

Abstract
Tumor-targeted 6-substituted pyrrolo[2,3-d]pyrimidine benzoyl compounds based on 2 were isosterically modified at the 4-carbon bridge by replacing the vicinal (C11) carbon by heteroatoms N (4), O (5) or S (6), or with an N-substituted formyl (7), trifluoroacetyl (8) or acetyl (9). Replacement with sulfur (6) afforded the most potent KB tumor cell inhibitor, ~6-fold better than the parent 2. In addition, 6 retained tumor transport selectivity via folate receptor (FR) α and -β over the ubiquitous reduced folate carrier (RFC). FRα-mediated cell inhibition for 6 was generally equivalent to 2, while the FRβ-mediated activity was improved by 16-fold over 2. N (4) and O (5) substitutions afforded similar tumor cell inhibitions as 2, with selectivity for FRα and -β over RFC. The N-substituted analogs 7–9 also preserved transport selectivity for FRα and -β over RFC. For FRα-expressing CHO cells, potencies were in the order of 8 > 7 > 9. Whereas 8 and 9 showed similar results with FRβ-expressing CHO cells, 7 was ~16-fold more active than 2. By nucleoside rescue experiments, all the compounds inhibited de novo purine biosynthesis, apparently at the step catalyzed by glycinamide ribonucleotide formyltransferase. Thus, heteroatom replacements of the CH2 in the bridge of 2 afford analogs with increased tumor cell inhibition that could provide advantages over 2, as well as tumor transport selectivity over clinically used antifolates including methotrexate and pemetrexed.
Keywords: pyrrolo[2,3-d]pyrimidine; heteroatom bridge; folate receptor; selective uptake; reduced folate carrier
Graphical Abstract
1. Introduction
Reduced folates (e.g. 5-methyltetrahydrofolate) play critical roles as cofactors in the biosynthesis of purines and pyrimidines.1–2 Mammalian cells lack the enzymatic machinery to synthesize folates de novo. As a result, it is necessary to obtain folates from dietary sources. Folate cofactors are bivalent anions and hydrophilic compounds that diffuse poorly across cell membranes. Reflecting this, specific transport systems are needed to achieve sufficient intracellular concentrations of folates to support cellular anabolism.3
Three specialized folate transport systems exist in mammalian cells that include the reduced folate carrier (RFC),4 folate receptors (FRs) α and β,5 and the proton-coupled folate transporter (PCFT).6–8 RFC and PCFT are facilitative folate transporters,4, 7 whereas FRs are glycosyl phosphatidylinositol-linked proteins that mediate uptake of folates into cells by receptor-mediated endocytosis.5 RFC is the major route for membrane transport of circulating folates into mammalian tissues and RFC is expressed in both normal and cancer cells, whereas FRα, FRβ, and PCFT exhibit narrower patterns of tissue expression and serve more specialized physiological roles.4–5, 7–8 RFC functions optimally at neutral pH (~7.4) associated with most normal tissues.4
PCFT is expressed at apical brush border membranes of the duodenum and proximal jejunum where it functions at low pH and is the major intestinal transporter for the absorption of dietary folates.9–10 Although PCFT can be detected in other tissues (e.g., liver, kidney),8 the requirement for an acidic pH (pH <7, optimum at pH 5–5.5) for optimal activity7, 10 precludes significant levels of PCFT transport in most normal tissues.6
For over 60 years, antifolates have served important therapeutic roles as anticancer, antimicrobial and immunomodulatory agents.11–13 Methotrexate (MTX), a dihydrofolate reductase inhibitor, is a well-known classical antifolate and continues to be used to treat acute lymphoblastic leukemia and rheumatoid arthritis.13 Other clinically used antifolates include pralatrexate, a DHFR inhibitor used for peripheral T-cell lymphoma,12 and the thymidylate synthase inhibitors raltitrexed (for colorectal cancer) and pemetrexed (PMX) (for non-small cell lung cancer (NSCLC) and malignant pleural mesothelioma) (Figure 1).14 PMX also inhibits secondary targets in one-carbon metabolism, including glycinamide ribonucleotide (GAR) and 5-aminoimidazole-4-carboxamide (AICA) formyltransferases (GARFTase and AICARFTase, respectively) in de novo purine biosynthesis, and DHFR.14 As all of these antifolates are excellent substrates for cellular uptake by the ubiquitously expressed RFC, they are comparatively nonselective for tumors versus normal tissues and show dose-limiting toxicities when used as cancer chemotherapeutic agents, resulting in tumor resistance and in some cases chemotherapeutic failures.4, 8, 15
Figure 1.
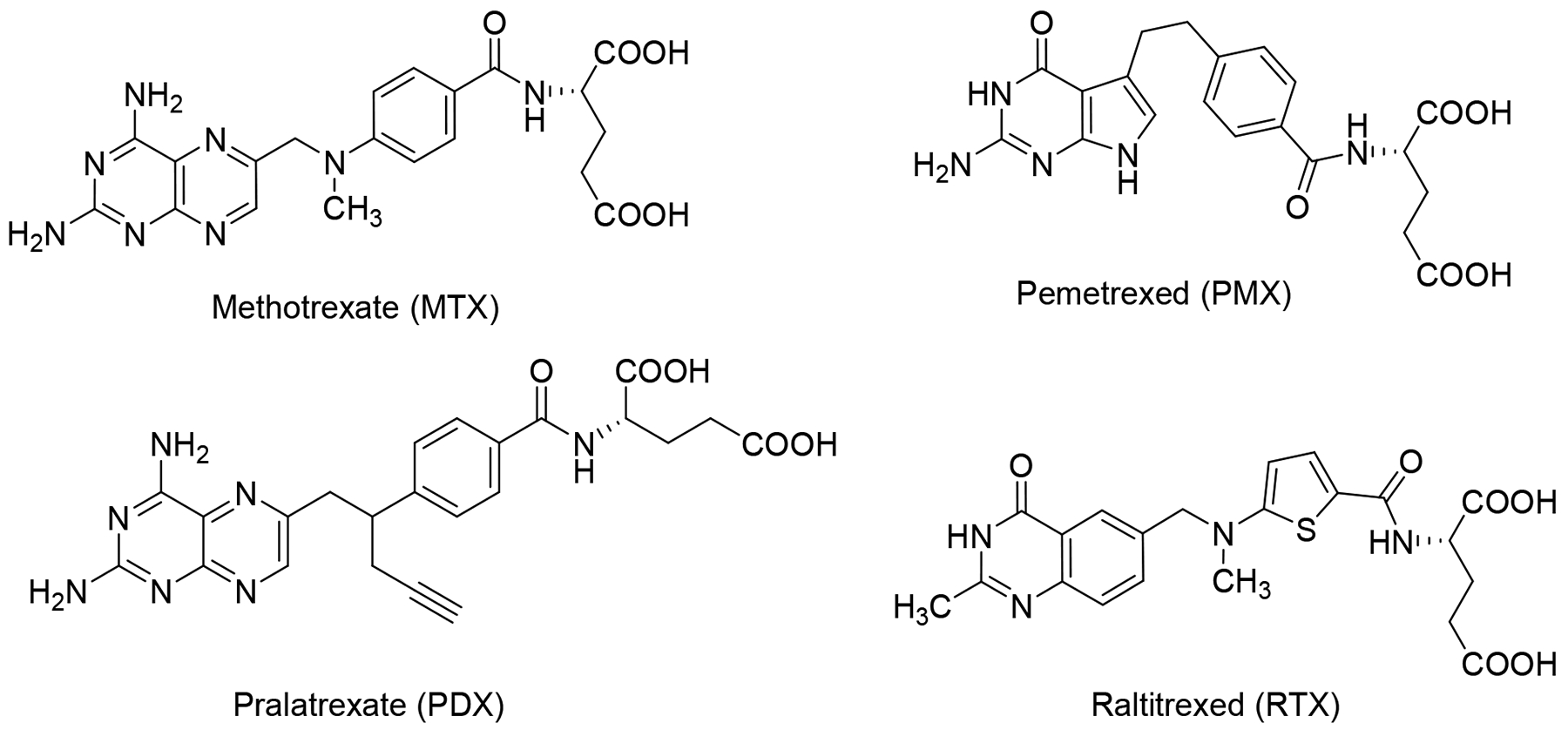
Structures of classicalantifolate drugs, including methotrexate (MTX), pemetrexed (PMX), pralatrexate (PDX), and raltitrexed (RTX).
There has been an increased recognition of human FRs as targets for the specific delivery of new classes of antifolates or folate conjugates to tumors or sites of inflammation.16–18 In humans, three genes encoding functional FRs termed FRα, FRβ, and FRγ have been recognized.5, 19–20 FRα and FRβ are anchored at the plasma membrane via a glycosyl-phosphatidylinositol (GPI)- anchor, whereas FRγ is secreted due to the lack of a signal sequence for GPI anchor attachment.5 FRα is detected on apical surfaces of several epithelial cells (e.g., kidney) where it is inaccessible to parenterally administered folates and related compounds.5 FRβ is detected in spleen, bone marrow, and thymus5 and is also expressed in activated macrophages.20
Moreover, in many types of cancer and inflammatory diseases, reflecting high expression levels of FRα or FRβ, FR-targeted therapies are expected to be effective.5, 17–18, 21–22 FR-targeted therapeutics are predicted to show modest toxicity in normal tissues because of the restricted expression of both FRα and FRβ, and for FRα, limited access to the circulation.5, 17–19, 23 FRα has been reported to be highly expressed in adenocarcinomas of the ovary, lung, uterus, breast, cervix, kidney, and colon, as well as testicular choriocarcinoma, ependymal brain tumors, and nonfunctioning pituitary adenocarcinoma.5, 17–19, 21, 24 For certain leukemias, such as chronic myelogenous leukemia and acute myelogenous leukemia, FRβ expression is elevated.25 FRβ is also expressed in activated synovial macrophages involved in the pathogenesis of rheumatoid arthritis26, and other inflammatory conditions including psoriasis and Crohn’s disease.20, 27 Finally, FRβ has been reported in gastric acid cancer and multiple myeloma, along with other human neoplastic tissues.24, 28–29
FR-targeted therapeutics based on FR-targeted antibodies, folate-conjugates, and antifolates are being pursued.18 A FRα-targeting antibody-drug conjugate mirvetuximab soravtansine (IMGN853; ImmunoGen)30 received “Fast Track” designation by the US Food and Drug Administration (FDA) and entered Phase III clinical trials. FRα-targeted strategies have used folate conjugates to deliver toxins, liposomes, and cytotoxic agents to FRα-expressing tumors.15 Clinically tested therapies include EC1456, a folic acid-tubulysin conjugate.17, 31 ONX-0801 (BGC 945), a thymidylate synthase (TS) inhibitor,16 completed a Phase I clinical trial with plans for a randomized biomarker pre-specified Phase II clinical trial.32
We discovered 6-substituted 2-amino-4-oxo pyrrolo[2,3-dpyrimidines as inhibitors of tumor cell proliferation with cellular uptake by FRs and PCFT and inhibition of de novo purine biosynthesis at the first folate-dependent step catalyzed by GARFTase.33–41 Among the most active analogs of the previously published series were compounds 1 and 3 (Figure 2), both three atom bridged compounds, with selective transport into certain tumor cells by FRs and PCFT (Table 1). Upon internalization, compounds 1 and 3 inhibited GARFTase in de novo purine nucleotide biosynthesis, resulting in depletion of purine nucleotides.33, 42 However, for compounds 1 and 3, selectivity over RFC was less than absolute.35, 42 The non-selective FR activity of these 3-atom bridged analogs may reflect their shorter bridge lengths, which are similar to those for the 2-atom bridged classic antifolate drugs such as MTX and PMX which are good RFC substrates and are poor (and non-selective) substrates for FRs.35, 42
Figure 2.
