

Abstract
Direct epoxidation of aromatic nuclei by cytochrome P450 monooxygenases is one of the major metabolic pathways of arenes in eukaryotes. The resulting arene oxides serve as versatile precursors to phenols, oxepines, or trans-dihydrodiol-based metabolites. Although such compounds have an important biological and chemical relevance, the lack of methods for their production has hampered access to their utility. Herein, we report a general arenophile-based strategy for the dearomative synthesis of arene oxides. The mildness of this method permits access to sensitive monocyclic arene oxides without any noticeable decomposition to phenols. Moreover, this method enables direct conversion of polycyclic arenes and heteroarenes into the corresponding oxepines. Finally, these studies provided direct connection between simple aromatic precursors and complex small organic molecules via arene oxides and oxepines.
Graphical Abstract

INTRODUCTION
One of the fundamental interests of biology, microbiology, and biochemistry is the metabolism of organic molecules by living organisms. This process often involves the incorporation of molecular oxygen into organic compounds catalyzed by oxidative enzymes.1 Aromatic compounds are subject to such oxidative degradation, and their metabolism by both eukaryotic and prokaryotic systems has been extensively studied.2 Notably, bacterial dioxygenases convert arenes into cis-dihydrodiols,3 whereas mammalian and fungal monooxygenases lead to arene oxides (Figure 1A).4 The latter intermediates are characterized by unique chemistry, including the valence tautomerization between arene oxide and oxepine forms.5 Moreover, arene oxides are known for their instability,6 often undergoing spontaneous and rapid [1,2]-hydride migration to phenols, known as the NIH shift.7 Arene oxides are also subject to nucleophilic epoxide opening, delivering trans-dihydrodiol derivatives that are important precursors involved in biogenesis of primary and secondary metabolites, environmental degradation, and drug metabolism (Figure 1B). For example, dihydrodiol 4 is a phase-I metabolite of the antiepileptic drug phenytoin (1) that is derived from the hydrolysis of the corresponding arene oxide.8 Gliotoxin (5) is an Aspergillus fumigatus produced mycotoxin that is derived from arene epoxidation of a phenylalanine precursor 2.9 Finally, perilloxin (6) is an oxepine-containing natural product that could be arising through arene oxidation of naphthalene 3.10
Figure 1.

(A) Representative arene oxidation pathways in Nature. (B) Selected metabolites derived from arene oxides.
Given the importance of biological arene oxidation processes, as well as the unique structural features of the resultant arene oxides that could be leveraged for further functionalization, significant efforts have been devoted to translating them to the field of synthetic chemistry. For example, the natural metabolic process of microbial arene oxidation has been implemented in laboratory settings utilizing whole-cell bacterial fermentations.11 The resulting cis-dihy-drodiols (cis-cyclohexadiene-1,2-diols) have proven to be versatile and key building blocks for the synthesis of many natural products and value-added intermediates.12 Yet, no practical chemoenzymatic arene epoxidations exist, likely because of the instability of arene oxides. Thus, the preparation of arene oxides and co-occurring oxepines has been limited to only a few synthetic strategies. For example, the most common preparation of monocyclic arene oxides involves the Birch reduction13 or bacterial arene oxidation as the initial dearomative step (Figure 2A).14 On the other hand, the preparation of oxepines is even less developed, as documented with only a few examples of the synthesis of 3-benzoxepine (Figure 2B). Thus, phthalaldehyde was converted to the corresponding 3-benzoxepine by means of a double Wittig reaction.15 The same product can be obtained also from photochemical isomerization of 7-oxabenzonorbornadiene,16 which can be prepared from benzyne and furan.
Figure 2.

(A) Established preparation of arene oxides. (B) Previous preparations of 3-benzoxepine. (C) This work: arenophile-mediated synthesis of arene oxides and oxepines.
More straightforward and mild methods for the generation of arene oxides would significantly expand their synthetic utility and general applicability.17 Considering the lack of methods for the direct epoxidation of arenes, we set out to develop an alternative preparation of arene oxides and oxepines based on an arenophile-mediated dearomative platform. Herein, we report the successful realization of this plan. Through the use of a Mn-catalyzed epoxidation as the olefin functionalization step (Figure 2C), a range of aromatic and heteroaromatic compounds delivered arene oxides and polycyclic oxepines, which are complementary to those obtained by other established strategies. Finally, the synthetic utility of this method was demonstrated through the preparation of several functionalized small molecules.
