

Abstract
Background
Although many studies have defined mechanisms of resistance to EGFR‐TKIs, acquired resistance remains the major limitation of monotherapy with EGFR‐TKIs.
Methods
Cell viability was analyzed using a Cell Counting Kit‐8 (CCK‐8) assay. EGFR T790M mutation was sequenced on a HiSeq 4000 platform. mRNAs from HCC827 and HCC827 gefitinib‐resistant (GR) cells were analyzed by genome analyzer‐based deep sequencing. The effect of anlotinib on apoptosis and cell cycle arrest of HCC827 GR was detected by fluorescence‐activated cell sorting (FACS) analysis. A mouse xenograft model was used to assess the effect of anlotinib on HCC827 GR cells.
Results
The T790M mutation was found in the PC‐9 GR cell line but not in the HCC827 GR cell line. Anlotinib could suppress the growth of HCC827 GR cells by inhibiting FGFR1 in vitro and in a mouse xenograft model. Moreover, FGFR1 was overexpressed in HCC827 GR cells, and the knockdown of FGFR1 reversed gefitinib resistance in HCC827 GR cells. Furthermore, anlotinib induced apoptosis and cell cycle arrest in HCC827 GR cells by increasing the activity of Caspase‐3.
Conclusions
FGFR1 overexpression could be the mechanism of EGFR‐TKI acquired resistance and anlotinib can suppresse the growth of EGFR‐TKI‐resistant NSCLC cells without T790M mutation.
Keywords: Anlotinib, EGFR‐TKI, FGFR1, non‐small cell lung cancer, resistance
Introduction
Lung cancer is the major cause of cancer‐related death throughout the world.1 Non‐small cell lung cancer (NSCLC) accounts for 85% of lung cancer. The five‐year survival rate for NSCLC is less than 15%.2 Epidermal growth factor receptor (EGFR)‐sensitizing mutations have been used for the selection of patients with advanced NSCLC for EGFR tyrosine kinase inhibitor (EGFR‐TKI) treatment.3 Numerous clinical studies have shown that EGFR‐TKIs exhibit a better clinical benefit than traditional therapeutic strategies, such as chemotherapy and radiotherapy.4 Nevertheless, all responding patients invariably acquire resistance following the initial response within 8–12 months of therapy.5 Several acquire resistance mechanisms which include secondary mutation in EGFR (T790M),6 KRAS mutation,7 PIK3CA mutation,8 MET amplification,9 epithelial‐mesenchymal transition (EMT) and small cell lung cancer (SCLC) transformation have been revealed.10 However, the possibility that other mechanisms are involved in resistance to EGFR‐TKIs cannot be excluded, and the need to develop effective therapeutic interventions to overcome acquired resistance is urgent.
The fibroblast growth factor receptor family (FGFR1‐4) belongs to the RTK family, and there are 18 different FGF ligands.11, 12 FGFR is involved in many physiological processes, including wound repair, embryogenesis and angiogenesis.13 Dysregulation of FGFR1 to tumorigenesis, transformation, and tumor progression has been reported in a broad range of malignancies.14, 15 The activation of FGFR1 can promote EMT in prostate cancer, breast cancer and FGFR1‐amplified lung cancer.16, 17, 18 FGF/FGFR signaling pathway alterations have been connected with chemotherapy resistance and poor clinical outcome.19, 20
Anlotinib is a multitarget receptor TKI in various cancers that has a broad spectrum of inhibitory actions against tumor growth and angiogenesis.21, 22 Numerous studies have indicated that anlotinib suppresses tumor growth via the inhibition of c‐Kit, Ret, Aurora‐B, c‐FMS and DDR1. Furthermore, anlotinib inhibits the angiogenesis of tumor cells by selectively targeting VEGFR (1, 2 and 3), PDGFR (α and β) and FGFR (1, 2, 3 and 4).22, 23, 24 Clinical trials have demonstrated that anlotinib therapy prolongs progression‐free survival (PFS) and overall survival (OS) in refractory advanced NSCLC patients subjected to third‐line or beyond third‐line therapy. One study reported that anlotinib therapy was beneficial for prolonging OS in NSCLC patients harboring EGFR mutations, especially patients harboring the EFGR T790M mutation.25, 26 Moreover, anlotinib inhibited angiogenesis in an H1975‐derived xenograft model via inhibiting CCL2.21 However, the effect of anlotinib on NSCLC patients with EGFR‐TKI resistance due to non‐T790M mutation and the mechanism of this effect remain unclear.
In this study, we demonstrated that anlotinib inhibited the growth of EGFR‐TKI‐resistant NSCLC cells without T790M mutation in vitro and in a mouse xenograft model. Furthermore, anlotinib induced apoptosis and cell cycle arrest in EGFR‐TKI‐resistant NSCLC cells by inhibiting FGFR1. These results provide novel alternatives and therapeutic strategies for patients with EGFR‐TKI acquired resistance.
Methods
Cell culture and reagents
HCC827 cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). To establish gefitinib‐resistant (GR) HCC827 (HCC827 GR) cell strain, HCC827 cells were exposed to gefitinib according to the methods of Koizumi et al.27 PC‐9 and GR PC9 (PC9 GR) cell lines were obtained from Professor Caicun Zhou as a gift. HCC827 and HCC827 GR cell lines were routinely cultured in RPMI‐1640 medium supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA). The PC‐9 and PC‐9 GR cell lines were cultured in DMEM containing 10% fetal bovine serum. All cells were cultured in a humidified incubator containing 5% CO2 at 37°C. Gefitinib was purchased from Selleck (Selleck Chemicals, Houston, TX, USA). Anlotinib was given as a gift from Chia Tai Tianqing Pharmaceutical Group Co., Ltd. (Jiangsu, China).
