

Abstract
Atrial fibrillation is a common cardiac arrhythmia with an increasing incidence rate. Particularly for the aging population, understanding the underlying mechanisms of atrial arrhythmia is important in designing clinical treatment. Recently, experiments have shown that atrial arrhythmia is associated with oxidative stress. In this study, an atrial cell model including oxidative-dependent Ca2+/calmodulin- (CaM-) dependent protein kinase II (CaMKII) activation was developed to explore the intrinsic mechanisms of atrial arrhythmia induced by oxidative stress. The simulation results showed that oxidative stress caused early afterdepolarizations (EADs) of action potentials by altering the dynamics of transmembrane currents and intracellular calcium cycling. Oxidative stress gradually elevated the concentration of calcium ions in the cytoplasm by enhancing the L-type Ca2+ current and sarcoplasmic reticulum (SR) calcium release. Owing to increased intracellular calcium concentration, the inward Na+/Ca2+ exchange current was elevated which slowed down the repolarization of the action potential. Thus, the action potential was prolonged and the L-type Ca2+ current was reactivated, resulting in the genesis of EAD. Furthermore, based on the atrial single-cell model, a two-dimensional (2D) ideal tissue model was developed to explore the effect of oxidative stress on the electrical excitation wave conduction in 2D tissue. Simulation results demonstrated that, under oxidative stress conditions, EAD hindered the conduction of electrical excitation and caused an unstable spiral wave, which could disrupt normal cardiac rhythm and cause atrial arrhythmia. This study showed the effects of excess reactive oxygen species on calcium cycling and action potential in atrial myocytes and provided insights regarding atrial arrhythmia induced by oxidative stress.
1. Introduction
Atrial fibrillation (AF) is the most common cardiac arrhythmia [1–3]. To design optimal treatment of AF, the mechanisms underlying AF need to be better understood. Both reactive oxygen species (ROS) and Ca2+/calmodulin- (CaM-) dependent protein kinase II (CaMKII) have been shown to be associated with the development of cardiac arrhythmias [4, 5]. The kinase CaMKII is ubiquitously expressed in the cardiomyocytes [4, 6]. It is involved in numerous cellular signaling cascades, such as phosphorylation of L-type Ca2+ channels [7–9], Na+ channels [10, 11], ryanodine receptors (RyRs) [12–18], and phospholamban (PLB) [12, 17, 19, 20]. Overexpression of CaMKII increases fractional sarcoplasmic reticulum (SR) Ca2+ release [13] and SR Ca2+ leakage [16], enhancing the activation of RyRs during both systole and diastole. As mentioned, it has been reported that CaMKII can also phosphorylate PLB [20]. In its unphosphorylated state, PLB acts as an endogenous inhibitor of sarco/endoplasmic reticulum Ca2+-ATPase (SERCA). Therefore, phosphorylation of PLB by CaMKII will enhance SERCA activity [21]. In addition to SR Ca2+ dynamics, many ion channels show CaMKII-dependent phosphorylation. Phosphorylation of L-type Ca2+ channels induced by CaMKII is associated with Ca2+-dependent Ca2+ current facilitation [22, 23]; CaMKII-dependent phosphorylation of Na+ channels may cause an increase in the late sodium current, which predisposes cardiomyocytes to arrhythmias [10, 11].
Conventionally, CaMKII is activated by Ca2+-bound calmodulin (Ca-CaM). Recently, a novel mechanism of CaMKII activation, which is ROS dependent, has been revealed [19, 24]. Previous studies have demonstrated that oxidative stress is closely associated with cardiac arrhythmias through alteration of the electrical activity and intracellular calcium dynamics of cardiac myocytes [25–27]. Oxidative stress is the main manifestation of cell metabolism disorders with excessive ROS. This oxidation-induced CaMKII activation is in relation to apoptosis [28], sinus node dysfunction [29], heart injury [30], and arrhythmias [31]. Although both ROS and CaMKII are associated with cardiac arrhythmias, the role of oxidative-dependent CaMKII activation in the development of atrial arrhythmias is not yet well understood.
In previous studies of cardiac arrhythmias, in vivo and in vitro experiments provide insights into mechanisms underlying arrhythmogenesis [5, 31]. However, these approaches have several limitations. For example, it is difficult to record the electrophysiological properties at different physical scales (from subcellular level to organ level) at the same time in one experiment. The electrophysiological properties recorded from different experiments, such as patch clamp and optical mapping, might be affected by gradual changes in cell or tissue properties. In addition to in vivo and in vitro experiments, a common method widely used in cardiac arrhythmia studies is in silico modeling. In this study, to simulate the effects of ROS-dependent CaMKII activation on regulations of intracellular Ca2+ and ionic currents, we first developed a Markov chain model of CaMKII, including both the autophosphorylation and oxidation pathways. Then, the effect of CaMKII on related proteins and ion channels was integrated to establish a computational model of the atrial cell to simulate changes in the electrophysiology under oxidative stress conditions, including ion currents, ion concentrations, calcium cycling, and transmembrane potential. Finally, the two-dimensional (2D) spiral wave was induced and the electrical excitation propagation was analyzed under normal and oxidative stress conditions.
2. Methods
2.1. CaMKII Model, including Both Autophosphorylation- and Oxidation-Dependent Activation
A six-state Markov chain model of CaMKII developed by our previous study of Zhang et al. [32] was incorporated into a human atrial model developed by Grandi et al. [33] to simulate autophosphorylation- and oxidation-dependent CaMKII activation.
The CaMKII monomer consists of three domains (an association domain, a regulatory domain, and a catalytic domain). When CaMCa4 (one calmodulin bound with four Ca2+) binds to the regulatory domain, the catalytic domain will be exposed, leading to CaMKII activation. In this state, CaMKII can be further autophosphorylated or oxidized, producing a long-lasting activation, even if CaMCa4 dissociates from CaMKII. Finally, CaMKII is fully deactivated by dephosphorylation with protein phosphatases, or reduction with methionine sulfoxide reductases (MsrA). Based on the four-state model of CaMKII developed by Chiba et al. [34], two oxidized states, with and without CaMCa4 binding with CaMKII, were added (see Figure S1 in the Supplementary Material for illustration), corresponding to the conformational change of oxidation-dependent CaMKII activation. Since there is no evidence to show that oxidation and autophosphorylation can occur at the same time, in this study, autophosphorylation and oxidation of CaMKII were treated as two independent processes [24]. The CaMKII autophosphorylation was based on the model developed by Chiba et al. [34]. The parameters of CaMKII oxidation were fitted to the experimental data recorded by Erickson et al. [24]. The detailed parameters of the CaMKII model are listed in Table S1 in the Supplementary Material.
2.2. Effects of CaMKII on Ion Channels
Previous studies showed that the ion channels influenced by CaMKII included the fast Na+ current (INa) and the L-type Ca2+ current (ICaL) [7–11]. According to the method developed by O'Hara et al. [35], the current of each ion channel (I) was divided into two parts:
| (1) |
where Ibase is the part of ion channel current not affected by CaMKII, ICaMK is the part of ion channel current affected by CaMKII, and Φ is the proportion affected by CaMKII using the following equations:
| (2) |
where CaMKactive is the fraction of CaMKII activation and KCaMKII is the Michaelis constant with the same value in O'Hara et al. [35]. For INa, the time constant of gate j was slowed down by 1.46-fold [32]. And for ICaL, experimental data showed that CaMKII activation produced an increased amplitude and a slowed inactivation of ICaL [36]. Therefore, in this study, the time constant of gate f was slowed down by 1.5-fold and the part of ICaL affected by CaMKII was increased by ΔICaL,CaMK shown in Equation (3).