RESULTS AND DISCUSSION
Optimization of Reaction Conditions
We have recently reported a series of dearomative functionalizations based on the arenophile 4-methyl-1,2,4-triazoline-3,5-dione (7, MTAD),18 which undergoes a visible-light-promoted para-cycloaddition with arenes. Subsequent in situ manipulation of the resulting cycloadducts gives rise to a diverse collection of dearomatized products.19 One of the possible functionalization strategies involves the application of olefin chemistry, providing bicycles that reveal dienes after [4 + 2]-cyclo-reversion of the arenophile moiety. Overall, this sequence can be formally seen as an arenophile-assisted isolation of a single π-component from an aromatic system. On the basis of this concept, we initially developed a dearomative dihydroxylation, a chemical method to complement arene dioxygenases.18 Interestingly, the corresponding epoxidation proved to be quite elusive despite a considerable number of attempts. The subjection of the arene-arenophile para-cycloadducts derived from benzene to numerous benchmark epoxidizing agents, such as peracetic acids and dioxiranes did not result in the detection of even trace amounts of the desired products (see Supporting Information for details). Ultimately, we evaluated transition-metal-mediated epoxidations and discovered that Stack’s Mn-catalyzed epoxidation methodology20 provided encouraging results.
Accordingly, our optimization commenced with screening of Mn-based epoxidation conditions with benzene-derived cycloadduct (Table 1; see also Supporting Information, Table S1, for full optimization details). The initial examination (entries 1–5) of commonly used ligands 10–12 for this process revealed that 1,10-phenanthroline (12) performed favorably. Though pyridine-oxazoline-type ligand 11 provided promising conversion (entry 3), further manipulations of reaction parameters to increase the yield of 8a failed, which was traced back to the overoxidation/decomposition of the ligand. Furthermore, we also observed the decomposition of the product under reaction conditions due to the low pH of the commercial samples of peracetic acid, likely because of the presence of sulfuric acid. This hurdle was surmounted by using a freshly prepared solution of peracetic acid that was free of any inorganic acids (entries 6–10). By screening different catalyst loadings and using an optimal 1:2 ratio of [Mn]/[12] (entries 6–10), we found that the addition of a manganese(II) perchlorate/phenanthroline complex (25/50 mol %) with peracetic acid (3.0 equiv) as an oxidant provided epoxidized product 8a in 45% yield, after chromatography and as a 3:1 mixture of diastereoisomers (entry 9).
Table 1.
Selected Optimization Studies for the Epoxidation of the Benzene-MTAD Cycloadducta
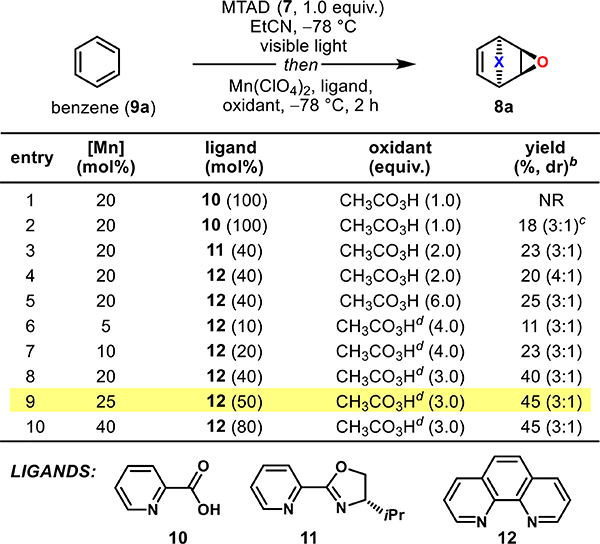 |
Standard reaction conditions: MTAD (7, 0.5 mmol, 1.0 equiv), benzene (9a, 5.0 mmol, 10 equiv), EtCN (0.10 M), visible light, −78 °C, 12 h, and then addition of a solution of catalyst [Mn(ClO4)2/ligand in MeCN], CH3CO3H, −78 °C, 2 h.
Reported yields are of isolated products and the ratio of diastereoisomers (in parentheses) was determined by 1H NMR of the crude reaction mixtures.
After addition of catalysts and oxidant, the reaction was warmed to 25 °C over the course of 2 h.
Freshly prepared, H2SO4-free solution of CH3CO3H was used.
With epoxidized cycloadduct 8a in hand, we turned our attention toward cycloreversion (Table 2). In our previous studies21 the urazole motif could be removed through a one-pot protocol involving hydrolysis to a cyclic hydrazine and oxidation to a cyclic diazine that readily extruded nitrogen to reveal the desired diene functionality. Unfortunately, the application of these established protocols to epoxidized cycloadduct 8a, using KOH or neat hydrazine at 100 °C followed by CuCl2 oxidation, resulted only in decomposition (entries 1 and 2). After several attempts, and with inspiration from the literature,22 we discovered that partial urazole hydrolysis/decarboxylation (KOH in i-PrOH, 40 °C) provided a stable cyclic semicarbazide intermediate 14, which could be further oxidized. Although CuCl2 was not a suitable oxidant, providing only a phenol (entry 3), this result inspired the search for a milder oxidant, as the phenol was derived from the desired arene oxide via NIH shift. After screening a variety of oxidants, we found that through the use of nickel(lIl) oxide, arene oxide 13a could be obtained, virtually free of phenol byproducts.