Next‐generation DNA sequencing
We profiled DNA from cells by using a capture‐based targeted sequencing panel. Human genomic regions 1.4 megabases in total size including selected exons and introns of 416 genes were captured using 120 base pair (bp) probes. DNA was fragmented into segments 200 to 250 bp in length, captured by the 120 bp probes, and sequenced by obtaining paired 2 × 150 bp reads. After DNA extraction using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany), DNA concentrations were measured by using the Qubit dsDNA assay (Invitrogen, Carlsbad, CA). The DNA quality was confirmed by ensuring that the A260/A280 ratio was 1.8:2.0. DNA was hybridized with the capture probes (the bait), selected using magnetic beads, and polymerase chain reaction (PCR) amplified. Then, a bioanalyzer (Qubit and Agilent 2100, Agilent Technologies, Santa Clara, CA) was used to perform high‐sensitivity assays assessing DNA quality and size range. All samples were sequenced on a HiSeq 4000 platform (Illumina, Inc., San Diego, CA); we obtained pair‐end reads.
Cell viability assay
Cells (3 × 103 cells/well) were seeded in 96‐well plates overnight. Anlotinib or gefitinib was added at different doses, and the cells were incubated for 72 hours. Then, a Cell Counting Kit‐8 (CCK‐8) assay kit (Boster, Wuhan, China) was used according to the manufacturer's instructions to assess cell viability. Each sample was plated in triplicate, three independent experiments were performed, and IC50 was defined as the concentration needed for a 50% reduction in the absorbance.
Quantitative real‐time PCR (qRT‐PCR)
Total RNA was reverse transcribed using reverse transcriptase M‐MLV (TaKaRa, Shiga, Japan) according to the manufacturer's protocol. qRT‐PCR was performed using SYBR Premix ExTaq (Takara). The primer sequences for qRT‐PCR of FGFR1 and GAPDH were as follows: AXL, sense: 5′‐TAATGGACTCTGTGGTGCCCTC‐3′, antisense: 5′‐ATGTGTGGTTGATGCTGCCG‐3′; GAPDH, sense: 5′‐TGCACCACCAACTGCTTAGC‐3′, antisense: 5′‐GGCATGGACTGTG GTCATGG‐3′. The PCR programme used was as follows: 50°C for two minutes and 95°C for 10 minutes followed by 45 cycles of 95°C for 15 seconds and 60°C for one minute. FGFR1 mRNA expression values were normalized to those of the internal control GAPDH. Relative expression was calculated using the cycle threshold (Ct) method.
Western blot analysis
Cells were lysed on ice with RIPA lysis buffer containing protease and phosphatase inhibitor for 20 minutes. The lysates were collected by high‐speed centrifugation at 12000 rpm for 15 minutes at 4°C. The protein concentration was measured using a BCA Protein Assay kit (Thermo Scientific, Rockford, IL, USA). The extracts were mixed with SDS sample buffer and subjected to 10% SDS‐PAGE. Following electrophoretic transfer onto nitrocellulose, the membrane was blocked with 5% skim milk in TBS for one hour and then incubated with primary antibodies at 4°C overnight. The membranes were washed thoroughly with Tris‐buffered saline with Tween‐20 (TBST) and then incubated with peroxidase‐conjugated secondary antibody for two hours at room temperature. All antibodies except anti‐FRFR1 and anti‐pFRFR1 were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti‐FRFR1 and anti‐pFRFR1 were purchased from Merck (Billerica, MA, USA).
Flow cytometry analysis
HCC827 GR cells were seeded in six‐well plates. Then, the cells were treated with anlotinib at different concentrations. As a control, cells were treated with the vehicle, DMSO. We analyzed cell apoptosis and the cell cycle status of the cells by using Annexin V‐FITC and propidium iodide (PI) (fluorescein isothiocyanate, R&D Systems) staining according to the manufacturer's protocol.
Cell transfection
Transfection was carried out using Lipofectamine 2000 transfection reagent (Invitrogen, Waltham, MA, USA) according to the manufacturer's protocols. All siRNAs were purchased from GenePharma Co. (Shanghai, China). The target siRNA sequences were as follows: si‐NC, sense: 5′‐UUCUCCGAACGUGUCACGU‐3′, antisense: 5′‐ACGUGACACGUUCGGAGAA‐3′; si‐FGFR1, sense: 5′‐CGGUCAUCGUCUACAAGAU‐3′, anti‐sense: 5′‐AUCUUGUAGACGAUGACCG‐3′.
Xenograft mouse model
BALB/c athymic nude mice (female, 3–4 weeks old and 16–20 g in weight) were purchased from the Experimental Animal Center of Soochow University and bred under pathogen‐free conditions. HCC827 GR cells (2.5 × 106) were subcutaneously injected into the fore‐axillary region. Tumor volume was calculated using the following formula: volume = length×width2/2. When tumor volumes reached approximately 200 mm3, the mice were randomly allocated into groups of six animals and administered anlotinib (2 mg/kg), gefitinib (4 mg/kg) or vehicle by oral gavage every two days for 12 days. Tumor volume was measured once a day. All animals were handled in strict accordance with good animal practice as defined by the Guide for the Care and Use of Experimental Animals from the Experimental Animal Center of Soochow University, and all protocols were approved (2017.165) by the Animal Ethical and Welfare Committee of the First Affiliated Hospital of Soochow University.