| (3) |
2.3. Effects of CaMKII on Ca2+ Cycling
Both RyR and PLB are primary regulatory proteins, controlling SR Ca2+ release and uptake; these are crucial processes, maintaining the balance of intracellular Ca2+. CaMKII activation-induced RyR phosphorylation can increase RyR opening probability and SR Ca2+ release. CaMKII activation-induced PLB phosphorylation can reduce SERCA inhibition, increasing SR Ca2+ uptake during diastole. The RyR and PLB models developed by Soltis and Saucerman [37] were used to simulate the effects of CaMKII activation on intracellular Ca2+ cycling. The rate constants controlling RyR opening (koSRCa) and SR leakage (kleak) were modified (Equations (4) and (5)) under CaMKII activation to increase RyR opening in both systolic and diastolic conditions. Meanwhile, the half maximal saturation constant of SERCA (Kmf) was modified (Equation (6)) to mimic the phenomenon of increased SERCA pump calcium sensitivity, which is induced by Thr17 phosphorylation of PLB under CaMKII activation.
| (4) |
| (5) |
| (6) |
2.4. Single Atrial Cell Model
The cell membrane of an atrial myocyte was mimicked as an electrical circuit. The AP in a human atrial cell was calculated using the following ordinary differential equation [33]:
| (7) |
where Cm is the cell capacitance, Vm is the transmembrane voltage, t is time, Iion is the total transmembrane ionic current, and Istim is the stimulus current. Iion was calculated as
| (8) |
where INa is the fast Na+ current, INabk is the background Na+ current, Ito is the transient outward K+ current, IKur is the ultrarapid delayed rectifier K+ current, IKr is the rapid activating K+ current, IKs is the slowly activating K+ current, IK1 is the inward rectifier K+ current, INaK is the Na+/K+ pump current, ICaL is the L-type Ca2+ current, ICabk is the background Ca2+ current, IpCa is the sarcolemmal Ca2+ pump current, INCX is the Na+/Ca2+ exchange current, IClCa is the Ca2+-activated Cl− current, and IClbk is the background Cl− current.
In this study, 0.5, 1, and 2 Hz stimulation frequencies were used to investigate the frequency dependency of CaMKII activation. Action potentials of atrial cells were produced by applying a stimulus current with an amplitude of −12.5 pA/pF and a duration of 5 ms. The time step used in the simulation was 0.1 ms using the CVODE solver of the SUite of Nonlinear and DIfferential/ALgebraic equation Solvers (SUNDIALS) to solve initial value problems for ordinary differential equation systems. These simulation results were consistent with the ones using forward Euler with a time step of 0.005 ms. Under the control condition, the value of H2O2 in the CaMKII model was 0 μM, and the one was 200 μM under the oxidative stress condition. To ensure that the model reached a quasistable steady state, the simulations were carried out for more than 50 s under the control condition and 150 s under the oxidative stress condition.
2.5. Excitation Wave Conduction in 2D Tissue
In this study, a monodomain model was used for 2D simulations in an ideal square tissue. The excitation wave propagation was simulated using the following equation:
| (9) |
where D is the diffusion tensor describing the conductivity of the tissue and ∇ is the spatial gradient operator.
In 2D simulations, the 2D ideal isotropic tissue was constructed as a sheet of 1000 × 1000 nodes to investigate the stability of the spiral waves, as used in [38]. The time step Δt was set at 0.005 ms, and the space step Δx was 0.1 mm along both direction axes, as used in [39]. The S1–S2 stimulation protocol was used to produce spiral waves. An S1 stimulus was applied to the five column nodes on the left side to induce a plane wave (Figure 1(a)). After the nodes in the middle line (the dashed line in Figure 1(b)) reached the end point of the refractory period, an S2 stimulus was applied in the left lower quadrant to induce a spiral wave (Figure 1(b)).
Figure 1.
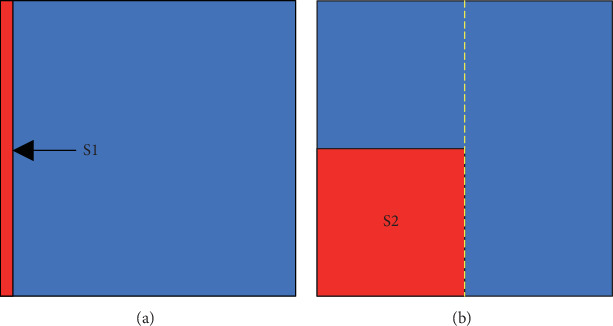
The S1–S2 protocol used in 2D simulations. The red bar in (a) and the red square in (b) represent the regions where S1 and S2 stimuli were applied.
The conduction velocity (CV) was measured using the following equation:
| (10) |
where x1 and x2 are the positions and t1 and t2 are the times when the excitation reaches the two recording points. In 2D simulation, the tissue was isotropic and the diffusion tensor (D) along both direction axes was set to 0.029 mm2/ms to produce a CV of 69 cm/s in control conditions, which was consistent with the CV (70 cm/s) previously used for human atrial tissue simulation [40–42].
3. Results
3.1. Electrophysiological Properties of a Single Atrial Cell under Control Conditions
The action potential (AP) in a single-cell model was generated by applying a series of 1 Hz stimuli. The time trace of the simulated AP is presented in Figure 2, along with other AP traces from previous mathematical models [33, 43, 44]. The resting membrane potential (RMP), maximum upstroke velocity (dV/dtmax), action potential amplitude (APA), and action potential duration at 90% repolarization (APD90) were measured as −75.2 mV, 160 mV/ms, 110 mV, and 312 ms, respectively. These parameters were consistent with previous models developed by Grandi et al. [33], Courtemanche et al. [43], and Nygren et al. [44], and APD90 was within the scope of previously reported experimental data [45–48], as listed in Table 1.
Figure 2.
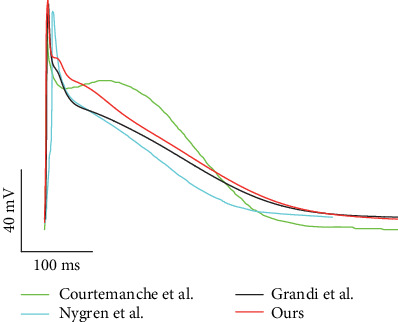
Action potential traces produced by our simulation and previous mathematical models and experiments at 1 Hz. Traces labeled “Courtemanche et al.,” “Nygren et al.,” and “Grandi et al.” were reproduced from mathematical models [33, 43, 44].
Table 1.
Comparison of action potential characteristics.
| Model and experimental data | RMP (mV) |
dV/dtmax (mV/ms) |
APA (mV) |
APD90 (ms) |
|---|---|---|---|---|
| Courtemanche et al. [43] | −79.5 | 216 | 106 | 297 |
| Nygren et al. [44] | −76.4 | 116 | 103.8 | 245 |
| Grandi et al. [33] | −74.5 | 135 | 105 | 294 |
| Our model | −75.2 | 160 | 110 | 312 |
| Dawodu et al. [45] | — | — | — | 361 ± 71 |
| Katoh et al. [46] | — | — | — | 255 ± 39 |
| Bosch et al. [47] | — | — | — | 255 ± 45 |
| Kim et al. [48] | — | — | — | 258 ± 25 |
To further validate the model, the ionic currents and intracellular calcium cycling process were investigated and compared with those from the model of Grandi et al. [33] at 1 Hz, as shown in Figure 3. These results demonstrated an elevation in the repolarization phase of the AP and, therefore, a longer action potential duration (APD) in our model (Figure 3(a)). This was attributed to the change in ICaL. In our model, ICaL showed a larger amplitude and a slowed inactivation (Figure 3(b)), which was consistent with the effect of CaMKII activation on ICaL [36]. In addition to the AP, the variation in ICaL altered intracellular calcium regulation, as ICaL was the main influx of intracellular Ca2+. The increase in ICaL caused a larger Ca2+ influx, leading to a larger SR Ca2+ release (Figure 3(c)) and thus a larger intracellular Ca2+ concentration (Figure 3(d)). The accumulation of intracellular Ca2+ finally caused an elevation in the concentration of SR Ca2+ (Figure 3(e)). Interestingly, the peak inward INCX in our model declined even when the intracellular Ca2+ concentration increased (Figure 3(f)). This may be explained by the fact that the elevation of the membrane potential during the repolarization phase suppressed the inward Na+ flux and decreased INCX.