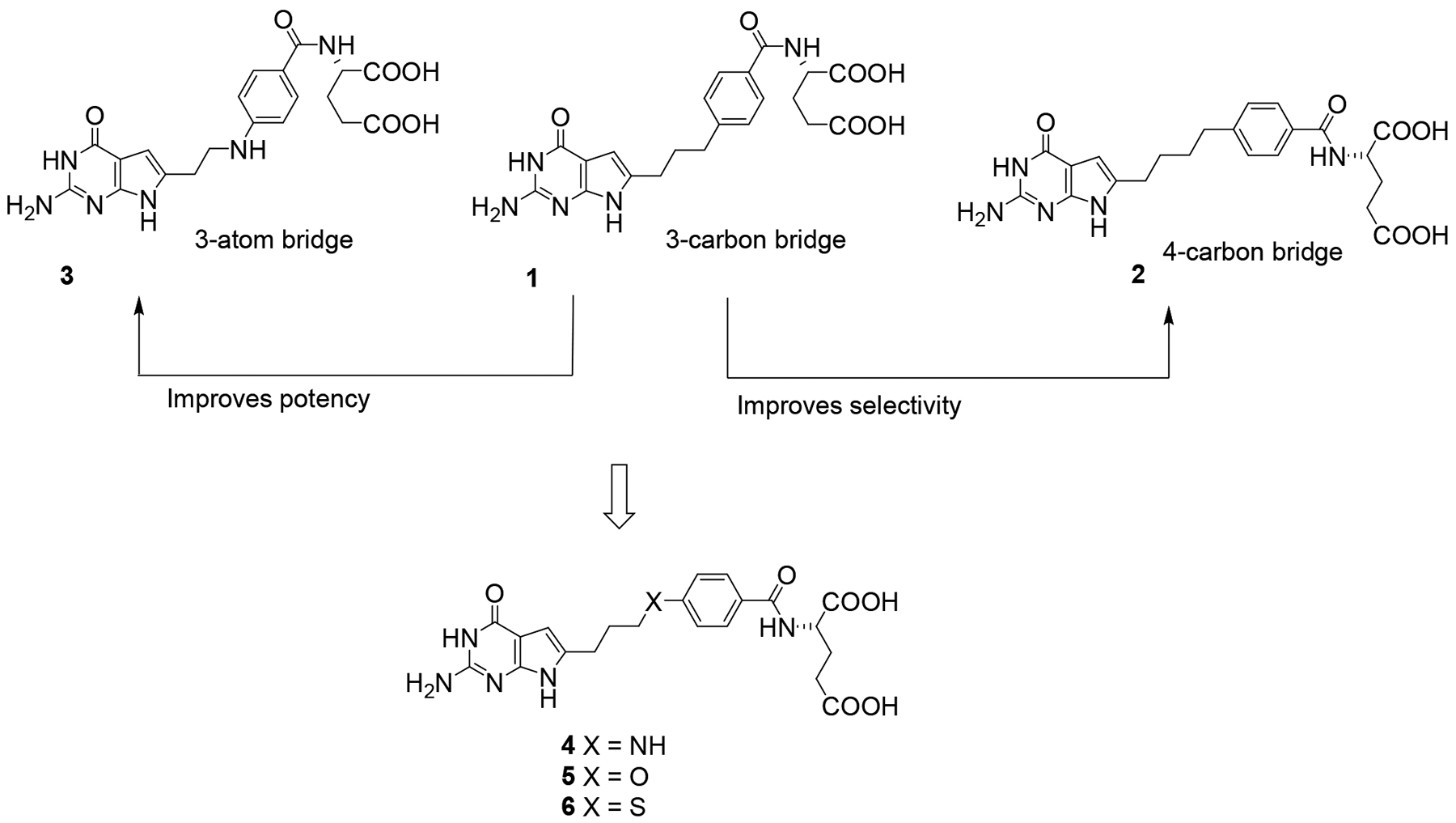
Design of compounds with optimized chain lengths.
Table 1. IC50s (in nM) for 6-substituted pyrrrolo[2,3-d]pyrimidine benzoyl antifolates with heteroatom replacements, and classical antifolates in RFC-, PCFT- and FR-expressing cell lines.
Proliferation assays were performed for CHO sublines engineered to express human RFC (PC43–10), FRα (RT16), FRβ (D4) or PCFT (R2/PCFT4), and transporter-null (R2) CHO cells, and KB human tumor cells (express RFC, FRα, and PCFT). For the experiments measuring FR-mediated effects, assays were performed in the presence or absence of 200 nM folic acid (results are shown only for KB cells). Results are presented as IC50 values, corresponding to the concentrations that inhibit growth by 50% relative to cells incubated without the drug. The data are mean values from 5–16 experiments (+/− standard errors in parentheses). Some of the data for 3, MTX, and PMX have been previously published.35, 51 Results are also summarized for KB cells for the protective effects of adenosine (60 μM), thymidine (10 μM), glycine (130 μM) or 5-aminoimidazole-4-carboxamide (320 μM). For compounds 2, 4, 5, 6, 7, 8, and 9, folic acid and nucleoside/AICA protection results are shown in Figure 7. Methods are summarized in the Experimental Section. Undefined abbreviations: Ade, adenosine; AICA, 5-aminoimidazole-4-carboxamide; FA, folic acid; gly, glycine; ND, not determined; and Thd, thymidine.
| Compounds | R2 | RFC | FRα | FRβ | PCFT | RFC/FRα/PCFT | ||
|---|---|---|---|---|---|---|---|---|
| PC43–10 | RT16 | D4 | R2/PCFT4 | KB | KB (+FA) | KB + Ade/Thd/AICA/gly | ||
| 1 | >1000 | 649(38) | 4.1(1.6) | 5.6(1.2) | 23.0(3.3) | 1.7(0.4) | >1000 | Ade/AICA |
| 2 | >1000 | >1000 | 6.3(1.6) | 10(2) | 213(28) | 1.9(0.7) | >1000 | Ade/AICA |
| 3 | >1000 | 510(90) | 3.04 (0.71) | 0.62(0.20) | 87.4(9.9) | 0.32(0.05) | 666(46) | Ade/AICA |
| 4 | 697(158) | 670(150) | 1.76(0.43) | 1.51(0.43) | 235(96) | 0.95(0.35) | >1000 | Ade/AICA |
| 5 | >1000 | >1000 | 4.6(1.3) | 5.6(1.4) | >1000 | 2.50(0.64) | >1000 | Ade/AICA |
| 6 | >1000 | >1000 | 2.56(0.73) | 0.60(0.12) | 593(204) | 0.30(0.04) | 757(41) | Ade/AICA |
| 7 | >1000 | >1000 | 35.63 (4.78) | 0.61 (0.11) | 893(161) | 13.20 (3.32) | >1000 | Ade/AICA |
| 8 | >1000 | >1000 | 5.78(1.45) | 3.87(0.54) | 487(171) | 5.35 (0.49) | >1000 | Ade/AICA |
| 9 | >1000 | >1000 | 57.24 (6.9) | 12.79 (1.90) | >1000 | 40.11 (9.34) | >1000 | Ade/AICA |
| MTX | 216(8.7) | 12(1.1) | 114(31) | 106(11) | 121(17) | 6.00(0.60) | 20(2.4) | Thd/Ade |
| PMX | 894(93) | 138(13) | 42(9) | 60 (8) | 13.2(2.4) | 68(12) | 327(103) | Thd/Ade |
Compound 2 is an analog of 1 but it has a 4-carbon bridge and showed better selectivity for FRs and PCFT than 1 or 3.35, 42 The length of the side chains in the carbon-bridged analogs was previously identified as an important structural determinant of potency and transport selectivity.35, 40, 42 For analogs with bridge lengths from 2–8 carbons, the most potent tumor inhibitors had 3 carbons (e.g., compound 1) (Figure 2). However, this was at the expense of transport selectivity.33, 42 For the 4-carbon bridge analogs like 2, there was an improvement in FR selectivity over RFC compared to 1 without loss of anti-tumor activity; however, this was accompanied by decreased PCFT activity.33, 42
We reported35 that the replacement of the benzylic carbon in 1 (3-carbon bridge) (Figure 2) with a heteroatom (N, O or S) provides a series of compounds that were more potent toward FRα-expressing KB human tumor cells, with substantial selectivity for FRs and PCFT over RFC.35 Analysis of the X-ray crystal structures of the bridge with a N-atom analog 3 (Figure 2) with human FRα and GARFTase showed that the bound conformations required flexibility to attach to both FRα (PDB 5IZQ) and GARFTase (PDB 5J9F).35 The X-ray crystal structure of 3 with GARFTase showed that the bridge heteroatoms of the bound inhibitor formed intermolecular interactions with GARFTase, mediated by an ordered water molecule (PDB 5J9F).35 Analogs of 3 with N-formyl (N-CHO), N-trifluoroacetyl (N-COCF3), or N-acetyl (N-COCH3) substitutions were designed to mimic the GARFTase substrate (N10-formyl tetrahydrofolate; 10-CHO-THF) (Figure 3) and to afford additional hydrogen bonding within the GARFTase active site.35 Compound 3, in addition to tumor cell inhibition, was also efficacious in vivo in mice bearing IGROV1 ovarian tumor xenografts.35
Figure 3.
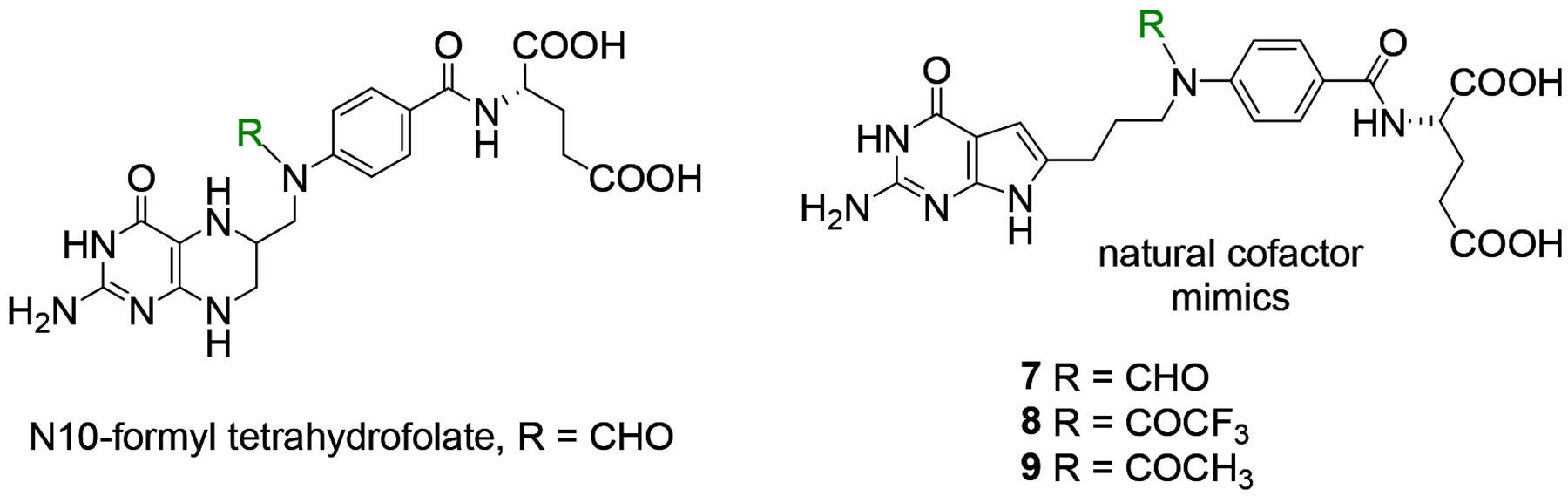
Structure of N10-formyl tetrahydrofolate and proposed mimics.