Statistical analysis
All obtained results are presented as the mean ± standard deviation (SD). Statistical significance was analyzed with a Student's t‐test, and P < 0.05 indicated significance. All statistical analyses were performed using SPSS 7.0 software (SPSS, Chicago, IL, USA) and GraphPad Prism 7.0 (GraphPad, San Diego, CA, USA).
Results
Gefitinib resistance in HCC827 GR cells is not due to EGFR T790M mutation
Our previous study found an activating deletion in exon 19 in the HCC827 and PC‐9 cell lines.28 The T790M mutation in EGFR is a known mechanism of acquired gefitinib resistance in lung cancer.6 To determine whether the GR strains had the T790M mutation, we first examined the sensitivity of HCC827 and PC‐9 parental and GR cells to gefitinib using a CCK‐8 assay. The viability of HCC827 GR and PC‐9 GR cells was unaffected by increasing concentrations of gefitinib up to 10 μM (IC50 > 10 μM), whereas HCC827 and PC‐9 cells were sensitive to gefitinib (Fig 1a,b). We then analyzed EGFR for DNA substitution corresponding to the T790M mutation. As shown in Fig 1c, the T790M mutation was found in the PC‐9 GR cell line, while we found no evidence for this mutation in the HCC827 GR cell line (Fig 1c and Table SS1). These results suggest that the T790M mutation is responsible for resistance to gefitinib in PC9 GR cells rather than HCC827 GR cells, and other mechanisms contribute to resistance to gefitinib in HCC827 GR cells.
Figure 1.
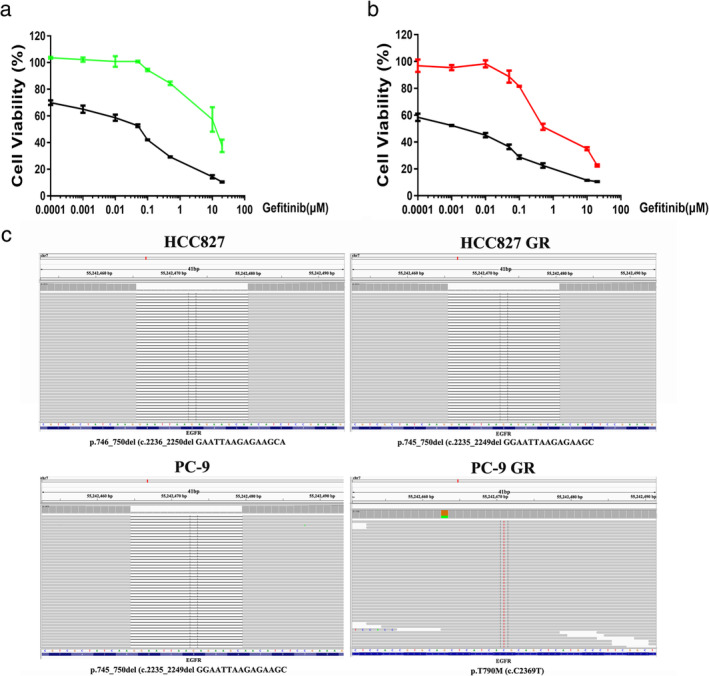
Gefitinib resistance in HCC827 GR cells is not due to EGFR T790M mutation. (a) and (b) HCC827, PC‐9, HCC827 GR and PC‐9 GR cells were treated with gefitinib at different concentrations, and CCK‐8 assays were used to assess cell viability (a:  HCC827 and
HCC827 and  HCC827 GR; b:
HCC827 GR; b:  PC‐9 and
PC‐9 and  PC‐9 GR). (c) Next‐generation DNA sequencing was used to analyze EGFR for the DNA substitution corresponding to the T790M mutation.
PC‐9 GR). (c) Next‐generation DNA sequencing was used to analyze EGFR for the DNA substitution corresponding to the T790M mutation.
FGFR1 expression is higher in HCC827 GR cells than in parental HCC827 cells
To explore the gefitinib resistance mechanism in HCC827 GR cells, mRNAs from HCC827 and HCC827 GR cells were submitted for RNA sequencing (RNA‐seq) analysis by genome analyzer‐based deep sequencing. The resulting analysis showed that the mRNA expression level of FGFR1 was higher in HCC827 GR cells than in parental HCC827 cells (Fig 2a). Western blot and qRT‐PCR analyses confirmed that FGFR1 expression was increased in HCC827 GR cells (Fig 2b). In contrast, FGFR1 expression was lower in PC‐9 GR cells than in parental PC‐9 cells (Fig. SS1). We next examined the inhibitory effect of anlotinib on the growth of HCC827 GR cells. As shown in Fig 2c, the inhibitory effect of anlotinib on HCC827 GR cells was stronger than that on parental HCC827 cells. To verify that anlotinib inhibited the growth of HCC827 GR cells through targeting FGFR1, we detected phosphorylated FGFR1 and FGFR1 in HCC827 GR cells treated with anlotinib at different concentrations by Western blot analysis. As shown in Fig 2d,e, anlotinib inhibited the phosphorylation of FGFR1 in HCC827 GR cells, and this inhibitory effect was stronger than that on parental HCC827 cells. In addition, anlotinib inhibited the phosphorylation of AKT and ERK in both HCC827 and HCC827 GR cells.