Figure 3.
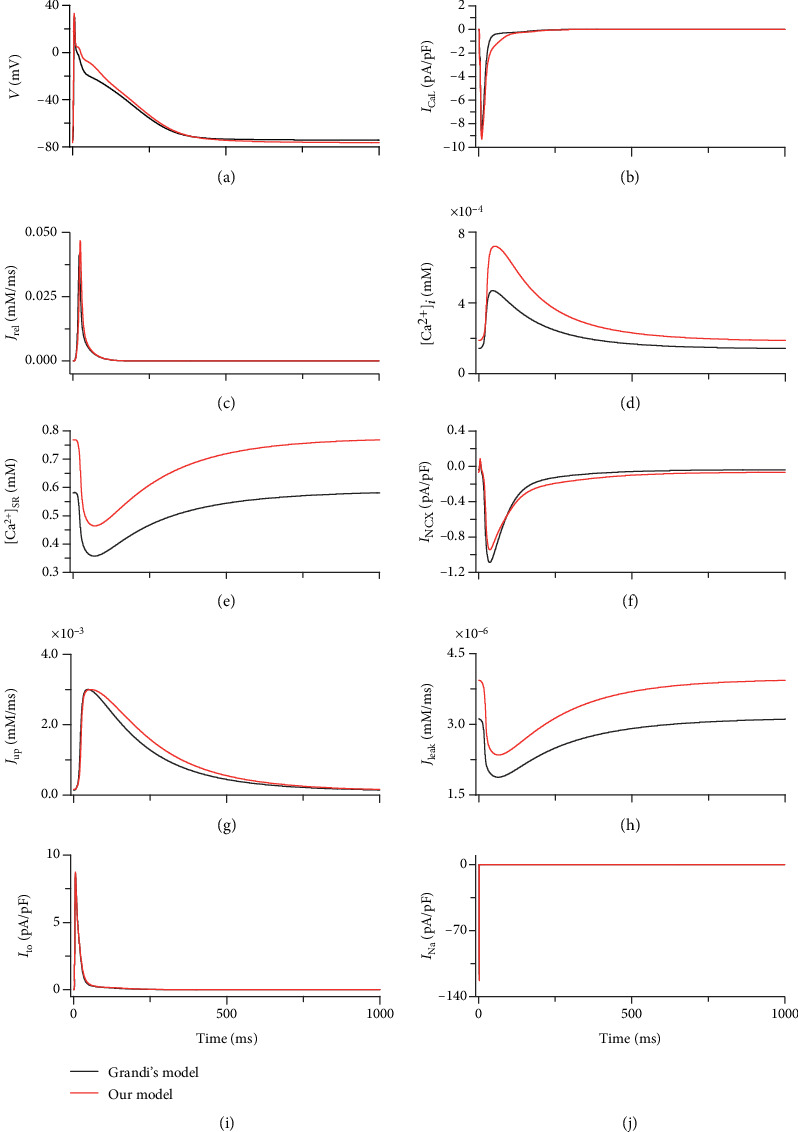
Traces of AP, ionic currents, and calcium cycling produced by our model (red) and the model of Grandi et al. [33] (black). (a) AP. (b) ICaL. (c) SR Ca2+ release (Jrel). (d) intracellular Ca2+ concentration ([Ca2+]i). (e) SR Ca2+ concentration ([Ca2+]SR). (f) INCX. (g) SR Ca2+ reuptake (Jup). (h) SR Ca2+ leakage (Jleak). (i) Ito. (j) INa.
To justify the increase of intracellular Ca2+ concentration in our model, the Ca2+ concentration was compared with previous studies. The calcium transient amplitude in our model was ~0.53 μM, which was consistent with the studies of Courtemanche et al. and Colman et al. (0.45–0.55 μM) [43, 49], although the Ca2+ transient amplitude in the study of Nygren et al. was reported to be even larger (~1.2 μM) [44].
The variations in ICaL and intracellular calcium cycling were caused by adding the effect of CaMKII activation in our model. Previous studies demonstrated that CaMKII activation was significantly influenced by pacing frequencies. In our simulation, CaMKII activation and intracellular Ca2+ dynamics were investigated at 0.5, 1, and 2 Hz. Figure 4(a)shows that the fraction of CaMKII activation increased with increasing pacing rate. Meanwhile, the time required for CaMKII activation was also decreased, indicating a faster activation at a higher pacing rate (Figure 4(a)). The frequency-dependent activation of CaMKII was mainly due to the intracellular Ca2+ concentration. With increasing pacing rate, both RyR Ca2+ release and intracellular Ca2+ concentration increased (Figure 4(b)), causing an increase in CaMCa4 and, therefore, augmenting CaMKII activation.
Figure 4.
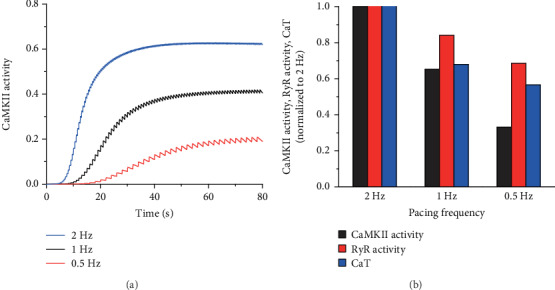
Frequency-dependent activation of CaMKII. (a) CaMKII activation curves at 0.5, 1, and 2 Hz in 80 s. (b) Normalized CaMKII activation, RyR calcium release (RyR Activity), and intracellular Ca2+ transient (CaT) to 2 Hz at 0.5, 1, and 2 Hz.
3.2. Electrophysiological Properties of a Single Atrial Cell under Oxidative Stress Conditions
The simulation results showed that an EAD was induced in the AP under conditions of oxidative stress at a pacing rate of 1 Hz (Figure 5(a)). In this case, the elevated ROS concentration (0.2 mM) induced a significant increase in CaMKII activation (Figure 5(b)). ROS-enhanced CaMKII activation further enlarged ICaL, INCX, fraction of RyR phosphorylation, and fraction of PLB phosphorylation (Figures 5(c)–5(f)). Under oxidative stress conditions, the intracellular Ca2+ dynamics were remarkably changed. Enhanced RyR and PLB phosphorylation caused significant increases in Jrel, Jup, and Jleak (Figures 5(g)–5(i)), which further induced a dramatic increase in intracellular Ca2+ concentration (Figure 5(j)) and a decrease in minimum SR Ca2+ (Figure 5(k)). However, there was no obvious change in the intracellular Na+ concentration (Figure 5(l)).
Figure 5.