Thus, it was of interest to synthesize 4-atom bridged compounds with heteroatoms (N, O, S, and N-CHO, N-COCF3, N-COCH3) (Figure 2 and 3) as hybrids of the 4-carbon bridged analog 2 that improves transport selectivity, and the heteroatom bridged analog 3 that increases potency towards tumor cells. These hybrid analogs were expected to afford improved antitumor potencies and transporter selectivities, compared to PMX. This is anticipated to overcome toxicity and potential resistance to PMX. The current work involves the synthesis of pyrrolo[2,3-d]pyrimidine analogs with the benzylic CH2 of the 4-carbon bridge of 2 replaced with NH (4), O (5), or S (6) (Figure 2). We measured the bond lengths and bond angles in compounds 4–6 and compared them with literature values.43 As flexibility is crucial for the binding to both FRα (PDB 5IZQ) and GARFTase (PDB 5J9F),35 the increased bond length of 6 (2.93 Å) compared to 2 (2.55 Å) (Figure 4) provides additional flexibility in the chain region to afford both FRα (PDB 5IZQ) selectivity over RFC and GARFTase potency. The bond angle of C-S-C in 6 (109.1°) is decreased compared to C-C-C (112.8°)44 in 2 (Figure 4), which isresponsible for a different conformation and orientation of the α and γ-carboxylates of the L-glutamate of 6 compared to 2. Compounds 4 and 5 have comparable bond lengths and bond angles to 2 and are predicted to mimic compound 2 (Figure 4). The purpose of this study is to determine the effects of heteroatom replacements of the vicinal (C11) carbon to the side chain phenyl ring in compound 2 by N, O, or S, and N-CHO, N-COCF3, or N-COCH3 moieties (7–9) on transport specificities by FRα, FRβ, and PCFT, over RFC, and on the inhibition of de novo purine nucleotide biosynthesis on cell proliferation and antitumor activity.
Figure 4.
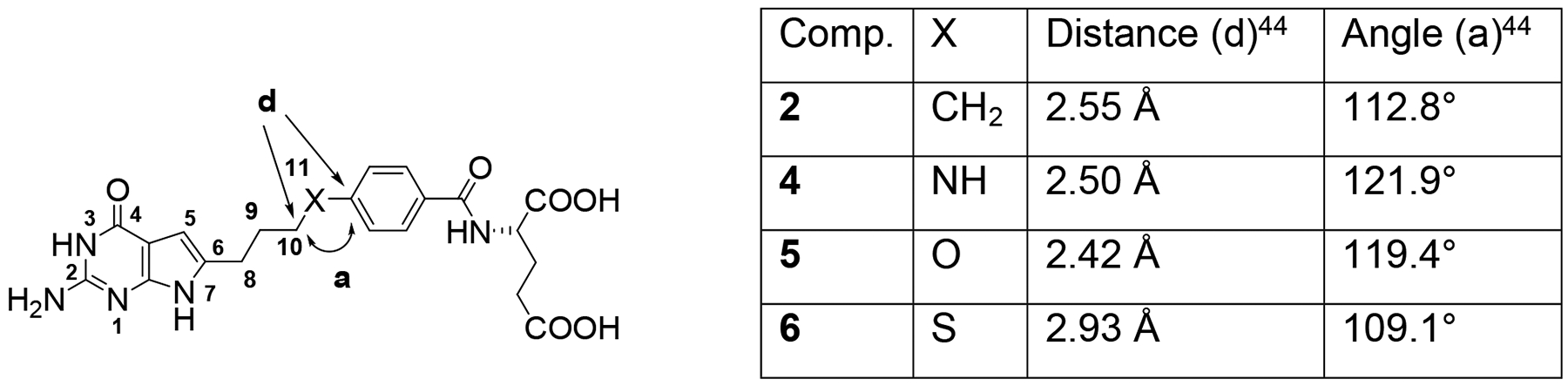
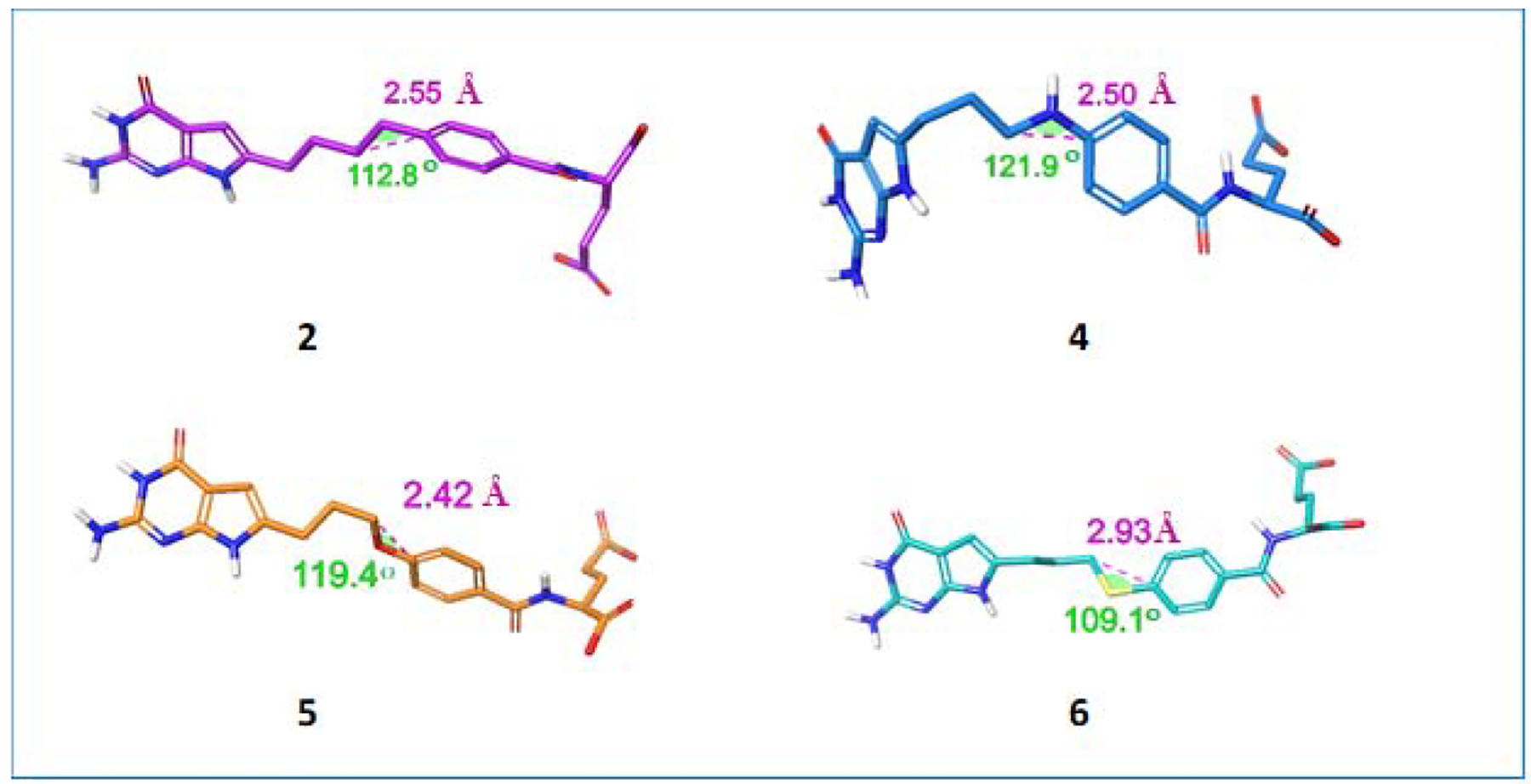
Distance and bond angle variations predicted by the nature of the bridge at the benzylic position (X). Distances and angles for X = NH, O, and S were measured using energy-minimized conformations of compounds with Maestro Schrödinger 2019–1.44
2. Molecular Modeling
To determine the ligand-protein interactions, docking studies were carried out with compounds 4–9 using X-ray crystal structures of human GARFTase (PDB 5J9F), FRα (PDB 5IZQ) and FRβ (PDB 4KN2).35, 37, 45,46Docking scores are provided in supporting information (Table 2S).
2.1. Docking studies of compounds 4–9 with Human GARFTase (PDB: 5J9F) (Figures 5A–5B):
Figure 5A.
