

Abstract
In the outer membrane of gram‐negative bacteria, O‐antigen segments of lipopolysaccharide (LPS) form a chemomechanical barrier, whereas lipid A moieties anchor LPS molecules. Upon infection, human guanylate binding protein‐1 (hGBP1) colocalizes with intracellular gram‐negative bacterial pathogens, facilitates bacterial killing, promotes activation of the lipid A sensor caspase‐4, and blocks actin‐driven dissemination of the enteric pathogen Shigella. The underlying molecular mechanism for hGBP1's diverse antimicrobial functions is unknown. Here, we demonstrate that hGBP1 binds directly to LPS and induces “detergent‐like” LPS clustering through protein polymerization. Binding of polymerizing hGBP1 to the bacterial surface disrupts the O‐antigen barrier, thereby unmasking lipid A, eliciting caspase‐4 recruitment, enhancing antibacterial activity of polymyxin B, and blocking the function of the Shigella outer membrane actin motility factor IcsA. These findings characterize hGBP1 as an LPS‐binding surfactant that destabilizes the rigidity of the outer membrane to exert pleiotropic effects on the functionality of gram‐negative bacterial cell envelopes.
Keywords: actin‐based motility, gram‐negative, guanylate binding proteins, lipopolysaccharide, O‐antigen
Subject Categories: Microbiology, Virology & Host Pathogen Interaction
Human Guanylate Binding Protein 1 (hGBP1) binds to LPS to destabilize gram‐negative outer membranes and thereby promoting antibacterial host defense pathways.
Introduction
Cell‐autonomous immunity describes the ability of individual cells within a multicellular organism to activate a wide range of cell‐intrinsic host defense programs directed at intracellular pathogens. These ancient, hard‐wired defense programs are commonly under spatial and temporal control and often require inducing signals to launch (Howard, 2007; Randow et al, 2013). Microbe‐associated molecular patterns such as the gram‐negative bacterial outer membrane molecule lipopolysaccharide (LPS) can act as such inducing signals in infected cells by activating specialized pattern recognition receptors that include the cytosolic LPS sensor caspase‐4 (Hagar et al, 2013; Kayagaki et al, 2013; Shi et al, 2014). Alternatively, proinflammatory cytokines released by sentinel and immune effector cells instruct host cells to transition into a state of heightened antimicrobial resistance. Perhaps the most potent inducer of cell‐autonomous immunity against bacterial pathogens is the lymphocyte‐derived cytokine gamma‐interferon (IFNγ), which controls the expression of hundreds of antimicrobial proteins encoded by IFN‐stimulated genes (ISGs). The specific functions of many of these ISGs are either unknown or only poorly characterized (MacMicking, 2012).
Among the most highly expressed ISGs are guanylate binding proteins (GBPs) which have important roles in antibacterial host defense (Coers, 2017; Man et al, 2017; Praefcke, 2018; Santos & Broz, 2018; Huang et al, 2019). GBPs promote the lysis of gram‐negative bacteria inside infected macrophages (Man et al, 2015; Meunier et al, 2015; Li et al, 2017; Liu et al, 2018) and aid in the activation of the LPS sensor caspase‐4 in response to infections, bacterial outer membrane vesicles or cytoplasmic LPS (Meunier et al, 2014; Pilla et al, 2014; Finethy et al, 2017; Lagrange et al, 2018; Santos et al, 2018). Human GBP1 also interferes with the ability of the cytosol‐invading bacterial pathogen Shigella flexneri to usurp the host actin polymerization machinery for intracellular bacterial motility and cellular dissemination (Piro et al, 2017; Wandel et al, 2017). The molecular mechanisms by which GBPs can exert these seemingly distinct cellular functions are undetermined; yet, they appear linked to the capacity of GBPs to specifically associate with intracellular bacterial pathogens.
Human GBP1 (hGBP1) colocalizes with the cytosol‐resident gram‐negative bacterial pathogens Burkholderia thailandensis and S. flexneri but not gram‐positive Listeria monocytogenes (Piro et al, 2017). Four of the six additional hGBP paralogs also associate with cytosolic S. flexneri, but in a strictly hGBP1‐dependent manner (Li et al, 2017; Piro et al, 2017; Wandel et al, 2017). Together, these reported observations hinted at the intriguing yet untested model that hGBP1 is unique among hGBPs in its ability to operate as a bona fide cytosolic receptor for gram‐negative bacteria. Here, we demonstrate that polymerizing hGBP1 attaches directly to gram‐negative bacteria via LPS binding. Following the initial attachment, hGBP1 transitions into a stable protein coat encapsulating exclusively “smooth” bacterial strains, i.e., bacteria expressing the outermost O‐antigen polysaccharide segment of LPS. We find that hGBP1 coating of bacteria disrupts the O‐antigen barrier protective against sublethal concentrations of the antimicrobial peptide polymyxin B, enables the recognition of lipid A by caspase‐4 on the bacterial surface, and also interferes with the function of the Shigella outer membrane protein IcsA critical for intracellular bacterial motility. Together, our observations designate hGBP1 as an LPS‐binding and LPS‐clustering surfactant that disrupts critical functions of the gram‐negative bacterial outer membrane and thereby promotes diverse antibacterial host defense pathways.
Results
Farnesylated hGBP1 binds directly to Shigella flexneri in a GTPase‐dependent manner
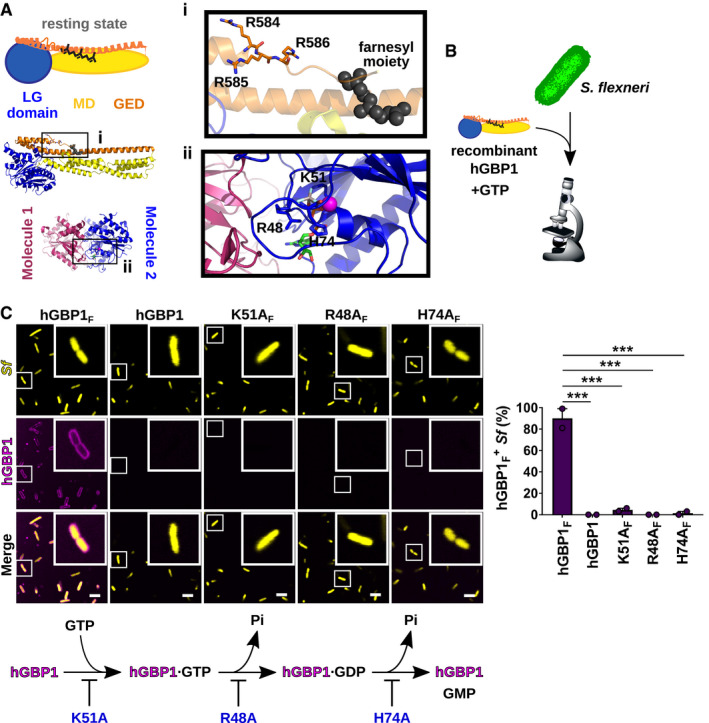
As typical for a dynamin superfamily protein (Daumke & Praefcke, 2016; Ramachandran & Schmid, 2018), hGBP1 consists of a large N‐terminal globular G domain (LG domain) and a helical C‐terminus that segregates into the middle domain (MD) and the GTPase effector domain (GED) (Fig 1A). In the presence of its substrate GTP, hGBP1 dimerizes (Ince et al, 2017) and binds to membranes via hydrophobic interactions mediated by its C‐terminal farnesyl moiety that is otherwise buried within a hydrophobic pocket when hGBP1 is nucleotide‐free (Fres et al, 2010; Shydlovskyi et al, 2017; Ji et al, 2019). Therefore, in order to test whether hGBP1 is able to bind to the outer membrane of the cytosolic gram‐negative bacterium S. flexneri, we mixed fluorescently labeled farnesylated hGBP1 (hGBP1F) with either formaldehyde‐fixed or live bacteria in the presence of guanine nucleotides and captured confocal images following varying incubation times (Fig 1B). We observed direct binding of hGBP1F to both fixed and live S. flexneri in the presence of GTP but not GDP (Fig 1C and Appendix Fig S1A) across a physiological range (Naschberger et al, 2006) of hGBP1 protein concentrations (Appendix Fig S1B). Consistent with colocalization studies performed in tissue culture (Li et al, 2017; Piro et al, 2017; Wandel et al, 2017), we found that mutations disrupting nucleotide binding (hGBP1F K51A), GTP hydrolysis (hGBP1F R48A) or GDP hydrolysis (hGBP1F H74A) (Praefcke et al, 2004), or lack of protein farnesylation stopped hGBP1 from binding to S. flexneri in vitro (Fig 1C). These data demonstrate that hGBP1F binds directly to S. flexneri through a GTP‐hydrolysis‐dependent process.
Figure 1. Farnesylated hGBP1 binds directly to Shigella flexneri in a GTPase‐dependent manner.
-
AStructure of full‐length, nucleotide‐free, farnesylated hGBP1 (PDB entry 6k1z) and GDP·AlFX‐bound LG‐domain dimer (PDB entry 2B92). Insert (i) shows the farnesyl moiety and the triple arginine stretch (3R = R584–586). Insert (ii) highlights residues required for nucleotide binding and hydrolysis.
-
BExperimental design: fluorescently labeled recombinant hGBP1 variants and nucleotides were mixed with broth‐cultured live or fixed bacteria; bacterial binding was monitored by confocal microscopy.
-
CConfocal images of formaldehyde‐fixed GFP+ S. flexneri following 20 min of incubation with 2 mM GTP and 10 μM Alexa‐Fluor647‐labeled protein. Bacteria associated with hGBP1 mutants after 20 min were quantified. Mean frequencies ± SEM of combined data from two independent experiments are shown. Significance was determined by one‐way ANOVA with Tukey's multiple comparison test. ***P ≤ 0.001. Scale bars equal 5 μm. Flow diagram depicts effects of K51A, R48A, and H74A hGBP1 mutations on nucleotide binding and hydrolysis.
Source data are available online for this figure.
hGBP1 polymerization is required for bacterial binding
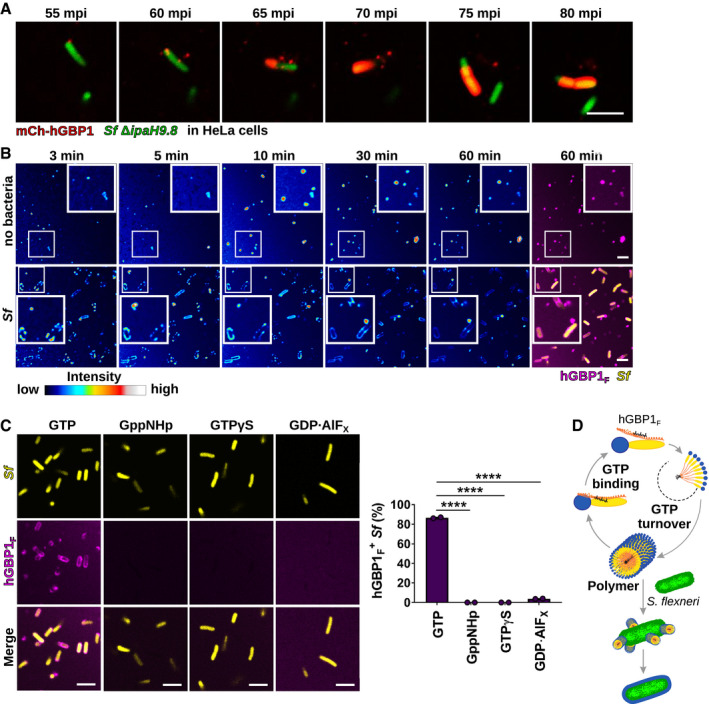
When expressed in cells, hGBP1 forms discrete granular structures (Britzen‐Laurent et al, 2010). In time‐lapse microscopy experiments using a S. flexneri mutant strain deficient for the bacterial hGBP1 antagonist IpaH9.8 shown to dramatically reduce hGBP1 recruitment to cytosolic bacteria (Li et al, 2017; Piro et al, 2017; Wandel et al, 2017), we observed the appearance of these hGBP1 granular structures in close proximity to cytosolic bacteria and furthermore recorded the transition of these granules into a hGBP1 protein coat encapsulating entire bacteria (Fig 2A and Movie EV1). Similar to the intracellular dynamics of hGBP1 targeting to S. flexneri, we found that upon GTP supplementation hGBP1F granular structures formed rapidly in vitro and associated with bacterial surfaces. In remarkable symmetry to our live cell imaging data, these bacteria‐associated hGBP1 granules then transformed into a protein coat encasing individual bacteria in vitro (Figs 2B and EV1A, and Movie EV2).
Figure 2. hGBP1 polymerization is required for bacterial binding.
-
ATranslocation of ectopically expressed mCherry‐hGBP1 to cytosolic GFP+ Shigella flexneri ΔipaH9.8 in HeLa hGBP1‐KO cells was monitored by time‐lapse microscopy. Individual time frames of Movie EV1 starting at 55 min post‐infection (mpi) are shown.
-
BConfocal time‐lapse microscopy was used to image 10 μM Alexa‐Fluor647‐hGBP1F supplemented with 2 mM GTP in the presence or absence of formaldehyde‐fixed GFP+ S. flexneri. Individual time frames of Movie EV2 depict hGBP1F fluorescence intensity. Merged images of hGBP1F and S. flexneri fluorescence are shown for the 60 min time points.
-
CImages were taken at 45 min after addition of 10 μM Alexa‐Fluor647‐hGBP1F to formaldehyde‐fixed GFP+ S. flexneri in the presence of indicated nucleotides (GTP, natural substrate; GppNHp, non‐hydrolysable GTP analog; GTPγS, slowly hydrolysable GTP analog; GDP·AlFX, GTP transition state analog). hGBP1‐associated bacteria after 45 min were quantified. Combined data from two independent experiments are shown as mean ± SEM. Significance was determined by one‐way ANOVA with Tukey's multiple comparison test. ****P ≤ 0.0001.
-
DModel: hGBP1 polymers bind to S. flexneri directly and transition into a bacterium‐encapsulating protein coat.
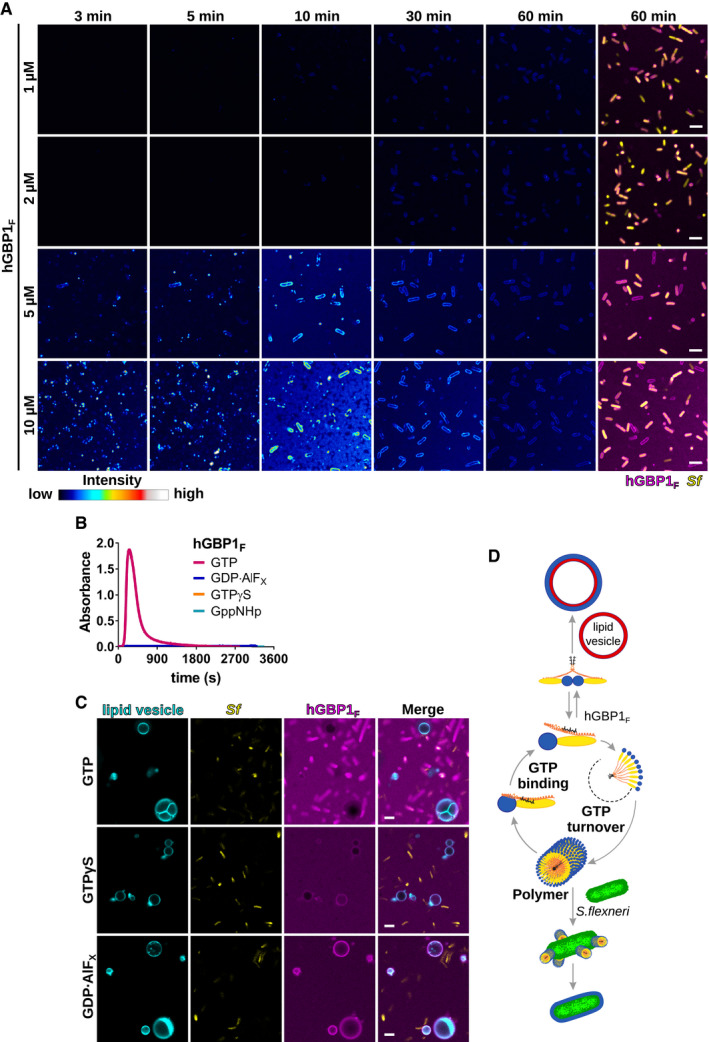
Figure EV1. Polymerizing hGBP1F binds directly to Shigella flexneri across a physiological range of hGBP1F protein concentrations.