Table 2.
Selected Cycloreversion Optimization Studies
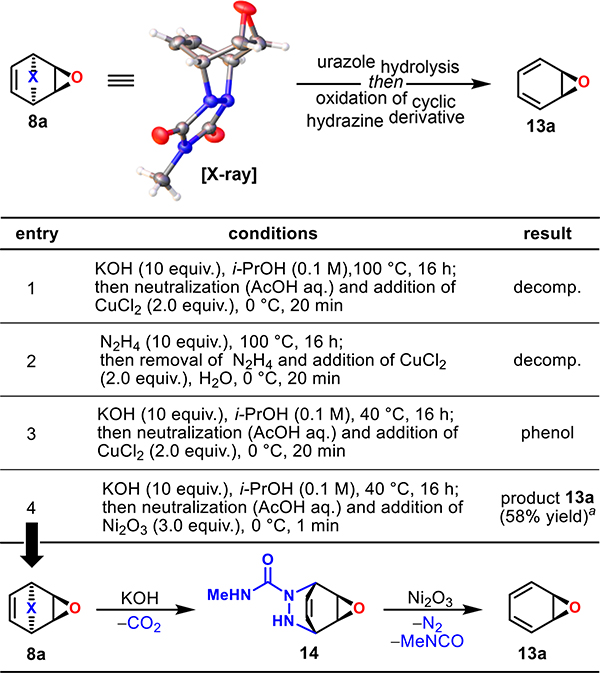 |
Reported yield is based on 1H NMR integration relative to the internal standard (MeNO2).
Synthesis of Arene Oxides
With both epoxidation and cycloreversion strategies developed for benzene (9a), we examined the general applicability of these conditions toward benzene derivatives (Table 3). First, the challenging epoxidation also proved viable for other monocyclic arenes, delivering products 8b–8f in 26–45% yields. Thus, benzene derivatives containing alkyl substituents such as tert-butyl (8b), a chlorinated alkyl side chain (8c), and a CF3 (8d) all provided the desired products. Although electron-deficient benzene derivatives do not undergo cycloaddition with MTAD (7), the corresponding ketal of acetophenone (8e) and the orthoester of benzoic acid (8f) successfully underwent cycloaddition and epoxidation. Overall, high chemoselectivity was observed, as only tert-butylbenzene (9b) gave a minor constitutional isomer (3:1). Importantly, though epoxidation of these substrates resulted in diastereoisomeric mixtures ranging from 3:1 to >20:1, this proved inconsequential for the next step involving the preparation of arene oxides. Thus, all monocyclic epoxidized precursors 8a–8f were rapidly converted to arene oxides 13a–13f with yields ranging from 58% to 82% based on 1H NMR (measured using an internal standard). Although it was not possible to isolate or purify these compounds, the corresponding solutions could be immediately used for further chemistry (see below).
Table 3.
Preparation of Monocyclic Arene Oxides (l3)a
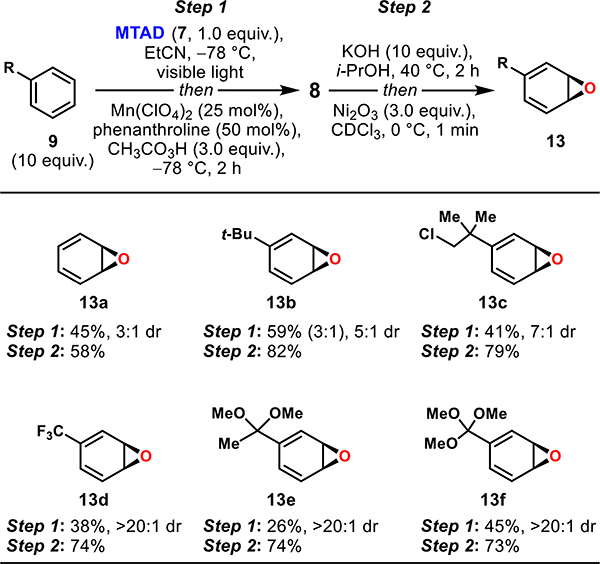 |
Standard reaction conditions. Step 1: MTAD (7, 1.0 mmol, 1.0 equiv), arene (9, 10 mmol, 10 equiv), EtCN (0.1 M), visible light, −78 °C, 12 h; then addition of Mn(ClO4)2/phenanthroline (25/50 mol %) in MeCN and CH3CO3H (3.0 mmol, 3.0 equiv), −78 °C, 2 h. Step 2: epoxide 8 (0.2 mmol, 1.0 equiv), KOH (2.0 mmol, 10 equiv), 40 °C, 2 h; then workup and exposure of crude to Ni2O3 (0.6 mmol, 3.0 equiv), CDO3 (0.1 M), 0 °C, 1 min. Reported yields of 8 (step 1) are of isolated products and the ratio of diastereoisomers and constitutional isomer (bracket) were determined by 1H NMR of the crude reaction mixtures. Reported yields of 13 (step 2) are based on 1H NMR integration relative to the internal standard (MeNO2).
