

Abstract
Tumor cells, especially drug-resistant cells, predominately support growth by glycolysis even under the condition of adequate oxygen, which is known as the Warburg effect. Glucose metabolism reprogramming is one of the main factors causing tumor resistance. Previous studies on Shenmai injection (SMI), a Chinese herbal medicine, have shown enhanced efficacy in the treatment of tumors in combination with chemotherapy drugs, but the mechanism is not clear. In this study, we investigated the effect of SMI combined with cisplatin on cisplatin-resistant lung adenocarcinoma A549/DDP cells. Our results showed that cisplatin-resistant A549/DDP cells exhibited increased glucose consumption, lactate production, and expression levels of key glycolytic enzymes, including hexokinase 2 (HK2), pyruvate kinase M1/2 (PKM1/2), pyruvate kinase M2 (PKM2), glucose transporter 1 (GLUT1), and lactate dehydrogenase A (LDHA), compared with cisplatin-sensitive A549 cells. SMI combined with cisplatin in A549/DDP cells, led to significantly lower expression levels of key glycolytic enzymes, such as HK2, PKM1/2, GLUT1, and pyruvate dehydrogenase (PDH). In addition, we found that the combination of SMI and cisplatin could inhibit cell proliferation and promote apoptosis by reducing the expression levels of p-Akt, p-mTOR, and c-Myc, and then, it reduced the glycolysis level. These results suggest that SMI enhances the antitumor effect of cisplatin via glucose metabolism reprogramming. Therefore, the combination of SMI and cisplatin may be a potential therapeutic strategy to treat cisplatin-resistant nonsmall cell lung cancer.
1. Introduction
The antitumor activities of cisplatin, such as induction of DNA damage and mitochondrial apoptosis, have been widely used in chemotherapy for many kinds of tumors, especially for advanced lung cancer [1]. Long-term cisplatin treatment partially leads to a variety of glucose metabolic pathways, including the glycolysis level and the expression of key enzymes, resulting in poor treatment with cisplatin, but the precise cisplatin resistance mechanism has not been completely understood [2, 3].
Shenmai injection (SMI) is derived from Shengmai San, the well-known Chinese medicine prescription, which consists of Radix Ginseng Rubra and Radix Ophiopogonis [4]. SMI is used to improve myocardial function and enhance immunity; recently, it has been found to increase the therapeutic effect combined with chemotherapy drugs in antitumor treatment [5, 6]. Recently, Liu reported that SMI enhances the cytotoxicity of chemotherapy drugs against colorectal cancer by improving the distribution of drugs in cells [7]. SMI has an obvious inhibitory effect on various tumors in mice, which effectively prolongs the survival time of tumor-bearing mice [8]. However, the exact antitumor mechanism of SMI is still unknown.
In this study, we first evaluated the difference in glycolysis metabolism between cisplatin sensitive cells (human lung adenocarcinoma cell line A549) and cisplatin-resistant cells (A549/DDP cells), and subsequently, we explored the antitumor mechanism of SMI in reversing cisplatin resistance in A549/DDP cells.
2. Materials and Methods
2.1. Cell Lines and Cell Culture
Human lung adenocarcinoma cell line (A549) was purchased from the Beijing Dingguo Changsheng Biotechnology Company (Beijing, China). Human lung adenocarcinoma cisplatin-resistant cell line (A549/DDP) was purchased from the Cancer Hospital of Chinese Academy of Medical Sciences (Beijing, China). The cells were cultured in Dulbecco's Modified Eagle Medium/High Glucose (DMEM/High Glucose) (Hyclone, Logan, UT, USA) containing 10% fetal bovine serum (Scitecher, Oxford, MS, USA), 100 U/mL penicillin, and 100 mg/mL streptomycin (Genview, Australia), and they were cultivated at 37°C in a 5% CO2 incubator. The A549/DDP cell medium contained 16.7 μM cisplatin to maintain drug resistance for 2 weeks, and no cisplatin for 2 weeks before the experiment, the stability of cisplatin resistance in A549/DDP cells can be maintained for one month.
2.2. Antibodies and Reagents
SMI (the concentration of crude drug was 200 mg/ml) was obtained from Shineway Pharmaceutical Co., Ltd. (Hebei, China). Cisplatin was purchased from Sigma Aldrich Company (St. Louis, MO, USA). The antibodies used in the experiment are shown in Table 1.
Table 1.
Antibodies used in the experiment.
