignment tools are not perfect and often leave considerable gaps between chromosomal sections, rendering true heterozygosity calls across entire chromosomes difficult. As an alternative, short-reads from each of two sequencing projects may be simultaneously mapped to a common reference sequence. Assuming equal numbers of reads from each haploid sequencing project, the resulting assembly should have generally comparable numbers of reads from both “parental” haploid sequencing projects at every locus; where disagreement occurs between the two sequenced samples, a consensus basecaller will indicate a heterozygous position. This method, which we have successfully employed, removes the need to curate error-prone chromosome alignments. The combined reads should not introduce large amounts of new variation in the genome assembly and should reliably reflect the mating of the two haploid lineages sampled. One caveat is that the haploid genomes should derive from the same interbreeding population (Mazet et al., 2015). Creating pseudodiploids from two isolates from separate breeding populations would violate the assumption that the sampled genome derives from a population close to mutation/drift equilibrium, which is at the core of demographic reconstruction programs.
Repetitive regions of the genome notoriously impede confident alignment of short reads (Treangen and Salzberg, 2011). These repetitive regions may derive from short tandem repeats (microsatellites), interspersed nuclear elements, transposable elements, segmental duplications, and a variety of other less common repeat types (Biscotti et al., 2015). A single duplication can quickly cause short-read alignment problems as the two copies diverge from each other within the same genome, making it difficult for algorithmic alignment. Misaligned reads can systematically inflate local estimates of heterozygosity. Consensus base calls rely on proper mapping of multiple reads to a specific homologous portion of the genome. If two duplicates have diverged by 10%, misalignment would introduce 3 heterozygotes for a 300 bp read. These incorrectly assigned heterozygotes may confuse demographic reconstruction programs by inflating the estimated occurrence of anciently-diverged alleles (characterized by elevated heterozygosity). PSMC treats short repetitive regions by scoring them as a single heterozygous recombination block. However, if the repetitive region extends for thousands of basepairs, multiple heterozygous blocks will provide erroneous evidence for portions of the genome harboring ancient variation. Overestimating the occurrence of such “ancient” sections would bias estimates of ancient population size.
We scrutinized the occurrence and impact of such misalignment errors first by algorithmic screening for known eukaryotic repetitive elements and then eliminating them from subsequent analyses by “masking” them. Repeat masking can be performed before or after aligning short-reads to the reference sequence, but should be accomplished prior to submitting the assembly to demographic reconstruction. Repetitive regions can also be suspected when a disproportionate number of reads maps to portions of the genome: too many reads will map to a “locus” if it has been condensed from repetitive paralogs. Elevated read depth therefore signals the possibility of local misassembly. Removing such suspect regions may provide less biased estimates of historical population size. (Some variation in read depth does occur even when repeats are not the cause; in practice, areas with greater than 2-fold difference in mean read depth might deserve to be pruned from an assembly prior to reconstructing demographic histories). A more conservative approach would remove all sections of the genome with greater than mean coverage, though this may sacrifice some areas of legitimate data harboring real biological signal. If genome sequences are long enough (longer than 1 million base pairs after unusual read-depth removal), the demographic signal should remain despite aggressive pruning.
Just such a need arose when reconstructing the demographic history of the potato which, like many related plants, underwent a tripling of the genome (Jiao et al., 2012). Anticipating that this would result in multiple misalignments of reads, we produced demographic reconstructions from the genome before and after removing regions suspected, on the basis of excess read coverage, as misaligned. In this case, we eliminated all sections of the genome assembly that had greater than mean read depth. By eliminating suspected duplicated regions, we drastically reduced estimates of ancient population size while not materially affecting the shape of the demographic curve. Our conclusions concerning the history of the potato and potato blight proved robust to this potential confounder.
5. Potential impact of inferences from comparative demography
Populations are limited by those factors that most constrain per-capita growth rate (Berryman, 2004). A combination of such limiting factors determines the maximum sustainable size to which a population can grow under prevailing environmental conditions, defining its realized niche. Identifying the contemporary limiting factors of population size and growth rate through demographic sensitivity analysis is one of the main contributions of population ecology to conservation, as it is assumed that the factors most limiting of a population’s growth are those most deserving of stewardship (Conroy and Brook, 2003, Gerber and Heppell, 2004). However, this defines only current constraints. It is not usually possible to account for compensatory changes in other parameters, such as mortality being shifted onto a different life stage or the antagonistic coevolution of a parasite (Halpern et al., 2005). However, feedback processes like these may have left their mark on the demographic response to analogous events in the past.
In time, the factors that limit instantaneous population size and growth rate drive demographic history, as change in one of them may cause growth or decline in the population. Comparative demographic history might be able to provide evidence to complement demographic sensitivity analysis, trading precision for a broader perspective. For example, Zhou et al. (2014) found that the population sizes of the giant panda and snub-nosed monkey have tracked the abundance of leaves and seeds in their native habitat for at least a million years. Bottom-up control by plant productivity would seem the most likely explanation. While an analysis of their modern population would also indicate that their population is chiefly limited by habitat/food availability, comparative demography shows that this relationship has persisted through significant environmental change, with the same limitation being in place even when their food was much more abundant, suggesting that there is much scope for improvement. In contrast, another species’ population size might be found to have tracked the abundance of their primary food only during periods of low productivity (e.g. glaciations), but instead tracked the inverse abundance of a predator during periods of higher productivity.