Synthesis of Oxepines
In addition to benzene derivatives, we questioned whether this process would be compatible with polycyclic arenes, such as naphthalenes (Table 4). Because of steric constraints and stabilization from the adjacent π-system, the initial naphthalene-2,3-oxide products should exclusively adopt the oxepine valence tautomer, which increases their stability significantly. Unfortunately, the developed epoxidation conditions (9 → 8) did not translate well to naphthalene (15a), leading only to trace product 16a. After screening a variety of conditions (see Supporting Information, Table S2), we found that through the use of manganese(II) perchlorate and picolinic acid (5/25 mol %) as the catalyst, the corresponding epoxidized MTAD-naphthalene adduct 16a was obtained in 84% yield from naphthalene (15a). Because polycyclic arene-MTAD cycloadducts are generally more thermally stable than their monocyclic congeners, the cycloaddition reactions could be conducted at −50 °C and epoxidations at −20 °C. Only with 1-substituted naphthalene derivatives and phenanthrene (15f–15i) did we observe greater thermal instability of MTAD-adducts, leading to poor yields under these conditions. Thus, a slight modification was developed, utilizing higher loadings of catalysts (Mn(ClO4)2/picolinic acid = 20/100 mol %) at −78 °C.
Table 4.
Preparation of Oxepines (17)a
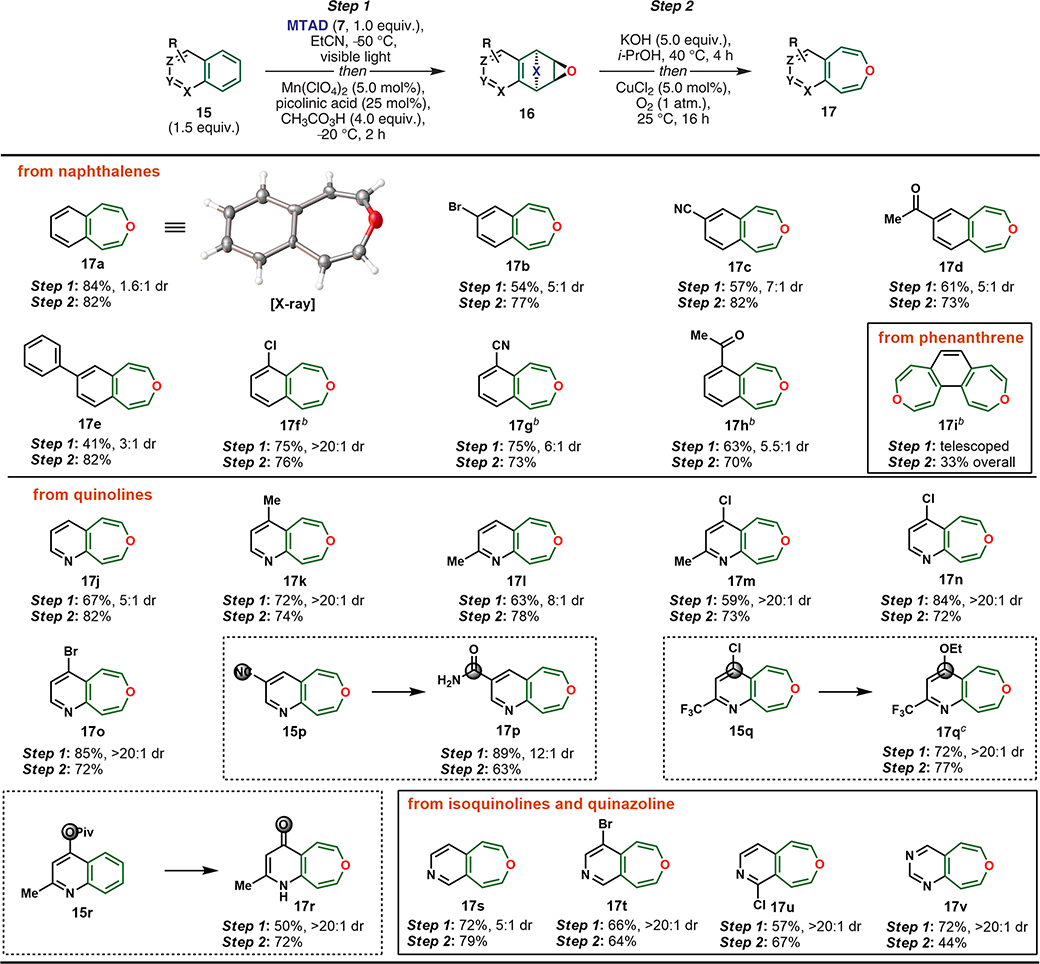 |
Standard reaction conditions. Step 1: MTAD (7, 1.0 mmol, 1.0 equiv), arene (15, 1.5 mmol, 1.5 equiv), EtCN (0.1 M), visible light, −50 °C, 12 h; then Mn(ClO4)2/picolinic acid (5/25 mol %) in MeCN and CH3CO3H (4.0 mmol, 4.0 equiv), −20 °C, 2 h. Reported yields are of isolated products. Step 2: epoxide 16 (0.2 mmol, 1.0 equiv), KOH (1.0 mmol, 5.0 equiv), i-PrOH (1.0 M), 40 °C, 4 h; then workup and exposure to CuCl2 (5.0 mol %) under O2 atm.