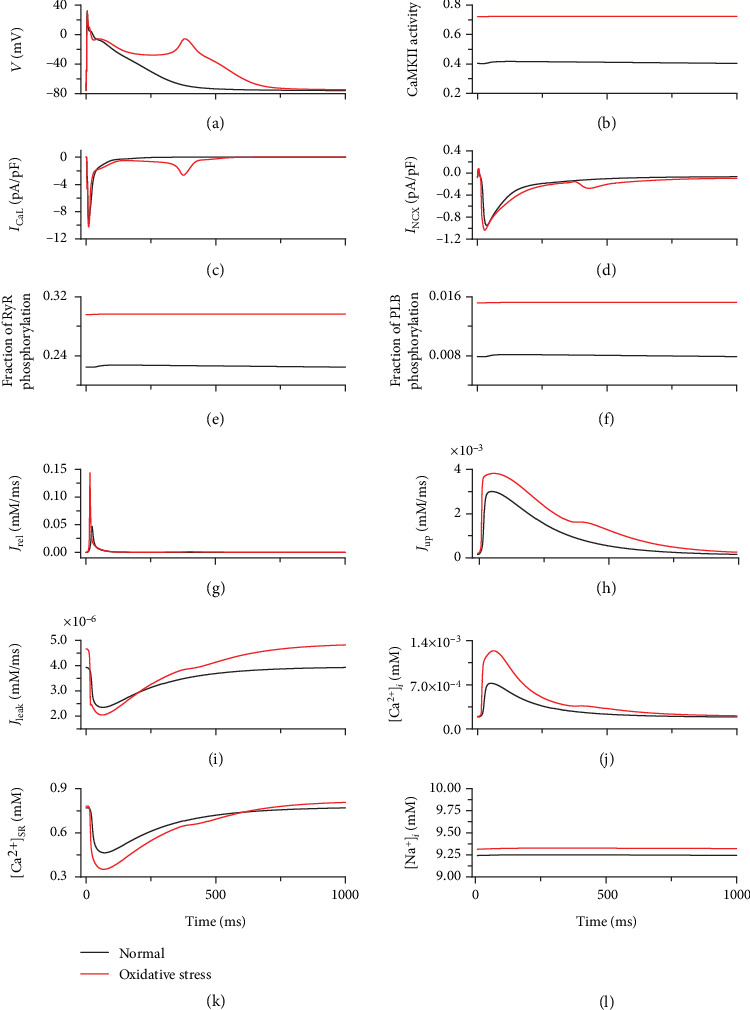
Traces of AP, ionic currents, and calcium cycling under normal (black) and oxidative stress (red) conditions. (a) AP. (b) CaMKII activity. (c) ICaL. (d) INCX. (e) Fraction of RyR phosphorylation. (f) Fraction of PLB phosphorylation. (g) Jrel. (h) Jup. (i) Jleak. (j) [Ca2+]i. (k) [Ca2+]SR. (l) Intracellular Na+ concentration ([Na+]i).
As CaMKII activation showed frequency-dependent behavior, ROS-induced EADs are rate dependent as well. Under oxidative stress conditions, the occurrence of EADs in 50 s became more prominent with decreasing pacing rate, as shown in Figure 6. Under control conditions, the CaMKII activation (without ROS-induced activation) gradually decreased with decreasing pacing rate (Figure 4(b)). This meant that more CaMKII could be activated by ROS under oxidative conditions at low pacing rates. Consequently, the CaMKII activation induced by ROS gradually increased with decreasing pacing rate (green bars in Figure 6). Therefore, under oxidative stress conditions, more EADs were induced at low pacing rates.
Figure 6.
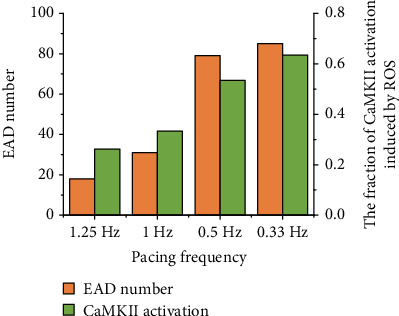
Number of EAD occurring in the same duration (orange) and the fraction of CaMKII activation induced by ROS (green) at different pacing rates under oxidative stress conditions.
3.3. Mechanisms Underlying the Genesis of EAD under Oxidative Stress Conditions
In the case of EAD, ICaL and INCX were the main currents significantly influenced by oxidative stress. Blocking ICaL at the point of ICaL reactivation abolished EAD (data not shown), implying that ICaL was a main factor contributing to the genesis of EAD. However, blocking INCX did not guarantee elimination of EAD. Figure 7 indicated the effects of blocking INCX at different times from the point of ICaL reactivation (70 ms) to the time of the peak of EAD (350 ms). With the delay in blocking INCX (Figure 7(a)), the reactivation of ICaL became more prominent (Figure 7(b)) and the APD gradually increased (Figure 7(c)). When blocking INCX in the interval 70–210 ms, the reactivation of ICaL did not induce AP depolarization and therefore abolished EAD. On blocking INCX during the interval 210–280 ms, the enlarged reactivation of ICaL started to transfer AP repolarization to depolarization. However, in this time interval, AP depolarization was not obvious and no EAD was induced. When blocking INCX during the interval 280–350 ms, the reactivation of ICaL was able to induce AP depolarization. Consequently, in this time interval, blocking INCX could not eliminate EAD (Figure 7(c)). Therefore, INCX played an important role in ICaL reactivation and thus in triggering EAD.
Figure 7.
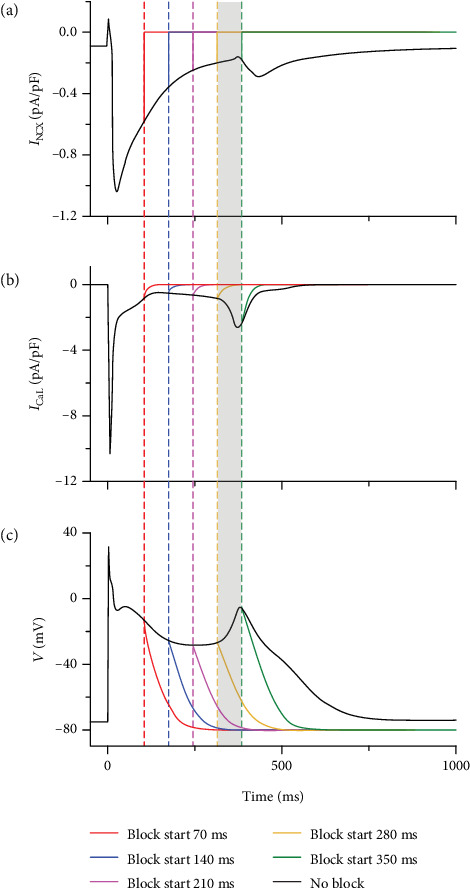
Effects of blocking INCX at different times on the genesis of EAD under oxidative stress condition: (a) INCX. (b) ICaL. (c) AP. The gray box represents the time interval during which EAD can occur while blocking INCX.
In our model, INCX was not directly regulated by ROS. As the main efflux of intracellular Ca2+, INCX was regulated by intracellular Ca2+ concentration; Ca2+ cycling was another important process affected by ROS-induced CaMKII activation. Three factors associated with intracellular cycling in our model were directly regulated by ROS-induced CaMKII activation, namely, ICaL, Jrel, and Jup. Under oxidative stress conditions, intracellular Ca2+ concentration significantly increased (Figure 5(j)). To investigate the role of [Ca2+]i regulation in the genesis of EAD, decreased ICaL, reduced Jrel, and enhanced Jup were independently applied to reduce the [Ca2+]i under oxidative stress conditions, as shown in Figure 8. Figure 8(a) shows that decreasing ICaL by 10% abolished EAD. This was due not only to [Ca2+]i decline but also to a decrease in ICaL reactivation. Reducing SR Ca2+ release postponed the occurrence of EAD but did not successfully eliminate EAD (Figure 8(b)). Although a reduction in SR Ca2+ release temporarily reduced [Ca2+]i, it caused Ca2+ accumulation in SR, which finally induced [Ca2+]i elevation (data not shown) and EAD. Increasing SR Ca2+ uptake by 10% had a similar effect to that of reducing SR calcium release (Figure 8(c)). Nonetheless, further increasing Jup by 20%–30% abolished EAD. In these two cases, the SR Ca2+ uptake and release reached a new balance and maintained [Ca2+]i and [Ca2+]SR at a steady state (data not shown).
Figure 8.