Figure 2.
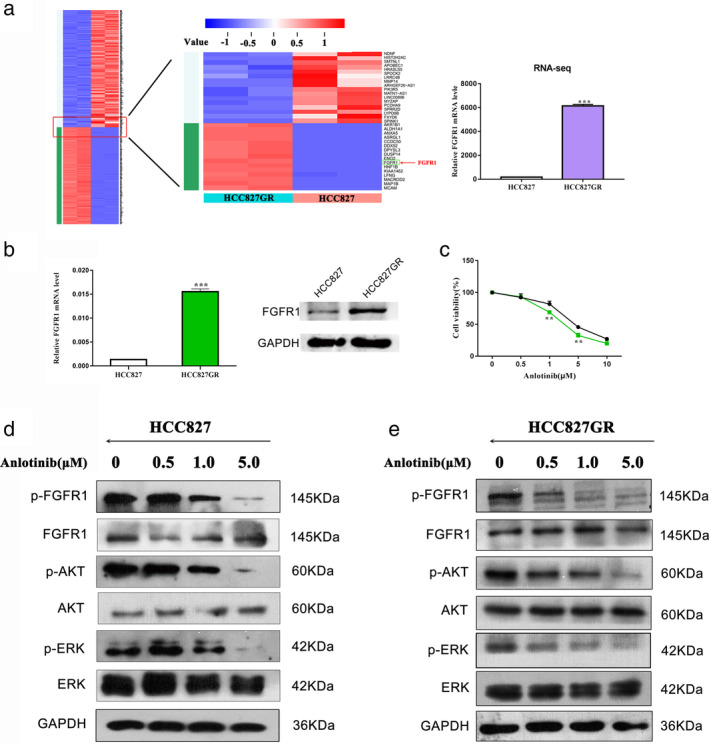
FGFR1 expression is higher in HCC827 GR cells than in parental HCC827 cells. (a) RNA sequencing analysis was used to analyze differences in mRNA expression levels between HCC827 and HCC827 GR cells. (b) The expression level of FGFR1 was determined by quantitative real‐time PCR and Western blot analysis. (c) CCK‐8 assays were performed to investigate the effects of anlotinib on HCC827 and HCC827 GR cells in vitro ( HCC827 and
HCC827 and  HCC827 GR). (d) and (e) Western blot analysis was used to detect the effect of anlotinib on p‐FGFR1, p‐AKT and p‐ERK in HCC827 and HCC827 GR cells. (** P < 0.01).
HCC827 GR). (d) and (e) Western blot analysis was used to detect the effect of anlotinib on p‐FGFR1, p‐AKT and p‐ERK in HCC827 and HCC827 GR cells. (** P < 0.01).
Anlotinib induces apoptosis and cell cycle arrest in HCC827 GR cells
We conducted fluorescence‐activated cell sorting (FACS) analysis to determine whether anlotinib can induce apoptosis and cell cycle arrest in HCC827 GR cells. HCC827 GR cells were treated with anlotinib at gradually increasing concentrations, As shown in Fig 3a,b, when the dose of anlotinib reached 5 μM, the proportion of HCC827 GR cells at G2/M phase and apoptosis increased significantly. Western blot analysis indicated that anlotinib induced a marked increase in the activity of Caspase‐3. Meanwhile Cyclin D1 showed similar protein level in HCC827 GR cells after Anlotinib treatment (Fig 3c).
Figure 3.
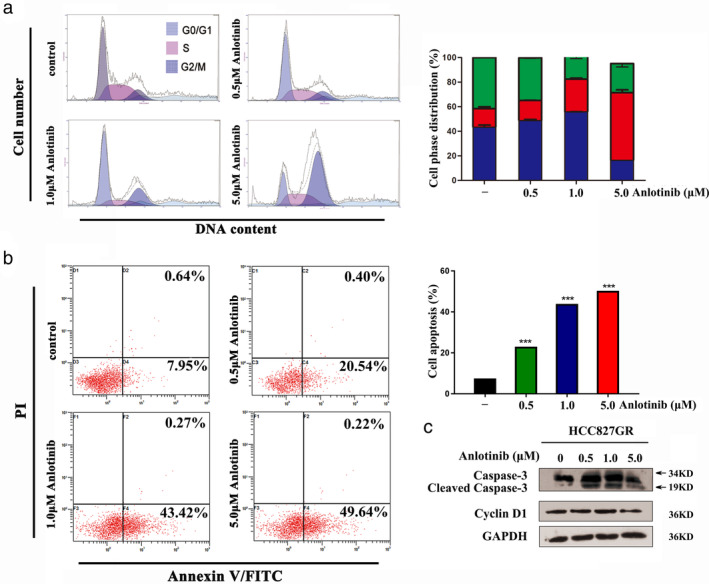
Anlotinib induces apoptosis and cycle arrest in HCC827 GR cells. (a) and (b) Fluorescence‐activated cell sorting (FACS) analysis was performed to investigate the effects of anlotinib on the cell cycle and apoptosis in HCC827 GR cells ( G0/G1,
G0/G1,  G2/M, and
G2/M, and  S). (c) Western blot analysis was used to detect the effect of anlotinib on Caspase‐3 and Cyclin D1 expression in HCC827 GR cells. (*** P < 0.001).