| Antibody name | Product code | Source | Host | Dilution |
|---|---|---|---|---|
| HK2 | #2867 | Cell signaling technology | Rabbit | 1 : 1000 |
| PFKP | #5412 | Cell signaling technology | Rabbit | 1 : 1000 |
| PKM2 | #4053 | Cell signaling technology | Rabbit | 1 : 1000 |
| PKM1/2 | #3186 | Cell signaling technology | Rabbit | 1 : 1000 |
| PDHX | #3205 | Cell signaling technology | Rabbit | 1 : 1000 |
| AKT | #4691 | Cell signaling technology | Rabbit | 1 : 500 |
| p-Akt (Thr308) | #2965 | Cell signaling technology | Rabbit | 1 : 1000 |
| p-Akt (Ser473) | 33281 M | Bioss | Mouse | 1 : 500 |
| mTOR | #2983 | Cell signaling technology | Rabbit | 1 : 1000 |
| p-mTOR (Ser2448) | #5536 | Cell signaling technology | Rabbit | 1 : 1000 |
| HIF-1α | #3716 | Cell signaling technology | Rabbit | 1 : 1000 |
| HK1 | ab65069 | Abcom | Rabbit | 1 : 500 |
| GLUT1 | ab652 | Abcom | Rabbit | 1 : 500 |
| LDHA | ab125683 | Abcom | Rabbit | 1 : 1000 |
| β-Actin | ab 179467 | Abcom | Rabbit | 1 : 1000 |
| Caspase 3/p17/19 | 19677-10-AP | Proteintech | Rabbit | 1 : 1000 |
| BAX | 50599-2-lg | Proteintech | Rabbit | 1 : 4000 |
| Bcl 2 | 26593-1-AP | Proteintech | Rabbit | 1 : 1000 |
| Goat anti-rabbit IgG | ZB-2301 | Zsbio | Goat | 1 : 5000 |
| Goat anti-mouse IgG | ZB-2305 | Zsbio | Goat | 1 : 5000 |
2.3. Inhibition Assay of SMI and Cisplatin
Inhibition rate assay of SMI alone and cisplatin alone is in A549/DPP cells. Briefly, 5 × 103 A549/DDP cells were seeded into a 96-well plate and cultured for 12 h. Then the cell culture medium was discarded and treated with various concentrations of SMI (4, 8, 16, 32, 64, and 128 mg/ml) or cisplatin (12, 24, 48, 96, and 192 μM) for 24 h. Further, 10 μL of cell counting kit-8 solution was added (Apexbio, Houston, TX, USA) to each plate, and the plate was incubated for 4 h. The absorbance of each individual experiment at 460 nm was detected at least three times. IC5, IC10, IC20, and IC50 values were calculated via using linear regression on the inhibition rate curve of SMI or cisplatin. Inhibition rate = (ODcontol − ODexperiment)/(ODcontol − ODblank) × 100%.
Inhibition rate of SMI combined with cisplatin in cisplatin-resistant A549/DPP cells. Briefly, 5 × 103 A549/DDP cells were seeded into a 96-well plate and cultured for 12 hours. The cell culture medium was discarded, and combined cisplatin and different concentrations of SMI at concentrations of 10 mg/mL, 15 mg/mL, and 20 mg/mL (equivalent to IC5, IC10, and IC20) were added to the culture for 24 h. Further, 10 μL of cell counting kit-8 solution was added to each plate, and the plate was incubated for 4 h. The measurement and calculation method was the same as above, no SMI as the control.
2.4. Glucose Consumption and Lactate Production Assay
Briefly, 2 × 105 A549/DDP cells were seeded into 6-well plates and cultured for 24 h. The culture medium and cells were collected separately, and glucose concentration and lactate production were measured using the Glucose assay kit (Applygen, Beijing, China) and the Lactic Acid assay (Jiancheng Bioengineering Institute, Nanjing, China) in accordance with the manufacturers' protocols, and the glucose consumption level or the lactate production level was divided by the number of cells.
2.5. 5-Ethynyl-2′-Deoxyuridine (EdU) Assay
Briefly, 2.5 × 104 A549/DDP cells were seeded into a 24-well plate. SMI at concentrations of IC5, IC10, and IC20 was combined with the cisplatin concentration of IC20 to treat A549/DDP cells for 24 h, and then, 500 μL 1×EdU solution (final concentration 10 μM) was added and incubated for 2 hours. Phosphate buffered saline (PBS) was used to wash the cells and 4% paraformaldehyde was used to fix the cells for 30 min. EdU staining and DNA staining were performed by the EdU Kit (Donghuan Biotech, Shanghai, China), and images were acquired by fluorescence microscopy (Zeiss, Germany).
2.6. Inhibitor Assay
Briefly, 2 × 105 A549/DDP cells were seeded on 6-well plates to adhere well. LY294002 (phosphorylated Akt inhibitor; Abmole Bioscience, Shanghai, China; 20 μM), rapamycin (phosphorylated mTOR inhibitor; Abmole Bioscience, Shanghai, China; 5 nM), and 10058-F4 (c-Myc inhibitor; Abmole Bioscience, Shanghai, China; 20 μM) were added to the cells for 12 h, and the same volume of dimethyl sulfoxide (DMSO) was used as the control. Then, the culture medium was removed, and mixed drugs containing 20 mg/ml SMI and 23.3 μM cisplatin were added, and 10% fetal bovine serum DMEM was used as a control and cultured for 24 h. Finally, the cells were collected and total proteins were extracted. The expression levels of related proteins were probed by western blot.
2.7. Real-Time PCR (qRT-PCR)
RNA was isolated with RNAiso plus (Takara, Dalian, China), and cDNA was reverse transcribed by Reverse Transcription Kit (Takara, Dalian, China). The expression of the target gene was quantitated by using SYBR Green Master Mix (Vazyme, Nanjing, China) according to the manufacturer's protocol. The primers used in the study are shown in Table 2.
Table 2.