Identical cycloaddition conditions to standard conditions; then Mn(ClO4)2/picolinic acid (20/100 mol %) in MeCN and CH3CO3H (6.0 mmol, 3.0 equiv), −78 °C, 2 h.
Ethanol was used instead of i-PrOH.
Cycloreversion proceeded in a similar manner as before, through the intermediacy of the semicarbazides. Gratifyingly, the pronounced stability of 3-benzoxepins permitted the use of aerobic oxidation conditions. For example, upon partial KOH-induced hydrolysis of the urazole moiety in 15a, the semicarbazide 16a underwent Cu-catalyzed oxidation (5 mol %) under an oxygen atmosphere, delivering 3-benzoxepine 17a in 82% yield. We were able to obtain a crystal structure of 17a in which a severe out-of-plane distortion relative to naphthalene was observed (Table 4, also see Supporting Information). To explore the substrate scope, a set of 1- and 2- substituted naphthalene derivatives were subjected to this process to deliver 3-benzoxepine analogs, with tolerated functionality including halogens (17b and 17f), nitriles (17c and 17g), and ketones (17d and 17h). Moreover, phenyl-substituted naphthalene 15e gave the desired oxepine 17e with exclusive site selectivity for dearomative oxidation at the less-substituted ring of naphthalene. Larger polycyclic arenes could be employed with this protocol, as demonstrated with phenanthrene, which underwent a double oxidation to bis-oxepine 17i.
In addition to arenes, we probed this dearomative oxidation strategy with heteroarenes such as benzopyridines and their derivatives, as the resulting azabenzoxepines are a largely unknown and unexplored class of heterocycles. Though at first glance they appear to be simple, containing only two fused rings and two heteroatoms, no general syntheses of such compounds have been reported to date.
Subjecting a range of quinolines to the described two-step sequence furnished the corresponding oxepino[4,5-b]pyridines 17j–17r, without any N-oxide formation, and with similar efficiency that was observed for hydrocarbon arenes. In addition to quinoline (17j), heteroarenes with different substituents and substitution patterns were tolerated, as exemplified with alkyl (17k–17m), halogen (17m–17o), and trifluoromethyl (l7q) substituted quinolines. Nevertheless, several functional groups did react under these conditions, during the basic KOH-hydrolysis/cycloreversion step, providing new functionalities. For example, the nitrile 15p hydrolyzed to yield amide 17p and 4-chloro-2-(trifluoromethyl)quinoline (l5q) was sufficiently electron-deficient to undergo SNAr with the solvent and base (EtOH/KOH) to yield compound 17q. Also, the pivalate group in substrate 15r underwent hydrolysis during the cycloreversion step, delivering 4-pyridone-fused oxepine 17r. Moreover, with use of isoquinolines as substrates, this method enabled access to the complementary oxepino[4,5-c]pyridines 17s–17u. Finally, benzo-fused heterocycles containing more than one nitrogen can be used, as exemplified with preparation of oxepino[4,5-d]pyrimidine (17v) from quinazoline. We were pleased to note that both epoxidation and cycloreversion could be performed on a gram scale, as demonstrated with compounds 16a, 16f, and 17a (see Supporting Information).
Derivatization of Products
The chemistry described herein can be extended greatly through the functionalization of the obtained reactive intermediates—arene oxides and oxepines—ultimately giving access to diverse and structurally elaborated small molecules (Figure 3). For example, benzene-derived bicyclic epoxide 8a underwent two complementary acid-mediated hydrolyses to provide aminocyclitol 18 or its dichloro derivative 19. Moreover, arene oxide 13b derived from tert-butylbenzene (9b, see Table 3) was directly converted to γ-hydroxyenone 20 or syn-1,4-diol 21 through the intermediacy of an endoperoxide.23
Figure 3.