When the generation time and mutation rate of a species can be estimated with confidence (and assuming these have not appreciably changed through time), it is possible to estimate when episodes of population growth or contraction took place. This implies that the reverse is also true: if the timing of a population’s growth or decline can be assumed a priori (driven, say, by the retreat of glaciers, the birth of a volcanic island, or the flourishing of a biological resource), then these methods provide a new means to estimate generation time and mutation rate. Assuming anthropogenic causes for the recent growth in Anopheline mosquitos provided just such an example, yielding a mutation rate estimate considerably consistent with prior empirical estimates (Hecht et al., 2018).
Given enough information, the comparative demography approach can also be used to estimate these parameters. Generation time and Ne/N are also related to demographic sensitivity and conservation outcomes because they reflect recruitment rate of juveniles, as well as the genetic resources of a population and the rate at which it can recover from disturbance or grow in response to a successful conservation intervention (Belmar-Lucero et al., 2012, Frankham, 1995, Palstra and Fraser, 2012, Romiguier et al., 2014).
6. Suggested avenues for the future
For the reasons outlined above, we believe much can be gained by comparing the inferred demographic histories among biological species. The purpose of this review is to provoke critics, inspire adherents, and otherwise incite the scholarly community into testing and improving upon this approach to better address pressing and as-yet unimagined questions.
We view C-PSMC as a step towards extracting ever more rigorous information on the history of biological dependencies and, more broadly, the concerted response of biological communities to changing conditions for life on the planet. We can imagine more formal approaches to modeling the hypothesized population dependencies among various groups of organisms. Methodological research might focus on developing models seeking to integrate demographic reconstruction with the detection of dependent population dynamics (say, in the form of a free parameter for correlation between effective population sizes). For example, we could imagine a Bayesian framework to evaluate whether, and to what extent, estimates of a given species’ history benefits by considering information derived from exogenous sources (such as the climate record or the demographic histories of other organisms). As increasing statistical sophistication is being integrated into efficient bioinformatic analyses of heretofore unimagined volumes of data, we hope comparative demography will be embraced as a concept worth exploring, refining, and improving.
Below are some questions we invite the community at large to consider as worthwhile endeavors.
6.1. Demographic history clustering analysis
We advocate developing methods to compare among multiple reconstructed demographic history plots to identify good matches among those derived from various interacting species. Using such a tool, one could test explicit a priori hypotheses regarding the longevity of species interactions. Such a tool could use any number of metrics to measure changes over time, whether it be slope, inflection points, or more complex statistical representations of shape and curvature. Ultimately this could be expanded to a likelihood algorithm that compares new demographic reconstructions to a database of all of those that have previously been published, returning a series of best “hits” in the database, like the BLAST algorithm used for DNA sequences (Altschul et al., 1990). Furthermore, a tool that could break down portions of demographic curves into different time segments could help delineate specific events that changed species relationships from independent to dependent, or vice versa. This could provide many interesting insights into species interactions and, by examining many curves, could provide supporting evidence for periods of great biological upheaval or unusual stability. Iterating over many curves from independent studies might resolve general patterns revealing how groups of species have jointly responded to common environmental perturbations. Establishing a tool that matches curves that span over 100,000 years in a database could provide tremendous insights into adaptability characteristics of extant species.
6.2. Landscape comparisons
Why stop at comparing a mere pair of demographic curves? The growth and contraction of entire landscapes (through i.e. glaciation, deforestation, desertification) has influenced the abundance and distribution of entire communities (Allen, 2003, Asner et al., 2004, Binney et al., 2017). The genomes of entire suites of organisms may retain a record of their common fate, as appears to be true for leaf-eating monkeys and pandas with overlapping habitats.
Would such a principal be evident on a far broader scale? Tropical rainforests, now critically threatened, are the home to the Earth's terrestrial biodiversity (Bradshaw et al., 2009). One reason for this is that the tropics have nurtured communities for longer (other explanations include more abundant sun and rainfall, greater habitat fragmentation, and greater resource competition) (Pianka, 1966, Rohde, 1992, Willig et al., 2003). Whereas temperate regions have undergone episodic climate extremes (for example, glacial advance and retreat), the tropics may be viewed as more enduring nurseries. Comparative demography holds promise for establishing whether species endemic to tropical rainforests have indeed experienced more stable populations, over longer intervals, than comparable species native to subtropical, temperate, or boreal regions, and whether species interactions (i.e. pollination, predation, parasitism) have endured longer.
6.3. Sessile vs vagile species
Species least capable of withstanding or escaping environmental extremes should be especially vulnerable to habitat loss and climate change (Foden et al., 2013). Their genomes may harbor evidence of this. Have such species undergone notably episodic growth and decline? Conversely, more resilient and more vagile species might experience greater demographic independence. Do actual data bear out this expectation? Comparative demography seems a natural approach to addressing such a question, especially if modified to jointly consider multiple demographic histories.
6.4. Community structure
Biological communities are structured by nutrient and energy flows from photosynthetic producers, herbivores, predators, and their parasites (Hairston and Hairston, 1993, Kuris et al., 2008, Odum, 1968). How does an organism's ecological role influence its long-term demographic responsiveness to changing circumstances? Have the primary producers in a given landscape responded, in concert, to changing climatological conditions? Are such effects mirrored, dampened, or exaggerated at higher trophic levels? And are such patterns characteristic of particular landscapes, or can more general patterns be discerned through a comparative demographic lens?