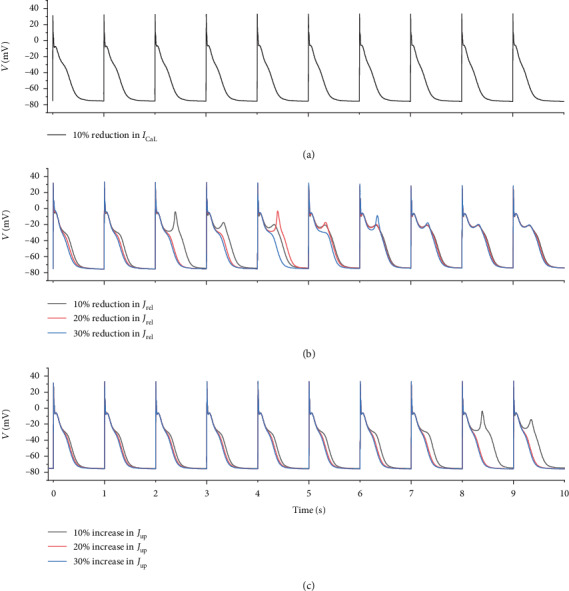
Effect of (a) reducing ICaL by 10%, (b) reducing Jrel by 10%, 20%, and 30%, and (c) increasing Jup by 10%, 20%, and 30% on the genesis of EAD under oxidative stress conditions.
3.4. Effect of Oxidative Stress on Excitation Wave Propagation in 2D Tissue
The excitation wave propagation was investigated in 2D ideal tissue under control and oxidative stress conditions. Spiral waves were induced using the S1–S2 protocol. Under control conditions with the S1–S2 interval of 550 ms, a stationary spiral wave was produced, with its tip anchored in the central region of the square tissue, as shown in Figure 9. Under oxidative stress conditions, the ROS concentration was set to 0.2 mM for all nodes in the tissue. In this case with the S1–S2 interval of 730 ms, a nonstationary spiral wave was induced. As the EAD was induced under this condition, the region to which the S2 stimulus was applied showed a prolonged depolarization (Figure 10, 1125 ms) and a second wavefront was induced at 1450 ms, as shown in Figure 10. When the wavefronts reached the region to which the S2 stimulus was applied (Figure 10, 1650 ms), the nodes were not fully repolarized, owing to the prolonged APD induced by EAD. Therefore, the excitation wave propagation was suppressed (Figure 10, 1650 ms). Finally, the tip of the spiral wave wandered across the tissue and produced a nonstationary spiral wave. When oxidative stress only occurred in the right third of the tissue, the spatial heterogeneity further aggravated the instability of the nonstationary spiral wave and gave rise to a breakup of the spiral wave (data not shown).
Figure 9.
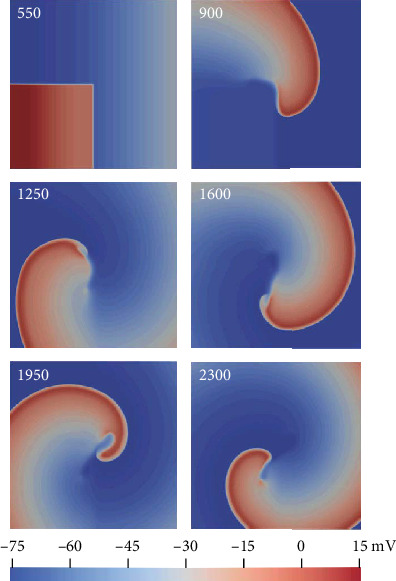
Spiral wave induced under control conditions using the S1–S2 protocol. The number in each panel represents the recording time.
Figure 10.

Spiral wave induced under oxidative stress conditions using the S1–S2 protocol. The number in each panel represents the recording time.
4. Discussion
In this study, the effects of ROS-dependent CaMKII activation on regulations of intracellular Ca2+ and ionic currents were investigated in silico using the updated human atrial cell model including the CaMKII model with autophosphorylation- and oxidation-dependent activation at the single-cell level and 2D tissue model. Our major findings follow: (i) A new Markov chain model of CaMKII, including both the autophosphorylation and oxidation pathways, was developed, and the effects of CaMKII on ion channels and Ca2+ cycling were incorporated into the computational model of the atrial myocyte. (ii) The mechanisms of oxidative stress-induced EADs in atrial cells were thoroughly examined which are helpful to further understand or investigate the mechanisms underlying oxidative stress-induced AF. The simulation results at the single-cell level illustrated that oxidative stress resulted in EADs of the AP at the normal pacing rate of 1 Hz. It was contributed by reactivation of ICaL and intracellular Ca2+ elevation induced by CaMKII activation under oxidative stress conditions. (iii) The 2D simulations provide insights into reentry in atrial tissue under the oxidative stress condition, which plays significant roles in AF mechanisms. In 2D simulation, oxidative stress aggravated the instability of the excitation wave and gave rise to a nonstationary spiral wave, owing to the EAD induced by oxidative stress hindering the electrical conduction.
4.1. Model Development of ROS-Dependent CaMKII Activation
Previous experimental studies have revealed that both ROS and CaMKII are associated with the development of atrial arrhythmia [4, 5]. In addition, it has recently been reported that CaMKII can keep persistent activity by oxidation [24]. However, the role of ROS-dependent CaMKII activation in the genesis of atrial fibrillation is not yet well understood. In this study, we developed a computational model of human atrial cell including both ROS-dependent and autophosphorylation-dependent CaMKII activation to investigate effects of CaMKII activation on atrial electrophysiology under oxidative stress condition. In this CaMKII model, a pathway of ROS-dependent CaMKII activation was introduced which was different from previous CaMKII models only including the autophosphorylation-dependent CaMKII activation [34, 35, 50]. Christensen et al. [39] also developed a model of CaMKII activity including oxidation and autophosphorylation activation pathways. However, different from their model, the oxidation and autophosphorylation activation pathways in our six-state CaMKII are mutual independence, implying that there is no such a state with both oxidation and autophosphorylation activation simultaneously. This fact is consistent with the experimental observations reported in [24].
The effect of CaMKII on INa, ICaL, RyR, and PLB was integrated into the human atrial myocyte model, producing action potential characteristics (such as RMP, dV/dtmax, APA, APD90, and the calcium transient amplitude) consistent with previous models and experimental data of human atrial myocytes [33, 43–49] (Figure 2 and Table 1).
4.2. Mechanisms Underlying the Genesis of EAD under Oxidative Stress Conditions
The generation of EADs associated with ectopic (triggered) activity can be contributed by CaMKII activation [51–55]. Animal experiments have shown that oxidative stress can promote EADs in the ventricle of guinea pigs and rabbits [56]. Therefore, this study explored the intrinsic mechanisms of EADs associated with atrial arrhythmia induced by oxidative stress, using the developed CaMKII model.
First, simulation results demonstrated that the characteristics of AP and calcium cycling generated by our model were consistent with previous computational and experimental studies [43–49]. After model validation, the role of oxidation-dependent CaMKII activation on the AP was investigated in a single atrial cell model. The simulation results demonstrated that, under oxidative stress conditions, increasing ROS concentrations in the cytoplasm enhanced CaMKII activation and consequently augmented ICaL, RyR phosphorylation, and PLB phosphorylation (Figures 5(c), 5(e), and (f)), inducing an enhanced Ca2+ influx, a larger SR Ca2+ release, and a promoted SR Ca2+ uptake (Figures 5(g) and 5(h)). These effects generated a remarkable elevation in intracellular Ca2+ concentration and therefore enlarged INCX via promoted Ca2+ extrusion (Figure 5(d)). INCX augmentation provided a depolarization component to counterbalance the repolarization reserve and prolong the APD. The elevated and prolonged AP in phase 3 gradually caused reactivation of ICaL (Figure 5(c)), which ultimately induced action potential depolarization and produced EAD (Figure 5(a)), consistent with previous studies [57–59].