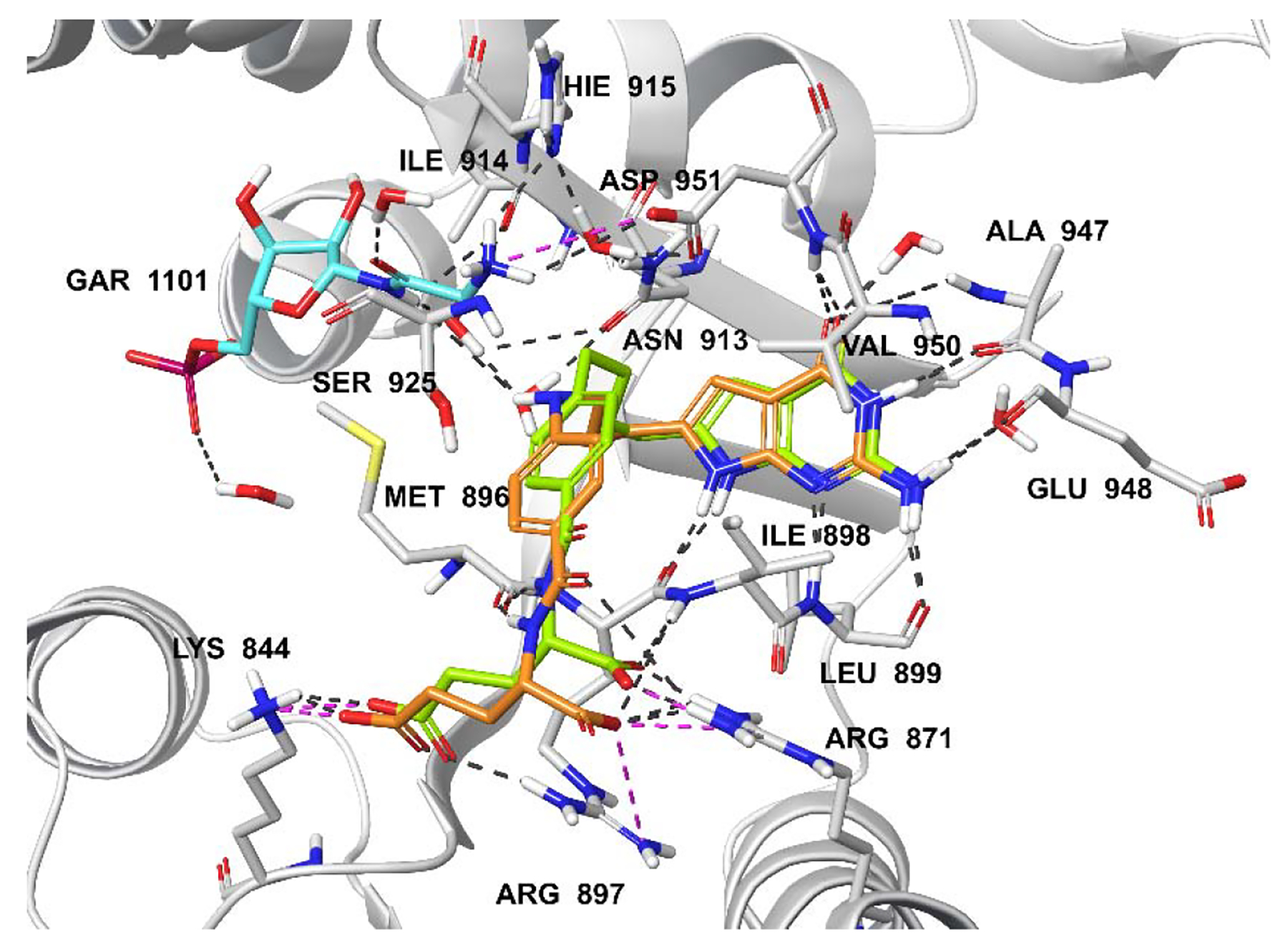
Molecular modeling studies with human GARFTase (PDB 5J9F).33 Superimposition of the docked pose of 2 (green), 4 (tan) and GAR (cyan). Docking scores of 2 and 4 are −14.02 and −14.29 kcal/mol, respectively. Hydrogen bond interactions, pi-pi interactions, and salt bridges are depicted as black, cyan and magenta dashed lines, respectively.
Figure 5B.
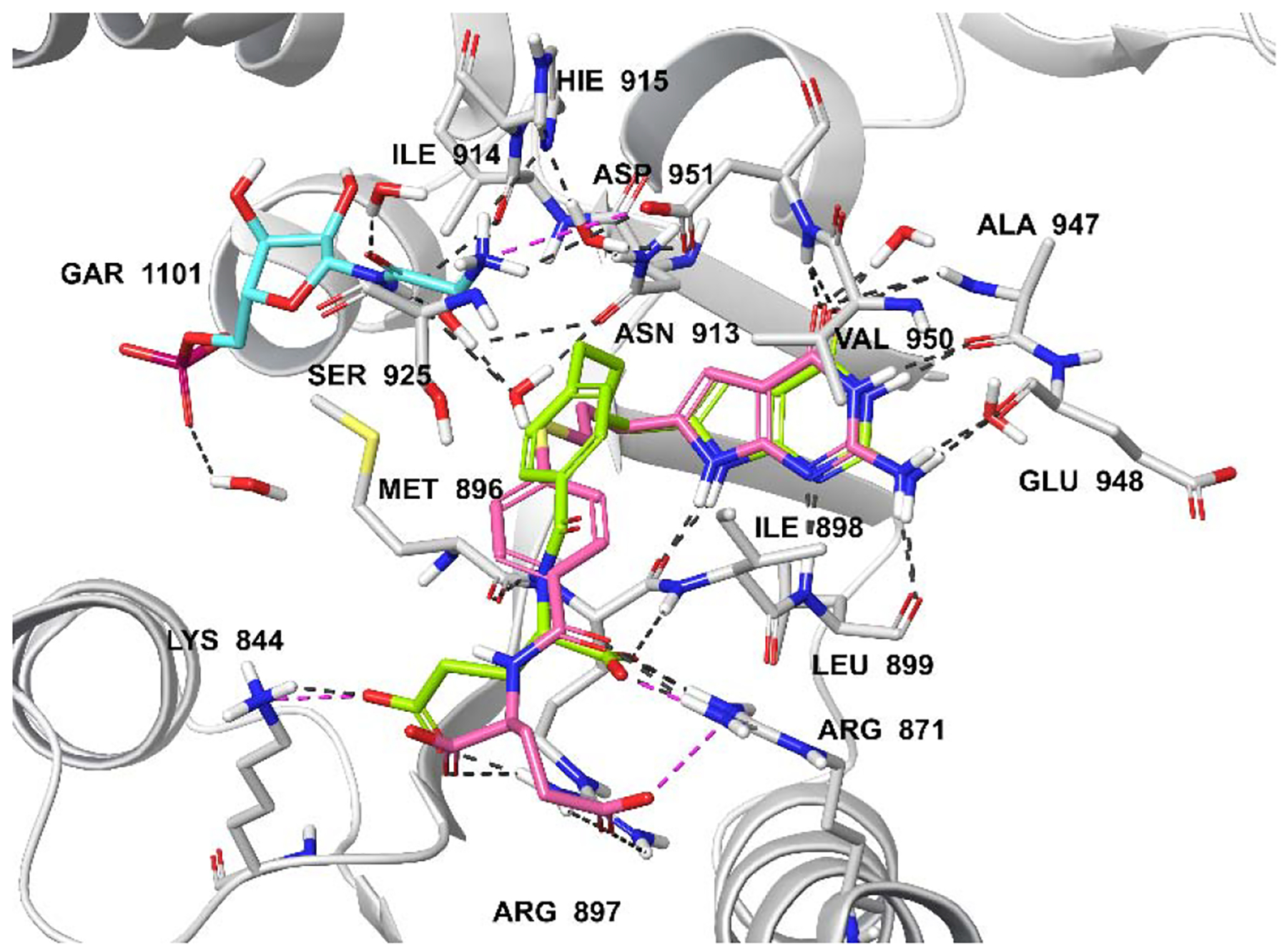
Molecular modeling studies with human GARFTase (PDB 5J9F).33 Superimposition of the docked pose of 2 (green), 6 (pink) and GAR (cyan). Docking scores of 2 and 6 are −14.02 and −14.53 kcal/mol, respectively. Hydrogen bond interactions, pi-pi interactions, and salt bridgesare depicted as black, cyan, and magenta dashed lines, respectively.
Docking results of compounds 2, 4, and 6 with human GARFTase (PDB 5J9F) are displayed in Figures 5A and 5B. The docked poses of 2, 4 and 6 indicate that the pyrrolo[2,3-d]pyrimidine scaffold is buried in the active site and occupies the same location as the bound ligand 335 (Figures 5A and 5B; bound ligand 3 not shown for clarity). The bicyclic pyrrolo[2,3-d]pyrimidine scaffold of 4 and 6 are stabilized by the hydrogen bonding interactions, including hydrogen bonds between the N1 nitrogen and the backbone amide -NH- of Leu899 and the 2-NH2 and the backbone carbonyls of Glu948 and Leu899. Other interactions include N3 and the backbone carbonyl of Ala947, and the 4-oxo and the backbone -NH- of Ala947 and Val950. The N11-H of compound 4 is hydrogen-bonded via a water-mediated hydrogen-bond with the substrate GAR (Figure 5A). Similar interactions are predicted to occur involving the heteroatom bridge of 5 (oxygen) (figure not shown). The flexible four-atom bridges of 4 and 6 orient the side chain phenyl ring into a hydrophobic cleft of Phe895 (not labeled), Met896, Leu899, and Ile898. The interactions involving the flexible glutamate side chains are similar to those reported for the glutamate side chain of 3.35 The α-carboxylic acid of 4 forms salt bridges with Arg871 and Arg897, and hydrogen bonds with the backbone of Ile898. The γ-carboxylic acid forms a salt bridge with Lys844 and Arg897 (Figure 5A). Compounds 2 and 4 had docked scores of −14.02 and −14.29 kcal/mol, respectively, in GARFTase, similar to that for 3 (−14.36 kcal/mol). Docking studies of 6 suggested an altered orientation of the phenyl ring and side-chain L-glutamate compared to 4. This is attributed to the larger size of the sulfur atom, resulting in a different set of interactions between the active site with the L-glutamate side chain of 6. The α-carboxylic acid forms a salt bridge with Arg897, while the γ-carboxylic acid of 6 forms a salt bridge with Arg871 (Figure 5B). The resulting docking score of 6 is −14.53 kcal/mol, somewhat better than 2–4, indicating an increased inhibition of GARFTase. The N11-CHO, COCF3, and COCH3 moieties in 7, 8, and 9, respectively, are oriented similarly to the N11-H of 4. Compounds 7–9 bind deeper into the formyl transfer region that is occupied by the formyl group of the natural GARFTase substrate, N10-formyl tetrahydrofolate (10-CHOTHF) (figure not shown). The docked scores of 5 and 7–9 (−14.38, −14.79, −14.87, and −14.12 kcal/mol, respectively), are equivalent to that for 4 (−14.29 cal/mol), which suggest that these compounds should be potent GARFTase inhibitors.
2.2. Docking studies of compound 4−9 with FRα (PDB 5IZQ) and FRβ (PDB 4KN2) (Figures 6A-6C):
Figure 6A.