-
ATime‐lapse microscopy frames of formaldehyde‐fixed GFP+ S. flexneri following admixture of varying concentrations of Alexa‐Fluor647‐labeled hGBP1F and 2 mM GTP. Individual time frames depict hGBP1F fluorescence intensity. Merged images of hGBP1F and S. flexneri fluorescence are shown for the 60 min time points.
-
BPolymerization of 10 μM hGBP1F monitored over time by absorption spectroscopy at 350 nm after addition of 2 mM (GTP, GppNHp, GTPγS) or 250 μM (GDP·AlFX) nucleotide.
-
CConfocal images taken of formaldehyde‐fixed GFP+ S. flexneri and rhodamine‐labeled lipid vesicles 20 min after addition of 10 μM Alexa‐Fluor647‐hGBP1F and 2 mM GTP, 2 mM GTPγS, or 250 μM GDP·AlFX.
-
DModel: In its GTP‐bound conformation, the C‐terminal farnesyl tail is released from its hydrophobic pocket allowing hGBP1 to bind to lipid vesicles in vitro and to host membranes inside human cells. Direct binding to bacteria on the other hand requires hGBP1 to self‐assemble over several GTP turnover cycles into polymers. These hGBP1 polymers bind to S. flexneri directly and then transition into a bacteria‐encapsulating protein coat.
Although hGBP1F granules detected inside cells were initially interpreted as membrane‐associated vesicle‐like structures (Britzen‐Laurent et al, 2010), our recent in vitro studies showed that hGBP1F not only binds to membranes but also self‐assembles into large polymers independent of exogenous lipids (Shydlovskyi et al, 2017; Sistemich et al, 2020). We therefore surmised that the hGBP1F granules that were forming both in the presence and in the absence of exogenously added bacteria in vitro (Fig 2B) constituted hGBP1F polymers. In support of this notion and in confirmation of previous work (Shydlovskyi et al, 2017), we found that the build‐up of supramolecular hGBP1F particles measured by spectroscopy occurred only in the presence of hydrolysable GTP (Fig EV1B). We further observed that the poorly hydrolysable analog GTPγS—while sufficient to direct the attachment of hGBP1F to artificial vesicles (Fig EV1C), as previously reported (Shydlovskyi et al, 2017)—failed to engender in vitro hGBP1F attachment to bacteria (Figs 2C and EV1C, and [Link], [Link]). Similarly, mimicking the hGBP1 GTP hydrolysis transition state through admixture of GDP·aluminum fluoride (GDP·AlFX) triggered hGBP1F binding to vesicles but not bacteria (Figs 2C and EV1C, and Movie EV5). Because hGBP1F forms irreversible short polymers in the presence of GDP·AlFX (Shydlovskyi et al, 2017; Sistemich et al, 2020), these results indicate that reversible hGBP1F polymerization is required for direct binding of hGBP1F to S. flexneri (Figs 2D and EV1D).
The C‐terminal hGBP1 polybasic motif is required for sustained binding to Shigella flexneri
Human GBP1 polymers have a ring‐like structure with individual hGBP1 molecules assembled around the hydrophobic core formed by their farnesyl moieties (Shydlovskyi et al, 2017). Immediately adjacent to the C‐terminal farnesyl group exists a short polybasic motif containing a stretch of three arginines (3R) (Fig 1A). We previously demonstrated that the C‐terminal polybasic motif containing the 3R stretch is unique to hGBP1 among the human GBP family and required for efficient colocalization of hGBP1 with intracytosolic S. flexneri (Piro et al, 2017). To determine the mechanisms by which 3R drives hGBP1 translocation to S. flexneri, we assessed the ability of recombinant hGBP1F R584‐586A, a mutant lacking the 3R stretch, to form polymers and to bind to bacteria in vitro. First, we monitored protein polymerization by absorption spectroscopy and found that hGBP1F R584‐586A could still form large particulates, albeit with delayed kinetics (Figs 3A and EV2A). Because hGBP1 undergoes polymerization‐accelerated cooperative hydrolysis (Praefcke et al, 2004; Shydlovskyi et al, 2017), the apparent slowdown in polymerization kinetics likely explains the moderately reduced rate of GTP hydrolysis that we observed in reactions with the hGBP1F R584‐586A mutant (Figs 3A and C, and EV2B). This conclusion is supported by our observation that the R584‐586A mutation has no impact on the GTP hydrolysis rates of non‐farnesylated and thus non‐polymerizing hGBP1 (Fig EV2C).
Figure 3. A C‐terminal hGBP1 polybasic motif is required for sustained binding to Shigella flexneri .
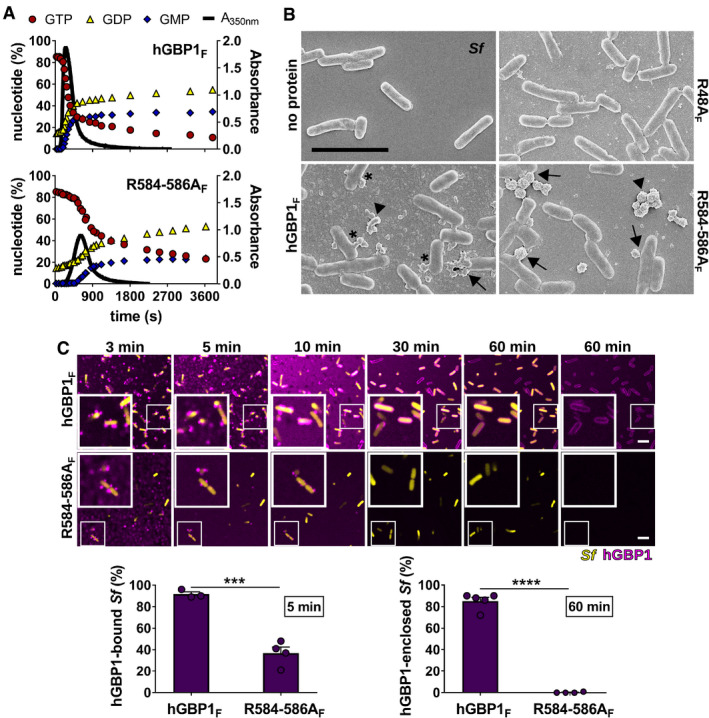
-
APolymerization of 10 μM hGBP1F and hGBP1F R584‐586A in the presence of 2 mM GTP was monitored over time by absorption spectroscopy at 350 nm. Absorbance signals were superimposed with nucleotide composition of the same solution analyzed at defined time points, revealing the characteristic first phase of slow polymer nucleation and the second phase of fast polymer growth and cooperative hydrolysis.
-
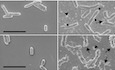
BScanning electron micrographs of live S. flexneri incubated with no protein or with 5 μM of either hGBP1F, hGBP1F R584‐586A, or hGBP1F R48A (non‐polymerizing mutant) in the presence of 2 mM GTP for 4 min. Arrowheads point to unattached hGBP1 polymers, arrows point to hGBP1 polymers attached to bacteria, and asterisks mark polymeric structures that appear to fuse with bacterial surfaces.
-
CConfocal time‐lapse microscopy frames of formaldehyde‐fixed GFP+ S. flexneri following admixture of 10 μM Alexa‐Fluor647‐labeled hGBP1F or hGBP1F R584‐586A and 2 mM GTP. Binding of hGBP1F polymers to bacteria at 5 min and enclosure of bacteria with hGBP1F protein coats after 60 min were quantified. Mean frequencies ± SEM of combined data from at least three independent experiments are shown. Significance was determined by unpaired t‐tests, two‐tailed. ***P ≤ 0.001; ****P ≤ 0.0001.
Figure EV2. hGBP1F polymerization is dependent on GTP, and subsequent GDP hydrolysis and its dynamics are modified by the C‐terminal 3R stretch.
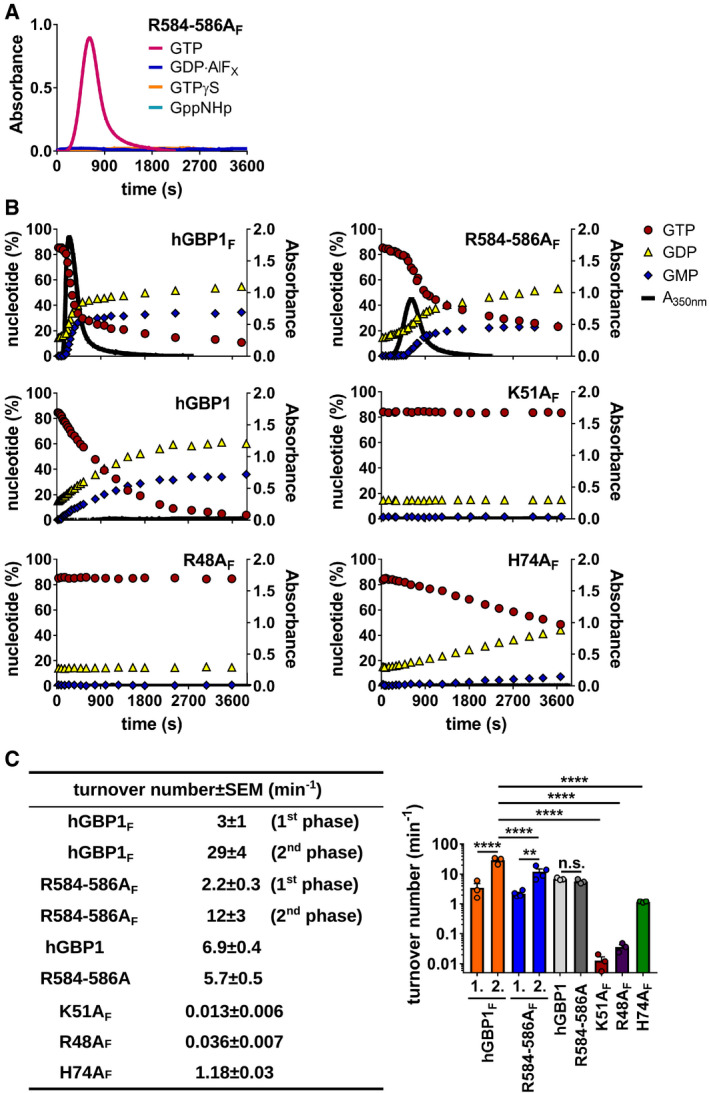
-
APolymerization of 10 μM hGBP1F R584‐586A was monitored over time by absorption spectroscopy at 350 nm after admixture of 2 mM (GTP, GppNHp, GTPγS) or 250 μM (GDP·AlFX) nucleotide.
-
BPolymerization of different hGBP1 variants (at 10 μM) induced by 2 mM GTP was monitored over time by absorption spectroscopy at 350 nm. Absorbance signals were superimposed with nucleotide composition analyzed at defined time points of the corresponding sample. Blots for hGBP1F and hGBP1F R584‐586A are the same as shown in Fig 3A.
-
CGTP turnover numbers were determined for different hGBP1 variants. Turnover numbers for both the slow and the fast phases of GTP hydrolysis (1st phase [1.], 2nd phase [2.]) were quantified during hGBP1F and hGBP1F R584‐586A polymerization. Combined data from three independent experiments are shown as mean turnover numbers ± SEM. Significance was determined by two‐way ANOVA with Tukey's multiple comparison test. n.s., not significant; **P ≤ 0.01; ****P ≤ 0.0001.
Source data are available online for this figure.
To interrogate the function of the C‐terminal polybasic motif further, we visualized hGBP1F polymer formation using scanning electron microscopy. After 4 min of incubation with GTP, we observed polymeric structures in the presence of hGBP1F or hGBP1F R584‐586A but not in the presence of the non‐polymerizing mutant hGBP1F R48A (Fig 3B). Notably, the polymers formed by hGBP1F R584‐586A appeared morphologically distinct from hGBP1F polymers. Moreover, hGBP1F but not hGBP1F R584‐586A polymers appeared to form continuous connections with the outer surface of S. flexneri (Fig 3B), suggesting that hGBP1F R584‐586A polymers could be functionally distinct from hGBP1F polymers. We therefore tested whether bacterial binding, or alternatively bacterial hGBP1 coating, was impacted by the R584‐586A mutation. We noticed that hGBP1F R584‐586A polymers were found in direct contact with bacteria at 5 min post‐hGBP1F R584‐586A and GTP supplementation, albeit with a two‐fold reduction in the total number of hGBP1‐bound bacteria compared to hGBP1F (Fig 3C). Although initial bacterial binding appeared relatively intact, hGBP1F R584‐586A failed to form a protein coat enclosing individual bacteria at any point during the 1 h incubation time (Fig 3C). Collectively, these observations suggest that the C‐terminal polybasic motif alters the dynamics of hGBP1F polymerization and is required for the tight association of hGBP1F with the bacterial surface.
Direct binding of hGBP1F to LPS mediates its association with gram‐negative bacteria
Our finding that hGBP1F polymers attached directly to S. flexneri led us to hypothesize that hGBP1F binds a non‐self substrate exposed on bacterial outer membranes. To test this hypothesis, we first monitored binding of hGBP1F to a diverse set of pathogenic bacterial species. We found that hGBP1F polymers bound not only to S. flexneri but also to other gram‐negative human bacterial pathogens that we tested, namely Salmonella enterica Typhimurium (ST), Legionella pneumophila (Lp), and uropathogenic Escherichia coli (UPEC). However, we did not observe any binding of hGBP1F to the gram‐positive bacteria Listeria monocytogenes (Lm) and Staphylococcus aureus (Sa) (Fig 4A). These results indicated that hGBP1F polymers recognized a molecule present in gram‐negative but not in gram‐positive bacterial cell envelopes. Because the surface‐exposed, lipidated sugar LPS is a highly expressed component of gram‐negative outer membranes but absent from gram positives (Simpson & Trent, 2019), we asked whether hGBP1F could bind directly to LPS. To test this hypothesis, we mixed fluorescently labeled, smooth (O‐antigen+) E. coli LPS (O55:B55) or rough (O‐antigen−) Salmonella minnesota LPS (SM) with hGBP1F in the presence of GTP and monitored colocalization of hGBP1F with LPS over time. As expected, GTP induced the formation of hGBP1F polymers, apparent as granular structures (Fig 4B). Notably, these hGBP1F granules colocalized with LPS clusters that formed simultaneously with hGBP1F granules and were also dependent on farnesylation as well as hGBP1F polymerization, since polymerization‐deficient hGBP1F mutants (K51AF, R48AF, H74AF) and non‐farnesylated hGBP1 did not induce LPS clusters (Figs 4B and EV3A, and Movie EV6). Notably, hGBP1 polymerization facilitated the clustering of both smooth LPS O55:B55 and rough LPS SM (Figs 4B and EV3A), demonstrating that O‐antigen is dispensable for LPS binding by polymerizing hGBP1. We next confirmed hGBP1F binding to LPS in dot‐blot assays (Fig EV3B). The polymerization‐competent but 3R‐deficient mutant hGBP1F R584‐586A was able to bind and cluster LPS in suspension (Fig 4B) but failed to adhere to nitrocellulose‐bound LPS in dot‐blot assays (Fig EV3B), further underscoring the importance of the 3R stretch in enabling sustained hGBP1F binding to an LPS‐decorated surface. To further characterize the dynamics of hGBP1F‐LPS interactions, we measured hGBP1F polymerization and GTP hydrolysis in the presence and absence of LPS O55:B5. We observed faster initiation of hGBP1F polymerization and accelerated GTP hydrolysis kinetics in the presence of LPS (Fig 4C), revealing a role for LPS as a potential nucleation‐promoting factor for hGBP1 polymerization. LPS is an amphipathic molecule that forms micelles in aqueous solution (Santos et al, 2003), and we can therefore conclude from the combined data that hGBP1F binds directly to LPS micelles, which results in accelerated hGBP1F polymerization as well as the assembly of large LPS aggregates in a polymerization‐dependent manner.
Figure 4. Direct binding of hGBP1F to LPS mediates its association with gram‐negative bacteria.