In this process, as ICaL was the direct depolarization current inducing EAD, blocking ICaL completely abolished EAD. However, blocking INCX at different time points had different effects on EAD. If INCX was blocked before ICaL reactivation could induce AP depolarization, EAD was eliminated (Figure 7). This phenomenon suggested that INCX augmentation acted as a trigger of ICaL reactivation. EAD can be induced only when the trigger is large enough. Under oxidative stress condition, ROS elevation did not affect INCX directly and the increase of [Ca2+]i primarily accounted for INCX augmentation. Therefore, the effects of ROS-induced CaMKII activation on intracellular calcium cycling also played a crucial role in the genesis of EAD.
To examine the role of ROS-induced [Ca2+]i elevation in the genesis of EAD, Ca2+ influx and SR Ca2+ release and uptake were modified to reduce ROS-induced [Ca2+]i elevation. First, ICaL was reduced by 10% to inhibit Ca2+ influx, causing a decline of [Ca2+]i. Together with the decreased reactivation of ICaL, the [Ca2+]i decline caused by reducing ICaL abolished EAD (Figure 8(a)). Second, partial inhibition of SR Ca2+ release (by 10%–30%) temporarily reduced [Ca2+]i. However, inhibition of SR Ca2+ release gradually caused SR Ca2+ accumulation and ultimately gave rise to further [Ca2+]i elevation, which accounted for the fact that partial inhibition of SR Ca2+ release could only postpone the occurrence of EAD but could not abolish EAD (Figure 8(b)). Increasing SR Ca2+ uptake by 10% had a similar effect to partial inhibition of SR Ca2+ release. Interestingly, on further increasing SR Ca2+ uptake by 20% and 30%, [Ca2+]i declined and EAD was abolished. This phenomenon might be attributed to a new balance of increased SR Ca2+ uptake and ROS-induced [Ca2+]i release (Figure 8(c)). Therefore, the dynamic balance between SR Ca2+ release and uptake is the key factor in maintaining [Ca2+]i at a steady state.
4.3. Effect of Rate-Dependent CaMKII Activation on EAD under Oxidative Stress Conditions
Under control conditions, CaMKII activation gradually decreased with a decrease in the pacing rate, as Ca2+ concentration gradually decreased at low pacing rates. However, under oxidative stress conditions, once CaMKII was activated, the high ROS concentration tended to maintain CaMKII activation. Therefore, under oxidative stress conditions, fractions of CaMKII activation at different pacing rates were similar and maintained at a high level (about 75%). This indicated that, at low pacing rates, ROS-induced CaMKII activation played a more important role. At low pacing rates, intracellular Ca2+ concentration could be dramatically increased by ROS-induced CaMKII activation and therefore could induce more EADs (Figure 6). Although the heart rate is not so slow such as 0.5 Hz and 0.33 Hz under normal conditions, it may be such slow under some pathological conditions, such as bradycardia [60]. The purpose of exploring up to such slow pacing frequencies is to investigate whether the occurrence of EAD is linear with the decrease of pacing frequency or not. The results showed that the occurrence of EAD was more dependent on ROS-induced CaMKII activation rather than the pacing frequencies.
4.4. Effect of Oxidative Stress on Excitation Wave Propagation in 2D Tissue
The spiral wave is a well-known approach to understanding reentry in cardiac tissue, which plays significant roles in AF mechanisms [61]. This study investigated the stability of the spiral wave in 2D ideal tissue under control and oxidative stress conditions. In tissue with a uniform distribution of oxidative stress (Figure 10), the region to which the S2 stimulus was applied had a prolonged depolarization phase, owing to the ROS-induced EAD, forming a spiral wave barrier. In addition, the ROS-induced EAD produced a second excitation wavefront during wave propagation. These effects finally produced a nonstationary spiral wave. In the case of nonuniform distribution of oxidative stress, a breakup of the spiral wave was induced, which might disrupt normal cardiac rhythm and cause atrial arrhythmia [62–64].
4.5. Model Limitations and Future Directions
The present model was based on models of Grandi et al. [33] and O'Hara et al. [35] and thus inherited the same limitations of both models. The main limitation of the current model, which will be addressed in the future, is that the ROS concentration is constant. In future versions, pathways of ROS production and scavenging should be added. Another issue for model development is to expand the model into real 2D heart tissue and 3D organ models, to explore electrical wave propagation induced by abnormal action potentials, and electrocardiography in real heart geometry.
Although the S1–S2 interval in 2D simulation to produce the spiral wave was dependent on the tissue size, it is reasonable to investigate the difference in the stability of the spiral wave using the same tissue size under control and oxidative stress conditions. Further work is required to measure the range of S1–S2 intervals for producing the spiral wave (vulnerable window) varying from control to oxidative stress conditions to further determine the proarrhythmic effect of oxidative stress.
5. Conclusions
In this study, we have investigated the role of oxidation-dependent CaMKII activation in the genesis of abnormal action potentials in atria. It was shown that, at the atrial cell level, oxidation-dependent CaMKII activation-induced ICaL reactivation and [Ca2+]i elevation contributed to EAD generation. Moreover, at the 2D tissue level, oxidative stress-induced EAD contributed to the instability of excitation waves, facilitating atrial arrhythmia. This study investigated the role of ROS-dependent CaMKII activation in the development of atrial arrhythmias, shedding light on the genesis of atrial arrhythmias under oxidative stress conditions.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant numbers 61601143, 81770328, and 61572152) and the Heilongjiang and China Postdoctoral Science Foundation (grant number 2015M581448).
Contributor Information
Qince Li, Email: qinceli@hit.edu.cn.
Henggui Zhang, Email: henggui.zhang@manchester.ac.uk.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Authors' Contributions
Qince Li, Henggui Zhang, and Na Zhao conceived the study and designed the simulations. Na Zhao and Haibo Sui developed the model and conducted the simulations. The paper was written by Qince Li. Qince Li and Henggui Zhang analyzed the simulation results. Qince Li and Na Zhao contributed equally to this work. All the authors read and approved the final manuscript.