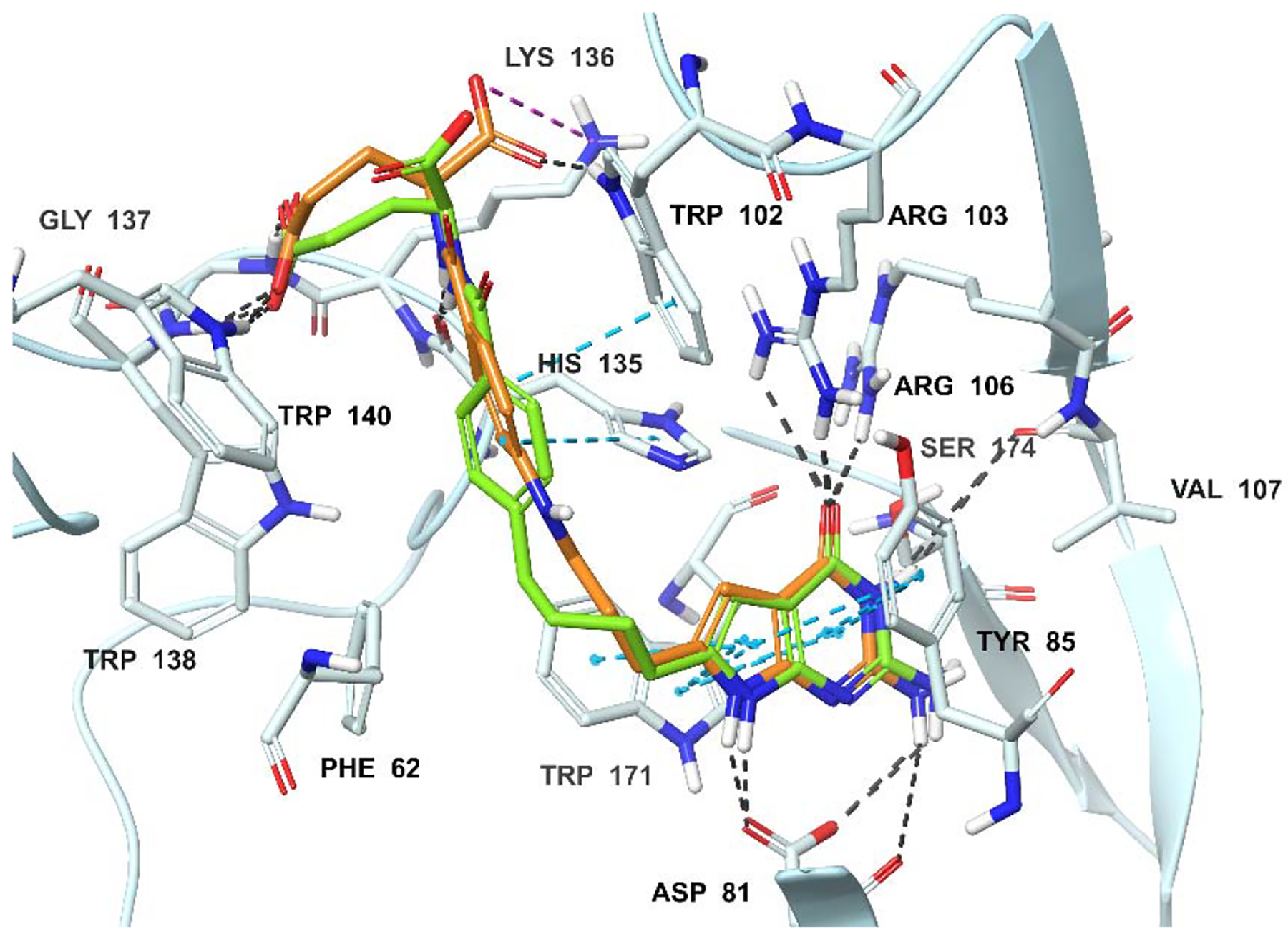
Molecular modeling studies using the human FRα (PDB 5IZQ)35, 46 crystal structures. Superimposition of the docked pose of 2 (green) with the docked pose of 4 (tan) in FRα. Docking scores of 2 and 4 are −11.72 and −12.02 kcal/mol, respectively. Hydrogen bonding interactions, pi-pi interactions, and salt bridges are depicted as black cyanand magenta dashed lines, respectively.
Figure 6C.
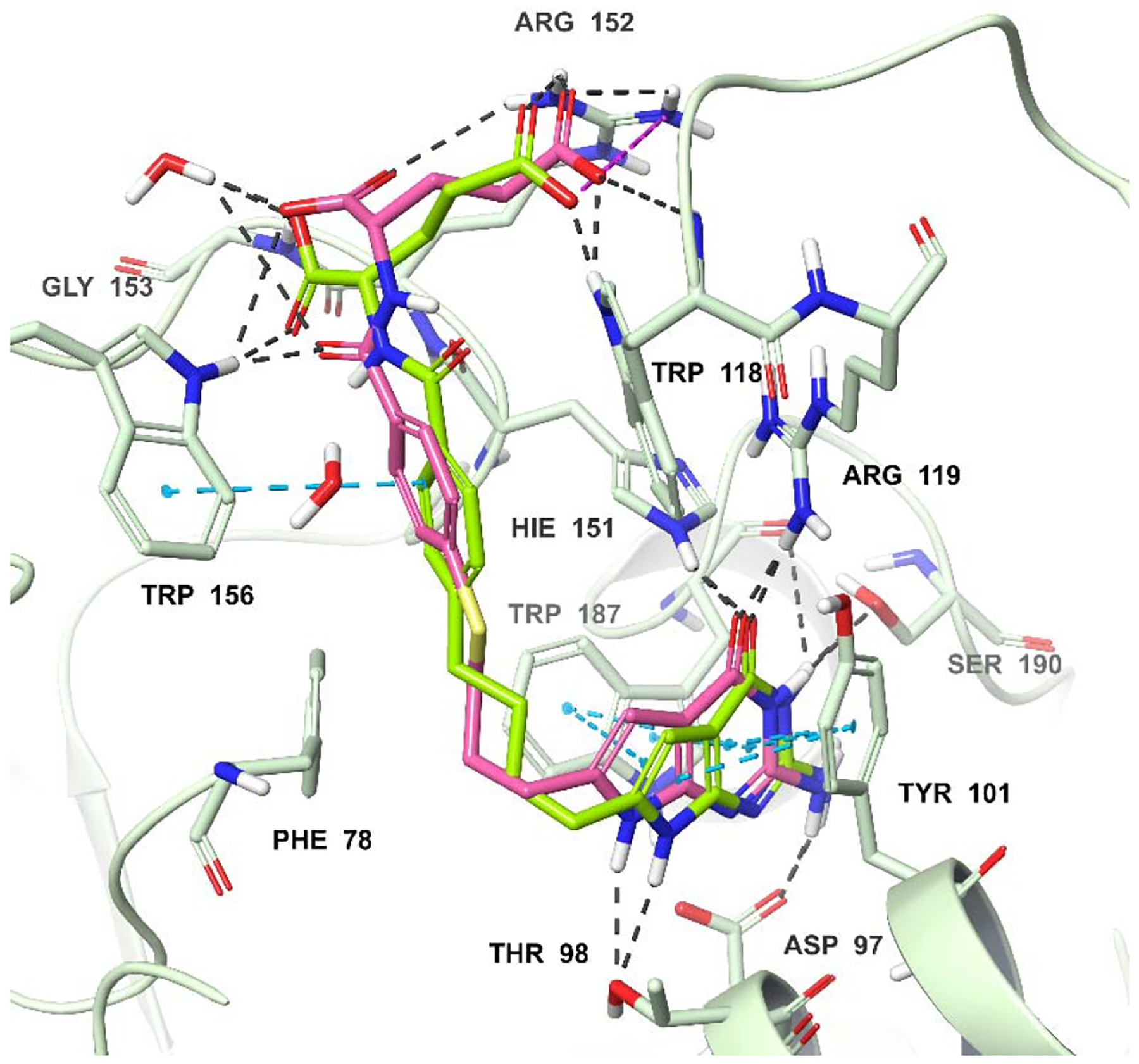
Molecular modeling studies using the human FRβ (PDB 4KN2)45 crystal structures. Superimposition of the docked pose of 2 (green) with the docked pose of 6 (pink) in FRβ. Docking scores of 2 and 6 are −13.92 and −14.84 kcal/mol, respectively. Hydrogen bonding interactions, pi-pi interactions, and salt bridgesare depicted as black, cyanand magenta dashed lines, respectively.
The docked poses of 2, 4 and 6 with the crystal structures of 3 in FRα (PDB 5IZQ)35, 46 and PMX in FRβ (PDB 4KN2)45 indicated that all three compounds are accommodated within a similar space as 3 and PMX, respectively (Figures 6A, 6B, 6C; 3 and PMX are not shown for clarity). Figures 6A and 6B display the docked poses of compounds 2, 4 and 6 in the X-ray crystal structures of human FRα. Molecular modeling revealed that the pyrrolo[2,3-d]pyrimidine scaffold of compounds 4 and 6 are stacked between the side chains of Tyr85 and Trp171, similar to the bicyclic scaffold of bound ligand 3 (Figures 6A, B; 3 not shown for clarity).35 The 2-NH2 and 7-NH of compounds 4 and 6 form hydrogen bonds with Asp81. Additional hydrogen bonds are formed between N3 and Ser174, and between the 4-oxo group and side chains of Arg103 and Arg106. The α-carboxylic acids of 4 and 6 form hydrogen bonds with the backbone NH of Gly137 and the pyrrole NH of Trp140, while the γ-carboxylic acid forms a salt bridge with Lys136. In comparison, with the 3-atom linker molecule 3 (not shown for clarity), the four atom linkers of 4 and 6 position the phenyl ring in the hydrophobic pocket (Tyr60, Trp102, His135, and Trp138) to make hydrophobic interactions (Figures 6A, B). The results revealed that the expanded chain length in the 4-atom chain compound (compared to 3) is vital for positioning the phenyl ring to the hydrophobic pocket for increased FRα selectivity.
Figure 6B.
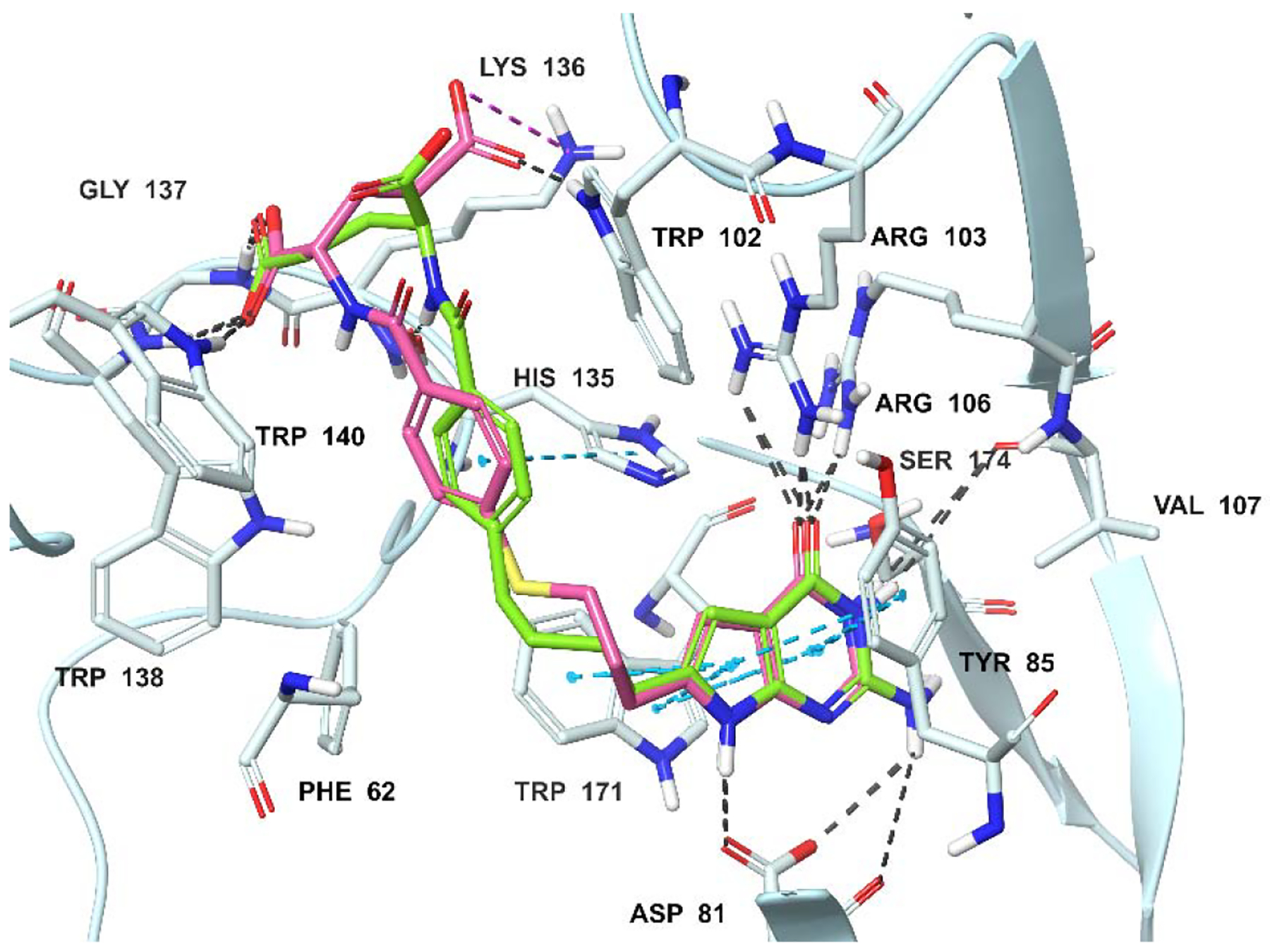
Molecular modeling studies using the human FRα (PDB 5IZQ)35, 46 crystal structures. Superimposition of the docked pose of 2 (green) with the docked pose of 6 (pink) in FRα. Docking scores of 2 and 6 are −11.72 and −12.07 kcal/mol, respectively. Hydrogen bondinteractions, pi-pi interactions, and salt bridges are depicted as black, cyanand magenta dashed lines, respectively.