6.5. Epidemiology
Changing transmission opportunities induce marked changes in pathogen abundance and distribution. Human pathogens, for example, reflect changes in human abundance, behavior, hygiene, population density, migration patterns, and trade (Araújo et al., 2009, Zanotto et al., 1996). Indeed, viruses have provided notably informative examples of the evolutionary dimensions of population growth and spread (Bush et al., 1999, Ferguson et al., 2003, Rambaut et al., 2004, Simmonds et al., 2019, Wertheim et al., 2019). We see much more opportunity in using a comparative demographic approach to assess outstanding questions in historical epidemiology and, for example, the history of host shifts (respecting the specific requirements of each method for demographic reconstruction). A comparative framework can contribute to understanding whether, for how long, and where a given pathogen's ancestors have depended on a given host. Similarly, we believe such tools could be used to better understand the impact of specific interventions (i.e. vaccines) or of general environmental conditions (i.e. hygiene) in minimizing the prevalence and impact of specified pathogens.
It is reasonable to suppose that predatory, pathogenic, or parasitic organisms may, in the short term, reduce the abundance of the organisms upon which they depend. As consumers generally benefit from growth in their resources, predators and parasites should flourish with the success of their prey and hosts over the longer term. Conversely, adverse conditions may undermine their collective welfare. Thus, population dynamics may be negatively correlated short-term but positively correlated in the long term. Indeed, such scale-dependent contrasts might provide signature evidence for enduring parasitism. (Persistent predation or resource competition might engender similar patterns). As discussed above, estimates of effective population size derive from harmonic means, driven more by long-term trends than by short-term fluctuations. Therefore, each time scale provides valuable information on the nature of antagonistic biological dependencies.
6.6. Human ecology and the Anthropocene
Our species is blessed with the ability, and cursed with the responsibility, to understand our impact on our surroundings. Every human economic activity directly or indirectly requires the use of natural resources, the harvesting of nature's bounty, the cultivation of domesticated crops and livestock for human consumption, and the alteration of the conditions of life for wildlife, pests, weeds, pathogens, and the like. Distinct events in the human story have unleashed intensified waves of anthropogenic impact, including the advent of agriculture, the advent of global seafaring, the industrial revolution, the green revolution, and the advance of hygiene, public health, and medicine.
We are surrounded by the evidence of our immense impact on the demography of countless other organisms, be it billions of chickens or zero dodo birds. In the history of life on the planet, human beings are now said to be causing its sixth global extinction crisis. Meanwhile, an estimated 11% of the world's land is under crop cultivation, approximately 80% of which feeds livestock. The biomass of our livestock outweighs our own biomass, and together outweigh all other vertebrates (excepting fish) by over twenty times; the globe's chickens collectively weigh more than three times the globe's wild birds (Bar-On et al., 2018). Overfishing, overhunting, habitat destruction, resource competition, climate forcing, and pathogen pollution have driven the most widely-recognized cases threatening species with extinction (such as whales, cheetahs, elephants, coral declines, the global die off of amphibians) but we remain only dimly aware of the extent and pace at which we drive the fate of life on the planet, writ large. We do not need comparative demography to understand these basic truths. But comparative demography does offer additional means to delineate the pace and origins of our impact. Perhaps, one day, it can be used to identify cases where we succeeded in reversing course.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by USDA projects 8042-32000-105-00-D and 8042-32420-007-00-D.
References
- Allen H.D. Response of past and present Mediterranean ecosystems to environmental change. Prog. Phys. Geogr. Earth Environ. 2003;27:359–377. [Google Scholar]
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Araújo J.M.G., Nogueira R.M.R., Schatzmayr H.G., de Zanotto P.M., A., Bello, G., Phylogeography and evolutionary history of dengue virus type 3. Infect. Genet. Evol. 2009;9:716–725. doi: 10.1016/j.meegid.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Armstrong J., Fiddes I.T., Diekhans M., Paten B. Whole-genome alignment and comparative annotation. Annu. Rev. Anim. Biosci. 2019;7:41–64. doi: 10.1146/annurev-animal-020518-115005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arneberg P., Skorping A., Grenfell B., Read A.F. Host densities as determinants of abundance in parasite communities. Proc. R. Soc. Lond. B Biol. Sci. 1998;265:1283–1289. doi: 10.1098/rspb.1998.0431. [DOI] [Google Scholar]