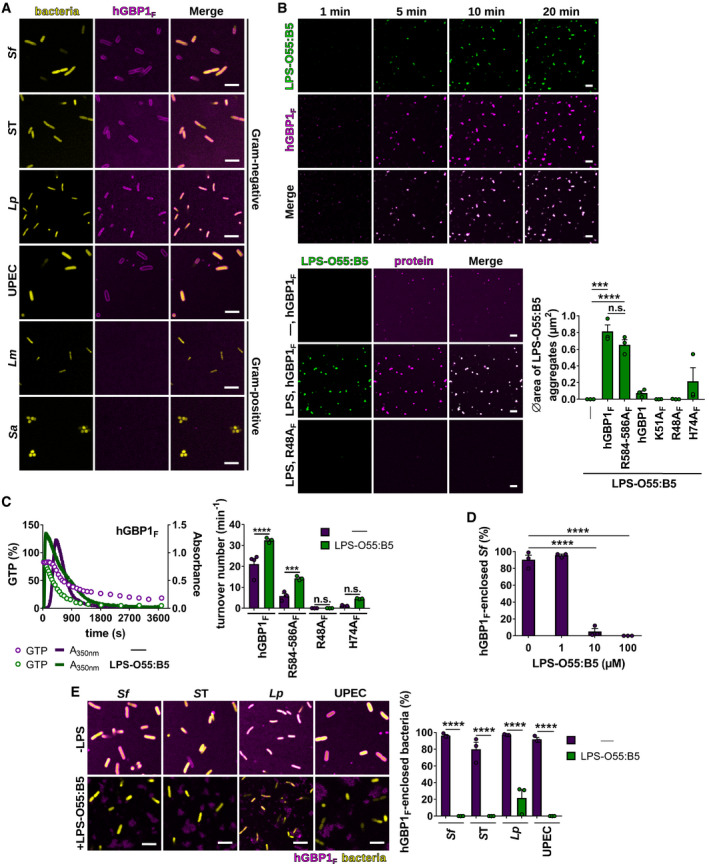
-
ARepresentative confocal images taken at 60 min after admixture of 2 mM GTP and 10 μM Alexa‐Fluor647‐ or Alexa‐Fluor488‐hGBP1F to formaldehyde‐fixed gram‐negative and gram‐positive bacteria expressing GFP (Salmonella enterica Typhimurium [ST], L. monocytogenes [Lm]), or RFP (Shigella flexneri [Sf], uropathogenic E. coli [UPEC]), or dsRed (L. pneumophila [Lp], S. aureus [Sa]).
-
B(Upper panel) Confocal time‐lapse microscopy frames of 5 μM Alexa‐Fluor488‐LPS‐O55:B5 after addition of 5 μM Alexa‐Fluor647‐hGBP1F supplemented with 2 mM GTP. (Lower panel) Confocal images taken of 5 μM Alexa‐Fluor488‐LPS‐O55:B5 20 min after addition of 5 μM Alexa‐Fluor647‐hGBP1F or Alexa‐Fluor647‐hGBP1F R48A supplemented with 2 mM GTP. Graphs depict average aggregate area of Alexa‐Fluor488‐LPS‐O55:B5 supplemented with 2 mM GTP and 5 μM of the indicated hGBP1 variant. Mean area ± SEM of combined data from three independent experiments. Significance was determined by one‐way ANOVA with Tukey's multiple comparison test. n.s., not significant; ***P ≤ 0.001; ****P ≤ 0.0001.
-
CPolymerization of 5 μM hGBP1F induced by addition of 2 mM GTP in the presence and absence of 5 μM LPS‐O55:B5 was monitored by absorption spectroscopy at 350 nm. The absorbance signal was superimposed with nucleotide composition of the same solution analyzed at defined time points. Maximal hydrolysis rates (turnover numbers) were determined for hGBP1F variants in the presence and absence of 5 μM LPS‐O55:B5. Graphs show mean turnover numbers ± SEM of combined data from at least three independent experiments. Significance was determined by two‐way ANOVA with Tukey's multiple comparison test. n.s., not significant; ***P ≤ 0.001; ****P ≤ 0.0001.
-
DFormaldehyde‐fixed GFP+ S. flexneri supplemented with varying LPS‐O55:B5 concentrations were mixed with 5 μM hGBP1F and 2 mM GTP. After 60 min, hGBP1F‐enclosed S. flexneri were quantified. Combined data from three independent experiments are shown as mean ± SEM. Significance was determined by one‐way ANOVA with Tukey's multiple comparison test. ****P ≤ 0.0001.
-
EConfocal images of formaldehyde‐fixed GFP‐, RFP‐, or dsRed‐expressing gram‐negative bacteria 60 min after addition of 5 μM Alexa‐Fluor647‐ or Alexa‐Fluor488‐hGBP1F and 2 mM GTP in the presence and absence of 100 μM LPS‐O55:B5. After 60 min, hGBP1F‐enclosed gram‐negative bacteria were quantified. Combined data from three independent experiments are shown as mean ± SEM. Significance was determined by two‐way ANOVA with Tukey's multiple comparison test. ****P ≤ 0.0001.
Figure EV3. Polymerizing hGBP1F binds to and clusters LPS, which acts as a competitive inhibitor blocking hGBP1F encapsulation of Shigella flexneri .
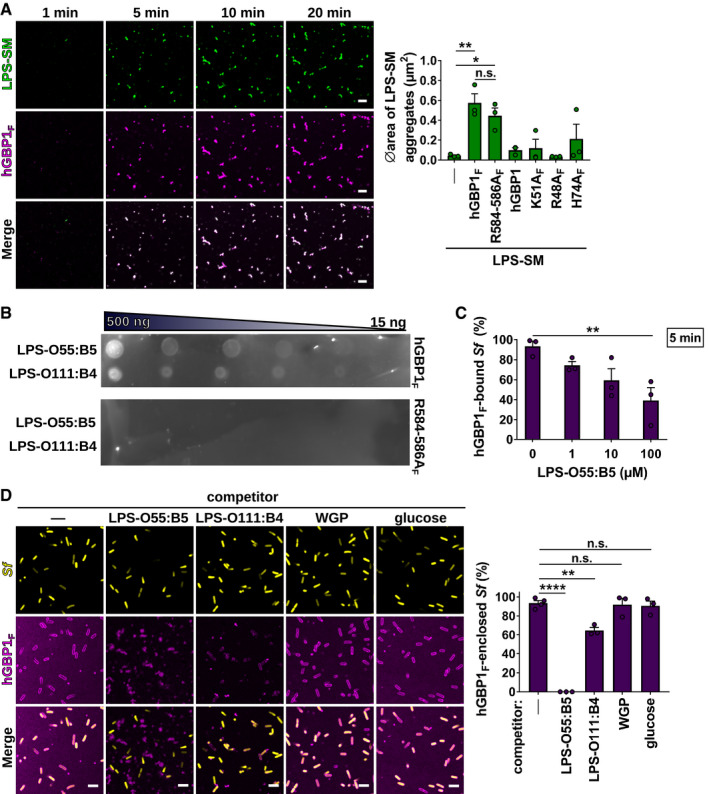
-
AConfocal time‐lapse microscopy frames of 5 μM Alexa‐Fluor488‐LPS derived from Salmonella minnesota (LPS‐SM) mixed with 5 μM Alexa‐Fluor647‐hGBP1F in the presence of 2 mM GTP. Graph depicts average aggregate area of Alexa‐Fluor488‐LPS‐SM supplemented with 2 mM GTP and 5 μM of the indicated hGBP1 variant. Mean area ± SEM of combined data from three independent experiments. Significance was determined by one‐way ANOVA with Tukey's multiple comparison test. n.s., not significant; *P ≤ 0.05; **P ≤ 0.01.
-
BNitrocellulose membrane dotted with 15–500 ng LPS‐O55:B5 and LPS‐O111:B4 spots was incubated with 2 μM Alexa‐Fluor647‐hGBP1F or Alexa‐Fluor647‐hGBP1F R584‐586A for 10 min in the presence of 1 mM GTP before measuring fluorescent signals.
-
CPercentage of hGBP1F‐bound formaldehyde‐fixed GFP+ S. flexneri after 5 min of incubation with 5 μM hGBP1F and 2 mM GTP in the presence of the indicated concentrations of LPS‐O55:B5. Mean percentages ± SEM of combined data from three independent experiments are shown. Significance was determined by one‐way ANOVA with Tukey's multiple comparison test. **P ≤ 0.01.
-
DConfocal images of formaldehyde‐fixed GFP+ S. flexneri supplemented with different carbohydrate species (LPS‐O55:B5, LPS‐O111:B4, glucose—100 μM; whole glucan particles (WGP) 1 mg/ml) taken 60 min after addition of 5 μM Alexa‐Fluor647‐hGBP1F and 2 mM GTP. After 60 min of incubation time, hGBP1F‐enclosed bacteria were quantified. Mean frequencies ± SEM of combined data from three independent experiments are shown. Significance was determined by one‐way ANOVA with Tukey's multiple comparison test. n.s., not significant; **P ≤ 0.01; ****P ≤ 0.0001.
Because hGBP1F binds directly to LPS, we tested whether LPS could act as a competitive inhibitor for hGBP1F attachment to bacteria. We found that E. coli LPS O55:B5 at concentrations of 10 μM or higher reduced initial docking of 5 μM hGBP1F to S. flexneri (Fig EV3C) and led to a near complete block in detectable hGBP1F on the surface of S. flexneri and other bacteria at 1 h post‐addition of hGBP1F and GTP (Fig 4D and E, and Movie EV7). A second LPS species (O111:B4) also inhibited hGBP1F anchoring to S. flexneri, albeit less efficiently than O55:B5 (Fig EV3D). Because O55:B5 forms smaller aggregates in aqueous solution than O111:B4 and thus features a larger effective surface area per mass (Risco et al, 1993; Bergstrand et al, 2006; Stenutz et al, 2006), O55:B5 is expected to sequester hGBP1 more effectively than O111:B4, thus explaining its superior performance as a competitive inhibitor. In contrast to these two LPS species, whole glucan particles (WGP) purified from yeast cell walls or monomeric glucose had no impact on the association of hGBP1F with S. flexneri (Fig EV3D). Together, these data show that hGBP1 is an LPS‐binding protein and that exogenous LPS disrupts the recruitment of hGBP1F polymers to the surface of gram‐negative bacteria.
Bacterial O‐antigen drives the transition from bacteria‐bound hGBP1F polymers into bacteria‐encasing hGBP1F protein coats
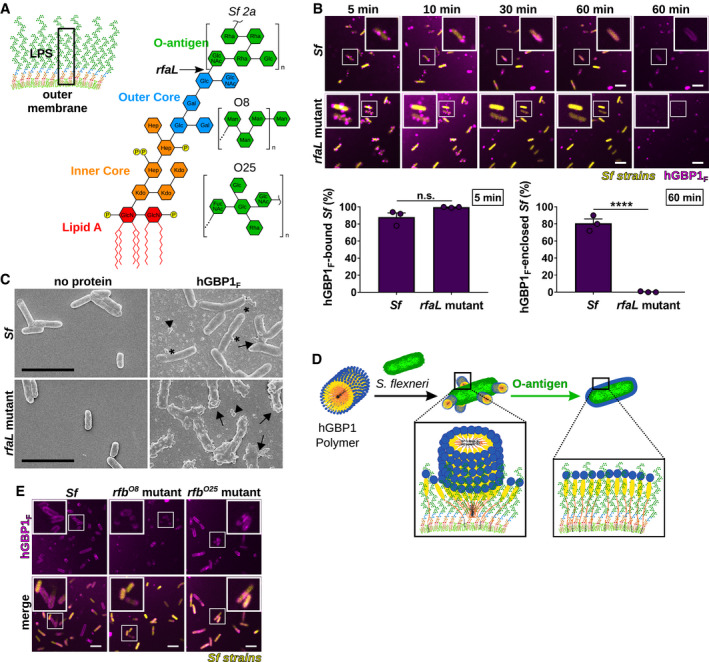
O‐antigen comprises the outward‐facing carbohydrate segment of membrane‐embedded LPS (Kalynych et al, 2014). We previously observed diminished intracytosolic targeting of hGBP1 to S. flexneri rough mutant strains such as rfaL (Piro et al, 2017), which lacks the O‐antigen portion of LPS but maintains the inner and outer carbohydrate cores attached to lipid A (Fig 5A). To investigate the mechanism by which O‐antigen impacts recruitment of hGBP1 to S. flexneri, we monitored in vitro hGBP1F binding to S. flexneri rfaL. We found that hGBP1F docked to the rfaL mutant as efficiently as to wild type S. flexneri at 5 min after initiation of the binding reaction (Fig 5B). However, hGBP1F failed to transition into a uniform protein coat enclosing S. flexneri rfaL; instead, granular structures were detectable on the surface of the rfaL mutant, both at early (Fig 5B and C) and late (Fig 5B and Appendix Fig S2A) incubation times. Similarly, we observed the formation of hGBP1F protein envelopes on the surface of smooth but not rough E. coli strains (Appendix Fig S2B). Thus, whereas hGBP1F binds to and aggregates LPS independent of O‐antigen (Fig 4B), O‐antigen promotes the transition of surface‐docked hGBP1F polymers into bacteria‐enveloping hGBP1F protein sheets (Fig 5D).
Figure 5. Bacterial O‐antigen drives the transition from bacteria‐bound hGBP1F polymers into bacteria‐encasing hGBP1F protein coats.
-
AGraphic depiction of the bacterial outer membrane composition and LPS structure of Shigella flexneri. Arrow indicates the LPS truncation site in the O‐antigen‐deficient rfaL mutant. O‐antigen oligosaccharide subunit composition of S. flexneri serotype 2a and E. coli serotypes O8 and O25 are shown.
-
BTime‐lapse microscopy of fixed GFP+ co‐isogenic S. flexneri wild type and rfaL mutant after adding 10 μM Alexa‐Fluor647‐hGBP1F and 2 mM GTP. Bacteria bound by hGBP1F at 5 min and hGBP1F‐enclosed bacteria after 60 min were quantified. Combined data from three independent experiments are shown as mean ± SEM. Significance was determined by unpaired t‐tests, two‐tailed. n.s., not significant; ****P ≤ 0.0001.
-
CScanning electron micrographs of live wild type and rfaL mutant strains incubated with no protein or with 5 μM of hGBP1F in the presence of 2 mM GTP for 4 min. Arrowheads point to unattached hGBP1 polymers, arrows point to hGBP1 polymers attached to bacteria, and asterisks mark polymeric structures that appear to fuse with bacterial surfaces.
-
DModel: O‐antigen drives the transition of surface‐docked hGBP1F polymers into bacteria‐enveloping hGBP1F protein sheets.
-
EConfocal images of formaldehyde‐fixed, co‐isogenic GFP+ wild type and mutant S. flexneri strains harboring rfb regions of E. coli serotypes O8 and O25 after 60 min of incubation time in the presence of 10 μM Alexa‐Fluor647‐hGBP1F and 2 mM GTP.
O‐antigen repeating units are highly variable among different bacterial species (Kalynych et al, 2014). To determine whether the specific sugar composition of O‐repeats impacts the transition of hGBP1F polymers into envelopes, we tested hGBP1F protein envelope formation on the surface of S. flexneri 2a strains harboring the E. coli serotypes O8 or O25 rfb region, which determines the O‐repeat sugar configuration (Sandlin et al, 1996). In spite of substantial variation in the O‐repeat biochemical composition (Fig 5A), bacterial encasement with hGBP1F was comparable between S. flexneri co‐isogenic rfb mutant strains (Fig 5E). These data indicate that the presence of O‐repeat side chains rather than the specific O‐repeat composition is a critical determinant enabling the stable association of hGBP1F with the bacterial outer membrane.