Supplementary Materials
Figure S1: The CaMKII model. B, CaMKII-CaMCa4 (bound state); BO, CaMKIIOx-CaMCa4 (oxidized and bound state); BP, CaMKIIP-CaMCa4 (phosphorylated and bound state); I, CaMKII (inactive state); O, CaMKIIOx (oxidized state); P, CaMKIIP (phosphorylated state). Table S1: Parameters of CaMKII model
References
- 1.Wyndham C. R. Atrial fibrillation: the most common arrhythmia. Texas Heart Institute Journal. 2000;27(3):257–267. [PMC free article] [PubMed] [Google Scholar]
- 2.Wijffels M. C. E. F., Kirchhof C. J. H. J., Dorland R., Power J., Allessie M. A. Electrical remodeling due to atrial fibrillation in chronically instrumented conscious goats. Circulation. 1997;96(10):3710–3720. doi: 10.1161/01.CIR.96.10.3710. [DOI] [PubMed] [Google Scholar]
- 3.Allessie M. A. Atrial electrophysiologic remodeling: another vicious circle? Journal of Cardiovascular Electrophysiology. 1998;9(12):1378–1393. doi: 10.1111/j.1540-8167.1998.tb00114.x. [DOI] [PubMed] [Google Scholar]
- 4.Wagner S., Rokita A. G., Anderson M. E., Maier L. S. Redox regulation of sodium and calcium handling. Antioxidants & Redox Signaling. 2013;18(9):1063–1077. doi: 10.1089/ars.2012.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swaminathan P. D., Purohit A., Hund T. J., Anderson M. E. Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circulation Research. 2012;110(12):1661–1677. doi: 10.1161/CIRCRESAHA.111.243956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang T., Brown J. H. Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovascular Research. 2004;63(3):476–486. doi: 10.1016/j.cardiores.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 7.Hudmon A., Schulman H., Kim J., Maltez J. M., Tsien R. W., Pitt G. S. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. The Journal of Cell Biology. 2005;171(3):537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dzhura I., Wu Y., Colbran R. J., Balser J. R., Anderson M. E. Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nature Cell Biology. 2000;2(3):173–177. doi: 10.1038/35004052. [DOI] [PubMed] [Google Scholar]
- 9.Blaich A., Welling A., Fischer S., et al. Facilitation of murine cardiac L-type Cav1.2 channel is modulated by calmodulin kinase II-dependent phosphorylation of S1512 and S1570. Proceedings of the National Academy of Sciences. 2010;107(22):10285–10289. doi: 10.1073/pnas.0914287107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner S., Dybkova N., Rasenack E. C. L., et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. The Journal of Clinical Investigation. 2006;116(12):3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagner S., Ruff H. M., Weber S. L., et al. Reactive oxygen species–activated Ca/calmodulin kinase IIδ is required for LateINaAugmentation leading to cellular Na and Ca overload. Circulation Research. 2011;108(5):555–565. doi: 10.1161/CIRCRESAHA.110.221911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maier L. S., Bers D. M. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation–contraction coupling in the heart. Cardiovascular Research. 2007;73(4):631–640. doi: 10.1016/j.cardiores.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Maier L. S., Zhang T., Chen L., DeSantiago J., Brown J. H., Bers D. M. Transgenic CaMKIIδCOverexpression uniquely alters cardiac myocyte Ca2+Handling. Circulation Research. 2003;92(8):904–911. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- 14.Wehrens X. H. T., Lehnart S. E., Reiken S. R., Marks A. R. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circulation Research. 2004;94(6):e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 15.Guo T., Zhang T., Mestril R., Bers D. M. Ca2+/calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circulation Research. 2006;99(4):398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- 16.Kohlhaas M., Zhang T., Seidler T., et al. Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circulation Research. 2006;98(2):235–244. doi: 10.1161/01.RES.0000200739.90811.9f. [DOI] [PubMed] [Google Scholar]
- 17.Sag C. M., Wolff H. A., Neumann K., et al. Ionizing radiation regulates cardiac Ca handling via increased ROS and activated CaMKII. Basic research in cardiology. 2013;108(6):p. 385. doi: 10.1007/s00395-013-0385-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho H.-T., Liu B., Snyder J. S., et al. Ryanodine receptor phosphorylation by oxidized CaMKII contributes to the cardiotoxic effects of cardiac glycosides. Cardiovascular Research. 2014;101(1):165–174. doi: 10.1093/cvr/cvt233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erickson J. R., He B. J., Grumbach I. M., Anderson M. E. CaMKII in the cardiovascular system: sensing redox states. Physiological Reviews. 2011;91(3):889–915. doi: 10.1152/physrev.00018.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simmerman H. K., Collins J. H., Theibert J. L., Wegener A. D., Jones L. R. Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. Journal of Biological Chemistry. 1986;261(28):13333–13341. [PubMed] [Google Scholar]
- 21.Brittsan A. G., Kranias E. G. Phospholamban and cardiac contractile function. Journal of Molecular and Cellular Cardiology. 2000;32(12):2131–2139. doi: 10.1006/jmcc.2000.1270. [DOI] [PubMed] [Google Scholar]
- 22.Anderson M. E., Braun A. P., Schulman H., Premack B. A. Multifunctional Ca2+/calmodulin-dependent protein kinase mediates Ca(2+)-induced enhancement of the L-type Ca2+ current in rabbit ventricular myocytes. Circulation Research. 1994;75(5):854–861. doi: 10.1161/01.RES.75.5.854. [DOI] [PubMed] [Google Scholar]
- 23.Yuan W., Bers D. M. Ca-dependent facilitation of cardiac Ca current is due to Ca-calmodulin-dependent protein kinase. American Journal of Physiology-Heart and Circulatory Physiology. 1994;267(3):H982–H993. doi: 10.1152/ajpheart.1994.267.3.h982. [DOI] [PubMed] [Google Scholar]
- 24.Erickson J. R., Joiner M.-l. A., Guan X., et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133(3):462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Q., Su D., O'Rourke B., Pogwizd S. M., Zhou L. Mitochondria-derived ROS bursts disturb Ca2+ cycling and induce abnormal automaticity in guinea pig cardiomyocytes: a theoretical study. American Journal of Physiology-Heart and Circulatory Physiology. 2015;308(6):H623–H636. doi: 10.1152/ajpheart.00493.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Q., Pogwizd S. M., Prabhu S. D., Zhou L. Xie L.-H., editor. Inhibiting Na+/K+ ATPase can impair mitochondrial energetics and induce abnormal Ca2+ cycling and automaticity in guinea pig cardiomyocytes. PLoS One. 2014;9(4):p. e93928. doi: 10.1371/journal.pone.0093928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zorov D. B., Juhaszova M., Sollott S. J. Mitochondrial ROS-induced ROS release: an update and review. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2006;1757(5–6):509–517. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 28.Palomeque J., Rueda O. V., Sapia L., et al. Angiotensin II–induced oxidative stress resets the Ca2+ dependence of Ca2+–calmodulin protein kinase II and promotes a death pathway conserved across different species. Circulation Research. 2009;105(12):1204–1212. doi: 10.1161/CIRCRESAHA.109.204172. [DOI] [PubMed] [Google Scholar]
- 29.Swaminathan P. D., Purohit A., Soni S., et al. Oxidized CaMKII causes cardiac sinus node dysfunction in mice. The Journal of Clinical Investigation. 2011;121(8):3277–3288. doi: 10.1172/JCI57833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He B. J., Joiner M.-l. A., Singh M. V., et al. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nature Medicine. 2011;17(12):1610–1618. doi: 10.1038/nm.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie L.-H., Chen F., Karagueuzian H. S., Weiss J. N. Oxidative stress–induced afterdepolarizations and calmodulin kinase II signaling. Circulation Research. 2009;104(1):79–86. doi: 10.1161/CIRCRESAHA.108.183475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang S., Li Q., Zhou L., Wang K., Zhang H. Development of a novel Markov chain model for oxidative-dependent CaMKIIδ activation. 2015 Computing in Cardiology Conference (CinC); September, 2015; Nice, France. pp. 881–884. IEEE. [DOI] [Google Scholar]