- Asner G.P., Elmore A.J., Olander L.P., Martin R.E., Harris A.T. Grazing systems, ecosystem responses, and global change. Annu. Rev. Environ. Resour. 2004;29:261–299. doi: 10.1146/annurev.energy.29.062403.102142. [DOI] [Google Scholar]
- Baalsrud H.T., Sæther B.-E., Hagen I.J., Myhre A.M., Ringsby T.H., Pärn H., Jensen H. Effects of population characteristics and structure on estimates of effective population size in a house sparrow metapopulation. Mol. Ecol. 2014;23:2653–2668. doi: 10.1111/mec.12770. [DOI] [PubMed] [Google Scholar]
- Bar-On Y.M., Phillips R., Milo R. The biomass distribution on Earth. Proc. Natl. Acad. Sci. 2018;115:6506–6511. doi: 10.1073/pnas.1711842115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmar-Lucero S., Wood J.L.A., Scott S., Harbicht A.B., Hutchings J.A., Fraser D.J. Concurrent habitat and life history influences on effective/census population size ratios in stream-dwelling trout. Ecol. Evol. 2012;2:562–573. doi: 10.1002/ece3.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkelmans R., van Oppen M.J.H. The role of zooxanthellae in the thermal tolerance of corals: a ‘nugget of hope’ for coral reefs in an era of climate change. Proc. R. Soc. Lond. B Biol. Sci. 2006;273:2305–2312. doi: 10.1098/rspb.2006.3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berryman A.A. Limiting factors and population regulation. Oikos. 2004;105:667–670. doi: 10.1111/j.0030-1299.2004.13381.x. [DOI] [Google Scholar]
- Binney H., Edwards M., Macias-Fauria M., Lozhkin A., Anderson P., Kaplan J.O., Andreev A., Bezrukova E., Blyakharchuk T., Jankovska V., Khazina I., Krivonogov S., Kremenetski K., Nield J., Novenko E., Ryabogina N., Solovieva N., Willis K., Zernitskaya V. Vegetation of Eurasia from the last glacial maximum to present: Key biogeographic patterns. Quat. Sci. Rev. 2017;157:80–97. doi: 10.1016/j.quascirev.2016.11.022. [DOI] [Google Scholar]
- Biscotti M.A., Olmo E., Heslop-Harrison J.S. Repetitive DNA in eukaryotic genomes. Chromosome Res. 2015;23:415–420. doi: 10.1007/s10577-015-9499-z. [DOI] [PubMed] [Google Scholar]
- Bradshaw C.J., Sodhi N.S., Brook B.W. Tropical turmoil: a biodiversity tragedy in progress. Front. Ecol. Environ. 2009;7:79–87. doi: 10.1890/070193. [DOI] [Google Scholar]
- Brooks D.R., Hoberg E.P. How will global climate change affect parasite–host assemblages? Trends Parasitol. 2007;23:571–574. doi: 10.1016/j.pt.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Buck J.C., Lutterschmidt W.I. Parasite abundance decreases with host density: evidence of the encounter-dilution effect for a parasite with a complex life cycle. Hydrobiologia. 2017;784:201–210. doi: 10.1007/s10750-016-2874-8. [DOI] [Google Scholar]
- Bush R.M., Bender C.A., Subbarao K., Cox N.J., Fitch W.M. Predicting the evolution of human influenza A. Science. 1999;286:1921–1925. doi: 10.1126/science.286.5446.1921. [DOI] [PubMed] [Google Scholar]
- Conroy S.D.S., Brook B.W. Demographic sensitivity and persistence of the threatened white- and orange-bellied frogs of Western Australia. Popul. Ecol. 2003;45:105–114. doi: 10.1007/s10144-003-0145-9. [DOI] [Google Scholar]
- Cooke D.E.L., Drenth A., Duncan J.M., Wagels G., Brasier C.M. A molecular phylogeny of phytophthora and related oomycetes. Fungal Genet. Biol. 2000;30:17–32. doi: 10.1006/fgbi.2000.1202. [DOI] [PubMed] [Google Scholar]
- Donnelly P., Tavare S. Coalescents and genealogical structure under neutrality. Annu. Rev. Genet. 1995;29:401–421. doi: 10.1146/annurev.ge.29.120195.002153. [DOI] [PubMed] [Google Scholar]
- Drummond A.J., Rambaut A., Shapiro B., Pybus O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005;22:1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- Eriksson A., Mehlig B., Rafajlovic M., Sagitov S. The total branch length of sample genealogies in populations of variable size. Genetics. 2010;186:601–611. doi: 10.1534/genetics.110.117135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson N.M., Galvani A.P., Bush R.M. Ecological and immunological determinants of influenza evolution. Nature. 2010;422:428–433. doi: 10.1038/nature01509. [DOI] [PubMed] [Google Scholar]
- Foden W.B., Butchart S.H.M., Stuart S.N., Vié J.-C., Akçakaya H.R., Angulo A., DeVantier L.M., Gutsche A., Turak E., Cao L., Donner S.D., Katariya V., Bernard R., Holland R.A., Hughes A.F., O’Hanlon S.E., Garnett S.T., Şekercioğlu Ç.H., Mace G.M. Identifying the world’s most climate change vulnerable species: a systematic trait-based assessment of all birds, amphibians and corals. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0065427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankham R. Effective population size/adult population size ratios in wildlife: a review. Genet. Res. 1995;66:95–107. doi: 10.1017/S0016672300034455. [DOI] [PubMed] [Google Scholar]
- Galbreath K.E., Hafner D.J., Zamudio K.R. When cold is better: climate-driven elevation shifts yield complex patterns of diversification and demography in an alpine specialist (american pika, Ochotona princeps) Evolution. 2009;63:2848–2863. doi: 10.1111/j.1558-5646.2009.00803.x. [DOI] [PubMed] [Google Scholar]