Binding of hGBP1F to bacteria disrupts the O‐antigen barrier function
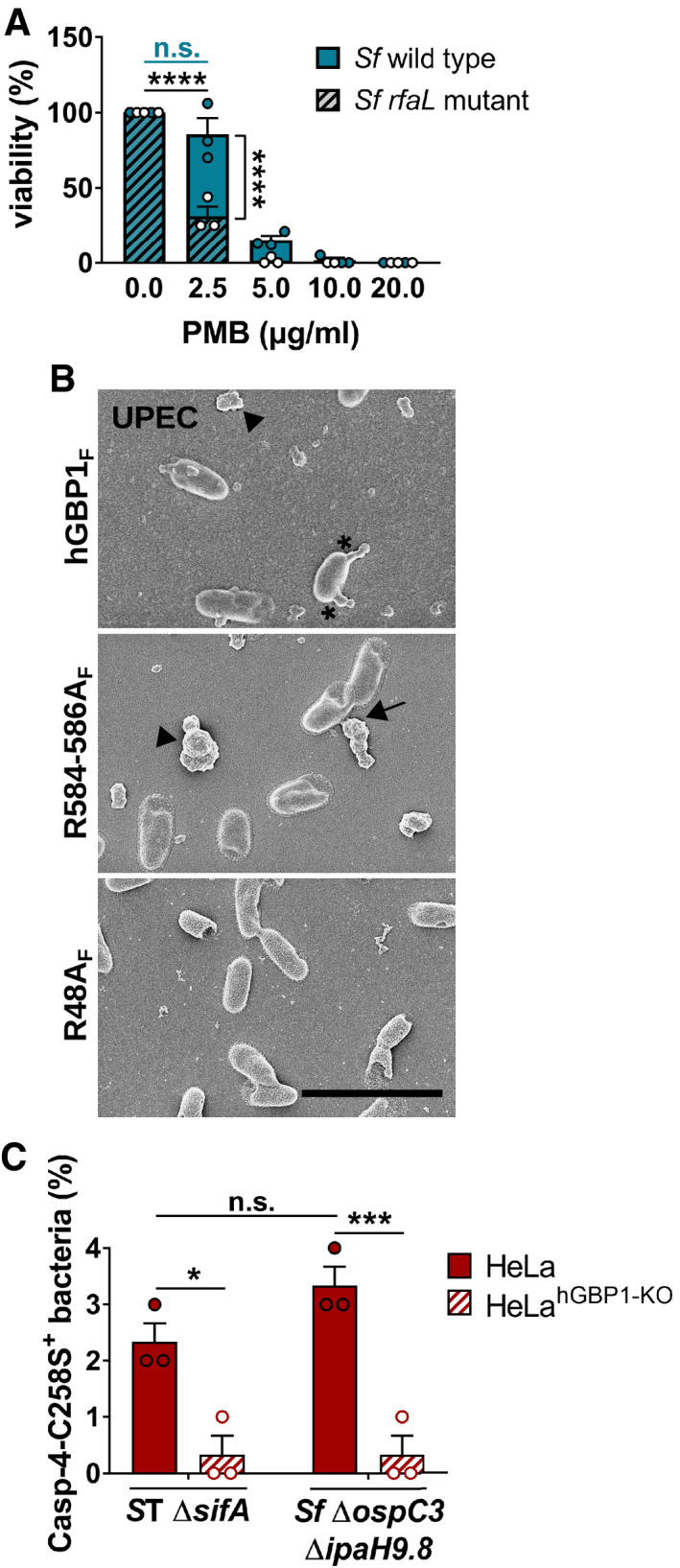
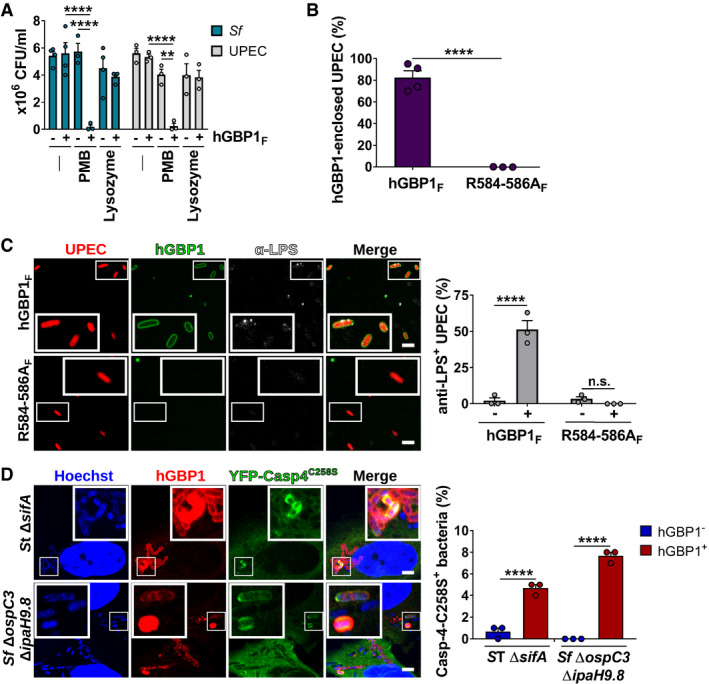
O‐antigen provides a non‐specific physical barrier to various substances including the antibiotic polymyxin B (PMB). PMB is a cationic antimicrobial peptide that binds with high affinity to the negatively charged phosphate groups of lipid A and disrupts bacterial membrane integrity via its hydrophobic tail (Moffatt et al, 2019). In agreement with previous observations made in other gram‐negative bacteria (Berry et al, 2009; Holzer et al, 2009), we found that the O‐antigen‐deficient S. flexneri rfaL mutant was hypersusceptible to PMB (Fig EV4A). Because hGBP1F has LPS‐aggregating properties (Fig 4), we hypothesized that hGBP1F bound to bacterial surfaces could function as a surfactant, dissolving the O‐antigen barrier. In support of this hypothesis, we found that in vitro binding of hGBP1F to wild type S. flexneri resulted in a one‐log reduction in bacterial colony‐forming units (CFUs) counts in the presence of an otherwise sublethal dose of PMB (Figs 6A and EV4A). Polybasic motif‐dependent encasement of UPEC with hGBP1F (Figs 6B and EV4B) similarly reduced bacterial viability (Fig 6A), whereas hGBP1F binding failed to promote bacterial killing by lysozyme (Fig 6A), an enzyme that degrades periplasmic peptidoglycan. These data suggested that hGBP1F binding to the bacterial surface of gram negatives could break down the O‐antigen barrier, thus permitting PMB to penetrate and to bind to lipid A. To test this hypothesis further, we stained UPEC bacteria with an antibody that binds to the inner core and lipid A of E. coli LPS. Incubation with hGBP1F but not the 3R mutant hGBP1F R584‐586A allowed anti‐LPS staining at distinct foci on the bacterial surface (Fig 6C). These data indicated that stable, 3R‐dependent engulfment of bacteria with hGBP1F led to localized disruptions of the O‐antigen barrier and thereby enabled antibody binding to the LPS inner core and lipid A. Lastly and in alignment with related observations made in human monocytes (Fisch et al, 2019), we found that hGBP1 facilitated the intracellular recruitment of the lipid A sensor caspase‐4 to gram‐negative bacteria invading the cytosol of infected HeLa cells (Figs 6D and EV4C). Together, these observations show that the formation of hGBP1F protein coat surrounding gram‐negative bacteria disrupts the O‐antigen barrier function and renders lipid A accessible to antimicrobial molecules and immune sensors alike.
Figure EV4. Lack of O‐antigen renders Shigella flexneri more susceptible to polymyxin B.
-
AFollowing incubation for 45 min with buffer, live S. flexneri wild type and rfaL mutant were treated for 30 min with varying concentrations of polymyxin B (PMB). The number of viable bacteria was subsequently determined by CFU counts. Graphs show mean CFUs ± SEM of combined data from three independent experiments. Significance was determined by two‐way ANOVA with Tukey's multiple comparison test. n.s., not significant; ****P ≤ 0.0001.
-
BScanning electron micrographs of live UPEC incubated for 4 min with 5 μM hGBP1F or equivalent concentration of either hGBP1F R584‐586A or hGBP1F R48A in the presence of 2 mM GTP. Scale bar equals 5 μm. Arrowheads point to unattached hGBP1 polymers, arrows point to hGBP1 polymers attached to bacteria, and asterisks mark polymeric structures that appear to fuse with bacterial surfaces.
-
CIFNγ‐primed wild type and hGBP1‐KO HeLa cells stably expressing YFP‐Caspase‐4C258S were infected with either Salmonella enterica Typhimurium mutant ΔsifA (MOI = 25) or with S. flexneri ΔospC3ΔipaH9.8 (MOI = 6). Cells were fixed at 4 hpi (ST ΔsifA) or at 2 hpi (Sf ΔospC3ΔipaH9.8). Percentage of YFP‐Caspase‐4C258S‐associated bacteria were quantified, and mean ± SEM of combined data from three independent experiments are shown. Significance was determined by two‐way ANOVA with Tukey's multiple comparison test. n.s., not significant; *P ≤ 0.05; ***P ≤ 0.001.
Source data are available online for this figure.
Figure 6. Binding of hGBP1F to bacteria disrupts the O‐antigen barrier function.
-
AFollowing incubation for 1.5 h with buffer in the absence or presence of 10 μM hGBP1F and 5 mM GTP, live bacteria were treated with antibiotics (2.5 μg/ml PMB or 5 mg/ml lysozyme) for 30 min or left untreated. The number of viable bacteria was subsequently determined by colony‐forming unit (CFU) counts. Graphs show mean CFUs ± SEM of combined data from at least three independent experiments. Significance was determined by two‐way ANOVA with Tukey's multiple comparison test. n.s., not significant; ****P ≤ 0.0001.
-
BFormaldehyde‐fixed RFP+ UPEC were mixed with 10 μM Alexa‐Fluor488‐hGBP1F or ‐hGBP1F R584‐586A supplemented with 2 mM GTP. Following 60 min of incubation, hGBP1F‐enclosed UPEC were quantified. Graphs depict mean ± SEM of combined data from three independent experiments. Significance was determined by unpaired t‐test, two‐tailed. ****P ≤ 0.0001.
-
CFormaldehyde‐fixed RFP+ UPEC were mixed with 10 μM Alexa‐Fluor488‐hGBP1F or ‐hGBP1F R584‐586A and 2 mM GTP. Samples were fixed after 60 min of incubation and stained with an antibody against the inner core/lipid A fragment of E. coli LPS (anti‐LPS). The frequency of anti‐LPS stained UPEC was quantified. Graphs show mean ± SEM of combined data from three independent experiments. Significance was determined by two‐way ANOVA with Tukey's multiple comparison test. n.s., not significant; ****P ≤ 0.0001.
-
DIFNγ‐primed wild type and hGBP1‐KO HeLa cells stably expressing inactive mutant YFP‐Caspase‐4C258S were infected with either cytosol‐entering Salmonella enterica Typhimurium mutant ΔsifA (MOI = 25) or with Shigella flexneri ΔospC3ΔipaH9.8 (MOI = 6), a mutant strain lacking Caspase‐4 antagonist OspC3 as well as hGBP1 antagonist IpaH9.8. Cells were fixed at 4 hpi (ST ΔsifA) or at 2 hpi (Sf ΔospC3ΔipaH9.8) and stained for endogenous hGBP1. YFP‐Caspase‐4C258S associated bacteria were quantified, and mean ± SEM of combined data from three independent experiments are shown. Significance was determined by two‐way ANOVA with Tukey's multiple comparison test. ****P ≤ 0.0001.
hGBP1 disrupts polar localization of Shigella IcsA and blocks the recruitment of the host actin polymerization machinery
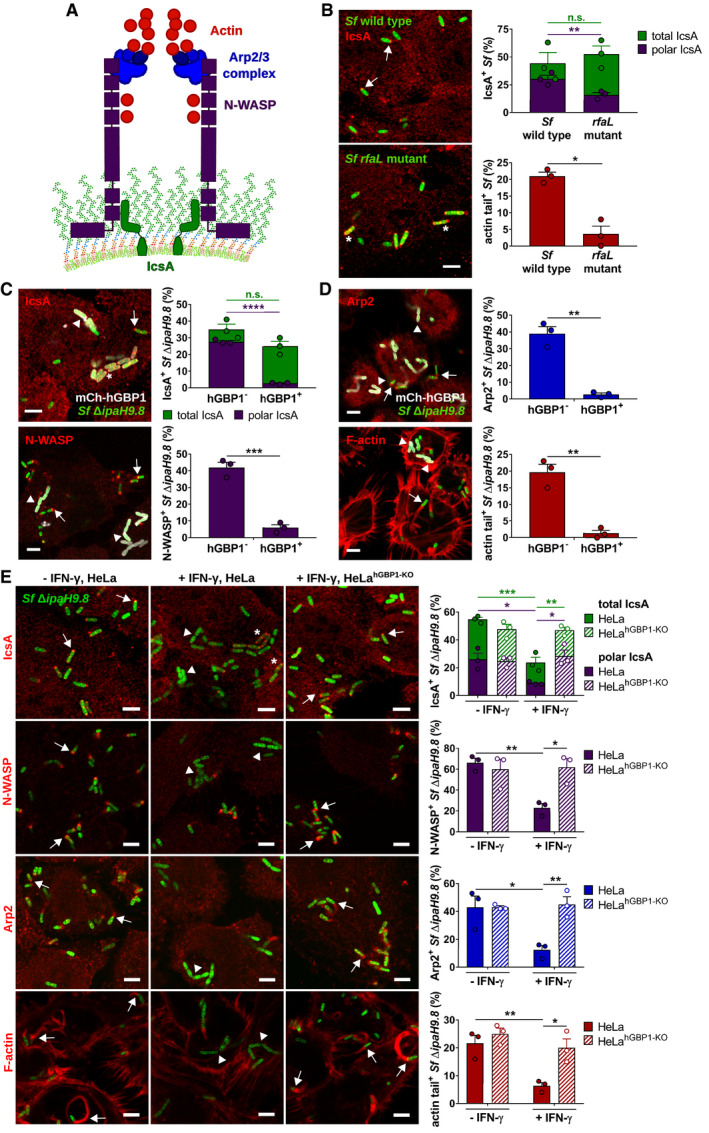
In addition to providing a physical barrier, O‐antigen controls other aspects of bacterial pathogenesis that include the regulation of actin‐based intracytosolic motility: Previous reports demonstrated that S. flexneri mutants lacking O‐antigen are defective for host actin assembly and consequently spread from cell to cell inefficiently (Sandlin et al, 1995, 1996; Hong & Payne, 1997; Van den Bosch et al, 1997). Polar surface expression of the Shigella autotransporter IcsA promotes the efficient bacterial actin‐based motility (Agaisse, 2016) (Fig 7A). We confirmed previous findings (Sandlin et al, 1996) that the rfaL mutant had diminished unipolar IcsA localization and frequently failed to form actin tails in the host cell cytosol (Fig 7B). Although the mechanism by which O‐antigen regulates polar IcsA localization and associated actin assembly has not been fully defined, one compelling model suggests that, following secretion of IcsA at the bacterial pole (Steinhauer et al, 1999; Charles et al, 2001), enhanced membrane fluidity in O‐antigen‐deficient bacteria leads to increased diffusion of IcsA away from the bacterial pole (Robbins et al, 2001). Based on this model as well as our data supporting a role for hGBP1 as an LPS‐binding surfactant (Fig 4), we hypothesized that binding of hGBP1 to bacteria could similarly increase membrane fluidity resulting in increased circumferential localization of IcsA. In support of this hypothesis, we noted an almost complete lack of unipolar IcsA localization and an accompanying dramatic reduction in the polar recruitment of the IcsA‐interaction partner N‐WASP and the actin nucleator Arp2/3 complex in hGBP1‐targeted S. flexneri, as well as the formation of actin tails (Fig 7C and D). The mislocalization of IcsA, the lack of N‐WASP and Arp2/3 recruitment, and actin tail formation were dependent on IFNγ‐induced hGBP1 expression, as evident by the reversal of these phenotypes in hGBP1‐deficient host cells (Fig 7E). Together, these findings not only provide a cellular mechanism for the previously reported hGBP1‐mediated inhibition of actin tail formation (Piro et al, 2017; Wandel et al, 2017) but also imply that hGBP1 exerts several of its diverse antimicrobial functions as a bacteriolytic, pro‐host cell death and antibacterial motility factor through a single molecular activity as a “detergent‐like” disruptor of the bacterial LPS surface layer (Fig EV5).
Figure 7. hGBP1 disrupts polar localization of Shigella IcsA and blocks the recruitment of the host actin polymerization machinery.
-
ASchematic depicts the molecular mechanism by which Shigella flexneri co‐opts the host actin polymerization machinery: The bacterial autotransporter IcsA localizes to one bacterial pole where it recruits and activates host actin nucleation‐promoting factor N‐WASP. N‐WASP then recruits and activates the host actin nucleator Arp2/3 complex to initiate actin polymerization.