Primer sequences used for RT-PCR.
| Gene | Primer | Sequence |
|---|---|---|
| HK1 | Forward | 5′-GCTCTCCGATGAAACTCTCATAG-3′ |
| Reverse | 5′-GGACCTTACGAATGTTGGCAA-3′ | |
|
| ||
| HK2 | Forward | 5′-CCCGTGCCCACAATGAGAC-3′ |
| Reverse | 5′-CCCGTGCCCACAATGAGAC-3′ | |
|
| ||
| PFKP | Forward | 5′-GCATGGGTATCTACGTGGGG-3′ |
| Reverse | 5′-CTCTGCGATGTTTGAGCCTC-3′ | |
|
| ||
| PKM1/2 | Forward | 5′-ATGTCGAAGCCCCATAGTGAA-3′ |
| Reverse | 5′-TGGGTGGTGAATCAATGTCCA-3′ | |
|
| ||
| PDHX | Forward | 5′-TTGGGAGGTTCCGACCTGT-3′ |
| Reverse | 5′-CAACCACTCGACTGTCACTTG-3′ | |
|
| ||
| GLUT1 | Forward | 5′-TCATCGTCGCTGAACTCTTCAG-3′ |
| Reverse | 5′-TCACACTTGGGAATCACCCCC-3′ | |
|
| ||
| LDHA | Forward | 5′-ATGGCAACTCTAAAGGATCAGC-3′ |
| Reverse | 5′-CCAACCCCAACAACTGTAATCT-3′ | |
2.8. Western Blot Analysis
Total cellular proteins were isolated with RIPA Lysis buffer (Beyotime, China) with a final phenylmethanesulfonyl fluoride (PMSF) concentration of 1 mM (Beyotime, China). Protein concentration was measured by the BCA protein assay (Leagene, Beijing, China) according to the manufacturers' protocols to ensure equal protein loading in each sample. The protein extracts (50 μg) were separated by 8–10% SDS-polyacrylamide gel electrophoresis and then transferred to a polyvinylidene fluoride membrane (Millipore) at 300 mA for 2 h. The membrane was blocked with skimmed milk overnight, and then, it was maintained with the primary antibody for 1 h at room temperature and with the secondary antibody for 1 h at room temperature. The membranes were maintained with enhanced chemiluminescence western blot detection kit (Vazyme, Nanjing, China) and detected using bio-imaging systems (DNR, Israel). The expression of β-actin was used as the internal control.
2.9. Statistical Analyses
All of the experiments were repeated three times, and the data are expressed as mean ± SD. A two-tailed Student's t-test was used to assess the differences between the two groups. An analysis of variance (ANOVA) test was used to assess the differences between multiple sets of data. P < 0.05 was considered to be significant. Data were analyzed using SPSS 19.0.
3. Results
3.1. A549/DDP Cells Exhibit Increased Aerobic Glycolysis
First, we measured the inhibition curves of A549 and A549/DDP cells at different concentrations of cisplatin, and the results showed that IC50 of A549 and A549/DDP to cisplatin were 37.8 μM and 63.3 μM, respectively. It has been reported that the aerobic glycolytic activity of cancer cells is abnormally elevated compared with that of normal cells, especially of drug-resistant cancer cells, which has become one of the main mechanisms of drug resistance [9]. In this study, we assessed the level of glucose metabolism, as shown in Figure 1(b); glucose consumption in A549/DDP cells was increased compared with that in A549 cells (P < 0.05). A549/DDP cells showed a similar increased trend in lactate production compared to A549 cells (P < 0.05), in Figure 1(c). Then, we analyzed the expression levels of key glycolytic enzymes at the protein and mRNA levels. The mRNA expression levels and protein expression levels of hexokinase 2 (HK2), pyruvate kinase M1/2 (PKM1/2), pyruvate kinase M2 (PKM2), glucose transporter 1 (GLUT1), and lactate dehydrogenase A (LDHA) were increased on comparison of A549/DDP cells with A549 cells (Figures 1(d) and 1(e)).
Figure 1.
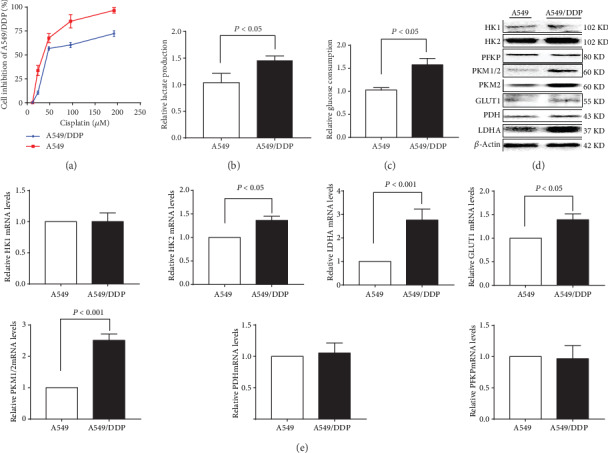
Glucose metabolism was upregulated in cisplatin-resistant A549/DDP cells. (a) Detection of cisplatin inhibition in cisplatin-sensitive or cisplatin-resistant A549 cells and A549/DDP cells. (b) Glucose consumption and (c) lactate production were measured in cisplatin-sensitive or cisplatin-resistant A549 cells and A549/DDP cells. Expression of glucose metabolism enzymes at the protein level or the mRNA level in cisplatin-sensitive or cisplatin-resistant A549 and A549/DDP cells by (d) western blot and (e) RT-PCR analysis. The results are presented as mean ± S.D. (n = 3); the experimental results are presented in triplicate. HK1, hexokinase 1; HK2, hexokinase 2; PFKP, platelet-type phosphofructokinase; PKM1/2, Pyruvate Kinase M1/2; PKM2, pyruvate kinase M2; GLUT1, glucose transporter 1; PDH, pyruvate dehydrogenase; LDHA, lactate dehydrogenase A.