- Galtier N., Depaulis F., Barton N.H. Detecting bottlenecks and selective sweeps from DNA sequence polymorphism. Genetics. 2000;155:981–987. doi: 10.1093/genetics/155.2.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber L.R., Heppell S.S. The use of demographic sensitivity analysis in marine species conservation planning. Biol. Conserv. 2004;120:121–128. doi: 10.1016/j.biocon.2004.01.029. [DOI] [Google Scholar]
- Gómez-Alpizar L., Carbone I., Ristaino J.B. An Andean origin of Phytophthora infestans inferred from mitochondrial and nuclear gene genealogies. Proc. Natl. Acad. Sci. 2007;104:3306–3311. doi: 10.1073/pnas.0611479104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenen M.A.M., Archibald A.L., Uenishi H., Tuggle C.K., Takeuchi Y., Rothschild M.F., Rogel-Gaillard C., Park C., Milan D., Megens H.-J., Li S., Larkin D.M., Kim H., Frantz L.A.F., Caccamo M., Ahn H., Aken B.L., Anselmo A., Anthon C., Auvil L., Badaoui B., Beattie C.W., Bendixen C., Berman D., Blecha F., Blomberg J., Bolund L., Bosse M., Botti S., Bujie Z., Bystrom M., Capitanu B., Carvalho-Silva D., Chardon P., Chen C., Cheng R., Choi S.-H., Chow W., Clark R.C., Clee C., Crooijmans R.P.M.A., Dawson H.D., Dehais P., De Sapio F., Dibbits B., Drou N., Du Z.-Q., Eversole K., Fadista J., Fairley S., Faraut T., Faulkner G.J., Fowler K.E., Fredholm M., Fritz E., Gilbert J.G.R., Giuffra E., Gorodkin J., Griffin D.K., Harrow J.L., Hayward A., Howe K., Hu Z.-L., Humphray S.J., Hunt T., Hornshøj H., Jeon J.-T., Jern P., Jones M., Jurka J., Kanamori H., Kapetanovic R., Kim J., Kim J.-H., Kim K.-W., Kim T.-H., Larson G., Lee K., Lee K.-T., Leggett R., Lewin H.A., Li Y., Liu W., Loveland J.E., Lu Y., Lunney J.K., Ma J., Madsen O., Mann K., Matthews L., McLaren S., Morozumi T., Murtaugh M.P., Narayan J., Truong Nguyen D., Ni P., Oh S.-J., Onteru S., Panitz F., Park E.-W., Park H.-S., Pascal G., Paudel Y., Perez-Enciso M., Ramirez-Gonzalez R., Reecy J.M., Rodriguez-Zas S., Rohrer G.A., Rund L., Sang Y., Schachtschneider K., Schraiber J.G., Schwartz J., Scobie L., Scott C., Searle S., Servin B., Southey B.R., Sperber G., Stadler P., Sweedler J.V., Tafer H., Thomsen B., Wali R., Wang Jian, Wang Jun, White S., Xu X., Yerle M., Zhang G., Zhang Jianguo, Zhang Jie, Zhao S., Rogers J., Churcher C., Schook L.B. Analyses of pig genomes provide insight into porcine demography and evolution. Nature. 2012;491:393–398. doi: 10.1038/nature11622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas B.J., Kamoun S., Zody M.C., Jiang R.H.Y., Handsaker R.E., Cano L.M., Grabherr M., Kodira C.D., Raffaele S., Torto-Alalibo T., Bozkurt T.O., Ah-Fong A.M.V., Alvarado L., Anderson V.L., Armstrong M.R., Avrova A., Baxter L., Beynon J., Boevink P.C., Bollmann S.R., Bos J.I.B., Bulone V., Cai G., Cakir C., Carrington J.C., Chawner M., Conti L., Costanzo S., Ewan R., Fahlgren N., Fischbach M.A., Fugelstad J., Gilroy E.M., Gnerre S., Green P.J., Grenville-Briggs L.J., Griffith J., Grünwald N.J., Horn K., Horner N.R., Hu C.-H., Huitema E., Jeong D.-H., Jones A.M.E., Jones J.D.G., Jones R.W., Karlsson E.K., Kunjeti S.G., Lamour K., Liu Z., Ma L., MacLean D., Chibucos M.C., McDonald H., McWalters J., Meijer H.J.G., Morgan W., Morris P.F., Munro C.A., O’Neill K., Ospina-Giraldo M., Pinzón A., Pritchard L., Ramsahoye B., Ren Q., Restrepo S., Roy S., Sadanandom A., Savidor A., Schornack S., Schwartz D.C., Schumann U.D., Schwessinger B., Seyer L., Sharpe T., Silvar C., Song J., Studholme D.J., Sykes S., Thines M., van de Vondervoort P.J.I., Phuntumart V., Wawra S., Weide R., Win J., Young C., Zhou S., Fry W., Meyers B.C., van West P., Ristaino J., Govers F., Birch P.R.J., Whisson S.C., Judelson H.S., Nusbaum C. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature. 2009;461:393–398. doi: 10.1038/nature08358. [DOI] [PubMed] [Google Scholar]
- Hafner M.S., Nadler S.A. Phylogenetic trees support the coevolution of parasites and their hosts. Nature. 1988;332:258–259. doi: 10.1038/332258a0. [DOI] [PubMed] [Google Scholar]
- Hafner M.S., Page R.D.M., Harvey P.H., Leigh Brown A.J., Smith J.M. Molecular phylogenies and host-parasite cospeciation: gophers and lice as a model system. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1995;349:77–83. doi: 10.1098/rstb.1995.0093. [DOI] [PubMed] [Google Scholar]
- Hafner M.S., Sudman P.D., Villablanca F.X., Spradling T.A., Demastes J.W., Nadler S.A. Disparate rates of molecular evolution in cospeciating hosts and parasites. Science. 1994;265:1087–1090. doi: 10.1126/science.8066445. [DOI] [PubMed] [Google Scholar]
- Hairston Nelson G., Hairston Nelson G. Cause-effect relationships in energy flow, trophic structure, and interspecific interactions. Am. Nat. 1993;142:379–411. doi: 10.1086/285546. [DOI] [Google Scholar]
- Halpern B.S., Gaines S.D., Warner R.R. Habitat size, recruitment, and longevity as factors limiting population size in stage-structured species. Am. Nat. 2005;165:82–94. doi: 10.1086/426672. [DOI] [PubMed] [Google Scholar]
- Hardie D.C., Hutchings J.A. Evolutionary ecology at the extremes of species’ ranges. Environ. Rev. 2010;18:1–20. doi: 10.1139/A09-014. [DOI] [Google Scholar]