Figure 6C shows the docked pose of compound 2 and 6 in the X-ray crystal structure of human FRβ (PDB 4KN2).45 In this pose, the orientation of the scaffold permits the 2-NH2 and 7-NH moieties of both 2 and 6 to form hydrogen bonds with Asp97, and the 4-oxo moiety to form hydrogen bonds with the side-chain nitrogenhydrogens of Arg119, His151 and Tyr101. Hydrogen bonding interaction with Ser190 and N3 of 2 and 6 were also observed. The pyrrolo[2,3-d]pyrimidine scaffold is stacked amid the hydrophobic aromatic side chains of Tyr101 and Trp187. The L-glutamate moiety occupies a similar binding space as the corresponding L-glutamate of the native ligand.35 The α-carboxylate forms a salt bridge with Arg152 and hydrogen bonds with the pyrrole NH of Trp118, while the γ-carboxylic acid of 2 and 6 forms hydrogen bonds with the pyrrole NH of Trp156 and a water-mediated hydrogen bond with the backbone of Trp154. The flexible four atom bridge with the sulfur atom of 2 and 6 helps to orient the side chain phenyl ring into a hydrophobic cleft of amino acids Phe78, Trp118, and Trp154. The extended chain length (4-atom chain) provides FRβ selectivity mediated by these hydrophobic interactions. These interactions are less apparent in the 3-atom chain compound 3 (3, not shown for clarity).
3. Chemistry
The reaction of β-propiolactone 10 (Scheme 1) and ethyl p-aminobenzoate 11 afforded 12.47 The carboxylic acid 12 was then treated with trifluoroacetic anhydride to give the protected amine 13.35 Compound 13 was converted to the acid chloride and immediately reacted with diazomethane, followed by treatment with silver acetate to provide the homologated carboxylic acid 14. Carboxylic acid 14 was then converted to the acid chloride and immediately reacted with diazomethane, followed by 48% HBr in water to afford the α-bromoketone 15. Condensation of 2,6-diamino-3H-pyrimidin-4-one with 15 in DMF at room temperature for 3 days provided the 6-substituted-2-amino-4-oxopyrrolo[2,3-d]pyrimidine 16. Hydrolysis of 16 resulted in the corresponding free acid 17. Subsequent coupling with L-glutamate dimethyl ester using 2-chloro-4,6-dimethoxy-1,3,5-triazine as the activating agent provided the diester 18. Saponification of the diester yielded the target 4. Compound 4 was then reacted with formic acid, trifluoroacetic anhydride or acetic anhydride to afford target compounds 7, 8 and 9, respectively.
Scheme 1.
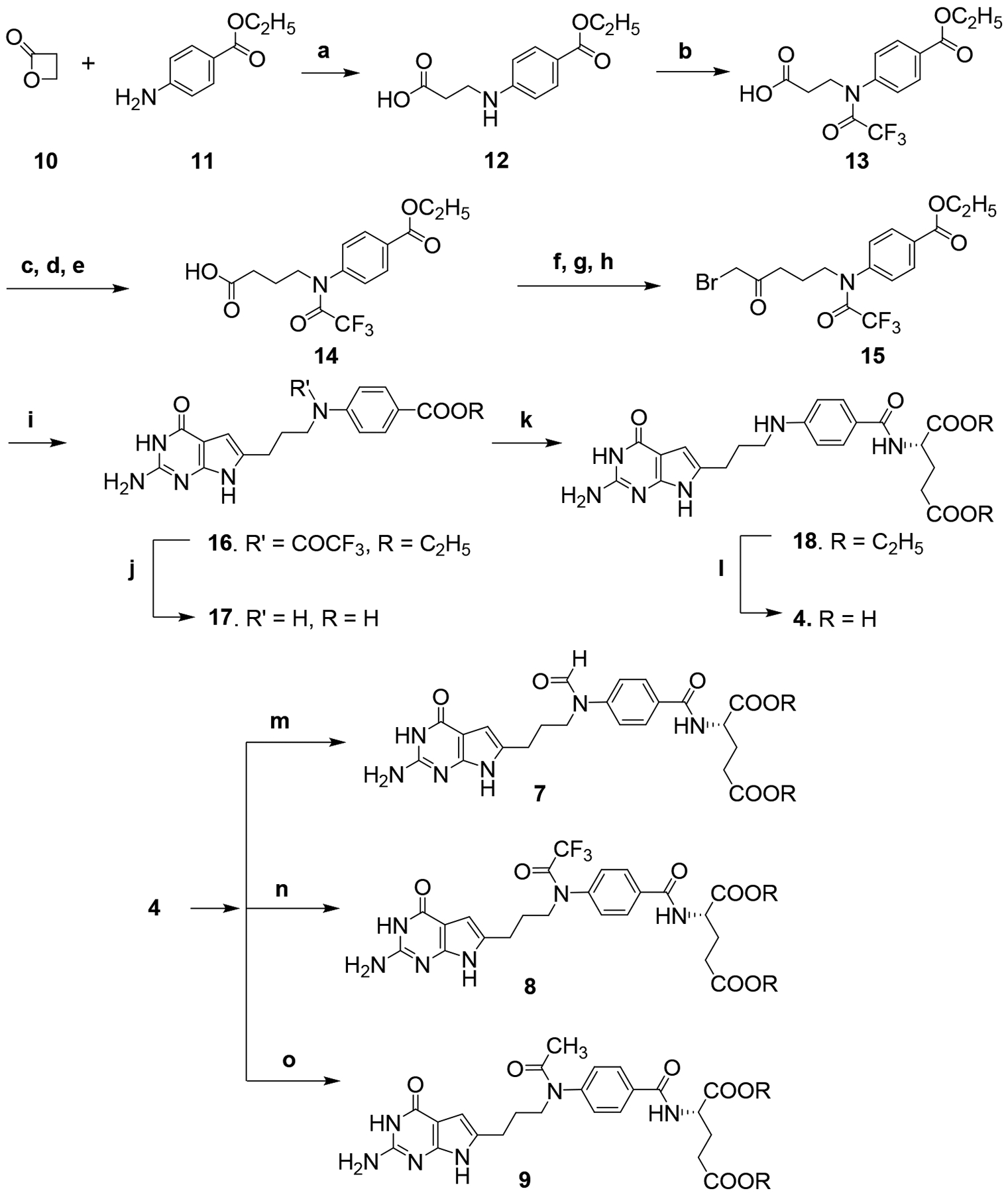
Reagents and conditions; (a) acetone, 4 h, reflux, 52%; (b) (CF3CO)2O, CH2Cl2, 2 h, rt, 88%; (c) oxalyl chloride, CH2Cl2, reflux, 1 h; (d) diazomethane, (CH3CH2)2O, rt, 1h; (e) AgCOOCH3, water, 1–4 dioxane, 1.5 h, reflux, 38%; (f) oxalyl chloride, CH2Cl2, reflux, 1 h; (g) diazomethane, (CH3CH2)2O, rt, 1h; (h) 48% HBr in water, reflux, 1h; (i) 2,6-diaminopyrimidin-4(3H)-one, DMF, rt, 3 d, 34%; (j) 1N NaOH, rt, 12 h, 88%;(k) N-methylmorpholine 2-chloro-4, 6-methoxy-1,3,5-triarzine, L-glutamate dimethyl ester, N-methyl morpholine, DMF, 12 h, 43%; (l) 1N NaOH, rt, 4 h, 88%; (m) HCOOH, (CH3CO)2O, reflux, 2 h, 42%; (n) (CF3CO)2O, 12 h, rt, 68%; (o) (CH3CO)2O, 12 h, rt, 74%.
The carboxylic acids 22 and 23 (Scheme 2) were obtained via nucleophilic substitution of the alkyl bromide 19 with 20 or 21, respectively, followed by deprotection of the tert-butyl ester with trifluoroacetic acid.48 Compounds 22 and 23 were then converted to the acid chloride and immediately reacted with diazomethane, followed by 48% HBr in water to provide the corresponding α-bromomethylketones 28 and 29, respectively.35 Condensation of 2,6-diamino-3H-pyrimidin-4-one with 28 and 29 in DMF at room temperature for 3 days afforded the 2-amino-4-oxo-6-substituted-pyrrolo[2,3-d]pyrimidines 30 and 31, respectively. Hydrolysis of 30 and 31 afforded the corresponding free acids 32 and 33, respectively. Subsequent coupling with L-glutamate ester using 2-chloro-4,6-dimethoxy-1,3,5-triazine as the activating agent provided the diesters 34 and 35. Saponification of the diesters yielded the target compounds 5 and 6.
Scheme 2.