3.2. SMI Enhances Cisplatin Cytotoxicity in A549/DDP Cells
SMI and cisplatin have direct inhibition on A549/DDP cell growth, and the IC50 of SMI was 40.00 mg/mL, and the IC5, IC10, and IC20 were 10.0, 15.0, and 20.0 mg/mL, respectively; meanwhile, the IC50 of cisplatin was 63.6 μM, and IC5, IC10, and IC20 were 6.6, 12.3, and 23.3 μM, respectively.
Cotreatment with SMI and cisplatin interfered with A549/DDP cells; with the increase in concentration of SMI, the inhibition rate of A549/DDP cells increased significantly (Figure 2(b)). By cotreatment with SMI at concentrations of 10.0, 15.0, and 20.0 mg/mL, the IC50 values of cisplatin were decreased from 63.6 μM to 21.0 μM, 20.3 μM, and 13.7 μM, respectively (Figure 2(c)).
Figure 2.
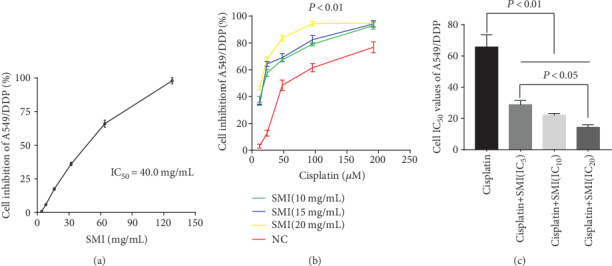
SMI enhances the inhibition of cisplatin in the proliferation of cisplatin-resistant A549/DDP cells. (a) Determination of cisplatin-resistant A549/DDP cell inhibition rate curve with SMI. (b) Inhibition rate curve of SMI at concentrations of IC5, IC10, and IC20 combined with cisplatin in cisplatin-resistant A549/DDP cells. (c) Comparison of cisplatin's IC50 combined with SMI at concentrations of IC5, IC10, and IC20 in cisplatin-resistant A549/DDP cells. The results represent the mean ± S.D. (n = 3); the experimental results are presented in triplicate. SMI: Shenmai injection; IC: inhibitory concentration.
3.3. Combination of SMI and Cisplatin Suppresses A549/DDP Cell Glycolysis and Proliferation
Cotreatment with SMI and cisplatin weakened A549/DDP cell glucose consumption and lactate production (Figures 3(a) and 3(b)). Notably, in the group of 20 mg/mL SMI combined with cisplatin (23.3 μM), glucose consumption was significantly decreased compared with that in the cisplatin alone group and the SMI alone group (P < 0.05). Similarly, the lactate production was significantly decreased in the group of SMI 15 mg/mL or 20 mg/mL combined with cisplatin 23.3 μM compared with the cisplatin alone group and the SMI alone group (P < 0.05). With the increase in SMI concentration, the expression levels of key glycolytic enzymes in A549/DDP cells were significantly inhibited, including HK2, PKM1/2, GLUT1, and PDH rather than HK1, PFKP, and LDHA, compared with the cisplatin alone group and the SMI alone group (Figure 3(c)).
Figure 3.
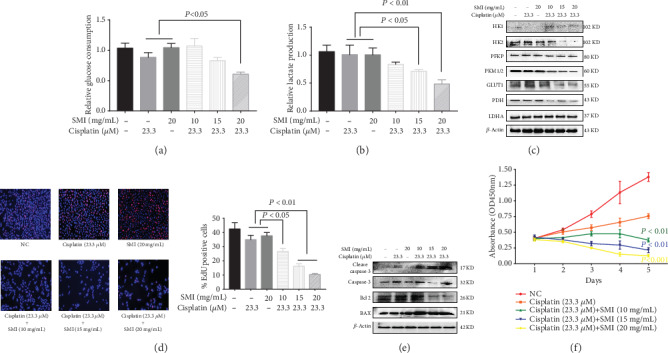
Combination of SMI and cisplatin suppresses A549/DDP cell glycolysis and proliferation and promotes apoptosis. (a) Glucose consumption and (b) lactate production were measured with SMI at concentrations of IC5, IC10, and IC20 combined with cisplatin (IC20) in cisplatin-resistant A549/DDP cells. (c) Expression levels of glycometabolic key enzymes were measured with SMI at concentrations of IC5, IC10, and IC20 combined with cisplatin (IC20) in cisplatin-resistant A549/DDP cells by western blot analysis. (d) Edu assay analyzed the proliferation of cisplatin-resistant A549/DDP cells with SMI at concentrations of IC5, IC10, and IC20 combined with cisplatin (IC20) for 24 h. (e) Western blot measured the expression of apoptosis-related proteins of cisplatin-resistant A549/DDP cells with SMI at concentrations of IC5, IC10, and IC20 combined with cisplatin (IC20) for 24 h. (f) The effects of SMI at concentrations of IC5, IC10, and IC20 combined with cisplatin (IC20) on the growth of cisplatin-resistant A549/DDP cells were measured at 1, 2, 3, 4, and 5 days, respectively. The results are expressed as the mean ± S.D. (n = 3); the experimental results are presented in triplicate. NC: negative control; SMI: Shenmai injection.