- 33.Grandi E., Pandit S. V., Voigt N., et al. Human atrial action potential and Ca2+ model. Circulation Research. 2011;109(9):1055–1066. doi: 10.1161/CIRCRESAHA.111.253955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiba H., Schneider N. S., Matsuoka S., Noma A. A Simulation Study on the Activation of Cardiac CaMKII _δ_ -Isoform and Its Regulation by Phosphatases. Biophysical Journal. 2008;95(5):2139–2149. doi: 10.1529/biophysj.107.118505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O'Hara T., Virág L., Varró A., Rudy Y. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS computational biology. 2011;7(5, article e1002061) doi: 10.1371/journal.pcbi.1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Christ T., Boknik P., Wöhrl S., et al. L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation. 2004;110(17):2651–2657. doi: 10.1161/01.CIR.0000145659.80212.6A. [DOI] [PubMed] [Google Scholar]
- 37.Soltis A. R., Saucerman J. J. Synergy between CaMKII substrates and β-adrenergic signaling in regulation of cardiac myocyte Ca2+ handling. Biophysical Journal. 2010;99(7):2038–2047. doi: 10.1016/j.bpj.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.ten Tusscher K. H. W. J., Panfilov A. V. Alternans and spiral breakup in a human ventricular tissue model. American Journal of Physiology-heart and Circulatory Physiology. 2006;291(3):H1088–H1100. doi: 10.1152/ajpheart.00109.2006. [DOI] [PubMed] [Google Scholar]
- 39.Christensen M. D., Dun W., Boyden P. A., Anderson M. E., Mohler P. J., Hund T. J. Oxidized calmodulin kinase II regulates conduction following myocardial infarction: a computational analysis. PLOS Computational Biology. 2009;5(12, article e1000583) doi: 10.1371/journal.pcbi.1000583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ni H., Whittaker D. G., Wang W., Giles W. R., Narayan S. M., Zhang H. Synergistic anti-arrhythmic effects in human atria with combined use of sodium blockers and acacetin. Frontiers in Physiology. 2017;8 doi: 10.3389/fphys.2017.00946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ni H., Adeniran I., Zhang H. _In-silico_ investigations of the functional impact of _KCNA5_ mutations on atrial mechanical dynamics. Journal of Molecular and Cellular Cardiology. 2017;111:86–95. doi: 10.1016/j.yjmcc.2017.08.005. [DOI] [PubMed] [Google Scholar]
- 42.Whittaker D. G., Colman M. A., Ni H., Hancox J. C., Zhang H. Human atrial arrhythmogenesis and sinus bradycardia in KCNQ1-linked short QT syndrome: insights from computational modelling. Frontiers in Physiology. 2018;9, article 1402 doi: 10.3389/fphys.2018.01402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Courtemanche M., Ramirez R. J., Nattel S. Ionic mechanisms underlying human atrial action potential properties: insights from a mathematical model. American Journal of Physiology-Heart and Circulatory Physiology. 1998;275(1):H301–H321. doi: 10.1152/ajpheart.1998.275.1.h301. [DOI] [PubMed] [Google Scholar]
- 44.Nygren A., Fiset C., Firek L., et al. Mathematical model of an adult human atrial cell. Circulation research. 1998;82(1):63–81. doi: 10.1161/01.RES.82.1.63. [DOI] [PubMed] [Google Scholar]
- 45.Dawodu A. A., Monti F., Iwashiro K., Schiariti M., Chiavarelli R., Puddu P. E. The shape of human atrial action potential accounts for different frequency- related changes in vitro. International Journal of Cardiology. 1996;54(3):237–249. doi: 10.1016/0167-5273(96)02605-8. [DOI] [PubMed] [Google Scholar]
- 46.Katoh H., Shinozaki T., Baba S., et al. Monophasic action potential duration at the crista terminalis in patients with sinus node disease. Circulation. 2005;69(11):1361–1367. doi: 10.1253/circj.69.1361. [DOI] [PubMed] [Google Scholar]
- 47.Bosch R. F., Zeng X., Grammer J. B., Popovic K., Mewis C., Kühlkamp V. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovascular Research. 1999;44(1):121–131. doi: 10.1016/S0008-6363(99)00178-9. [DOI] [PubMed] [Google Scholar]
- 48.Kim B.-S., Kim Y.-H., Hwang G.-S., et al. Action potential duration restitution kinetics in human atrial fibrillation. Journal of the American College of Cardiology. 2002;39(8):1329–1336. doi: 10.1016/S0735-1097(02)01760-6. [DOI] [PubMed] [Google Scholar]
- 49.Colman M. A., Aslanidi O. V., Kharche S., et al. Pro-arrhythmogenic effects of atrial fibrillation-induced electrical remodelling: insights from the three-dimensional virtual human atria. The Journal of Physiology. 2013;591(17):4249–4272. doi: 10.1113/jphysiol.2013.254987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hund T. J., Rudy Y. Rate dependence and regulation of action potential and calcium transient in a canine cardiac ventricular cell model. Circulation. 2004;110(20):3168–3174. doi: 10.1161/01.CIR.0000147231.69595.D3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anderson M. E., Braun A. P., Wu Y., et al. KN-93, an inhibitor of multifunctional Ca++/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. Journal of Pharmacology and Experimental Therapeutics. 1999;287(3):996–1006. [PubMed] [Google Scholar]
- 52.Wu Y., MacMillan L. B., McNeill R. B., Colbran R. J., Anderson M. E. CaM kinase augments cardiac L-type Ca2+ current: a cellular mechanism for long Q-T arrhythmias. American Journal of Physiology-heart and Circulatory Physiology. 1999;276(6):H2168–H2178. doi: 10.1152/ajpheart.1999.276.6.h2168. [DOI] [PubMed] [Google Scholar]
- 53.Wu Y., Temple J., Zhang R., et al. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106(10):1288–1293. doi: 10.1161/01.CIR.0000027583.73268.E7. [DOI] [PubMed] [Google Scholar]
- 54.Bers D. M., Morotti S. Ca2+ current facilitation is CaMKII-dependent and has arrhythmogenic consequences. Frontiers in Pharmacology. 2014;5, article 144 doi: 10.3389/fphar.2014.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heijman J., Voigt N., Wehrens X. H. T., Dobrev D. Calcium dysregulation in atrial fibrillation: the role of CaMKII. Frontiers in Pharmacology. 2014;5, article 30 doi: 10.3389/fphar.2014.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song Y., Shryock J. C., Wagner S., Maier L. S., Belardinelli L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. Journal of Pharmacology and Experimental Therapeutics. 2006;318(1):214–222. doi: 10.1124/jpet.106.101832. [DOI] [PubMed] [Google Scholar]
- 57.Qi X. Y., Yeh Y.-H., Chartier D., et al. The calcium/calmodulin/kinase system and arrhythmogenic afterdepolarizations in bradycardia-related acquired long-QT syndrome. Circulation. Arrhythmia and Electrophysiology. 2009;2(3):295–304. doi: 10.1161/CIRCEP.108.815654. [DOI] [PubMed] [Google Scholar]
- 58.Burashnikov A., Antzelevitch C. Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarization–induced triggered activity. Circulation. 2003;107(18):2355–2360. doi: 10.1161/01.CIR.0000065578.00869.7C. [DOI] [PubMed] [Google Scholar]
- 59.Patterson E., Lazzara R., Szabo B., et al. Sodium-calcium exchange initiated by the Ca2+ transient. Journal of the American College of Cardiology. 2006;47(6):1196–1206. doi: 10.1016/j.jacc.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 60.Kusumoto F. M., Schoenfeld M. H., Barrett C., et al. 2018 ACC/AHA/HRS guideline on the evaluation and management of patients with bradycardia and cardiac conduction delay: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines and the Heart Rhythm Society. Journal of the American College of Cardiology. 2019;74(7):e51–e156. doi: 10.1016/j.jacc.2018.10.044. [DOI] [PubMed] [Google Scholar]
- 61.Nattel S., Xiong F., Aguilar M. Demystifying rotors and their place in clinical translation of atrial fibrillation mechanisms. Nature Reviews Cardiology. 2017;14(9):509–520. doi: 10.1038/nrcardio.2017.37. [DOI] [PubMed] [Google Scholar]
- 62.Jalife J., Berenfeld O., Mansour M. Mother rotors and fibrillatory conduction: a mechanism of atrial fibrillation. Cardiovascular Research. 2002;54(2):204–216. doi: 10.1016/S0008-6363(02)00223-7. [DOI] [PubMed] [Google Scholar]
- 63.Kneller J., Zou R., Vigmond E. J., Wang Z., Leon L. J., Nattel S. Cholinergic atrial fibrillation in a computer model of a two-dimensional sheet of canine atrial cells with realistic ionic properties. Circulation Research. 2002;90(9):E73–E87. doi: 10.1161/01.res.0000019783.88094.ba. [DOI] [PubMed] [Google Scholar]
- 64.Kneller J., Kalifa J., Zou R., et al. Mechanisms of atrial fibrillation termination by pure sodium channel blockade in an ionically-realistic mathematical model. Circulation Research. 2005;96(5):e35–e47. doi: 10.1161/01.RES.0000160709.49633.2b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: The CaMKII model. B, CaMKII-CaMCa4 (bound state); BO, CaMKIIOx-CaMCa4 (oxidized and bound state); BP, CaMKIIP-CaMCa4 (phosphorylated and bound state); I, CaMKII (inactive state); O, CaMKIIOx (oxidized state); P, CaMKIIP (phosphorylated state). Table S1: Parameters of CaMKII model
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.