- Harmon L.J., Andreazzi C.S., Débarre F., Drury J., Goldberg E.E., Martins A.B., Melián C.J., Narwani A., Nuismer S.L., Pennell M.W., Rudman S.M., Seehausen O., Silvestro D., Weber M., Matthews B. Detecting the macroevolutionary signal of species interactions. J. Evol. Biol. 2019;32:769–782. doi: 10.1111/jeb.13477. [DOI] [PubMed] [Google Scholar]
- Hecht L.B.B., Thompson P.C., Rosenthal B.M. Comparative demography elucidates the longevity of parasitic and symbiotic relationships. Proc R Soc B. 2018;285:20181032. doi: 10.1098/rspb.2018.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heled J., Drummond A.J. Bayesian inference of population size history from multiple loci. BMC Evol. Biol. 2008;8:289. doi: 10.1186/1471-2148-8-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller R., Chikhi L., Siegismund H.R. The confounding effect of population structure on Bayesian skyline plot inferences of demographic history. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0062992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S.Y.W., Shapiro B. Skyline-plot methods for estimating demographic history from nucleotide sequences. Mol. Ecol. Resour. 2011;11:423–434. doi: 10.1111/j.1755-0998.2011.02988.x. [DOI] [PubMed] [Google Scholar]
- Hoberg E.P., Brooks D.R. Evolution in action: climate change, biodiversity dynamics and emerging infectious disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015;370:20130553. doi: 10.1098/rstb.2013.0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoberg E.P., Brooks D.R. A macroevolutionary mosaic: episodic host-switching, geographical colonization and diversification in complex host–parasite systems. J. Biogeogr. 2008;35:1533–1550. doi: 10.1111/j.1365-2699.2008.01951.x. [DOI] [Google Scholar]
- Hubbard M.J., Baker A.S., Cann K.J. Distribution of Borrelia burgdorferi s.l. spirochaete DNA in British ticks (Argasidae and Ixodidae) since the 19th Century, assessed by PCR. Med. Vet. Entomol. 1998;12:89–97. doi: 10.1046/j.1365-2915.1998.00088.x. [DOI] [PubMed] [Google Scholar]
- Hudson R.R., Kaplan N.L. The coalescent process in models with selection and recombination. Genetics. 1988;120:831–840. doi: 10.1093/genetics/120.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y., Leebens-Mack J., Ayyampalayam S., Bowers J.E., McKain M.R., McNeal J., Rolf M., Ruzicka D.R., Wafula E., Wickett N.J., Wu X., Zhang Yong, Wang J., Zhang Yeting, Carpenter E.J., Deyholos M.K., Kutchan T.M., Chanderbali A.S., Soltis P.S., Stevenson D.W., McCombie R., Pires J.C., Wong G.K.-S., Soltis D.E., dePamphilis C.W. A genome triplication associated with early diversification of the core eudicots. Genome Biol. 2012;13:R3. doi: 10.1186/gb-2012-13-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K.P., Adler F.R., Cherry J.L. Genetic and phylogenetic consequences of island biogeography. Evolution. 2000;54:387–396. doi: 10.1111/j.0014-3820.2000.tb00041.x. [DOI] [PubMed] [Google Scholar]
- Keane R.M., Crawley M.J. Exotic plant invasions and the enemy release hypothesis. Trends Ecol. Evol. 2002;17:164–170. doi: 10.1016/S0169-5347(02)02499-0. [DOI] [Google Scholar]
- Kendall Prendergast, Bjørnstad, The macroecology of population dynamics: taxonomic and biogeographic patterns in population cycles. Ecol. Lett. 1998;1:160–164. doi: 10.1046/j.1461-0248.1998.00037.x. [DOI] [Google Scholar]
- Kimura M. Cambridge University Press; 1983. The Neutral Theory of Molecular Evolution. [Google Scholar]
- Kliman R., Sheehey B., Schultz J. Genetic drift and effective population size. Nat. Education. 2008;1(3):3. [Google Scholar]
- Kuhner M.K., Yamato J., Felsenstein J. Maximum likelihood estimation of population growth rates based on the coalescent. Genetics. 1998;149:429–434. doi: 10.1093/genetics/149.1.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuris A.M., Hechinger R.F., Shaw J.C., Whitney K.L., Aguirre-Macedo L., Boch C.A., Dobson A.P., Dunham E.J., Fredensborg B.L., Huspeni T.C., Lorda J., Mababa L., Mancini F.T., Mora A.B., Pickering M., Talhouk N.L., Torchin M.E., Lafferty K.D. Ecosystem energetic implications of parasite and free-living biomass in three estuaries. Nat. Lond. 2008;454:515–518. doi: 10.1038/nature06970. [DOI] [PubMed] [Google Scholar]
- Li H., Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Hansen M.M. PSMC (pairwise sequentially Markovian coalescent) analysis of RAD (restriction site associated DNA) sequencing data. Mol. Ecol. Resour. 2017;17:631–641. doi: 10.1111/1755-0998.12606. [DOI] [PubMed] [Google Scholar]
- Liu S., Hansen M.M., Jacobsen M.W. Region-wide and ecotype-specific differences in demographic histories of threespine stickleback populations, estimated from whole genome sequences. Mol. Ecol. 2016;25:5187–5202. doi: 10.1111/mec.13827. [DOI] [PubMed] [Google Scholar]
- MacArthur R.H., Wilson E.O. Princeton University Press; 2001. The Theory of Island Biogeography. [Google Scholar]
- Marshall W.F., Telford S.R., Rys P.N., Rutledge B.J., Mathiesen D., Malawista S.E., Spielman A., Persing D.H. Detection of Borrelia burgdorferi DNA in museum specimens of Peromyscus. J. Infect. Dis. 1994;170:1027–1032. doi: 10.1093/infdis/170.4.1027. [DOI] [PubMed] [Google Scholar]