The proliferation of A549/DDP cells was significantly inhibited with the increase in concentration of SMI with the same cisplatin concentration after 24 h (Figure 3(d)). In addition, the expression levels of apoptosis-related proteins, such as Caspase 3, cleaved Caspase 3, and Bcl 2, were significantly increased, and the expression level of BAX was significantly decreased in the group of SMI combined with cisplatin 23.3 μM compared with the cisplatin alone group and the SMI alone group (Figure 3(e)). The proliferation of A549/DDP cells was also significantly inhibited after treatment of SMI combined with cisplatin for 5 days (Figure 3(f)).
3.4. SMI Enhances Cisplatin-Suppressed Expression Levels of P-AKT, P-mTOR, and c-Myc
Numerous studies have suggested that AKT/mTOR is involved in the Warburg effect by activating HIF-1α or c-Myc in tumor cells. Our results showed that cotreatment with SMI and cisplatin significantly inhibited the expression levels of p-AKT (Ser473), p-AKT (Thr308), p-mTOR (Ser2448), and c-Myc in A549/DDP cells; further, the higher the concentration of SMI, the more obvious the inhibition. However, HIF-1α expression levels did not change significantly (Figures 4(a) and 4(b)).
Figure 4.
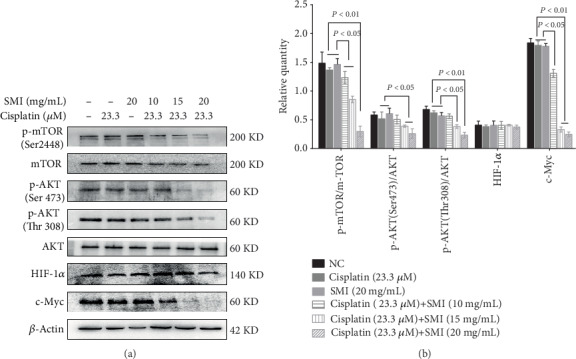
Combination of SMI and cisplatin inhibition effect on expression levels of p-AKT, p-mTOR, and c-Myc in cisplatin-resistant A549/DDP cells and HIF-1α in cisplatin-resistant A549/DDP cells. (a) Western blot analysis of protein expression levels of AKT, p-AKT, mTOR, p-mTOR, c-Myc, and HIF-1α. (b) Quantification of relative protein expression levels of p-AKT, p-mTOR, c-Myc, and HIF-1α. The results represent the mean ± S.D. (n = 3); the experimental results are presented in triplicate. AKT: AKT serine/threonine kinase; mTOR: mechanistic target of rapamycin; p: phosphorylated; HIF-1α: hypoxia-inducible factor-1α; c-Myc: cellular-myelocytomatosis viral oncogene.
3.5. SMI Enhances the Inhibitory Effect of Cisplatin on Glucose Metabolism via the AKT-mTOR-c-Myc Pathway
After pretreatment with LY294002 (inhibitor of PI3K), A549/DDP cells were incubated with the mixed drugs (SMI 20 mg/mL + cisplatin 23.3 μM). In these cells, the expression levels of p-AKT (Ser473), p-mTOR, c-Myc, HK2, and PKM1/2 were significantly reduced compared with those in the single mixed drug treatment group and the LY294002 alone treatment group, but the expression of p-AKT (Thr308) was not changed (Figure 5(a)). Similarly, after treatment with rapamycin (mTOR inhibitor) and mixed drugs (SMI 20 mg/mL + cisplatin 23.3 μM), the expression levels of p-mTOR, c-Myc, HK2, and PKM1/2 in A549/DDP cells were significantly lower than those in the mixed drug treatment group and the rapamycin treatment group alone, respectively, and the expression levels of p-AKT (Ser473) and p-AKT (Thr308) remained unchanged (Figure 5(b)). Finally, A549/DDP cells were incubated with 10058-F4 (c-Myc inhibitor) and mixed drugs successively; the expression levels of c-Myc, HK2, and PKM1/2 were significantly reduced compared to those in the mixed drug treatment group alone and the 10058-F4 treatment group alone, but the expression levels of p-AKT (Ser473), p-AKT (Thr308), and p-mTOR were not changed (Figure 5(c)). It was concluded that SMI combined with cisplatin suppressed the expression level of cell glucose metabolism genes through the AKT-mTOR-c-Myc signaling pathway.
Figure 5.