S). (c) Western blot analysis was used to detect the effect of anlotinib on Caspase‐3 and Cyclin D1 expression in HCC827 GR cells. (*** P < 0.001).
Knockdown of FGFR1 reverses gefitinib resistance in HCC827 GR cells
We evaluated the effect of FGFR1 on gefitinib resistance in HCC827 GR cells by the siRNA‐mediated knockdown of FGFR1. First, we succeeded in knocking down FGFR1 with siRNA in HCC827 GR cells (Fig 4a). The growth rate of the FGFR1 interference group was slower than that of the control group (Fig 4b). The CCK‐8 assay indicated that HCC827 GR cells transfected with si‐FGFR1 were more sensitive to gefitinib and more resistant to anlotinib than the control group (Fig 4e,f). Transfection of si‐FGFR1 in HCC827 GR cells enhanced the gefitinib‐induced downregulation of p‐EGFR (Fig 4c,d).
Figure 4.
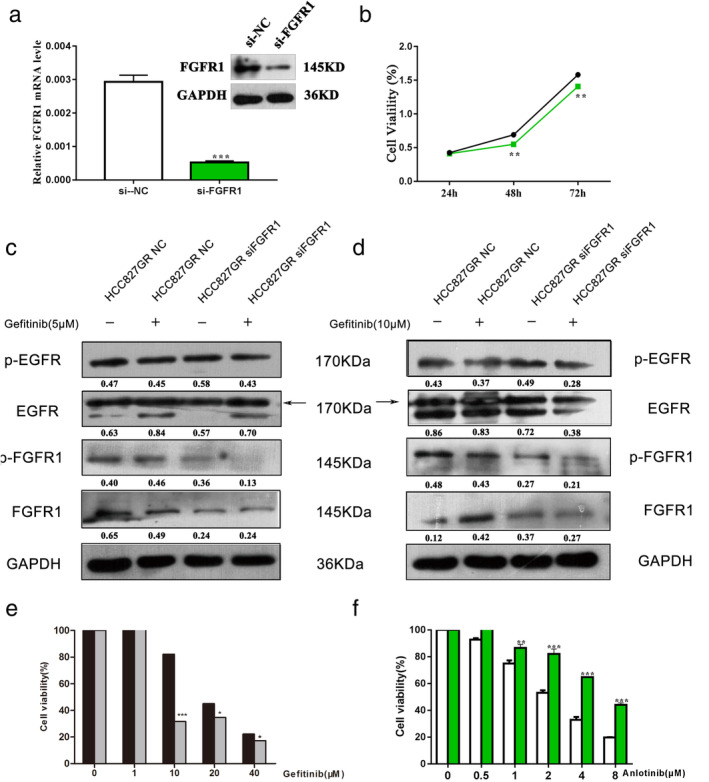
Knockdown of FGFR1 reverses gefitinib resistance in HCC827 GR cells. (a) qRT‐PCR and western blot analysis were used to detect the expression of FGFR1 in HCC827 GR cell lines after transfection with si‐FGFR1. (b) The cell viability of HCC827 GR cells transfected with si‐FGFR1 or si‐NC was detected by CCK‐8 assays ( si‐NC and
si‐NC and  si‐FGFR1). (c) The cell viability of HCC827 GR cells transfected with si‐FGFR1 or si‐NC treated with gefitinib or anlotinib was detected by CCK‐8 assays. (e) and (f) Western blot analysis was used to detect the effect of anlotinib on the gefitinib‐induced downregulation of p‐EGFR, p‐AKT and p‐ERK in HCC827 GR cells transfected with si‐FGFR1 or si‐NC (e:
si‐FGFR1). (c) The cell viability of HCC827 GR cells transfected with si‐FGFR1 or si‐NC treated with gefitinib or anlotinib was detected by CCK‐8 assays. (e) and (f) Western blot analysis was used to detect the effect of anlotinib on the gefitinib‐induced downregulation of p‐EGFR, p‐AKT and p‐ERK in HCC827 GR cells transfected with si‐FGFR1 or si‐NC (e:  HCC827GR NC and
HCC827GR NC and  HCC827GR siFGFR1; f:
HCC827GR siFGFR1; f:  si‐NC and
si‐NC and  si‐FGFR1).
si‐FGFR1).
Anlotinib inhibits the growth of HCC827 GR cells in vivo
To investigate whether anlotinib inhibits the growth of HCC827 GR cells in vivo, we inoculated athymic nude mice with HCC827 GR cells and then administered 2 mg/kg anlotinib, 4 mg/kg gefitinib or vehicle control. As shown in Fig 5(a), the administration of anlotinib had the strongest inhibitory effect on tumor growth, and the tumors began to shrink on the third day, whereas the tumors in the gefitinib‐administered group began to grow on the fifth day. In addition, compared to those in the gefitinib‐administered group, the tumors in the anlotinib‐administered group were significantly smaller and lighter after treatment for 12 days (Fig 5b,c). Resected tissues from the treated xenograft tumors were analyzed by western blot to confirm that anlotinib inhibited the phosphorylation of FGFR1, AKT and ERK (Fig 5d).
Figure 5.