- Martin F.N., Blair J.E., Coffey M.D. A combined mitochondrial and nuclear multilocus phylogeny of the genus Phytophthora. Fungal Genet. Biol. 2014;66:19–32. doi: 10.1016/j.fgb.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Mattila E., Kuitunen M.T. Nutrient versus pollination limitation in Platanthera bifolia and Dactylorhiza incarnata (Orchidaceae) Oikos. 2000;89:360–366. doi: 10.1034/j.1600-0706.2000.890217.x. [DOI] [Google Scholar]
- Mazet O., Rodríguez W., Chikhi L. Demographic inference using genetic data from a single individual: Separating population size variation from population structure. Theor. Popul Biol. 2015;104:46–58. doi: 10.1016/j.tpb.2015.06.003. [DOI] [PubMed] [Google Scholar]
- Mazet O., Rodríguez W., Grusea S., Boitard S., Chikhi L. On the importance of being structured: instantaneous coalescence rates and human evolution—lessons for ancestral population size inference? Heredity. 2016;116:362–371. doi: 10.1038/hdy.2015.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller W., Schuster S.C., Welch A.J., Ratan A., Bedoya-Reina O.C., Zhao F., Kim H.L., Burhans R.C., Drautz D.I., Wittekindt N.E., Tomsho L.P., Ibarra-Laclette E., Herrera-Estrella L., Peacock E., Farley S., Sage G.K., Rode K., Obbard M., Montiel R., Bachmann L., Ingólfsson Ó., Aars J., Mailund T., Wiig Ø., Talbot S.L., Lindqvist C. Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proc. Natl. Acad. Sci. 2012;109:E2382–E2390. doi: 10.1073/pnas.1210506109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris S.C. Parasites and the fossil record. Parasitology. 1981;82:489–509. doi: 10.1017/S0031182000067020. [DOI] [Google Scholar]
- Nadachowska-Brzyska K., Burri R., Smeds L., Ellegren H. PSMC analysis of effective population sizes in molecular ecology and its application to black-and-white Ficedula flycatchers. Mol. Ecol. 2016;25:1058–1072. doi: 10.1111/mec.13540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odum E.P. Energy flow in ecosystems: a historical review. Integr. Comp. Biol. 1968;8:11–18. doi: 10.1093/icb/8.1.11. [DOI] [Google Scholar]
- Palstra F.P., Fraser D.J. Effective/census population size ratio estimation: a compendium and appraisal. Ecol. Evol. 2012;2:2357–2365. doi: 10.1002/ece3.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellmyr O. Evolution of insect pollination and angiosperm diversification. Trends Ecol. Evol. 1992;7:46–49. doi: 10.1016/0169-5347(92)90105-K. [DOI] [PubMed] [Google Scholar]
- Peñalver E., Arillo A., Delclòs X., Peris D., Grimaldi D.A., Anderson S.R., Nascimbene P.C., Pérez-de la Fuente R. Ticks parasitised feathered dinosaurs as revealed by Cretaceous amber assemblages. Nat. Commun. 2017;8:1–13. doi: 10.1038/s41467-017-01550-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persing D.H., Telford S.R., Rys P.N., Dodge D.E., White T.J., Malawista S.E., Spielman A. Detection of Borrelia burgdorferi DNA in museum specimens of Ixodes dammini ticks. Science. 1990;249:1420–1423. doi: 10.1126/science.2402635. [DOI] [PubMed] [Google Scholar]
- Pianka E.R. Latitudinal gradients in species diversity: a review of concepts. Am. Nat. 1966;100:33–46. doi: 10.1086/282398. [DOI] [Google Scholar]
- Poinar G. Trends in the evolution of insect parasitism by nematodes as inferred from fossil evidence. J. Nematol. 2003;35:129–132. [PMC free article] [PubMed] [Google Scholar]
- Poinar G., Boucot A.J. Evidence of intestinal parasites of dinosaurs. Parasitology. 2006;133:245–249. doi: 10.1017/S0031182006000138. [DOI] [PubMed] [Google Scholar]
- Pybus O.G., Rambaut A., Harvey P.H. An integrated framework for the inference of viral population history From reconstructed genealogies. Genetics. 2000;155:1429–1437. doi: 10.1093/genetics/155.3.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A., Posada D., Crandall K.A., Holmes E.C. The causes and consequences of HIV evolution. Nat. Rev. Genet. 2004;5:52–61. doi: 10.1038/nrg1246. [DOI] [PubMed] [Google Scholar]
- Rohde K. Latitudinal gradients in species diversity: the search for the primary cause. Oikos. 1992;65:514–527. doi: 10.2307/3545569. [DOI] [Google Scholar]
- Romiguier J., Gayral P., Ballenghien M., Bernard A., Cahais V., Chenuil A., Chiari Y., Dernat R., Duret L., Faivre N., Loire E., Lourenco J.M., Nabholz B., Roux C., Tsagkogeorga G., Weber A., a.-T., Weinert, L.A., Belkhir, K., Bierne, N., Glémin, S., Galtier, N., Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature. 2014;515:261–263. doi: 10.1038/nature13685. [DOI] [PubMed] [Google Scholar]
- Rosenberg N.A., Nordborg M. Genealogical trees, coalescent theory and the analysis of genetic polymorphisms. Nat. Rev. Genet. 2002;3:380–390. doi: 10.1038/nrg795. [DOI] [PubMed] [Google Scholar]
- Schiffels S., Durbin R. Inferring human population size and separation history from multiple genome sequences. Nat. Genet. 2014;46:919–925. doi: 10.1038/ng.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraiber J.G., Akey J.M. Methods and models for unravelling human evolutionary history. Nat. Rev. Genet. 2015;16:727–740. doi: 10.1038/nrg4005. [DOI] [PubMed] [Google Scholar]