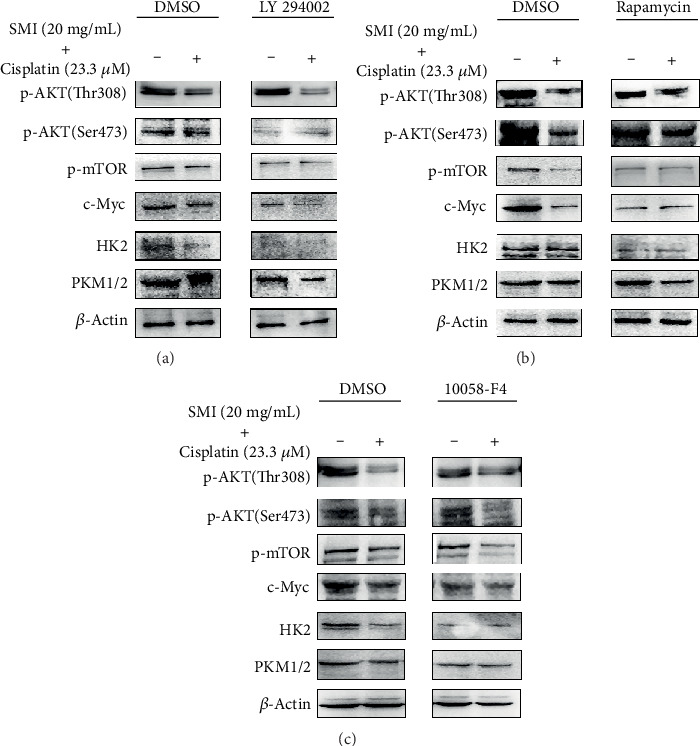
SMI enhances cisplatin cytotoxicity in cisplatin-resistant A549/DDP cells via the AKT-mTOR-c-Myc signaling pathway. (a) LY294002 (PI3K inhibitor), (b) rapamycin (mTOR inhibitor), and (c) 10058-F4 (c-Myc inhibitor) were treated or not treated in cisplatin-resistant A549/DDP cells via western blot analysis of protein expression levels of genes involved in cell glucose metabolism.
4. Discussion
Numerous studies have shown that most cancer cells exhibit increased glycolysis even in the presence of oxygen, including increased glucose consumption and lactate production [10, 11]. Abnormal metabolic shifts in cancer cells are frequently related to tumor growth and increased drug resistance, in which some rate-limiting enzymes directly regulate the glycolysis level to promote drug resistance of cancer cells [12]. HK2 is the first rate-limiting enzyme in the glycolysis process, which promotes cisplatin resistance by enhancing drug-induced and ERK-mediated autophagy in ovarian cancer [13]. HK1 is also a member of the hexokinase family, and it has been found to be related to cell activity and proliferation [14]. The second rate-limiting enzyme in the glycolysis process is phosphofructokinase (PFK), which is responsible for the phosphorylation of fructose-6-phosphate to fructose-1,6-diphosphate. A study showed that rectal cancer cells, highly resistant to 5-fluorouracil and oxaliplatin, have enhanced glucose uptake and significantly higher levels of lactic acid due to increased activity of HK and PFK [15]. In addition, pyruvate kinase (PK) is the last rate-limiting enzyme in the glycolysis process, which is involved in the conversion of phosphoenol pyruvate into pyruvate. Cheng et al. found that the expression of PKM2 was increased in two drug-resistant colon cancer cell lines, vincristine resistant HCT-8 cell line, and oxaliplatin resistant HCT116 cell line [16]. In addition, it was found that some enzymes are also involved in the development of drug resistance. For example, GLUT1 is responsible for regulating glucose transport inside and outside the cells, and it has been found to be upregulated in insulin-resistant liver cancer cells [17]. It was found that lactate dehydrogenase (LDH) activity was enhanced in tumor cells to produce a large amount of lactic acid, which placed the tumor cells in an acidic microenvironment to promote their growth. Studies have shown that the effects of chemotherapeutic drugs can be enhanced by inhibiting the activity of LDH [18, 19]. In this study, we also found increased glucose consumption and lactic acid accumulation, as well as increased expression levels of HK1, HK2, PKM1/2, PKM2, GLUT1, and LDHA in cisplatin-resistant A549/DDP cells compared to A549 cells, which suggested that glycolysis induced drug resistance in nonsmall cell lung carcinoma cells.
In the traditional Chinese medicine (TCM) theory, qi is considered to be the basic substance of human activities and life energy, including physical and spiritual levels. We have shown that some TCMs with the functionality of invigorating qi, such as Bu-Zhong-Yi-Qi decoction and Shenqi Fuzheng injection, might increase chemotherapy sensitivity in cisplatin resistance of NSCLCs by activating apoptosis and the autophagy pathway and inducing cell cycle arrest [20]. SMI is the precise TCM, which has the function of invigorating qi. Previous studies have found that SMI combined with chemotherapeutic drugs improves the clinical efficacy and relieves the adverse reactions, including incidence of leukopenia and gastrointestinal reactions in gastric and breast cancer patients [21]. At present, most of the studies on SMI are based on a network meta-analysis, and its antitumor mechanism is still unclear. Our results showed that SMI combined with cisplatin reduced the glucose consumption and lactate production in A549/DDP cells by inhibiting the expression levels of key glycolytic enzymes, including HK2, PKM1/2, GLUT1, and PDH, thereby enhancing the toxic effect of cisplatin on A549/DDP cells. Furthermore, we detected the expression levels of Caspase 3, Bcl 2, and BAX by Edu proliferation and western blot, and the results showed that SMI could enhance the inhibitory effect of cisplatin on A549/DDP cell proliferation and promote cell apoptosis.