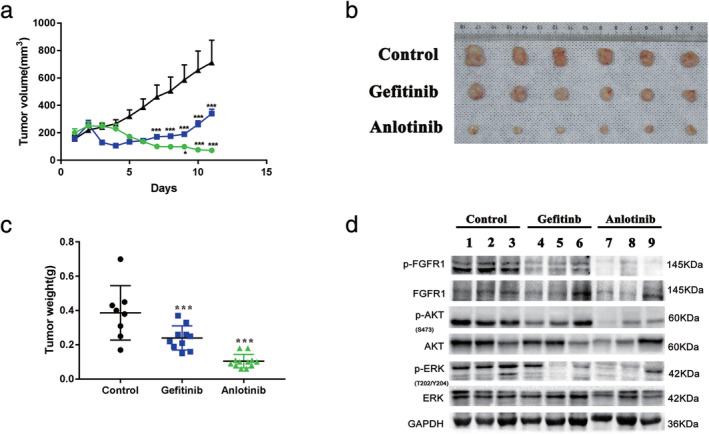
Anlotinib inhibits the growth of HCC827 GR cells in vivo. (a) The tumor volumes were measured at the indicated time intervals and calculated. See the Methods section for details ( Anlotinib,
Anlotinib,  Gefitinb, and
Gefitinb, and  Control). (b) and (c) At the end of treatment, the tumors were excised, photographed as indicated and weighed. (d) Western blotting was used to analyze the levels of p‐FGFR, p‐AKT and p‐ERK in tumor tissues from different treatment groups. (*** P < 0.001).
Control). (b) and (c) At the end of treatment, the tumors were excised, photographed as indicated and weighed. (d) Western blotting was used to analyze the levels of p‐FGFR, p‐AKT and p‐ERK in tumor tissues from different treatment groups. (*** P < 0.001).
Discussion
Almost all NSCLC patients treated with EGFR‐TKI inevitably develop acquired resistance.29, 30 Although many studies have defined mechanisms of resistance to EGFR‐TKIs, including secondary mutations in EGFR (T790M),6 KRAS mutations,7 PIK3CA mutations,8 MET amplification,9 EMT and SCLC transformation,10 acquired resistance remains the major limitation of monotherapy with EGFR‐TKI. Therefore, there is an urgent need to develop effective therapeutic interventions to overcome acquired resistance. Osimertinib monotherapy is the currently recommended second‐line treatment for EGFR T790M mutation‐positive NSCLC. However, there is no drug to treat NSCLC with other mechanisms leading to acquired resistance. In this study, we found that anlotinib inhibited the growth of EGFR‐TKI‐resistant NSCLC cells without harboring T790M mutation in vitro and in a mouse xenograft model. Furthermore, anlotinib induced apoptosis and cell cycle arrest in EGFR‐TKI‐resistant NSCLC cells by inhibiting FGFR1. These results provide novel alternatives and therapeutic strategies for non‐T790M‐mutated, EGFR‐TKI‐resistant NSCLC patients.
As an oral multitargeted TKI, anlotinib exhibits efficacy in various cancers.31, 32, 33 The clinical trials have indicated that anlotinib therapy prolongs PFS and OS in refractory advanced NSCLC subjected to third‐line or beyond third‐line therapy.31, 34, 35 The underlying mechanism might be attributed to anlotinib‐induced blockade of angiogenesis. One study reported that anlotinib therapy was beneficial for prolonging OS in NSCLC patients harboring EGFR mutations, especially patients harboring the EGFR T790M mutation.25, 26 Moreover, anlotinib inhibited angiogenesis in an H1975‐derived xenograft model via inhibiting CCL2.21 However, the effect of anlotinib on NSCLC patients with EGFR‐TKI resistance due to non‐T790M mutation and the mechanism of this effect remain unclear.
To examine the inhibitory effect of anlotinib on EGFR‐TKI‐resistant NSCLC cells without T790M mutation, we exposed HCC827 and PC‐9 cells to gefitinib to establish the GR cell lines HCC827 GR and PC‐9 GR. We then analyzed EGFR for the DNA substitution corresponding to the T790M mutation. The T790M mutation was found in the PC‐9 GR cell line but not in the HCC827 GR cell line. In addition, anlotinib inhibited the growth of HCC827 GR cells in vitro and in a mouse xenograft model. Although the xenograft study indicated that HCC827 GR cells inhibited by gefitinib as well as anlotinib significantly, perhaps the difference between the in vivo and in vitro and the complexity of the in vivo environment have led to the inhibitory effect of gefitinib on HCC827 GR cells. However, in the last few days of in vivo experiments, we observed that gefitinib hardly inhibited the growth of HCC827GR cells. It may be that with the prolongation of the treatment, the growth inhibition of HCC827GR cells by gefitinib is less obvious. Furthermore, anlotinib induced apoptosis and cell cycle arrest in HCC827 GR cells by increasing the activity of Caspase‐3. However, anlotinib mainly caused G2/M arrest, which may be due to abnormal expression of Cyclin B1 and CDK1 induced by anlotinib, These results suggested that anlotinib suppresses the growth of EGFR‐TKI‐resistant NSCLC without T790M mutation.