- Shea K., Chesson P. Community ecology theory as a framework for biological invasions. Trends Ecol. Evol. 2002;17:170–176. doi: 10.1016/S0169-5347(02)02495-3. [DOI] [Google Scholar]
- Shrimpton J.M., Heath D.D. Census vs. effective population size in chinook salmon: large- and small-scale environmental perturbation effects. Mol. Ecol. 2003;12:2571–2583. doi: 10.1046/j.1365-294X.2003.01932.x. [DOI] [PubMed] [Google Scholar]
- Simmonds P., Aiewsakun P., Katzourakis A. Prisoners of war — host adaptation and its constraints on virus evolution. Nat. Rev. Microbiol. 2019;17:321–328. doi: 10.1038/s41579-018-0120-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M., Hudson R.R. Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics. 1991;129:555–562. doi: 10.1093/genetics/129.2.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence J.P., Steinrücken M., Terhorst J., Song Y.S. Inference of population history using coalescent HMMs: review and outlook. Curr. Opin. Genet. Dev. Genetics of Human Origins. 2018;53:70–76. doi: 10.1016/j.gde.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staerk J., Conde D.A., Ronget V., Lemaitre J.-F., Gaillard J.-M., Colchero F. Performance of generation time approximations for extinction risk assessments. J. Appl. Ecol. 2019;56:1436–1446. doi: 10.1111/1365-2664.13368. [DOI] [Google Scholar]
- Terhorst J., Kamm J.A., Song Y.S. Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat. Genet. 2017;49:303–309. doi: 10.1038/ng.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchin M.E., Lafferty K.D. In: Biological Invasions in Marine Ecosystems: Ecological, Management, and Geographic Perspectives, Ecological Studies. Rilov G., Crooks J.A., editors. Springer; Berlin, Heidelberg: 2009. Escape from Parasites; pp. 203–214. [DOI] [Google Scholar]
- Torchin M.E., Lafferty K.D., Dobson A.P., McKenzie V.J., Kuris A.M. Introduced species and their missing parasites. Nature. 2003;421:628–630. doi: 10.1038/nature01346. [DOI] [PubMed] [Google Scholar]
- Torchin M.E., Lafferty K.D., Kuris A.M. Release from parasites as natural enemies: increased performance of a globally introduced marine crab. Biol. Invasions. 2001;3:333–345. doi: 10.1023/A:1015855019360. [DOI] [Google Scholar]
- Treangen T.J., Salzberg S.L. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 2011;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trillmich F., Limberger D. Drastic effects of El Niño on Galapagos pinnipeds. Oecologia. 1985;67:19–22. doi: 10.1007/BF00378445. [DOI] [PubMed] [Google Scholar]
- Turner T.F., Osborne M.J., Moyer G.R., Benavides M.A., Alò D. Life history and environmental variation interact to determine effective population to census size ratio. Proc. R. Soc. B Biol. Sci. 2006;273:3065–3073. doi: 10.1098/rspb.2006.3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells B.K., Santora J.A., Henderson M.J., Warzybok P., Jahncke J., Bradley R.W., Huff D.D., Schroeder I.D., Nelson P., Field J.C., Ainley D.G. Environmental conditions and prey-switching by a seabird predator impact juvenile salmon survival. J. Mar. Syst. 2017;174:54–63. doi: 10.1016/j.jmarsys.2017.05.008. [DOI] [Google Scholar]
- Wertheim J.O., Oster A.M., Switzer W.M., Zhang C., Panneer N., Campbell E., Saduvala N., Johnson J.A., Heneine W. Natural selection favoring more transmissible HIV detected in United States molecular transmission network. Nat. Commun. 2019;10:1–10. doi: 10.1038/s41467-019-13723-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willig M.R., Kaufman D.M., Stevens R.D. Latitudinal gradients of biodiversity: pattern, process, scale, and synthesis. Annu. Rev. Ecol. Evol. Syst. 2003;34:273–309. doi: 10.1146/annurev.ecolsys.34.012103.144032. [DOI] [Google Scholar]
- Wilson E.O., Bossert W.H. edition. ed. Sinauer Associates is an imprint of Oxford University Press; Sunderland, Mass: 1971. A Primer of Population Biology, 1. [Google Scholar]
- Wolfe Nathan, Dunavan CP, Diamond Jared. Origins of major human infectious diseases. Nature. 2007;447(7142):279–283. doi: 10.1038/nature05775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotto P.M., Gould E.A., Gao G.F., Harvey P.H., Holmes E.C. Population dynamics of flaviviruses revealed by molecular phylogenies. Proc. Natl. Acad. Sci. U. S. A. 1996;93:548–553. doi: 10.1073/pnas.93.2.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Guang X., Sun D., Xu S., Li M., Seim I., Jie W., Yang L., Zhu Q., Xu J., Gao Q., Kaya A., Dou Q., Chen B., Ren W., Li S., Zhou K., Gladyshev V.N., Nielsen R., Fang X., Yang G. Population genomics of finless porpoises reveal an incipient cetacean species adapted to freshwater. Nat. Commun. 2018;9:1–8. doi: 10.1038/s41467-018-03722-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Wang B., Pan Q., Zhang J., Kumar S., Sun X., Liu Z., Pan H., Lin Y., Liu G., Zhan W., Li Mingzhou, Ren B., Ma X., Ruan H., Cheng C., Wang D., Shi F., Hui Y., Tao Y., Zhang C., Zhu P., Xiang Z., Jiang W., Chang J., Wang H., Cao Z., Jiang Z., Li B., Yang G., Roos C., Garber P.A., Bruford M.W., Li R., Li Ming. Whole-genome sequencing of the snub-nosed monkey provides insights into folivory and evolutionary history. Nat. Genet. 2014;46:1303–1310. doi: 10.1038/ng.3137. [DOI] [PubMed] [Google Scholar]