Diversification of products.
These examples demonstrate that the arenophile-based preparation of arene oxides can be applied for downstream chemistry, bypassing the highly unstable nature of these compounds. In addition, benzoxepines could serve as intermediates for further derivatization, as confirmed with substrates 17a and 17o. Thus, partial or full hydrogenation of benzoxepine 17a provided 22 and 24. The dihydrobenzox-epine 22 was further elaborated via bromohydrin chemistry to the functionalized ether 23. Also, benzoxepine 17a could serve as a viable cycloaddition partner, undergoing nitrile oxide mediated [3 + 2] dipolar cycloaddition24 to give product 25 (7:1 ratio of constitutional isomers). Finally, further derivatization was also possible by conducting a Suzuki reaction on the brominated oxepinopyridine (17o → 26), highlighting the ability to utilize these unexplored heterocycles as practical building blocks for medicinal chemistry.
CONCLUSION
We have developed a dearomative strategy for epoxidation of simple arenes. Our approach features an arenophile-based cycloaddition and epoxidation, followed by cycloreversion to reveal arene oxides and 3-benzoxepines. Several benzene derivatives were converted to the corresponding arene oxides with complementary chemoselectively to known methods and without any noticeable decomposition to phenols. Moreover, this protocol converted polycyclic arenes and heteroarenes into the corresponding oxepines with a broad functional group tolerability. Importantly, the described chemistry enables formal epoxidation of naphthalenes at positions 2 and 3, a biomimetic epoxidation for which no chemical equivalent exists. Given the lack of chemical methods for molecular editing of this type, as well as practical gram-scale feasibility and further options for elaboration, we anticipate the application of this dearomative strategy in the preparation of high-value intermediates.
Supplementary Material
ACKNOWLEDGMENTS
Financial support for this work was provided by the University of Illinois, the National Science Foundation (CAREER Award No. CHE-1654110), and the NIH/National Institute of General Medical Sciences (R01 GM122891). Amgen, Eli Lilly, and Bristol-Myers Squibb are also acknowledged for unrestricted research support. W.C.W. acknowledges support from Springborn Fund and NSF (GRFP). We also thank Dr. D. Olson and Dr. L. Zhu for NMR spectroscopic assistance, Dr. D. L. Gray, Dr. T. Woods, and Mr. A. Shved for X-ray crystallographic analysis assistance, and F. Sun for mass spectrometric assistance.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c02724.
Full experimental procedures and characterization data for all new compounds; 1H and 13C NMR spectra and crystallographic information for 8a, 16f, 16q, and 17a (PDF)
(Crystallographic data for 8a CIF)
(Crystallographic data for 16f CIF)
(Crystallographic data 16q CIF)
(Crystallographic data 17a CIF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c02724
Contributor Information
Zohaib Siddiqi, Roger Adams Laboratory, Department of Chemistry, University of Illinois, Urbana, Illinois 61801, United States.
William C. Wertjes, Roger Adams Laboratory, Department of Chemistry, University of Illinois, Urbana, Illinois 61801, United States
David Sarlah, Roger Adams Laboratory, Department of Chemistry, University of Illinois, Urbana, Illinois 61801, United States.
REFERENCES
- (1).(a) Boll M; Fuchs G; Heider J Anaerobic Oxidation of Aromatic Compounds and Hydrocarbons. Curr. Opin. Chem. Biol. 2002, 6, 604–611. [DOI] [PubMed] [Google Scholar]; (b) Horvath RS. Microbial Co-Metabolism and the Degradation of Organic Compounds in Nature. Bacteriol. Rev. 1972, 36, 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).For reviews on this topic, see:Goodwin BL. In Handbook of biotransformations of aromatic compounds; CRC Press LLC: Boca Raton, FL, 2005; pp 109–124.Fuchs G Anaerobic Metabolism of Aromatic Compounds. Ann. N. Y. Acad. Sci. 2008, 1125, 82–99.