The FGFR family belongs to the RTK family, and there are 18 different FGF ligands.11, 12 FGFR is involved in many physiological processes, including wound repair, embryogenesis and angiogenesis.13 Activation of the FGF/FGFR signaling pathway promotes cancer progression and enhances the angiogenic potential of the tumor microenvironment.36, 37, 38 Dysregulation of FGFR1 leading to tumorigenesis, transformation, and tumor progression has been reported in a broad range of malignancies.14, 15 The activation of FGFR1 can promote EMT in prostate cancer, breast cancer and FGFR1‐amplified lung cancer.16, 17, 18 FGF/FGFR signaling alterations have been connected with chemotherapy resistance and poor clinical outcome.19, 20 We found that FGFR1 was overexpressed in HCC827 GR cells and that knockdown of FGFR1 reversed gefitinib resistance in HCC827 GR cells. Moreover, anlotinib inhibited FGFR1 in HCC827 GR cells in vitro and in a mouse xenograft model. These results suggested that the overexpression of FGFR1 is one of the mechanisms of EGFR‐TKI acquired resistance and that anlotinib inhibits the FGFR1 signaling pathway in HCC827 GR cells.
There are some limitations to our study. First, the anti‐tumor effect of anlotinib was confirmed in only EGFR‐TKI‐resistant HCC827 cells. However, our findings should be validated in various EGFR‐TKI‐resistant lung cancers. Second, because anlotinib is a multitarget receptor TKI, whether anlotinib inhibits the growth of EGFR‐TKI‐resistant NSCLC cells through other targets should be further investigated in future studies. Third, the conclusions in our research have not been well supported, and we need to improve and design the experiment more fully. For example, we need to silence FGFR1 to detect changes in DNA content and apoptosis by FACS analysis to verify whether G2/M arrest and apoptosis induced by anlotinib are caused by irregular spindle assembly due to abnormal expression of cyclin B1 and CDK1. Similarly, after anlotinib treatment, we also need to detect the cleaved caspase‐3 and PARP level in HCC827GR cells with or without TNF‐alpha and CHX treatment. Other apoptosis‐associated protein like Bcl‐2, Bax should also be tested. In addition, we need to re‐examine the p‐AKT and p‐ERK protein level in HCC827GR cells under FGFR1 knockdown condition. Finally, we should determine that the molecular mechanism by which anlotinib inhibits the growth of EGFR‐TKI‐resistant NSCLC cells in animal and cellular models. To expand the application of anlotinib as a preclinical cancer drug, we will need to further verify the effect of anlotinib in patient‐derived xenograft (PDX) models of EGFR‐TKI‐resistant NSCLC.
In summary, we demonstrate that the overexpression of FGFR1 is one of the mechanisms of EGFR‐TKI acquired resistance and that anlotinib suppresses the growth of EGFR‐TKI‐resistant NSCLC cells without T790M mutation in vitro and in a mouse xenograft model by inhibiting FGFR1. Our study suggests anlotinib as a rational therapeutic drug for non‐T790M‐mutated, EGFR‐TKI‐resistant NSCLC patients.
Disclosure
The authors declare that they have no competing interests.
Supporting information
Figure S1 (a) and (b) The mRNA and protein expression levels of FGFR1 were lower in PC‐9 GR cells than in PC‐9 cells.
Table S1 HiSeq 4000 second‐generation high‐throughput gene sequencing showed the acquisition of a secondary T790M mutation in PC9 GR cells but not in HCC827 GR cells.
Acknowledgments
This work was supported by grants from the Science and Technology Plan Project of Suzhou (No. SYS201749), the Suzhou Key Laboratory for Respiratory Medicine (No. SZS201617), the Clinical Medical Center of Suzhou (No. Szzx201502), the Jiangsu Provincial Key Medical Discipline (No. ZDXKB2016007), the Beijing medical and health foundation (No. F2035E) and the Clinical Key Specialty Project of China.
Contributor Information
Zeyi Liu, Email: liuzeyisuda@163.com.
Jian‐an Huang, Email: huang_jian_an@163.com.
References
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018; 68: 394–424. [DOI] [PubMed] [Google Scholar]
- 2. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med 2008; 359: 1367–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lynch TJ, Bell DW, Sordella R et al Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 4. Pao W, Chmielecki J. Rational, biologically based treatment of EGFR‐mutant non‐small‐cell lung cancer. Nat Rev Cancer 2010; 10: 760–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ettinger DS, Wood DE, Aisner DL et al Non‐small cell lung cancer, version 5.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw 2017; 15: 504–35. [DOI] [PubMed] [Google Scholar]
- 6. Pao W, Miller VA, Politi KA et al Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLOS Med 2005; 2: e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pao W, Wang TY, Riely GJ et al KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLOS Med 2005; 2: e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Engelman JA, Mukohara T, Zejnullahu K et al Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR‐amplified lung cancer. J Clin Invest 2006; 116: 2695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Engelman JA, Zejnullahu K, Mitsudomi T et al MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316: 1039–43. [DOI] [PubMed] [Google Scholar]
- 10. Sequist LV, Waltman BA, Dias‐Santagata D et al Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011; 3: 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol 2015; 4: 215–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beenken A, Mohammadi M. The FGF family: Biology, pathophysiology and therapy. Nat Rev Drug Discov 2009; 8: 235–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang K, Ji W, Yu Y et al FGFR1‐ERK1/2‐SOX2 axis promotes cell proliferation, epithelial‐mesenchymal transition, and metastasis in FGFR1‐amplified lung cancer. Oncogene 2018; 37: 5340–54. [DOI] [PubMed] [Google Scholar]