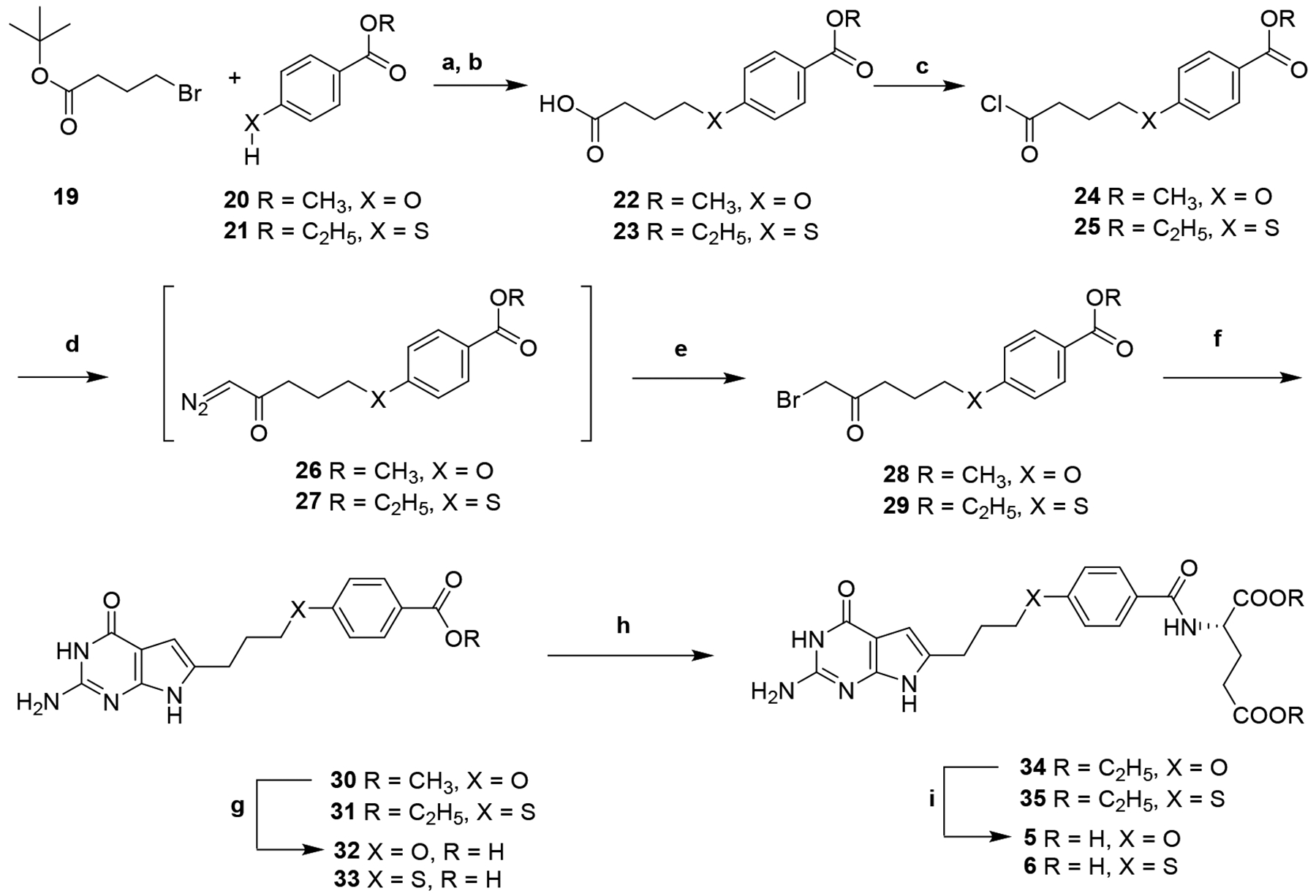
Reagent and conditions; (a) Cs2CO3, tetrabutyl ammonium chloride, DMF, 1 h, 100 °C, 47–65%; (b) CF3COOH, CH2Cl2, 84–86%; (c) oxalyl chloride, CH2Cl2, reflux, 1 h, 79%; (d) diazomethane, (CH3CH2)2O, rt, 1h; (e) 48% HBr in water, 80 °C, 2 h, 86–94% (crude yield); (f) 2,6-diaminopyrimidin-4(3H)-one, DMF, rt, 3 d, 31–34%; (g) 1N NaOH, rt, 12 h, 66–88%; (h) N-methylmorpholine 2-chloro-4, 6-methoxy-1,3,5-triarzine, L-glutamate diethyl ester, DMF, 12 h, 43–45%; (i) 1N NaOH, rt, 4 h, 85–88%.
4. Biological Evaluation and Discussion
Anti-proliferative effects of 6-substituted pyrrolo[2,3-d]pyrimidine benozyl analogs with heteroatom bridge substitutions in relation to mechanisms of folate transport.
As a part of the study of structure-activity relationships for tumor-targeted antitumor compounds with specificities for the individual transporters, we measured the impact of isosteric heteroatom replacements, including N, O, and S (compounds 4, 5, and 6, respectively), of CH2 at position-11 (compound 2) (Figure 2). Additional N-substituted analogs related to 4 with formyl (7), trifluoroacetyl (8), and acetyl (9) substitutions (Figure 3), as a mimic of the natural substrate, N10-formyl tetrahydrofolate cofactor for GARFTase were also tested.
To screen for antiproliferative activities, we used a panel of isogenic Chinese hamster ovary (CHO) sublines engineered from RFC-, FR- and PCFT-null MTXRIIOuaR2–4 CHO cells49 (R2) cells to individually express FRα (RT16 cells), FRβ (D4), RFC (PC43–10), or PCFT (R2/PCFT4).33, 42, 50–51 The CHO cells were cultured with a range of drug concentrations for up to 96 h and cell viabilities were measured to calculate IC50 values, corresponding to the concentrations that inhibit growth by 50%.42 Results are summarized in Table 1 and are compared to the inhibition of FRα-expressing KB human tumor cells.
The 6-substituted pyrrolo[2,3-d]pyrimidine analogs with heteroatom bridge replacements, 4 (N), 5 (O), and 6 (S), all showed potent growth inhibition toward FRα-expressing RT16 cells and toward FRβ-expressing D4 cells with IC50 values generally less than those previously reported for 2 (Table 1).42 Compounds 4 (N) and 6 (S) displayed 3- and 2-fold better activities respectively than the lead analog 2 in FRα-expressing RT16 cells. The impact of N-substitutions on compound 4 including 7 (N-CHO), 8 (N-COCF3) and 9 (N-COCH3) were disparate. Compound 8 showed comparable potency to 2 with RT16 cells, whereas 7 and 9 were much less inhibitory. The N-substituted compounds 7 (N-CHO), 8 (N-COCF3), and 9 (N-COCH3) showed activity in the rank order of 8>7>9 for FRα (RT16). Compounds 4, 5 and 6, were highly active (6-, 2-, and 16-fold, respectively) toward D4 cells (expresses FRβ) compared to compound 2. For the N-substituted compounds with D4 cells, activity was comparable (9) or exceeded (~16-fold for 7; ~3-fold for 8) that for 2.
Relative inhibition by the heteroatom compounds toward KB human tumor cells generally paralleled that in FRα-expressing RT16 cells. Compounds 4 (N) and 6 (S) exhibited sub-nanomolar potencies (IC50= 0.95 and 0.30 nM, respectively) in KB tumor cells which were ~2- and ~6-fold, respectively, greater than for the lead compound 2.
From the results with the engineered CHO sublines, compounds 4–9 were all more selective toward FRs compared to PCFT and RFC. Anti-proliferative activities toward PCFT- (R2/PCFT4) and RFC- (PC43–10) expressing cells ranged from modest to undetectable and were similar to those for compound 2. These results contrast with those for the classic antifolates (MTX, PMX) which inhibit cells regardless of the expressed transporter (Table 1).
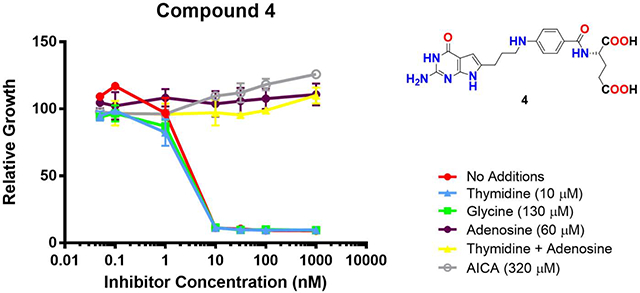
We previously reported that compound 2 was an inhibitor of de novo purine biosynthesis at the reaction catalyzed by GARFTase.42 We evaluated inhibition of de novo purine biosynthesis by compounds 4–9 by comparing the extent of growth of KB cells in the presence of adenosine (60 μM) to that with thymidine (10 μM).35, 42 We also evaluated the protective effects of glycine (130 μM) to assess the potential for inhibition of C1 metabolism in mitochondria.52 Results for the nucleoside/glycine protection experiments are shown in Figure 7.
Figure 7.
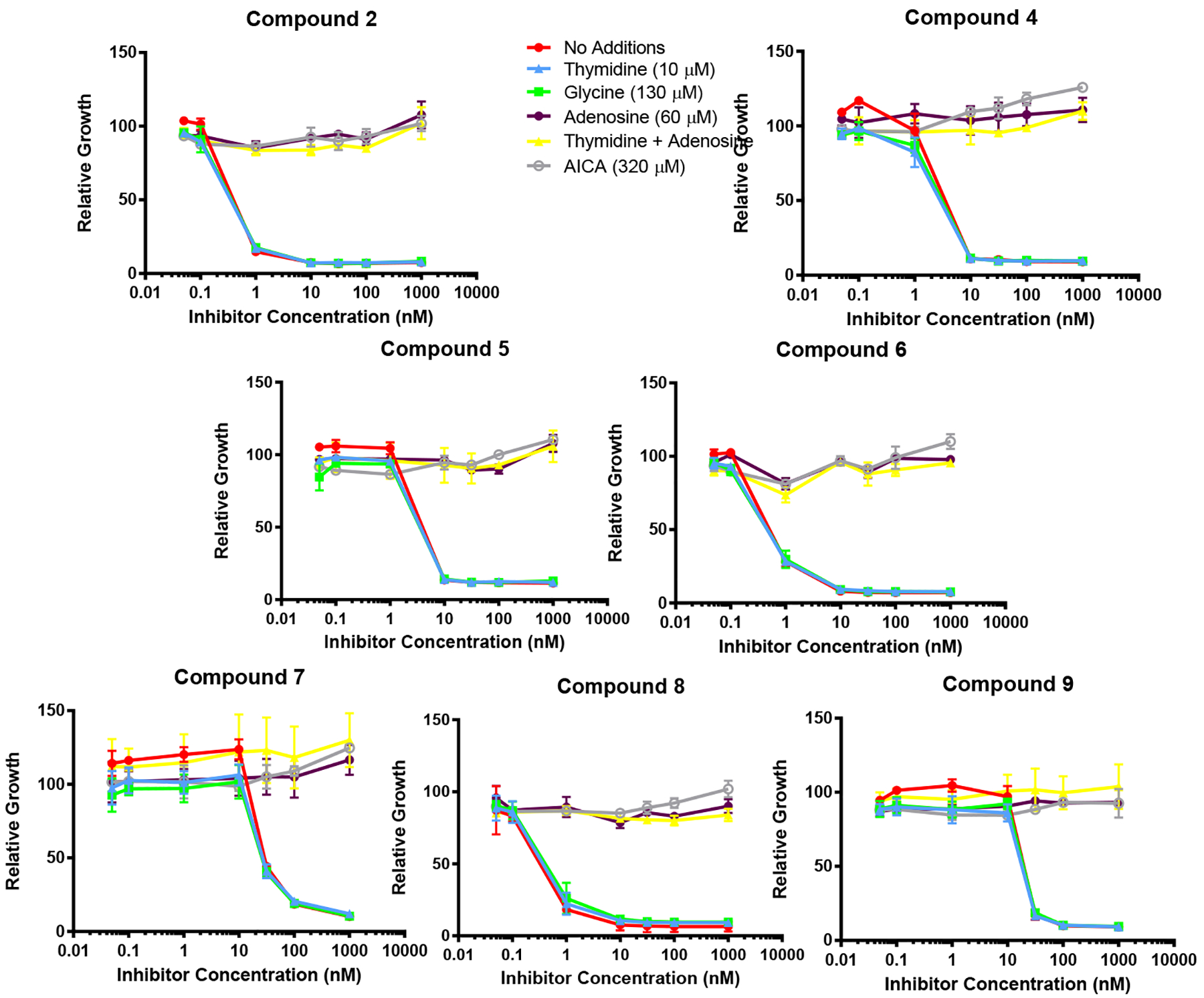
Identification of the intracellular target by protection by nucleosides, glycine and AICA. KB cells were incubated with drugs in folate-, nucleoside-, and glycine-free RPMI 1640 medium with 10% dialyzed fetal bovine serum (FBS), antibiotics, L-glutamine, and 2 nM leucovorin (LCV) with a range of drug concentrations in the presence of folic acid (200 nM), adenosine (60 μM), thymidine (10 μM), glycine (130 μM) or AICA (320 μM). Cell proliferation was assayed with Cell Titer Blue™ (Promega) using a fluorescence plate reader42. Data are mean values for at least triplicate experiments. Error bars represent standard errors. Experimental details are summarized in the Experimental Section.