Previous studies have found that the AKT/mTOR signaling pathway is one of the most important signaling pathways in cancer, and it plays an important role in cell proliferation and survival. The Akt/mTOR pathway is involved in regulating the expression of HIF-1α. Recent research has found that HIF-1α is activated via the Akt/mTOR pathway, enhancing the migration and invasion of human glioblastoma U87 cells [22]. Furthermore, the AKT/mTOR signaling pathway can also regulate c-Myc to affect the metabolism and growth of cancer cells [23]. Under hypoxic conditions, HIF-1α mainly regulates the glycolysis-related target genes, whereas c-Myc regulates the same genes under normoxic conditions [11, 24]. In this study, we conducted experiments under normoxic conditions, and we found that the combination of SMI and cisplatin downregulated the levels of p-AKT (Ser473), p-AKT (Thr308), p-mTOR (Ser2448), and c-Myc of the AKT/mTOR/c-Myc signaling pathway in A549/DDP cells, but the expression of HIF-1α was not changed. Reduced AKT/mTOR/c-Myc signaling pathway suppressed the expression levels of key enzymes, such as HK1, HK2, PKM1/2, PKM2, GLUT1, and PDH, in the glycolysis pathway. In addition, we also interfered with several key points of the Akt/mTOR/c-Myc signaling pathway by adding inhibitors. The results showed that inhibition of Akt phosphorylation was inhibited by adding LY294002, and there was inhibition of the expression of p-Akt, p-mTOR, c-Myc, and key enzymes of glucose metabolism by SMI combined with cisplatin; mTOR phosphorylation was inhibited by adding rapamycin, and SMI combined with cisplatin could still reduce the expression of p-Akt, but it could not reduce the expression of p-mTOR, c-Myc, and key enzymes of glucose metabolism; the same 10058-f4 could inhibit the expression of c-Myc; SMI combined with cisplatin could still reduce the expression of p-Akt and p-mTOR, but it could not reduce the expression of c-Myc and key enzymes of glucose metabolism in the pathway. In conclusion, these results suggest that SMI is through Akt/mTOR/c-Myc signaling pathway sensitized cisplatin to cisplatin-resistant A549/DDP cells.
Recently, a new study confirmed that SMI can inhibit the function and expression of P-gp through the MAPK/NF-κB signaling pathway and enhance the sensitivity of breast cancer cells to chemotherapy [25]. Combined with our study, SMI can also reprogram glucose metabolism, inhibit cell proliferation, and promote cell apoptosis through the Akt/mTOR/c-Myc signaling pathway, enhance the sensitivity of lung cancer cells to chemotherapy drugs, and improve the mechanism of SMI sensitization towards chemotherapy drugs. However, more in-depth and extensive investigations are needed.
5. Conclusions
The present study suggests that the addition of SMI could increase the cytotoxicity of cisplatin by inhibition of the glycolysis metabolism through the AKT/mTOR/c-Myc pathway, which might provide a theoretical basis for the treatment of cisplatin-resistant nonsmall cell lung cancer.
Acknowledgments
This work was financially supported by the National Natural Science Fund of China (grants 81573856, 81774184, and 81973735) and the Natural Science Foundation of Liaoning Province (grant 20180550548). We thank LetPub (http://www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Conflicts of Interest
There were no conflicts of interest in this study.
References
- 1.Galluzzi L., Senovilla L., Vitale I., et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 2.Feng X., Liu H., Zhang Z., Gu Y., Qiu H., He Z. Annexin A2 contributes to cisplatin resistance by activation of JNK-p53 pathway in non-small cell lung cancer cells. Journal of Experimental & Clinical Cancer Research. 2017;36(1):123–136. doi: 10.1186/s13046-017-0594-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chakraborty P. K., Mustafi S. B., Xiong X., et al. MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nature Communications. 2017;8(1, article 14634) doi: 10.1038/ncomms14634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou Q., Sun Y., Tan W., et al. Effect of Shenmai injection on preventing the development of nitroglycerin-induced tolerance in rats. PLoS One. 2017;12(4, article e0176777) doi: 10.1371/journal.pone.0176777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X., Ye L., Wang L. Protective mechanism of shenmai on myocardial ischemia-reperfusion through the energy metabolism pathway. American Journal of Translational Research. 2019;11(7):4046–4062. [PMC free article] [PubMed] [Google Scholar]
- 6.Fang T., Li J., Wu X. Shenmai injection improves the postoperative immune function of papillary thyroid carcinoma patients by inhibiting differentiation into Treg cells via miR-103/GPER1 axis. Drug Development Research. 2018;79(7):324–331. doi: 10.1002/ddr.21459. [DOI] [PubMed] [Google Scholar]
- 7.Liu W. Y., Zhang J. W., Yao X. Q., et al. Shenmai injection enhances the cytotoxicity of chemotherapeutic drugs against colorectal cancers via improving their subcellular distribution. Acta Pharmacologica Sinica. 2017;38(2):264–276. doi: 10.1038/aps.2016.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu L. M., Qian H., Chen Z., Lin S. Y., Wang Z. S. Preliminary experimental study on antineoplastic effect of Shenmai Injection. Chinese Journal of Experimental Traditional Medical Formulae. 1996;2(4):11–14. [Google Scholar]