-
B–ECells were infected with poly‐d‐lysine treated GFP+ Shigella flexneri strains at an MOI of 6. Cells were stained for indicated proteins, and Z‐stacks were recorded using confocal fluorescence microscopy. Actin tails were classified as tails when ≥ 2.5 μm. All graphs show mean values ± SEM of combined data from three independent experiments. All scale bars are 5 μm. Arrows point to bacteria associated with indicated proteins, arrowheads point to bacteria lacking indicated proteins, and asterisks mark bacteria lacking unipolar IcsA localization. (B) Unprimed HeLa cells were infected with wild type or rfaL S. flexneri. Total and unipolar localization of IcsA as well as the actin tail formation were quantified at 1 hpi. Significance was determined by unpaired two‐tailed t‐tests. n.s., not significant; *P ≤ 0.05; **P ≤ 0.01. (C, D) mCherry‐hGBP1 was expressed in HeLa hGBP1‐KO cells infected with S. flexneri ΔipaH9.8 and stained for IcsA, N‐WASP, Arp2, and F‐actin at 2.5 hpi. Colocalization of hGBP1 and indicated proteins on bacteria was quantified. Significance was determined by unpaired t‐tests, two‐tailed. n.s., not significant; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. (E) IFNγ‐primed and unprimed wild type and hGBP1‐KO HeLa cells were infected with S. flexneri ΔipaH9.8, and subcellular localization of IcsA, N‐WASP, Arp2, and F‐actin was assessed and quantified at 2.5 hpi. Significance was determined by two‐way ANOVA with Tukey's multiple comparison test. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Source data are available online for this figure.
Figure EV5. Model: hGBP1 acts as an LPS‐binding surfactant and thereby exerts pleiotropic effects on the functionality of gram‐negative bacterial cell envelopes.
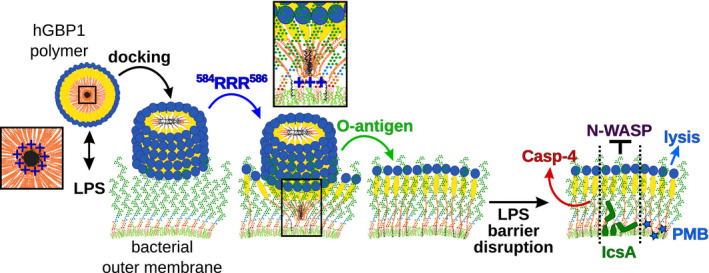
Model: Polymerizing hGBP1 binds to LPS and docks to the surface of gram‐negative bacteria. The C‐termini of polymerized hGBP1 molecules contain three arginines (584RRR586, 3R stretch) which form a positively charged ring around the polymer's hydrophobic core. This positively charged ring promotes stable binding of hGBP1 polymers to the bacterial surface through two non‐mutually exclusive mechanisms: (i) attractive electrostatic interactions between hGBP1's C‐terminal 3R stretch and negative charges on the surface of the bacterial outer membrane (phosphate groups present at the inner core and lipid A segments of LPS) pull the polymer's hydrophobic core toward the bacterial membrane; and (ii) accelerated depolymerization releases the farnesyl tails from the polymer's hydrophobic core to anchor farnesylated hGBP1 inside the outer leaflet of bacterial outer membranes. The depolymerization process leads to the formation of a seemingly uniform hGBP1 protein coat encasing entire bacteria. The transition from docked hGBP1 polymers into a stable hGBP1 protein coat is dependent on O‐antigen, the outer polysaccharide structure of bacterial LPS. We propose that interactions with O‐antigen maintain individual hGBP1 molecules in their outstretched conformation, as these proteins become anchored inside the bacterial outer membrane via their farnesyl tails. Inserted hGBP1 polymers as well as embedded individual hGBP1 proteins act as surfactants and disrupt LPS barrier functions. By clustering LPS molecules together, hGBP1 exerts pleiotropic effects on the functionality of gram‐negative bacterial cell envelopes: Binding of hGBP1 to the bacterial surface unmasks lipid A for attack by caspase‐4 or the antimicrobial peptide polymyxin B. The “detergent‐like” activity of hGBP1 further disturbs the polar localization and O‐antigen‐dependent function of the Shigella virulence protein IcsA, thereby interfering with the pathogen's ability to form actin tails required for bacterial dissemination.
Discussion
Intracellular defense programs are indispensable for effective host immunity. A critical catalyst for the recognition, entrapment, and elimination of intracellular gram‐negative bacteria is hGBP1, which was shown to facilitate bacterial killing (Tietzel et al, 2009; Al‐Zeer et al, 2013; Li et al, 2017; Liu et al, 2018), to promote activation of the LPS sensor caspase‐4 (Lagrange et al, 2018; Fisch et al, 2019), and to block actin‐based bacterial motility (Piro et al, 2017; Wandel et al, 2017). The molecular mechanism by which hGBP1 or its murine homologs execute such distinct cellular functions has remained an enigma. Here, we provide evidence that hGBP1 functions as an LPS‐binding and LPS‐clustering surfactant that disrupts the physicochemical properties of the outward‐facing LPS layer, thereby promoting the recruitment of the lipid A sensor caspase‐4 to the bacterial surface and also enhancing the efficacy of the lipid A‐targeting antimicrobial PMB. In further support of the “surfactant model”, we demonstrate that binding of hGBP1 to S. flexneri leads to increased circumferential rather than unipolar localization of the outer membrane protein IcsA, likely due to increased outer membrane fluidity (Herrmann et al, 2015) in the presence of “detergent‐like” hGBP1. Our studies therefore provide a novel conceptual framework to define the molecular mechanisms that underlie the antibacterial activities of hGBP1 and likely of related GBP family members in other host species.
How does hGBP1 function as a surfactant? The core of hGBP1 polymers consists of hydrophobic farnesyl groups extending from the positively charged protein C‐termini (Shydlovskyi et al, 2017). Therefore, the hGBP1 polymer core is predicted to provide amphiphilic properties typical of surfactants. Although we characterized the molecular mechanisms of hGBP1 polymerization previously (Shydlovskyi et al, 2017; Sistemich et al, 2020), we had not yet reported its biological function prior to this current work. The present studies assign such a biological function to hGBP1 polymerization by demonstrating that this process is essential for hGBP1 binding to LPS and attachment to the surface of gram‐negative bacteria. We further show that hGBP1 polymerization kinetics and GTP hydrolysis rates are accelerated in the presence of LPS, and thus have identified LPS as its physiological lipid template. While the precise structure of LPS‐bound hGBP1 polymers will require further investigation, we can already conclude that both the biochemical features of the hGBP1 polymer core as well as our in vitro binding studies are consistent with the proposed “surfactant model”.
Following the initial attachment of polymeric hGBP1 structures to the bacterial surface, the polymers seemingly disassemble and transition into a uniform hGBP1 protein coat completely encasing gram‐negative bacteria. At this time, we can only speculate how individual hGBP1 molecules are arranged within this hGBP1 protein coat: We predict that the farnesyl tail is released from the core of hGBP1 polymers and inserted into the outer leaflet of the bacterial outer membrane (Fig EV5). We further predict that the C‐terminal polybasic motif of hGBP1 undergoes attractive electrostatic interactions with phosphate groups attached to lipid A and the LPS inner core (Simpson & Trent, 2019), thus explaining at least in part the requirement for the polybasic motif to form a stable hGBP1 coat on the bacterial surface. Lastly, we conjecture that hGBP1 molecules maintain the outstretched conformation of their polymeric state (Shydlovskyi et al, 2017; Sistemich et al, 2020) and intersperse with the O‐antigen segment of LPS, thereby disrupting O‐antigen‐mediated LPS‐LPS interactions that contribute to outer membrane stiffness (Rojas et al, 2018) and diminishing the O‐antigen barrier function.
As the exposed surface structure of many gram‐negative envelopes, O‐antigen is a direct and common target of host immune responses. Because the O‐antigen segment of LPS is highly variable between bacterial serotypes and species, the adaptive immune system detects an extensive O‐antigen repertoire via highly specific antibodies, whereas the innate immune system can only detect subsets of O‐antigens via genome‐encoded lectins (Mey et al, 1996; Kawabata & Iwanaga, 1999; Lerouge & Vanderleyden, 2002; Wesener et al, 2015, 2017). Unexpectedly, our data indicate that the innate immune protein hGBP1 detects the presence of O‐antigen on the bacterial surface rather than a specific biochemical configuration of O‐antigen. Insights into the underlying molecular mechanism can be derived from our findings that hGBP1 polymers bind to the bacterial surface in an O‐antigen‐independent manner but require the presence of O‐antigen to effectively transition from the bacteria‐bound polymeric state into a bacteria‐encapsulating protein coat. We propose that hGBP1 polymers depolymerize at their bacteria‐attached ends to insert individual hGBP1 molecules into the bacterial membrane (Fig EV5) and, further, that interactions between O‐antigen segments and individual hGBP1 molecules support this process, for example by maintaining individual hGBP1 proteins in their outstretched confirmation.
Our studies also reveal a critical role for the C‐terminal polybasic motif of hGBP1 in promoting depolymerization, a process self‐evidently essential for the transition from polymers into a non‐polymeric protein coat. Whereas much additional work is required to characterize this transition process in detail, data presented here demonstrate that hGBP1 evolved a complex, multi‐step mechanism to form a tightly associated protein coat on the surface of gram‐negative bacteria. The specificity of this process is founded in the ability of hGBP1 to recognize surface‐exposed smooth LPS as a unique molecular pattern that consists of polysaccharides attached to a negatively charged lipid membrane. While our studies identify LPS as the primary bacterial pattern that facilitates the stable binding of hGBP1 to the surface of gram‐negative bacteria, we cannot exclude that hGBP1 also recognizes other microbial molecules that may aid in its recruitment to gram‐negative bacteria or facilitate its targeting to other classes of intracellular microbial pathogens. In addition, hGBP1 may also recognize host damage‐associated molecular patterns. In support of this hypothesis, we previously demonstrated that hGBP1 translocates to sterilely damaged endosomes (Feeley et al, 2017; Piro et al, 2017) The recognition of cytosolically exposed host sugars—normally confined to the luminal side of intact vesicles—is also likely to be involved in the recruitment of hGBP1 to bacteria‐containing vacuoles (Feeley et al, 2017). Similarly, albeit speculative at this point, low‐affinity but broad specificity interactions of hGBP1 polymers with the highly variable sugars that constitute the outer envelope of gram‐negative bacteria may mediate the initial docking reaction of the polymer to the bacterial surface.
We propose that hGBP1 acts as an LPS‐binding surfactant. This model gives rise to several testable hypotheses. For instance, GBPs promote the lytic destruction of cytosolic bacteria in macrophages but not in epithelial cells (Man et al, 2015; Meunier et al, 2015; Li et al, 2017; Piro et al, 2017; Wandel et al, 2017; Liu et al, 2018). We demonstrate here that binding alone of hGBP1 to gram‐negative bacteria has neither bactericidal nor bacteriostatic effects but instead renders bacteria more susceptible to the antimicrobial peptide PMB. We therefore propose that GBP‐dependent bacteriolysis in macrophages is mediated by the synergistic activity of bacteriolytic proteins robustly expressed in the cytosol of macrophages but not of epithelial cells. GBPs were also shown to accelerate caspase‐4 activation in response to smooth as well as rough LPS delivered into the host cell cytoplasm (Pilla et al, 2014; Finethy et al, 2015; Lagrange et al, 2018; Santos et al, 2018). Our current study demonstrates that hGBP1 can aggregate both smooth and rough LPS in vitro. We therefore propose that hGBP1 and potentially other GBPs interact with LPS in the host cell cytoplasm to form high molecular weight LPS aggregates, the predicted physiological binding substrate of caspase‐4 (Wacker et al, 2017; An et al, 2019). Lastly, this work shows that hGBP1 disrupts the function of the Shigella outer membrane protein IcsA, providing for the first time a molecular model for the previously reported hGBP1‐mediated inhibition of actin‐based motility (Piro et al, 2017; Wandel et al, 2017). We predict that incorporation of hGBP1 into bacterial outer membranes may impact the function of additional bacterial outer membrane proteins that, similar to IcsA, are sensitive to changes in membrane fluidity.
Previous studies had already implicated a potential role for GBPs in direct LPS sensing. We showed that mouse GBPs accelerate the kinetics of caspase‐4 activation in response to LPS transfection of murine bone marrow‐derived macrophages (Pilla et al, 2014), and a later study expanded these observations to human GBPs (Lagrange et al, 2018). In follow‐up work, we demonstrated that mouse GBPs promote caspase‐4 activation in response to bacterial outer membrane vesicles (OMVs)—LPS‐studded vesicles released by bacteria. We found that mouse Gbp2, the closest murine ortholog of hGBP1, colocalized with LPS inside OMV‐treated macrophages (Finethy et al, 2017), arguing in favor of direct interaction between mouse Gbp2 and LPS. A separate study showed that mouse Gbp5 colocalized with transfected LPS in macrophages (Santos et al, 2018). Collectively, these studies gave rise to the hypothesis that one or more GBPs could directly sense LPS. In support of this hypothesis, our study identified hGBP1 as a bona fide LPS receptor.
The mammalian host cell cytosol is therefore surveyed by at least two distinct classes of LPS sensors: hGBP1 and caspase‐4/‐5. These two sensors operate synergistically: Whereas caspase‐4 binds to LPS aggregates (Shi et al, 2014; Wacker et al, 2017; An et al, 2019), our studies demonstrate that hGBP1 initially interacts with LPS micelles and then generates LPS aggregates that are much larger in size than the ones self‐assembling in aqueous solution. Future studies will need to determine the structural configuration of the hGBP1‐LPS complex, yet it is intriguing to speculate that hGBP1 disrupts the integrity of LPS micelles, as it does with the bacterial outer membrane, and consequentially binds to monomeric LPS molecules as the LPS‐hGBP1 complex is being formed. This disruption of LPS micelles and LPS‐containing bacterial membranes may play an important role in the activation of the non‐canonical inflammasome. Indeed, confirming previous observations (Fisch et al, 2019), we show that hGBP1 facilitates the recruitment of caspase‐4 to the bacterial surface; a process that we propose is mediated by hGBP1‐driven partial solubilization of the LPS layer. By the same principal mechanism, we expect that hGBP1 renders gram‐negative bacteria more susceptible to a range of antimicrobials, as already shown here for the antibiotic PMB. Therefore, interventions that promote hGBP1 binding to bacterial outer membranes, e.g., in the case of Shigella infections through the inhibition of the bacterial hGBP1 antagonist IpaH9.8 (Li et al, 2017; Piro et al, 2017; Wandel et al, 2017), could provide therapeutic benefits that include improved efficacies of select antibiotics.