- (3).(a) Gibson DT; Cardini GE; Maseles FC; Kallio RE Oxidative Degradation of Aromatic Hydrocarbons by Microorganisms. IV. Incorporation of Oxygen-18 into Benzene by Pseudomonas Putida. Biochemistry 1970, 9, 1631–1635. [DOI] [PubMed] [Google Scholar]; (b) Boyd DR; Sheldrake GN The Dioxygenase-Catalysed Formation of Vicinal Cis-Diols. Nat. Prod. Rep. 1998, 15, 309–324. [Google Scholar]
- (4).(a) Daly J; Witkop B; Zaltzman-Nirenberg P; Udenfriend S Role of the Arene Oxide-Oxepine System in the Metabolism of Aromatic Substrates: I. In Vitro Conversion of Benzene Oxide to a Premercapturic Acid and a Dihydrodiol. Arch. Biochem. Biophys. 1968, 128, 176–183. [Google Scholar]; (b) Jerina DM; Kaubisch N; Daly JW Arene Oxides as Intermediates in the Metabolism of Aromatic Substrates: Alkyl and Oxygen Migrations during Isomerization of Alkylated Arene Oxides. Proc. Natl. Acad. Sci. U. S. A. 1971, 68, 2545–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Vogel E; Günther H Benzene Oxide-Oxepine Valence Tautomerism. Angew. Chem. Int. Ed. Engl. 1967, 6, 385–401. [Google Scholar]; (b) Boyd D R; Sharma, N. D. The Changing Face of Arene Oxide–Oxepine Chemistry. Chem. Soc. Rev. 1996, 25, 289–296. [Google Scholar]
- (6).(a) Kasperek GJ; Bruice TC Mechanism of the Aromatization of Arene Oxides. J. Am. Chem. Soc. 1972, 94, 198–202. [Google Scholar]; (b) Bruice TC; Bruice PY Solution Chemistry of Arene Oxides. Acc. Chem. Res. 1976, 9, 378–384. [Google Scholar]
- (7).(a) Guroff G; Renson J; Udenfriend S; Daly JW; Jerina DM; Witkop B Hydroxylation-Induced Migration: The NIH Shift: Recent Experiments Reveal an Unexpected and General Result of Enzymatic Hydroxylation of Aromatic Compounds. Science 1967, 157, 1524–1530. [DOI] [PubMed] [Google Scholar]; (b) Jerina DM; Daly JW Arene Oxides: A New Aspect of Drug Metabolism. Science 1974, 185, 573–582. [DOI] [PubMed] [Google Scholar]
- (8).Maguire JH; Kraus BL; Butler TC; Dudley KH Determination of 5-(3,4-Dihydroxy-1,5-Cyclohexadien-1-Yl)-5-Phe-nylhydantoin (Dihydrodiol) and Quantitative Studies of Phenytoin Metabolism in Man. Ther. Drug Monit. 1979, 1, 359–370. [DOI] [PubMed] [Google Scholar]
- (9).(a) Suhadolnik RJ; Chenoweth RG Biosynthesis of Gliotoxin. I.1 Incorporation of Phenylalanine-1- and −2-C14. J. Am. Chem. Soc. 1958, 80, 4391–4392. [Google Scholar]; (b) For a recent review, see: Scharf DH; Heinekamp T; Remme N; Hortschansky P; Brakhage AA; Hertweck C Biosynthesis and Function of Gliotoxin in Aspergillus Fumigatus. Appl Microbiol. Biotechnol 2012, 93, 467–472. [DOI] [PubMed] [Google Scholar]
- (10).Liu J; Steigel A; Reininger E; Bauer R Two New Prenylated 3-Benzoxepine Derivatives as Cyclooxygenase Inhibitors from Perilla Frutescens Var. Acuta. J. Nat. Prod. 2000, 63, 403–405. [DOI] [PubMed] [Google Scholar]
- (11).Johnson R A. Microbial Arene Oxidations. Org. React. 2004, 63, 117–264. [Google Scholar]
- (12).(a) Lewis SE Applications of Biocatalytic Arene Ipso,Ortho Cis-Dihydroxylation in Synthesis. Chem. Commun. 2014, 50, 2821–2830. [DOI] [PubMed] [Google Scholar]; (b) Hudlicky T Benefits of Unconventional Methods in the Total Synthesis of Natural Products. ACS Omega 2018, 3, 17326–17340. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lan P; Ye S; Banwell MG The Application of Dioxygenase-Based Chemoenzymatic Processes to the Total Synthesis of Natural Products. Chem. - Asian J. 2019, 14, 4001–4012. [DOI] [PubMed] [Google Scholar]
- (13).(a) Vogel E; Schubart R; Böll WA Synthesis of an Oxepine Derivative. Angew. Chem., Int. Ed. Engl. 1964, 3, 510–510. [Google Scholar]; (b) Vogel E; Böll WA; Günther H Oxepin-benzoloxyd-valenztautomerie. Tetrahedron Lett. 1965, 6, 609–615. [Google Scholar]
- (14).Boyd DR; Sharma ND; Dalton H; Clarke DA Chemoenzymatic Synthesis of Arene Oxides and Trans-Dihydrodiols from Cis-Dihydrodiols of Monosubstituted Benzenes. Chem. Commun. 1996, 45–46. [Google Scholar]
- (15).(a) Dimroth K; Pohl G 3-Benzoxepin. Angew. Chem. 1961, 73, 436. [Google Scholar]; (b) Dimroth K; Pohl G; Follmann H Die Synthese von Derivaten Des 3-Oxepins Und Des Furans Durch Eine Zweifache Wittig-Reaktion. Chem. Ber. 1966, 99, 634–641. [Google Scholar]
- (16).Ziegler GR; Hammond GS Photorearrangement of 1,4-Epoxy-1,4-Dihydronaphthalene to Benz[f]Oxepin. J. Am. Chem. Soc. 1968, 90, 513–514. [Google Scholar]
- (17).For selected applications of benzene oxide in synthesis, see:Foster CH; Berchtold GA. Synthesis of Trans-Benzene Trioxide. J. Am. Chem. Soc. 1972, 94, 7939–7939.Rastetter WH Sym-Oxepin Oxide. J. Am. Chem. Soc. 1976, 98, 6350–6353.Bertozzi F; Crotti P; Del Moro F; Feringa BL; Macchia F; Pineschi M Unprecedented Catalytic Enantioselective Trapping of Arene Oxides with Dialkylzinc Reagents. Chem. Commun. 2001, 2606–2607.Matías DM; Johnson JS Synthesis and Desymmetrization of Meso Tricyclic Systems Derived from Benzene Oxide. J. Org. Chem. 2018, 83, 4859–4866.Da Silva Pinto S; Davies SG; Fletcher AM; Roberts PM; Thomson JE Synthesis of (–)-Conduramine A1, (–)-Conduramine A2 and (–)-Conduramine E2 in Six Steps from Cyclohexa-1,4-Diene. Org. Lett. 2019, 21, 7933–7937.