- 9.Zhou Y., Zheng X., Lu J., Chen W., Li X., Zhao L. Ginsenoside 20(S)-Rg3 inhibits the warburg effect via modulating DNMT3A/ MiR-532-3p/HK2 pathway in ovarian cancer cells. Cellular Physiology and Biochemistry. 2018;45(6):2548–2559. doi: 10.1159/000488273. [DOI] [PubMed] [Google Scholar]
- 10.Apicella M., Giannoni E., Fiore S., et al. Increased lactate secretion by cancer cells sustains non-cell-autonomous adaptive resistance to MET and EGFR targeted therapies. Cell Metabolism. 2018;28(6):848–865.e6. doi: 10.1016/j.cmet.2018.08.006. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X., Ai Z., Chen J., et al. Glycometabolic adaptation mediates the insensitivity of drug-resistant K562/ADM leukaemia cells to adriamycin via the AKT-mTOR/c-Myc signalling pathway. Molecular Medicine Reports. 2017;15(4):1869–1876. doi: 10.3892/mmr.2017.6189. [DOI] [PubMed] [Google Scholar]
- 12.Cheng C., Xie Z., Li Y., Wang J., Qin C., Zhang Y. PTBP1 knockdown overcomes the resistance to vincristine and oxaliplatin in drug-resistant colon cancer cells through regulation of glycolysis. Biomedicine & Pharmacotherapy. 2018;108:194–200. doi: 10.1016/j.biopha.2018.09.031. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X. Y., Zhang M., Cong Q., et al. Hexokinase 2 confers resistance to cisplatin in ovarian cancer cells by enhancing cisplatin-induced autophagy. The International Journal of Biochemistry & Cell Biology. 2018;95(2):9–16. doi: 10.1016/j.biocel.2017.12.010. [DOI] [PubMed] [Google Scholar]
- 14.Zhou Y., Liu K., Liu Y., Tan L. Microrna-34a inhibit hepatocellular carcinoma progression by repressing hexokinase-1. Journal of Cellular Biochemistry. 2018;120(5):7147–7153. doi: 10.1002/jcb.27988. [DOI] [PubMed] [Google Scholar]
- 15.Mou J. J., Peng J., Shi Y. Y., et al. Mitochondrial dna content reduction induces aerobic glycolysis and reversible resistance to drug-induced apoptosis in sw480 colorectal cancer cells. Biomedicine & Pharmacotherapy. 2018;103:729–737. doi: 10.1016/j.biopha.2018.04.099. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y. L., Song J. J., Chen X. C., et al. Mechanisms of pyruvate kinase M2 isoform inhibits cell motility in hepatocellular carcinoma cells. World Journal of Gastroenterology. 2015;21(30):9093–9102. doi: 10.3748/wjg.v21.i30.9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mokashi P., Khanna A., Pandita N. Flavonoids from _Enicostema littorale_ blume enhances glucose uptake of cells in insulin resistant human liver cancer (HepG2) cell line via IRS-1/PI3K/Akt pathway. Biomedicine & Pharmacotherapy. 2017;90(6):268–277. doi: 10.1016/j.biopha.2017.03.047. [DOI] [PubMed] [Google Scholar]
- 18.Fu J., Jiang H., Wu C., Jiang Y., Xiao L., Tian Y. Overcoming cetuximab resistance in Ewing's sarcoma by inhibiting lactate dehydrogenase-A. Molecular Medicine Reports. 2016;14(1):995–1001. doi: 10.3892/mmr.2016.5290. [DOI] [PubMed] [Google Scholar]
- 19.Li X., Zhao H., Zhou X., Song L. Inhibition of lactate dehydrogenase A by microRNA-34a resensitizes colon cancer cells to 5-fluorouracil. Molecular Medicine Reports. 2015;11(1):577–582. doi: 10.3892/mmr.2014.2726. [DOI] [PubMed] [Google Scholar]
- 20.Yu N., Xiong Y., Wang C. Bu-Zhong-Yi-Qi Decoction, the Water Extract of Chinese Traditional Herbal Medicine, Enhances Cisplatin Cytotoxicity in A549/DDP Cells through Induction of Apoptosis and Autophagy. BioMed Research International. 2017;2017:9. doi: 10.1155/2017/3692797.3692797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L., Huang X. E., Cao J. Clinical study on safety of cantharidin sodium and shenmai injection combined with chemotherapy in treating patients with breast cancer postoperatively. Asian Pacific Journal of Cancer Prevention. 2014;15(14):5597–5600. doi: 10.7314/APJCP.2014.15.14.5597. [DOI] [PubMed] [Google Scholar]
- 22.Huang W., Ding X., Ye H., Wang J., Shao J., Huang T. Hypoxia enhances the migration and invasion of human glioblastoma u87 cells through pi3k/akt/mtor/hif-1α pathway. Neuroreport. 2018;29(18):1578–1585. doi: 10.1097/WNR.0000000000001156. [DOI] [PubMed] [Google Scholar]
- 23.Andrews F. H., Singh A. R., Joshi S., et al. Dual-activity PI3K-BRD4 inhibitor for the orthogonal inhibition of MYC to block tumor growth and metastasis. Proceedings of the National Academy of Sciences of the United States of America. 2017;114(7):E1072–E1080. doi: 10.1073/pnas.1613091114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeh Y. H., Hsiao H. F., Yeh Y. C., Chen T. W., Li T. K. Inflammatory interferon activates HIF-1α-mediated epithelial-to-mesenchymal transition via PI3K/AKT/mTOR pathway. Journal of Experimental & Clinical Cancer Research. 2018;37(1):70–84. doi: 10.1186/s13046-018-0730-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang L., Zhang C., Chen J., et al. Shenmai injection suppresses multidrug resistance in MCF-7/ADR cells through the MAPK/NF-κB signalling pathway. Pharmaceutical Biology. 2020;58(1):276–285. doi: 10.1080/13880209.2020.1742167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.