Materials and Methods
Reagents and Tools table
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti‐E. coli J5 LPS | RayBiotech | DS‐MB‐01267 |
| Rabbit monoclonal anti‐hGBP1 | Abcam | ab131255 |
| Rabbit polyclonal anti‐IcsA | Goldberg et al (1993) | N/A |
| Rabbit monoclonal anti‐N‐WASP | Cell Signaling | 4848 |
| Mouse monoclonal anti‐Arp2 | Abcam | ab49674 |
| Alexa‐Fluor 568‐conjugated goat anti‐mouse IgG | Thermo Fisher Scientific | A11004 |
| Alexa‐Fluor 660‐conjugated goat anti‐mouse IgG | Thermo Fisher Scientific | A21054 |
| Alexa‐Fluor 568‐conjugated donkey anti‐rabbit IgG | Thermo Fisher Scientific | A10042 |
| Alexa‐Fluor 660‐conjugated goat anti‐rabbit IgG | Thermo Fisher Scientific | A21073 |
| Bacterial and virus strains | ||
| BL21‐CodonPlus (DE3)‐RIL | Stratagene | N/A |
| Rosetta (DE3) pLysS | Stratagene | N/A |
| Veggie NovaBlue Singles (Novagen) | Millipore Sigma | 71251 |
| pInducer‐mCherry‐hGBP1 | Piro et al (2017) | N/A |
| pMX‐CMV‐YFP‐CASP4C258S | Fisch et al (2019) | N/A |
| Shigella flexneri 2457T | Sandlin et al (1995) | N/A |
| Shigella flexneri 2457T pEGFPmut2 | Piro et al (2017) | N/A |
| Shigella flexneri 2457T rfaL mutant | Kohler et al (2002) | BS520 |
| Shigella flexneri 2457T rfaL mutant pEGFPmut2 | Piro et al (2017) | N/A |
| Shigella flexneri 2457T rfbO8 mutant | Sandlin et al (1996) | BS515 |
| Shigella flexneri 2457T rfbO8 mutant pEGFPmut2 | This study | N/A |
| Shigella flexneri 2457T rfbO25 mutant | Sandlin et al (1996) | BS525 |
| Shigella flexneri 2457T rfbO25 mutant pEGFPmut2 | This study | N/A |
| Shigella flexneri 2457T ΔipaH9.8 | Piro et al (2017) | N/A |
| Shigella flexneri 2457T ΔipaH9.8 pEGFPmut2 | Piro et al (2017) | N/A |
| Shigella flexneri 2457T ΔospC3ΔipaH9.8 | This study | N/A |
| Salmonella enterica Typhimurium SL1344 pGFP | Valdivia and Falkow (1997) | N/A |
| Salmonella enterica Typhimurium 14028s StrR phoN::Tn10dCm ΔsifA | Freeman et al (2003) | N/A |
| Uropathogenic Escherichia coli CFT073 pLRFP‐C1 | Welch et al (2002) | N/A |
| Uropathogenic Escherichia coli CI5 pLRFP‐C1 | Abraham et al (1985); Song et al (2009) | N/A |
| Legionella pneumophila serogroup 1 strain LP01 rpsL pMMB207‐dsRed | Feeley et al (2017) | N/A |
| Listeria monocytogenes 10403S DH‐L1039 | Shen and Higgins (2005) | N/A |
| Staphylococcus aureus RN4220 pSRFPS1 | Rodriguez et al (2017) | N/A |
| Escherichia coli DH10B pEGFPmut2 | This study | N/A |
| Escherichia coli DH5α pEGFPmut2 | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Alexa‐Fluor 660 Phalloidin | Thermo Fisher Scientific | A22285 |
| Farnesyl pyrophosphate (FPP) | Cayman Chemical | 63250 |
| Isopropyl β‐d‐1‐thiogalactopyranoside (IPTG) | Carl Roth | CN08.4 |
| Phenylmethylsulfonyl fluoride (PMSF) | Carl Roth | 6367.2 |
| Guanosine triphosphate (GTP) solution | Thermo Fisher Scientific and Jena Bioscience | R0461 and NU‐1012 |
| Guanosine diphosphate (GDP) | Abcam and Jena Bioscience | ab146529 and NU‐1172 |
| Guanosine 5′‐O‐[gamma‐thio]triphosphate (GTPγS) | Abcam and Jena Bioscience | ab146662 and NU‐412 |
| 5′‐Guanylyl imidodiphosphate (GppNHp) | Abcam and Jena Bioscience | ab146659 and NU‐401 |
| Alexa‐Fluor 488 C5 Maleimide (Alexa‐Fluor488) | Thermo Fisher Scientific | A10254 |
| Alexa‐Fluor 647 C2 Maleimide (Alexa‐Fluor647) | Thermo Fisher Scientific | A20347 |
| Lipopolysaccharides from Escherichia coli Serotype 055:B5 (LPS‐O55:B5), Alexa‐Fluor 488 Conjugate | Thermo Fisher Scientific | L23351 |
| Lipopolysaccharides from Salmonella Minnesota (LPS‐SM), Alexa‐Fluor 488 Conjugate | Thermo Fisher Scientific | L23356 |
| Polymyxin B (PMB) solution | Millipore Sigma | 81271 |
| Lysozyme | Thermo Fisher Scientific | 89833 |
| Lipopolysaccharides from E. coli O55:B5 (LPS‐O55:B5) | Invivogen | tlrl‐pb5lps |
| Lipopolysaccharides from E. coli O111:B4 (LPS‐O111:B4) | Invivogen | tlrl‐eblps |
| Whole glucan particles (WGP) from S. cerevisiae | Invivogen | tlrl‐wgps |
| Brain Polar Lipid (BPL) Extract (porcine) | Avanti Polar Lipids | 141101 |
| Dulbecco's Modified Eagle Medium (Gibco) | Thermo Fisher Scientific | 11995040 |
| Fetal Bovine Serum (FBS) | Corning and Omega | 35‐010‐CV and FB‐01 |
| Non‐essential amino acid (NEAA, Gibco) | Thermo Fisher Scientific | 11140050 |
| β‐mercaptoethanol (β‐ME, Gibco) | Thermo Fisher Scientific | 21985023 |
| HisPur Cobalt resin | Thermo Fisher Scientific | 89965 |
| 16:0 Liss Rhod PE (Rho‐PE) | Avanti Polar Lipids | 810158 |
| KOD Hot Start DNA Polymerase | Millipore Sigma | 71086 |
| Dithiothreitol (DTT) | Carl Roth | 6908.2 |
| Nitrocellulose membrane, 0.2 μm | Bio‐Rad | 1620112 |
| Tris(2‐carboxyethyl)phosphine hydrochloride (TCEP‐HCl) | Thermo Fisher Scientific | 20490 |
| QIAprep Spin Miniprep Kit | Qiagen | 27104 |
| 1 × HBSS | Thermo Fisher Scientific | 14025092 |
| 1 × PBS | Thermo Fisher Scientific | 10010023 |
| Poly‐d‐Lysine hydrobromide | Millipore Sigma | P6407 |
| Interferonγ (IFNγ) | Millipore Sigma | IF002 |
| Anhydrotetracycline (aTc) | Takara | 631310 |
| Mowiol 4‐88 | Millipore Sigma | 81381 |
| p‐Phenylenediamine (PPD) | Millipore Sigma | P1519 |
| Experimental models: cell lines | ||
| HeLa cells with inserted Cas9 gen (HeLa wild type cells) | Piro et al (2017) | N/A |
| hGBP1‐deficient HeLa cells (HeLa hGBP1‐KO cells) | Piro et al (2017) | N/A |
| hGBP1‐deficient HeLa cells pInducer‐mCherry‐hGBP1 | Piro et al (2017) | N/A |
| Oligonucleotides | ||
| K51A‐F: GGC CTC TAC CGC ACA GGC GCA TCC TAC CTG ATG AAC AAG C | This study | N/A |
| K51A‐R: GCT TGT TCA TCA GGT AGG ATG CGC CTG TGC GGT AGA GGC C | This study | N/A |
| Q577A‐F: GC AGA ATA ATG AAA AAT GAG ATA TGC GAT CTC CAG ACG AAA ATG AGA C | This study | N/A |
| Q577A‐R: C GTC TCA TTT TCG TCT GGA GAT CGC ATA TCT CAT TTT TCA TTA TTC TG | This study | N/A |
| R584‐586A‐F: C CAG ACG AAA ATG GCA GCG GCC AAG GCA TGT ACC ATA AGC | This study | N/A |
| R584‐586A‐R: GCT TAT GGT ACA TGC CTT GGC CGC TGC CAT TTT CGT CTG G | This study | N/A |
| Recombinant DNA | ||
| pRSF‐Duet1‐His6‐FTase | Fres et al (2010) | N/A |
| pQE‐80L‐His6‐hGBP1 | Ince et al (2017) | N/A |
| pQE‐80L‐His6‐hGBP1‐R48A‐Q577C | This study | N/A |
| pQE‐80L‐His6‐hGBP1‐K51A | This study | N/A |
| pQE‐80L‐His6‐hGBP1‐H74A‐Q577C | This study | N/A |
| pQE‐80L‐His6‐hGBP1‐R584‐586A | This study | N/A |
| pEGFPmut2 | Cormack et al (1996) | N/A |
| Software and algorithms | ||
| ChromPass HPLC software 1.8.6.1 | Jasco | |
| Fiji | Schindelin et al (2012) | https://imagej.net/Fiji |
| Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| PyMOL 1.7.2.1 | Schrödinger | https://pymol.org |
| Other | ||
| HiLoad 26/60 Superdex 200 prep grade | GE Healthcare Life Sciences | 28989336 |
| HiPrep Butyl FF 16/10 | GE Healthcare Life Sciences | 28‐9365‐47 |
| Ultra‐filtration column Vivaspin 20, 10,000 MWCO, PES | Sartorius | VS2002 |
| Ultra‐filtration column Vivaspin Turbo 4, 10,000 MWCO, PES | Sartorius | VS04T01 |
| Glass bottom microwell dish | MatTek | P35G‐1.5‐10‐C |
Methods and Protocols
Materials availability
There are no restrictions on the availability of materials and reagents mentioned in this work.
Experimental model and subject details
Cell lines
Parental (wild type) and previously described hGBP1‐deficient (hGBP1‐KO) HeLa cells (Piro et al, 2017) were cultivated in Dulbecco's Modified Eagle Medium supplemented with 10% FBS, 1% NEAA, and 55 μM β‐ME at 37°C and 5% CO2. HeLa hGBP1‐KO cells were stably transduced with an anhydrotetracycline (aTc)‐inducible gene expression systems to drive the expression of mCherry‐hGBP1. Wild type and hGBP1‐KO HeLa cells were stably transduced with pMX‐CMV‐YFP‐CASP4C258S (Fisch et al, 2019) to drive the expression of YFP‐caspase‐4‐C258S. All cell lines were screened regularly for mycoplasma contamination.
Bacterial strains and bacterial culture conditions
Bacterial strains were grown on tryptic soy broth (TSB) agar supplemented with Congo Red (S. flexneri strains), TSB agar (S. aureus), Luria Broth (LB)‐Miller agar (E. coli strains, S. enterica Typhimurium strains), or brain heart infusion (BHI) agar (L. monocytogenes), supplemented with antibiotics, as needed (50 μg/ml carbenicillin, 30 μg/ml kanamycin, 34 μg/ml chloramphenicol, 100 μg/ml streptomycin, 10 μg/ml trimethoprim) for 12–16 h at 37°C. L. pneumophila was grown on N‐(2‐Acetamido)‐2‐aminoethanesulfonic acid (ACES)‐buffered charcoal‐yeast extract agar supplemented with 150 μg/ml FeNO3, 400 μg/ml cysteine, 10 μg/ml chloramphenicol, and 100 μg/ml streptomycin for 2–3 days. For in vitro binding assays, electron microscopy, viability assays, and infections, bacteria were cultivated overnight at 37°C and 250 rpm in respective broth cultures. Overnight cultures of S. flexneri strains, S. aureus, E. coli strains, S. enterica Typhimurium (supplemented with 10 mM MgCl2), and L. monocytogenes were diluted 1:30 in fresh broth and grown over 1–2.5 h to an optical density at 600 nm (OD600) of 0.3–0.7. Overnight cultures of L. pneumophila with an OD600 of 2–3 were used without further dilution. For infection studies, overnight cultures of S. flexneri and S. enterica Typhimurium ΔsifA were diluted 1:30 in fresh broth and were grown either for 1–1.5 h to an OD600 of 0.3–0.7 (S. flexneri strains) or for 2.5 h to an OD600 of 1.6–2 (S. enterica Typhimurium ΔsifA). Bacteria were harvested at 2,400 × g for 3 min, washed once with 1 × PBS, and were either treated with 100 ng/ml poly‐d‐lysine in 1 × PBS for 15 min (S. flexneri wild type, rfaL mutant, and ΔipaH9.8) or left untreated and used directly for infection (S. flexneri ΔospC3ΔipaH9.8, S. enterica Typhimurium ΔsifA).
Method details
Shigella flexneri strain construction
To transform S. flexneri with plasmids, bacterial strains were prepared for electroporation as described (Warren, 2011). Briefly, overnight bacterial cultures were diluted 1:100 in a volume of 200 ml modified SOB media (2% Bacto Tryptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, pH 7.0) and grown to an OD600 of 0.4–0.6. Bacterial cultures were centrifuged at 2,000 × g for 15 min at 4°C. Bacteria were resuspended in 50 ml ice‐cold 4% glycerol/1.5% mannitol, subjected to density gradient centrifugation and then resuspended in 200 μl of ice‐cold 4% glycerol/1.5% mannitol. Bacteria were transformed with pEGFPmut2 (Cormack et al, 1996). Strain ΔipaH9.8 was previously reported (Piro et al, 2017). To generate ΔospC3ΔipaH9.8, the KANR cassette from ∆ipaH9.8::FRT‐KANR‐FRT (Piro et al, 2017) was removed by expressing Flp recombinase via cell transfection with plasmid pCP20 (Cherepanov & Wackernagel, 1995). Next, the FRT::KANR::FRT from ∆ospC3::FRT‐KANR‐FRT (Mou et al, 2018) was PCR‐amplified and introduced into ∆ipaH9.8 using the lambda red recombination system (Datsenko & Wanner, 2000).
Bacterial strains for cloning, mutagenesis and recombinant protein expression
Escherichia coli strains Veggie NovaBlue Singles, BL21 CodonPlus (DE3) RIL, and Rosetta (DE3) pLysS were grown on LB‐Miller agar supplemented with 100 μg/ml ampicillin overnight at 37°C. For plasmid purification E. coli strains were cultivated in LB‐Miller broth overnight at 37°C in a shaker at 250 rpm.
Site‐directed mutagenesis
Point mutations were introduced into pQE‐80L‐His6‐hGBP1 using QuikChange site‐directed mutagenesis with KOD Hot Start high‐fidelity DNA Polymerase and oligonucleotides introducing the desired mutations. Following mutagenesis, plasmids were transformed into chemically competent E. coli Veggie NovaBlue Singles and purified with Qiaprep spin miniprep columns. Mutations were verified by DNA Sanger sequencing with a 3130×l sequencer (Applied Biosystems).
Protein expression, purification, and farnesylation
Recombinant hGBP1 wild type and variants were expressed, purified, and farnesylated essentially as described previously (Ince et al, 2017; Sistemich et al, 2020). N‐terminally His6‐tagged hGBP1 wild type protein and point mutants were expressed from bacterial vector pQE‐80L in E. coli strain BL21 CodonPlus (DE3) RIL. N‐terminal His6‐tagged farnesyltransferase (FTase) was expressed in E. coli strain Rosetta (DE3) pLysS from pRSF‐Duet1 vector. Bacteria were cultivated in terrific broth media (supplemented with 0.2 mM ZnCl2 for FTase expression) and grown at 37°C and 90 rpm to an OD600 of 0.4–0.6. The temperature was decreased to either 20°C (hGBP1 constructs) or to 25°C (FTase), and protein expression was induced with 100 μM IPTG. For FTase expression, 0.5 mM ZnCl2 was added to the culture. Bacteria were harvested after 16–18 h at 1,960 g for 15 min at 4°C (Sorvall LYNX 6000 centrifuge, F9‐6x1000 LEX rotor, Thermo Fisher Scientific).
Buffer compositions for the purification of recombinant hGBP1 and FTase only differed in the use of 50 mM HEPES, pH 7.8 (for FTase) instead of 50 mM Tris–HCl, pH 7.9 (for hGBP1). Harvested bacteria were resuspended in buffer A (50 mM Tris–HCl, pH 7.9, 500 mM NaCl and 5 mM MgCl2) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) and lysed by sonication (Ultrasonic homogenizer Sonoplus HD 2200, Bandelin). Cell debris was removed by centrifugation at 34,310 × g and 4°C for 45–60 min (Sorvall LYNX 6000 centrifuge, F21‐8x50y rotor, Thermo Fisher Scientific). Supernatant containing soluble fractions of hGBP1 constructs or FTase were purified by immobilized metal affinity chromatography (IMAC) exploiting the N‐terminal His6‐tags of the proteins followed by size‐exclusion chromatography (SEC) to remove non‐specific protein aggregates. IMAC and SEC columns were connected to ÄKTA Purifier or ÄKTA Prime systems (GE Healthcare Life Sciences). Following loading of soluble proteins, IMAC column (packed with 30 ml HisPur Cobalt Resin) was sequentially washed with 5–10 column volumes (CVs) buffer A and 3–4 CVs buffer B10 (50 mM Tris–HCl, pH 7.9, 150 mM NaCl, 5 mM MgCl2, and 10 mM imidazole). Protein was eluted with 2 CVs buffer B150 (50 mM Tris–HCl, pH 7.9, 150 mM NaCl, 5 mM MgCl2, and 150 mM imidazole). hGBP1 containing fractions from IMAC were pooled and precipitated by adding 3 M (NH4)2SO4. (NH4)2SO4 protein precipitates were dissolved in buffer C (50 mM Tris–HCl, pH 7.9, 150 mM NaCl, and 5 mM MgCl2) and loaded on a buffer C‐equilibrated SEC column (Superdex 200 26/60, 320 ml) to remove (NH4)2SO4 from purified protein. FTase containing fractions from IMAC were pooled, concentrated via ultra‐filtration using Vivaspin 20 centrifugal columns, and loaded on the SEC column to isolate monomeric protein.