- (18).Southgate EH; Pospech J; Fu J; Holycross DR; Sarlah D Dearomative Dihydroxylation with Arenophiles. Nat. Chem. 2016, 8, 922–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Okumura M; Sarlah D Arenophile-Mediated Dearomative Functionalization Strategies. Synlett 2018, 29, 845–855. [Google Scholar]
- (20).(a) Murphy A; Dubois G; Stack TDP Efficient Epoxidation of Electron-Deficient Olefins with a Cationic Manganese Complex. J. Am. Chem. Soc. 2003, 125, 5250–5251. [DOI] [PubMed] [Google Scholar]; (b) Murphy A; Pace A; Stack TDP Ligand and pH Influence on Manganese-Mediated Peracetic Acid Epoxidation of Terminal Olefins. Org. Lett. 2004, 6, 3119–3122. [DOI] [PubMed] [Google Scholar]; (c) Moretti RA; Du Bois J; Stack TDP Manganese(II)/Picolinic Acid Catalyst System for Epoxidation of Olefins. Org. Lett. 2016, 18, 2528–2531. [DOI] [PubMed] [Google Scholar]
- (21).(a) Okumura M; Nakamata Huynh SM; Pospech J; Sarlah D Arenophile-Mediated Dearomative Reduction. Angew. Chem. Int. Ed. 2016, 55, 15910–15914. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Reference 15. [Google Scholar]
- (22).(a) Heyman M; Bandurco VT; Snyder JP Azoalkane Synthesis by Direct Oxidation of Semicarbazides with Copper Halide. J. Chem. Soc. D 1971, 297–298. [Google Scholar]; (b) Jösel R; Schröder G N-Phenyltriazolindionaddukte des Bicyclo[4.2.2]decatetraens und Tricyclo[3.3.2.02,8]decatriens (Bullvalen). Liebigs Annalen der Chemie 1980, 1980, 1428–1437. [Google Scholar]; (c) Jain R; Sponsler MB; Coms FD; Dougherty DA Cyclobutanediyls: A New Class of Localized Biradicals. Synthesis and EPR Spectroscopy. J. Am. Chem. Soc. 1988, 110, 1356–1366. [Google Scholar]
- (23).(a) Kornblum N; DeLaMare HE The Base Catalyzed Decomposition of a Di-alkyl Peroxide. J. Am. Chem. Soc. 1951, 73, 880–881. [Google Scholar]; (b) Highly regioselective openings of unsymmetrical endoperoxides have been observed before. For examples, see: Gesinski MR; Brenzovich WE; Staben ST; Srinilta DJ; Toste FD. A Divergent/Convergent Approach to Dolabriferol: The Kornblum-DeLaMare Enantiomeric Resolution. Tetrahedron Lett. 2015, 56, 3643–3646. [Google Scholar]; (c) Palframan MJ; Kociok-Köhn G; Lewis SE Photooxygenation of a Microbial Arene Oxidation Product and Regioselective Kornblum-DeLaMare Rearrangement: Total Synthesis of Zeylenols and Zeylenones. Chem. - Eur. J. 2012, 18, 4766–4774. [DOI] [PubMed] [Google Scholar]
- (24).Vyas DM; Chiang Y; Doyle TW A Short, Efficient Total Synthesis of (±) Acivicin and (±) Bromo-Acivicin. Tetrahedron Lett. 1984, 25, 487–490. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.