- 14. Konermann S, Brigham MD, Trevino AE et al Genome‐scale transcriptional activation by an engineered CRISPR‐Cas9 complex. Nature 2015; 517: 583–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vad‐Nielsen J, Lin L, Bolund L, Nielsen AL, Luo Y. Golden Gate assembly of CRISPR gRNA expression array for simultaneously targeting multiple genes. Cell Mol Life Sci 2016; 73: 4315–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weiss J, Sos ML, Seidel D et al Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med 2010; 2: 62ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Malchers F, Dietlein F, Schottle J et al Cell‐autonomous and non‐cell‐autonomous mechanisms of transformation by amplified FGFR1 in lung cancer. Cancer Discov 2014; 4: 246–57. [DOI] [PubMed] [Google Scholar]
- 18. Facchiano A, Russo K, Facchiano AM et al Identification of a novel domain of fibroblast growth factor 2 controlling its angiogenic properties. J Biol Chem 2003; 278: 8751–60. [DOI] [PubMed] [Google Scholar]
- 19. Turner N, Pearson A, Sharpe R et al FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res 2010; 70: 2085–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matsumoto K, Arao T, Hamaguchi T et al FGFR2 gene amplification and clinicopathological features in gastric cancer. Br J Cancer 2012; 106: 727–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu J, Zhong H, Chu T et al Role of anlotinib‐induced CCL2 decrease in anti‐angiogenesis and response prediction for nonsmall cell lung cancer therapy. Eur Respir J 2019; 53: 1801562. [DOI] [PubMed] [Google Scholar]
- 22. Lin B, Song X, Yang D, Bai D, Yao Y, Lu N. Anlotinib inhibits angiogenesis via suppressing the activation of VEGFR2, PDGFRbeta and FGFR1. Gene 2018; 654: 77–86. [DOI] [PubMed] [Google Scholar]
- 23. Xie C, Wan X, Quan H et al Preclinical characterization of anlotinib, a highly potent and selective vascular endothelial growth factor receptor‐2 inhibitor. Cancer Sci 2018; 109: 1207–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taurin S, Yang CH, Reyes M et al Endometrial cancers harboring mutated fibroblast growth factor receptor 2 protein are successfully treated with a new small tyrosine kinase inhibitor in an orthotopic mouse model. Int J Gynecol Cancer 2018; 28: 152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou M, Chen X, Zhang H et al China National Medical Products Administration approval summary: Anlotinib for the treatment of advanced non‐small cell lung cancer after two lines of chemotherapy. Cancer Commun 2019; 39: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Si X, Zhang L, Wang H et al Management of anlotinib‐related adverse events in patients with advanced non‐small cell lung cancer: Experiences in ALTER‐0303. Thorac Cancer 2019; 10: 551–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koizumi F, Shimoyama T, Taguchi F, Saijo N, Nishio K. Establishment of a human non‐small cell lung cancer cell line resistant to gefitinib. Int J Cancer 2005; 116: 36–44. [DOI] [PubMed] [Google Scholar]
- 28. Zhang N, Zeng Y, Du W et al The EGFR pathway is involved in the regulation of PD‐L1 expression via the IL‐6/JAK/STAT3 signaling pathway in EGFR‐mutated non‐small cell lung cancer. Int J Oncol 2016; 49: 1360–8. [DOI] [PubMed] [Google Scholar]
- 29. Paez JG, Janne PA, Lee JC et al >EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–500. [DOI] [PubMed] [Google Scholar]
- 30. Mok TS, Wu YL, Thongprasert S et al Gefitinib or carboplatin‐paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361: 947–57. [DOI] [PubMed] [Google Scholar]
- 31. Han BH, Li K, Wang QM et al Efficacy and safety of third‐line treatment with anlotinib in patients with refractory advanced non‐small‐cell lung cancer (ALTER‐0303): A randomised, double‐blind, placebo‐controlled phase 3 study. Lancet Oncol 2017; 18: S3–3. [Google Scholar]
- 32. Sun Y, Chi Y, Tan P et al Phase II study of anlotinib for treatment of advanced medullary thyroid carcinoma. ASCO Publ 2016; 34: 6015. [Google Scholar]
- 33. Chi Y, Sun Y, Cai J et al Phase II study of anlotinib for treatment of advanced soft tissues sarcomas. ASCO Publ 2016; 34: 11005. [Google Scholar]
- 34. Han BH, Li K, Zhao YZ et al Anlotinib as a third‐line therapy in patients with refractory advanced non‐small‐cell lung cancer: A multicentre, randomised phase II trial (ALTER0302). Br J Cancer 2018; 118: 654–61. 10.1038/bjc.2017.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Han BH, Li K, Wang QM et al Effect of anlotinib as a third‐line or further treatment on overall survival of patients with advanced non–small cell lung cancer the ALTER 0303 phase 3 randomized clinical trial. JAMA Oncol 2018; 4: 1569–75. 10.1001/jamaoncol.2018.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Turner N, Grose R. Fibroblast growth factor signalling: From development to cancer. Nat Rev Cancer 2010; 10: 116–29. [DOI] [PubMed] [Google Scholar]
- 37. Knights V, Cook SJ. De‐regulated FGF receptors as therapeutic targets in cancer. Pharmacol Ther 2010; 125: 105–17. [DOI] [PubMed] [Google Scholar]
- 38. Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. Biochem J 2011; 437: 199–213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (a) and (b) The mRNA and protein expression levels of FGFR1 were lower in PC‐9 GR cells than in PC‐9 cells.
Table S1 HiSeq 4000 second‐generation high‐throughput gene sequencing showed the acquisition of a secondary T790M mutation in PC9 GR cells but not in HCC827 GR cells.