Monomeric hGBP1 constructs were incubated for 16 h at 4°C in a glass vial with farnesyl pyrophosphate (FPP) and FTase in buffer D (50 mM Tris–HCl, pH 7.9, 5 mM MgCl2, 150 mM NaCl, 10 μM ZnCl2). The ratio of hGBP1:FPP:FTase was 1:2.5:0.02. The reaction mixture was supplemented with (NH4)2SO4 to a final concentration of 1.25 M and loaded on a hydrophobic interaction chromatography (HIC) column (Butyl FF 16/10, 20 ml), previously equilibrated with buffer E (50 mM Tris–HCl, pH 7.9, 5 mM MgCl2, 1.2 M (NH4)2SO4). Following loading, the HIC column was sequentially washed with 2 CVs buffer E and with 2 CVs of buffer E with its initial (NH4)2SO4 concentration decreased to 60%. Farnesylated protein was separated from non‐farnesylated protein by decreasing the (NH4)2SO4 concentration further in a continuous gradient over 3 CVs from 60% to 45% (NH4)2SO4 (elution of farnesylated hGBP1) followed by a continuous gradient over 3.75 CVs from 45% to 25% (NH4)2SO4 (elution of non‐farnesylated hGBP1). Fractions with farnesylated hGBP1 were pooled, concentrated via ultra‐filtration using Vivaspin 20 centrifugal columns and further purified by SEC to isolate monomeric protein. Following the SEC purification of monomeric target proteins (FTase, farnesylated and non‐farnesylated hGBP1), protein preparations were concentrated via ultra‐filtration using Vivaspin 20 columns, frozen in liquid nitrogen, and stored at −80°C. Concentrations of proteins were calculated according to Lambert–Beer law, using absorption at 280 nm in buffer C and respective molar absorption coefficients (FTase 157,110 M−1cm−1, hGBP1 45,840 M−1cm−1).
Labeling of proteins with fluorescent dyes
After exchanging buffer C to buffer G (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, and 5 mM MgCl2), proteins were incubated with Alexa‐Fluor maleimide dyes on ice for 10–15 min. The molar ratio of protein:dye was 1:1. Labeling reactions were stopped by changing buffer G to buffer C supplemented with 2 mM DTT via ultra‐filtration using Vivaspin Turbo 4 centrifugal columns. Concentrations of proteins and labeling efficiencies were calculated according to Lambert–Beer law, using absorptions at 280 nm, 491 nm, and 651 nm in buffer C, respective molar absorption coefficients (hGBP1 45,840 M−1cm−1, Alexa‐Fluor488 71,000 M−1cm−1, Alexa‐Fluor647 268,000 M−1cm−1), and correction factors for fluorescent dyes (Alexa‐Fluor488 0.11, Alexa‐Fluor647 0.03). Cysteine Q577C was introduced into pQE‐80L‐His6‐hGBP1‐R48A and ‐H74A for efficient labeling of hGBP1‐R48AF and ‐H74AF. Labeling efficiencies for Alexa‐Fluor488‐labeled proteins ranged from 46% to 63%. Labeling efficiencies for Alexa‐Fluor647‐labeled proteins ranged from 10% to 41%.
Absorbance‐based polymerization assay and analysis of nucleotide composition
Absorbance‐based measurements were performed with a Specord200 UV/Vis spectrophotometer (Analytik Jena) as described previously (Shydlovskyi et al, 2017). Proteins were diluted in buffer C supplemented with 50 μM BSA and incubated for 5 min in a temperature‐controlled cuvette at 25°C. Nucleotides were injected into the cuvette at final concentrations of 1 mM (GTP, GppNHp, GTPγS) or 250 μM (GDP·AlFX). Polymerization of hGBP1 was followed as absorbance signal at 350 nm over time. In experiments with GTP, the nucleotide composition of the sample was analyzed at defined time points. To do so, 5 μl aliquots were taken from the cuvette and GTPase hydrolysis was immediately stopped by addition of 10 μl of 10% H3PO4, followed by neutralization with 30 μl of 0.77 M K2HPO4. Nucleotide composition was analyzed via separation of GTP, GDP, and GMP by reversed‐phase high‐performance liquid chromatography (HPLC) using a Chromolith Performance RP‐18 endcapped column (Merck) connected to a BT4100 HPLC‐pump (Shimadzu). Retention times of nucleotides were detected via monitoring the absorption at 254 nm with a MD‐2010 Plus multi wavelength detector (Jasco). To quantify the concentration of GMP, GDP, and GTP, peak areas corresponding to the respective nucleotide were integrated with the ChromPass software (Jasco).
Lipid vesicle preparation
Rhodamine‐labeled lipid vesicles were generated by hydration of dry lipid films in an oscillating electric field. Brain polar lipids (BPL) and Rhod‐PE were diluted in chloroform to a final concentration of 2 mg/ml and 40 μg/ml, respectively, and desiccated under 250 mbar vacuum onto platinum electrodes for 20 min. Electrodes were then sealed with a Teflon cap and filled with a sucrose solution adjusted to the same osmolarity as measuring buffer C. Lipid vesicles were formed by applying a sine voltage of 1.3 V and 12 Hz for 4 h. Afterwards, vesicles were detached from electrodes by applying a sine voltage of 2.0 V and 4 Hz for 30 min.
In vitro binding assay
Bacteria were harvested at 2,400 × g for 3 min, washed once with 1 × PBS, and were either (i) fixed with 4% formaldehyde in 1 × PBS, pH 7.4 for 20 min followed by two washes with 1 × PBS and resuspended in 1 × PBS supplemented with 0.03% NaN3 (formaldehyde‐fixed bacteria), or (ii) washed once with 1 × PBS and resuspended in buffer C (live bacteria). Live or formaldehyde‐fixed bacteria expressing fluorescent proteins or Alexa‐Fluor488‐conjugated LPS were diluted in buffer C supplemented with 50 μM BSA, and the dilution was applied to the coverslide of a glass bottom 10 mm microwell dish. After centrifugation for 1 min at 3,000 × g bacteria or LPS were incubated for 5 min at 25°C on the temperature‐controlled microscope stage. In experiments with lipid vesicles, vesicles were gently added to bacteria and the mix was incubated an additional 5 min at 25°C. Alexa‐Fluor‐labeled hGBP1 wild type or mutant protein was diluted in buffer C supplemented with 50 μM BSA. Following admixture of nucleotides, hGBP1 was directly added to bacteria or LPS at t = 0 min. Final concentrations for all in vitro binding experiments were 105–3 × 106 bacteria/ml, 5 μM LPS, 1–25 μM protein, and 2 mM (GTP, GppNHp, GTPγS) or 250 μM (GDP·AlFX) nucleotide, unless stated otherwise in the figure legends. Next, the samples were gently mixed and subjected to time‐lapse imaging. Images were collected every 1 min or 1.5 min. After recording time‐lapse images for 60 min (bacteria experiments) or 20 min (LPS experiments) different field of views were imaged for quantification. Imaging was performed either on a Zeiss 780 Inverted Confocal or a Zeiss 880 Airyscan Fast Inverted Confocal on Axio Observer Z1 microscopes using Zeiss Plan‐Apochromat 63×/1.4 oil objectives. Images were processed with Fiji.
Scanning electron microscopy
Live bacteria in buffer C at a concentration of ~ 108 bacteria/ml were applied as 5 μl drops to isopropanol cleaned silicon wafers. Following incubation for 20 min at room temperature (RT), bacteria were gently mixed with either 5 μl buffer C or 5 μl buffer C supplemented with 5 μM hGBP1F, or hGBP1F R584‐586A, or hGBP1F R48A, and 2 mM GTP. Samples were fixed after 4 min and 45 min incubation time with 10 μl of 2 × concentrated fixative (8% formaldehyde, 4% glutaraldehyde in 1 × PBS) for 20 min. Samples were washed once with 1 × PBS and twice with purified water and then air dried. Dried samples were coated with gold for 200 s using a Desk V sputter coater (Denton). Scanning electron micrographs were acquired with an Apreo S scanning electron microscope (FEI, Thermo Fisher Scientific) operating at 2 kV.
Dot‐Blot assay
0.5 μl aliquots of LPS‐O55:B5 and LPS‐O111:B4 two‐fold serial dilutions with concentrations ranging from 1 mg/ml to 0.03 mg/ml were dotted on nitrocellulose membranes and then air dried. Membranes were incubated 1 h with blocking buffer H (50 mM Tris–HCl, pH 7.9, 5 mM MgCl2, 150 mM NaCl, 10 mg/ml BSA, 2 mM TCEP‐HCl) and then incubated with 2 μM Alexa‐Fluor647‐hGBP1F or ‐hGBP1F R584‐586A and 1 mM GTP diluted in buffer H for 10 min at RT. Fluorescence of proteins on membranes was detected with a ChemiDoc MP Imaging System (Bio‐Rad) after a brief wash with buffer H supplemented with 1 mM GTP. Since hGBP1F R584‐586A showed slower polymerization and GTP hydrolysis kinetics, hGBP1F R584‐586A was incubated for 10 min with GTP prior to addition to LPS‐loaded membranes.
Bacterial colony‐forming unit assay
Live bacteria were washed and resuspended to a concentration of 5 × 105 bacteria/ml in buffer C supplemented with 50 μM BSA and 5 mM GTP. Bacteria were then incubated with or without 10 μM hGBP1F at 25°C for 1.5 h. Next, where indicated, antimicrobials were added at a final concentration of 2.5 μg/ml (PMB) or 5 mg/ml (lysozyme), and bacteria were incubated for an additional 30 min at 25°C. Serial dilutions of bacteria were plated on LB‐Miller agar plates, and CFU counts were determined following overnight incubation at 37°C. Viability assays performed with S. flexneri wild type and rfaL mutant were performed in a similar fashion but the incubation time prior to PBM treatment was shortened to 45 min.
Bacterial cell culture infections
Cell lines were cultivated on glass coverslides in 24‐well plates and were either treated with 200 U/ml IFNγ, or with 1–2 μg/ml aTc, or left untreated for 20–22 h. Cells were then infected at an MOI of 6 (S. flexneri strains) or at an MOI of 25 (S. enterica Typhimurium ΔsifA). Poly‐d‐lysine treated or non‐treated bacteria were resuspended in prewarmed cell culture media and spun on cells for 10 min at 700 × g. Infected cells were incubated for 45 min (most S. flexneri strains) or 30 min (S. flexneri ΔospC3ΔipaH9.8, S. enterica Typhimurium ΔsifA) at 37°C and 5% CO2. Following two washes with 1× Hank's Balanced Salt Solution (HBSS), cells were incubated with cell culture media containing 25 μg/ml gentamycin. Cells were incubated at 37°C and 5% CO2 until 1 hpi (S. flexneri wild type and rfaL mutant), 2 hpi (S. flexneri ΔospC3ΔipaH9.8), 2.5 hpi (S. flexneri ΔipaH9.8), or 4 hpi (S. enterica Typhimurium ΔsifA). Cells were fixed with 4% formaldehyde at RT and washed three times with 1 × PBS for immunofluorescence staining.
Immunofluorescence microscopy
Endogenous hGBP1, Arp2, N‐WASP, F‐actin, and bacterial LPS and IcsA were stained with antibodies. Cells were first permeabilized with 0.25% Triton X‐100 in 1 × PBS for 10 min at RT and then washed three times with 1 × PBS. Permeabilized cells were blocked with buffer J (1 × PBS, 50 mg/ml BSA, 300 mM glycine) for 45 min–1 h. Cells were treated with primary antibodies diluted in buffer G either for 1 h at RT or overnight (o.n.) at 4°C. Specific antibody dilutions/concentrations and incubation conditions were as follows: anti‐hGBP1, 1:150, 1 h RT; anti‐Arp2, 7 μg/ml, o.n. 4°C; anti‐N‐WASP, 1:50, 1 h RT; and anti‐IcsA, 1:25, o.n. 4°C. Next, cells were washed with 0.05% Triton X‐100 in 1 × PBS three times for 5 min and then treated with 1:1,000 dilution of anti‐mouse or anti‐rabbit IgG conjugated with Alexa‐Fluor568 or Alexa‐Fluor660 in buffer G for 1 h at RT. F‐actin was stained with Alexa‐Fluor660‐phalloidin (1:40 in buffer G) for 1 h at RT. Following three washes with 0.05% Triton X‐100 for 5 min, cells were mounted on microscopy slides with mounting media (100 mM Tris–HCl, pH 8.5, 25% glycerol, 125 μg/ml Mowiol) diluted 9:1 with 0.1 mg/ml PPD. UPEC was stained with anti‐LPS (1:50, 1 h RT) in glass bottom microwell dishes without mounting. Processed slides were imaged with either a Zeiss 780 Inverted Confocal or a Zeiss 880 Airyscan Fast Inverted Confocal on Axio Observer Z1 microscopes using Zeiss Plan‐Apochromat 63×/1.4 oil objectives, or a Leica STED and Confocal on Leica DMi8 motorized Inverted microscope using a Zeiss Plan‐Apochromat 40×/1.3 oil objective. Z‐stacks were acquired with an interval of 0.5 μm. Images were processed with Fiji.
Time‐lapse microscopy of infected cells
HeLa hGBP1‐KO pInducer‐mCherry‐hGBP1 cells were plated on glass bottom 10 mm microwell dishes and treated with 2 μg/ml aTc for 20‐22 h to stimulate mCherry‐hGBP1 expression. Cells were infected with poly‐d‐lysine treated GFP‐expressing S. flexneri ΔipaH9.8 as described above. Time‐lapse images of infected cells were acquired every 1 min with a Zeiss 780 inverted confocal on an Axio Observer Z1 microscope, with stage incubator set to 37°C and 5% CO2 buffering, using Zeiss Plan‐Apochromat 63×/1.4 oil objectives. Images were processed with Fiji.
Author contribution
MK and JC conceived the project. MK and LS conducted the experiments. MK and JC wrote the manuscript. All authors contributed to manuscript reviewing and editing. CFL and MBG provided reagents. JC and CH supervised the project. JC, CH, CFL, and MBG acquired funding related to the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We are grateful to Drs. Clare Smith and David Tobin as well as members of the Coers laboratory for critical reading of the manuscript. We thank Dr. Anthony Maurelli for sharing S. flexneri strains with us; Drs. Soman Abraham, Meta Kuehn, and Edward Miao for sharing UPEC, Staphylococcus, and Salmonella strains with us; Drs. Avinash Shenoy and Eva Frickel for sharing the pMX‐CMV‐YFP‐CASP4C258S plasmid; and Dr. Gerrit Praefcke for sharing the pRSF‐Duet1‐FTase plasmid. This work was supported by National Institutes of Health grants AI139425 (to J.C.), and AI28360 (C.F.L.) and AI081724 (to M.B.G.); by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Research Fellowship 427472513 (to M.K.) and research grant HE2679/6‐1 (to C.H.); and by a Harvard Medical School Dean's Innovation Award (to M.B.G.). J.C. holds an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. This work was performed in part at the Duke University Light Microscopy Core Facility (LMCF), and at the Duke University Shared Materials Instrumentation Facility (SMIF), a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), which is supported by the National Science Foundation (Grant ECCS‐1542015) as part of the National Nanotechnology Coordinated Infrastructure (NNCI). We thank LMCF members Drs. Benjamin Carlsen, Yasheng Gao, Lisa Cameron, and SMIF member Michelle Plue for providing training.
EMBO Journal (2020) 39: e104926
Data availability
This study includes no data deposited in external repositories.
References
- Abraham SN, Babu JP, Giampapa CS, Hasty DL, Simpson WA, Beachey EH (1985) Protection against Escherichia coli‐induced urinary tract infections with hybridoma antibodies directed against type 1 fimbriae or complementary D‐mannose receptors. Infect Immun 48: 625–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agaisse H (2016) Molecular and cellular mechanisms of Shigella flexneri dissemination. Front Cell Infect Microbiol 6: 29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Zeer MA, Al‐Younes HM, Lauster D, Abu Lubad M, Meyer TF (2013) Autophagy restricts Chlamydia trachomatis growth in human macrophages via IFNG‐inducible guanylate binding proteins. Autophagy 9: 50–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J, Kim SH, Hwang D, Lee KE, Kim MJ, Yang EG, Kim SY, Chung HS (2019) Caspase‐4 disaggregates lipopolysaccharide micelles via LPS‐CARD interaction. Sci Rep 9: 826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrand A, Svanberg C, Langton M, Nyden M (2006) Aggregation behavior and size of lipopolysaccharide from Escherichia coli O55:B5. Colloids Surf B Biointerfaces 53: 9–14 [DOI] [PubMed] [Google Scholar]
- Berry MC, McGhee GC, Zhao Y, Sundin GW (2009) Effect of a waaL mutation on lipopolysaccharide composition, oxidative stress survival, and virulence in Erwinia amylovora. FEMS Microbiol Lett 291: 80–87 [DOI] [PubMed] [Google Scholar]
- Britzen‐Laurent N, Bauer M, Berton V, Fischer N, Syguda A, Reipschlager S, Naschberger E, Herrmann C, Sturzl M (2010) Intracellular trafficking of guanylate‐binding proteins is regulated by heterodimerization in a hierarchical manner. PLoS ONE 5: e14246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles M, Perez M, Kobil JH, Goldberg MB (2001) Polar targeting of Shigella virulence factor IcsA in Enterobacteriacae and Vibrio. Proc Natl Acad Sci USA 98: 9871–9876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov PP, Wackernagel W (1995) Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp‐catalyzed excision of the antibiotic‐resistance determinant. Gene 158: 9–14 [DOI] [PubMed] [Google Scholar]
- Coers J (2017) Sweet host revenge: galectins and GBPs join forces at broken membranes. Cell Microbiol 19: e12793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormack BP, Valdivia RH, Falkow S (1996) FACS‐optimized mutants of the green fluorescent protein (GFP). Gene 173: 33–38 [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL (2000) One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proc Natl Acad Sci USA 97: 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daumke O, Praefcke GJ (2016) Invited review: Mechanisms of GTP hydrolysis and conformational transitions in the dynamin superfamily. Biopolymers 105: 580–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeley EM, Pilla‐Moffett DM, Zwack EE, Piro AS, Finethy R, Kolb JP, Martinez J, Brodsky IE, Coers J (2017) Galectin‐3 directs antimicrobial guanylate binding proteins to vacuoles furnished with bacterial secretion systems. Proc Natl Acad Sci USA 114: E1698–E1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finethy R, Jorgensen I, Haldar AK, de Zoete MR, Strowig T, Flavell RA, Yamamoto M, Nagarajan UM, Miao EA, Coers J (2015) Guanylate binding proteins enable rapid activation of canonical and noncanonical inflammasomes in Chlamydia‐infected macrophages. Infect Immun 83: 4740–4749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finethy R, Luoma S, Orench‐Rivera N, Feeley EM, Haldar AK, Yamamoto M, Kanneganti TD, Kuehn MJ, Coers Coers J (2017) Inflammasome activation by bacterial outer membrane vesicles requires guanylate binding proteins. mBio 8: e01188‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisch D, Bando H, Clough B, Hornung V, Yamamoto M, Shenoy AR, Frickel EM (2019) Human GBP1 is a microbe‐specific gatekeeper of macrophage apoptosis and pyroptosis. EMBO J 38: e100926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman JA, Ohl ME, Miller SI (2003) The Salmonella enterica serovar typhimurium translocated effectors SseJ and SifB are targeted to the Salmonella‐containing vacuole. Infect Immun 71: 418–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fres JM, Muller S, Praefcke GJ (2010) Purification of the CaaX‐modified, dynamin‐related large GTPase hGBP1 by coexpression with farnesyltransferase. J Lipid Res 51: 2454–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MB, Barzu O, Parsot C, Sansonetti PJ (1993) Unipolar localization and ATPase activity of IcsA, a Shigella flexneri protein involved in intracellular movement. Infect Agents Dis 2: 210–211 [PubMed] [Google Scholar]
- Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA (2013) Cytoplasmic LPS activates caspase‐11: implications in TLR4‐independent endotoxic shock. Science 341: 1250–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann M, Schneck E, Gutsmann T, Brandenburg K, Tanaka M (2015) Bacterial lipopolysaccharides form physically cross‐linked, two‐dimensional gels in the presence of divalent cations. Soft Matter 11: 6037–6044 [DOI] [PubMed] [Google Scholar]
- Holzer SU, Schlumberger MC, Jackel D, Hensel M (2009) Effect of the O‐antigen length of lipopolysaccharide on the functions of Type III secretion systems in Salmonella enterica. Infect Immun 77: 5458–5470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Payne SM (1997) Effect of mutations in Shigella flexneri chromosomal and plasmid‐encoded lipopolysaccharide genes on invasion and serum resistance. Mol Microbiol 24: 779–791 [DOI] [PubMed] [Google Scholar]
- Howard JC (2007) Introduction: cell‐autonomous immunity. Microbes Infect 9: 1633–1635 [DOI] [PubMed] [Google Scholar]
- Huang S, Meng Q, Maminska A, MacMicking JD (2019) Cell‐autonomous immunity by IFN‐induced GBPs in animals and plants. Curr Opin Immunol 60: 71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince S, Kutsch M, Shydlovskyi S, Herrmann C (2017) The human guanylate‐binding proteins hGBP‐1 and hGBP‐5 cycle between monomers and dimers only. FEBS J 284: 2284–2301 [DOI] [PubMed] [Google Scholar]
- Ji C, Du S, Li P, Zhu Q, Yang X, Long C, Yu J, Shao F, Xiao J (2019) Structural mechanism for guanylate‐binding proteins (GBPs) targeting by the Shigella E3 ligase IpaH9.8. PLoS Pathog 15: e1007876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalynych S, Morona R, Cygler M (2014) Progress in understanding the assembly process of bacterial O‐antigen. FEMS Microbiol Rev 38: 1048–1065 [DOI] [PubMed] [Google Scholar]
- Kawabata S, Iwanaga S (1999) Role of lectins in the innate immunity of horseshoe crab. Dev Comp Immunol 23: 391–400 [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi‐Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A et al (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341: 1246–1249 [DOI] [PubMed] [Google Scholar]
- Kohler H, Rodrigues SP, McCormick BA (2002) Shigella flexneri Interactions with the basolateral membrane domain of polarized model intestinal epithelium: role of lipopolysaccharide in cell invasion and in activation of the mitogen‐activated protein kinase ERK. Infect Immun 70: 1150–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagrange B, Benaoudia S, Wallet P, Magnotti F, Provost A, Michal F, Martin A, Di Lorenzo F, Py BF, Molinaro A et al (2018) Human caspase‐4 detects tetra‐acylated LPS and cytosolic Francisella and functions differently from murine caspase‐11. Nat Commun 9: 242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerouge I, Vanderleyden J (2002) O‐antigen structural variation: mechanisms and possible roles in animal/plant‐microbe interactions. FEMS Microbiol Rev 26: 17–47 [DOI] [PubMed] [Google Scholar]
- Li P, Jiang W, Yu Q, Liu W, Zhou P, Li J, Xu J, Xu B, Wang F, Shao F (2017) Ubiquitination and degradation of GBPs by a Shigella effector to suppress host defence. Nature 551: 378–383 [DOI] [PubMed] [Google Scholar]
- Liu BC, Sarhan J, Panda A, Muendlein HI, Ilyukha V, Coers J, Yamamoto M, Isberg RR, Poltorak A (2018) Constitutive interferon maintains GBP expression required for release of bacterial components upstream of pyroptosis and Anti‐DNA responses. Cell Rep 24: 155–168 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking JD (2012) Interferon‐inducible effector mechanisms in cell‐autonomous immunity. Nat Rev Immunol 12: 367–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Karki R, Malireddi RK, Neale G, Vogel P, Yamamoto M, Lamkanfi M, Kanneganti TD (2015) The transcription factor IRF1 and guanylate‐binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat Immunol 16: 467–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Place DE, Kuriakose T, Kanneganti TD (2017) Interferon‐inducible guanylate‐binding proteins at the interface of cell‐autonomous immunity and inflammasome activation. J Leukoc Biol 101: 143–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, Roose‐Girma M, Bumann D, Kayagaki N, Takeda K et al (2014) Caspase‐11 activation requires lysis of pathogen‐containing vacuoles by IFN‐induced GTPases. Nature 509: 366–370 [DOI] [PubMed] [Google Scholar]
- Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, Ruhl S, Dussurgey S, Dick MS, Kistner A, Rigard M et al (2015) Guanylate‐binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida . Nat Immunol 16: 476–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mey A, Leffler H, Hmama Z, Normier G, Revillard JP (1996) The animal lectin galectin‐3 interacts with bacterial lipopolysaccharides via two independent sites. J Immunol 156: 1572–1577 [PubMed] [Google Scholar]
- Moffatt JH, Harper M, Boyce JD (2019) Mechanisms of polymyxin resistance. Adv Exp Med Biol 1145: 55–71 [DOI] [PubMed] [Google Scholar]
- Mou X, Souter S, Du J, Reeves AZ, Lesser CF (2018) Synthetic bottom‐up approach reveals the complex interplay of Shigella effectors in regulation of epithelial cell death. Proc Natl Acad Sci USA 115: 6452–6457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naschberger E, Lubeseder‐Martellato C, Meyer N, Gessner R, Kremmer E, Gessner A, Sturzl M (2006) Human guanylate binding protein‐1 is a secreted GTPase present in increased concentrations in the cerebrospinal fluid of patients with bacterial meningitis. Am J Pathol 169: 1088–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K, Ernst RK, Yamamoto M, Miao EA, Coers J (2014) Guanylate binding proteins promote caspase‐11‐dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci USA 111: 6046–6051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piro AS, Hernandez D, Luoma S, Feeley EM, Finethy R, Yirga A, Frickel EM, Lesser CF, Coers J (2017) Detection of cytosolic Shigella flexneri via a C‐terminal triple‐arginine motif of GBP1 inhibits actin‐based motility. mBio 8: e01979‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praefcke GJ, Kloep S, Benscheid U, Lilie H, Prakash B, Herrmann C (2004) Identification of residues in the human guanylate‐binding protein 1 critical for nucleotide binding and cooperative GTP hydrolysis. J Mol Biol 344: 257–269 [DOI] [PubMed] [Google Scholar]
- Praefcke GJK (2018) Regulation of innate immune functions by guanylate‐binding proteins. Int J Med Microbiol 308: 237–245 [DOI] [PubMed] [Google Scholar]
- Ramachandran R, Schmid SL (2018) The dynamin superfamily. Curr Biol 28: R411–R416 [DOI] [PubMed] [Google Scholar]
- Randow F, MacMicking JD, James LC (2013) Cellular self‐defense: how cell‐autonomous immunity protects against pathogens. Science 340: 701–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risco C, Carrascosa JL, Bosch MA (1993) Visualization of lipopolysaccharide aggregates by freeze‐fracture and negative staining. J Electron Microsc (Tokyo) 42: 202–204 [PubMed] [Google Scholar]
- Robbins JR, Monack D, McCallum SJ, Vegas A, Pham E, Goldberg MB, Theriot JA (2001) The making of a gradient: IcsA (VirG) polarity in Shigella flexneri . Mol Microbiol 41: 861–872 [DOI] [PubMed] [Google Scholar]
- Rodriguez MD, Paul Z, Wood CE, Rice KC, Triplett EW (2017) Construction of stable fluorescent reporter plasmids for use in Staphylococcus aureus . Front Microbiol 8: 2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas ER, Billings G, Odermatt PD, Auer GK, Zhu L, Miguel A, Chang F, Weibel DB, Theriot JA, Huang KC (2018) The outer membrane is an essential load‐bearing element in Gram‐negative bacteria. Nature 559: 617–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandlin RC, Lampel KA, Keasler SP, Goldberg MB, Stolzer AL, Maurelli AT (1995) Avirulence of rough mutants of Shigella flexneri: requirement of O antigen for correct unipolar localization of IcsA in the bacterial outer membrane. Infect Immun 63: 229–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandlin RC, Goldberg MB, Maurelli AT (1996) Effect of O side‐chain length and composition on the virulence of Shigella flexneri 2a. Mol Microbiol 22: 63–73 [DOI] [PubMed] [Google Scholar]
- Santos NC, Silva AC, Castanho MA, Martins‐Silva J, Saldanha C (2003) Evaluation of lipopolysaccharide aggregation by light scattering spectroscopy. ChemBioChem 4: 96–100 [DOI] [PubMed] [Google Scholar]
- Santos JC, Broz P (2018) Sensing of invading pathogens by GBPs: at the crossroads between cell‐autonomous and innate immunity. J Leukoc Biol 104: 729–735 [DOI] [PubMed] [Google Scholar]
- Santos JC, Dick MS, Lagrange B, Degrandi D, Pfeffer K, Yamamoto M, Meunier E, Pelczar P, Henry T, Broz P (2018) LPS targets host guanylate‐binding proteins to the bacterial outer membrane for non‐canonical inflammasome activation. EMBO J 37: e98089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen A, Higgins DE (2005) The 5′ untranslated region‐mediated enhancement of intracellular listeriolysin O production is required for Listeria monocytogenes pathogenicity. Mol Microbiol 57: 1460–1473 [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514: 187–192 [DOI] [PubMed] [Google Scholar]
- Shydlovskyi S, Zienert AY, Ince S, Dovengerds C, Hohendahl A, Dargazanli JM, Blum A, Gunther SD, Kladt N, Sturzl M et al (2017) Nucleotide‐dependent farnesyl switch orchestrates polymerization and membrane binding of human guanylate‐binding protein 1. Proc Natl Acad Sci USA 114: E5559–E5568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson BW, Trent MS (2019) Pushing the envelope: LPS modifications and their consequences. Nat Rev Microbiol 17: 403–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sistemich L, Kutsch M, Hamisch B, Zhang P, Shydlovskyi S, Britzen‐Laurent N, Sturzl M, Huber K, Herrmann C (2020) The molecular mechanism of polymer formation of farnesylated human guanylate‐binding protein 1. J Mol Biol 432: 2164–2185 [DOI] [PubMed] [Google Scholar]
- Song J, Bishop BL, Li G, Grady R, Stapleton A, Abraham SN (2009) TLR4‐mediated expulsion of bacteria from infected bladder epithelial cells. Proc Natl Acad Sci USA 106: 14966–14971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhauer J, Agha R, Pham T, Varga AW, Goldberg MB (1999) The unipolar Shigella surface protein IcsA is targeted directly to the bacterial old pole: IcsP cleavage of IcsA occurs over the entire bacterial surface. Mol Microbiol 32: 367–377 [DOI] [PubMed] [Google Scholar]
- Stenutz R, Weintraub A, Widmalm G (2006) The structures of Escherichia coli O‐polysaccharide antigens. FEMS Microbiol Rev 30: 382–403 [DOI] [PubMed] [Google Scholar]
- Tietzel I, El‐Haibi C, Carabeo RA (2009) Human guanylate binding proteins potentiate the anti‐chlamydia effects of interferon‐gamma. PLoS ONE 4: e6499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia RH, Falkow S (1997) Fluorescence‐based isolation of bacterial genes expressed within host cells. Science 277: 2007–2011 [DOI] [PubMed] [Google Scholar]
- Van den Bosch L, Manning PA, Morona R (1997) Regulation of O‐antigen chain length is required for Shigella flexneri virulence. Mol Microbiol 23: 765–775 [DOI] [PubMed] [Google Scholar]
- Wacker MA, Teghanemt A, Weiss JP, Barker JH (2017) High‐affinity caspase‐4 binding to LPS presented as high molecular mass aggregates or in outer membrane vesicles. Innate Immun 23: 336–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandel MP, Pathe C, Werner EI, Ellison CJ, Boyle KB, von der Malsburg A, Rohde J, Randow F (2017) GBPs inhibit motility of Shigella flexneri but are targeted for degradation by the bacterial ubiquitin ligase IpaH9.8. Cell Host Microbe 22: 507–518 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren DJ (2011) Preparation of highly efficient electrocompetent Escherichia coli using glycerol/mannitol density step centrifugation. Anal Biochem 413: 206–207 [DOI] [PubMed] [Google Scholar]
- Welch RA, Burland V, Plunkett G III, Redford P, Roesch P, Rasko D, Buckles EL, Liou SR, Boutin A, Hackett J et al (2002) Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli . Proc Natl Acad Sci USA 99: 17020–17024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesener DA, Wangkanont K, McBride R, Song X, Kraft MB, Hodges HL, Zarling LC, Splain RA, Smith DF, Cummings RD et al (2015) Recognition of microbial glycans by human intelectin‐1. Nat Struct Mol Biol 22: 603–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesener DA, Dugan A, Kiessling LL (2017) Recognition of microbial glycans by soluble human lectins. Curr Opin Struct Biol 44: 168–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
This study includes no data deposited in external repositories.