Adenosine completely reversed the drug effects of all the compounds, whereas thymidine and glycine were ineffective alone and they did not augment the effects of adenosine in combination (not shown). Thus, de novo purine biosynthesis rather than thymidylate synthase or mitochondrial C1 metabolism is the targeted pathway for compounds 4–9. AICA (320 μM) has been used to identify GARFTase inhibitors as this is metabolized to AICA ribonucleotide (ZMP) to circumvent the GARFTase step.35, 42 As AICA was completely protective for compounds 4–9, we conclude that GARFTase is the primary cellular target for this series.
5. Summary
In this report, we identified novel heteroatom substituted pyrrolo[2,3-d]pyrimidine compounds with N, O, or S replacements for CH2 at position-11. Our results document remarkably increased in vitro antiproliferative activity resulting from heteroatom bridge substitutions toward CHO cell lines engineered to express human FRα or FRβ, and FRα-expressing human tumor cells, over the corresponding CH2 analog 2. All these compounds have substantial selectivities toward FRα and FRβ compared to RFC. PCFT-targeted activity for this series was modest. All compounds inhibited de novo purine biosynthesis, most likely at GARFTase. Compounds 6 and 7 displayed ~16-fold increased activity over the CH2 analog (2) and selectivity for FRβ compared to FRα, along with 6-fold increased activity against KB tumor cells for 6 over 2. In addition to tumor targeting, these results suggest potential applications for targeting FRβ expressing hematopoietic cancers and activated macrophages.5, 20 As such, dual therapeutic targeting FRα-expressing tumors (e.g., epithelial ovarian cancer), along with FRβ-expressing tumor-associated macrophages, would be highly impactful. Collectively, these compounds provide an exciting platform for continued discovery of new and highly selective FR-targeted therapeutics for cancer, as well as other diseases.
6. Experimental Section
A rotary evaporator was used to carry out evaporation in vacuo. Final compounds and intermediates were dried in a CHEM-DRY drying apparatus over P2O5 at 80 °C. A MELTEMP II melting point with a FLUKE 51 K/J electronic thermometer apparatus was used and uncorrected to record melting points. A Bruker WH-400 [400-megahertz (MHz)] spectrometer or a Bruker WH-500 (500 MHz) spectrometer was used to record proton nuclear magnetic resonance spectra (1H NMR). Tetramethylsilane was used as an internal standard to express the chemical shift in ppm (parts per million): s, singlet; d, doublet; t, triplet; q, quartet; quin, quintet m, multiplet; and bs, broad singlet. Chemical names follow International Union of Pure and Applied Chemistry (IUPAC) nomenclature. Whatman Sil G/UV254 silica gel plates with a fluorescent indicator were used for performing thin-layer chromatography (TLC), and the spots were visualized under 254 and 365 nm illumination. All analytical samples were homogeneous on TLC in three different solvent systems. Solvents used for TLC were measured in volume. Columns of silica gel (230–400 mesh) (Fisher, Somerville, NJ) were used for chromatography. In spite of 24–48 h of drying in vacuo, fractional moles of water found in the analytical samples of antifolates could not be prevented and were confirmed by their presence in the 1H NMR spectra. Chemicals and solvents were purchased from Aldrich Chemical Co. or Fisher Scientific Co. and were used as received. Elemental analysis (C, H, N, F, and S) was performed by Atlantic Microlab, Inc. (Norcross, GA). Element compositions were within 0.4% of the calculated values and confirmed >95% purity for all the compounds submitted for biological evaluation.
6.1. Synthesis
3-((4-(ethoxycarbonyl)phenyl)amino)propanoic acid (12).
A mixture of ethyl paminobenzoate (11) (1.65 g, 10 mmol) and β-propiolactone (10) (0.72 g, 10 mmol) in 15 mL of acetone was refluxed for 4 hours. After evaporation of the solvent under reduced pressure, MeOH (10 mL) was added, followed by silica gel (5 g). The resulting plug was loaded on to a silica gel column and eluted with ethyl acetate: hexane 1:10. Fractions with Rf = 0.34 (hexane: EtOAc 1:1) were pooled and evaporated to afford 12 as yellow semisolid (1.23 g, yield; 52%). TLC Rf = 0.34 (1:1 hexane:EtOAc); 1H NMR (CDCl3) δ 1.26–1.29 (t, 3H, OCH2CH3, J = 7 Hz), 2.51–2. 2.53 (t, 2H, CH2CH2NH, J = 6.8 Hz), 3.30–3.32 (m, 2H, CH2CH2NH), 4.18–4.24 (q, 2H, OCH2CH3, J = 7 Hz), 6.59–6.61 (d, 2H, Ar-CH, J = 7.2 Hz), 7.68–7.70 (d, 2H, Ar-CH, J = 7.2 Hz).
3-(N-(4-(ethoxycarbonyl)phenyl)-2,2,2-trifluoroacetamido)propanoic acid (13).
A solution of compound 12 (1.18 g, 5 mmol) in anhydrous CH2Cl2 (20 mL) was treated with triethylamine (1 mL) and trifluoroacetic anhydride (5 mL) under the anhydrous condition for 2 hours. After removal of the excess of solvent under reduced pressure at 40–45 °C, the yellowish oily residue was dissolved in dichloromethane (20 mL) followed by silica gel (3 g). The resulting plug was loaded on to a silica gel column and eluted with 10% ethyl acetate in hexane. Fractions with and Rf = 0.59 (1:1 hexane: EtOAc) were pooled and evaporated to afford 13 (1.47 g, yield; 88%) as yellow semisolid. TLC Rf = 0.59 (1:1 hexane:EtOAc); 1H NMR (CDCl3) δ 1.40–1.44 (t, 3H, OCH2CH3, J = 7.2 Hz), 2.68–2.72 (t, 2H, CH2CH2NCOCF3, J = 7.2 Hz), 4.07–4.10 (t, 2H, CH2CH2NCOCF3, J = 7.2 Hz), 4.39–4.44 (q, 2H, OCH2CH3, J = 7.2 Hz), 7.33–7.35 (d, 2H, Ar-CH, J = 8.4 Hz ), 8.13–8.15 (d, 2H, CH2, Ar-CH, J = 8.4 Hz). Anal. calcd (C14H14F3NO5): C, 50.46; H, 4.23; N, 4.20; F, 17.10. Found: C, 50.50; H, 4.22; N, 4.19; F, 16.87.
4-(N-(4-(ethoxycarbonyl)phenyl)-2,2,2-trifluoroacetamido)butanoic acid (14).
Compound 13 (1.33 g, 4 mmol) was dissolved in 20 mL anhydrous dichloromethane and oxalyl chloride (2 mL, 23.30 mmol) was added. The resulting solution was refluxed for 1 hour and then cooled to room temperature. After evaporating the solvent under reduced pressure, the residue was dissolved in 20 mL of diethyl ether. The resulting solution was added dropwise to ice-cooled diazomethane (generated in situ from 10 g of Diazald® by using Aldrich mini Diazald® Apparatus) in an ice bath over 10 min. The resulting mixture was allowed to stand for 30 min and then stirred for an additional 1 hour, excess diazomethane was decomposed with l mL of acetic acid, and the solution evaporated to dryness to afford diazo compound. To a stirred suspension of silver acetate (1.2 g) in 30 mL of water was added a solution of the diazo compound in 30 mL of 1,4-dioxane. The reaction mixture was heated to reflux for 1.5 hours, and then 1.2 g of sodium carbonate was added. The solid obtained was filtered and extracted with chloroform (3 × 25 mL). The combined chloroform extract was dried (Na2SO4), filtered, and the solvent was evaporated under reduced pressure. The residue was recrystallized from ethyl acetate to afford 14 (527 mg, yield over three steps 38%) as yellow semi-solid. TLC Rf = 0.64 (1:1 hexane:EtOAc); 1H NMR (CDCl3) δ 1.41–1.44 (t, 3H, OCH2CH3, J = 7.2 Hz), 1.89–1.96 (quin, 2H, CH2CH2CH2NCOCF3, J = 7.2 Hz), 2.42–2.46 (t, 2H, CH2CH2CH2NCOCF3, J = 7.2 Hz), 3.82–3.86 (t, 2H, CH2CH2CH2NCOCF3, J = 7.2 Hz), 4.40–4.45 (q, 2H, OCH2CH3, J = 7.2 Hz), 7.31–7.34 (d, 2H, Ar-CH, J = 8.4 Hz ), 8.14–8.16 (d, 2H, CH2, Ar-CH, J = 8.4 Hz). Anal. calcd (C15H16NF3O5): C, 51.88; H, 4.64; N, 4.03; F, 16.41. Found: C, 51.82; H, 4.83; N, 3.99; F, 16.26.
4-((3-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)propyl)amino) benzoic acid (17).
To 14 (2.4 g, 10 mmol) in a 250 mL flask was added oxalyl chloride (5.14 mL, 60 mmol) and anhydrous CH2Cl2 (20 mL). The resulting solution was refluxed for 1 hour and then cooled to room temperature. After the solvent was evaporated under reduced pressure, the residue was dissolved in 20 mL of diethyl ether. The resulting solution was added dropwise to ice-cooled diazomethane (generated in situ from 15 g of Diazald® by using Aldrich Mini Diazald® apparatus) in an ice bath over 10 min. The resulting mixture was allowed to stand for 30 min and then stirred for an additional 1 hour. To this solution was added 48% HBr in water (20 mL). The resulting mixture was refluxed for 1.5 hours. After the mixture was cooled to room temperature, the organic layer was separated, and the aqueous layer was extracted with Et2O (2 × 200 mL). The combined organic layer and Et2O extract was washed with two portions of 10% Na2CO3 solution and dried over Na2SO4. Evaporation of the solvent afforded 15 in 94% yield. To a suspension of 2,6-diaminopyrimidin-4-one (1.26 g, 10 mmol) in anhydrous DMF (25 mL) was added 15 (9.4 mmol). The resulting mixture was stirred under N2 at room temperature for 3 days. After evaporation of the solvent under reduced pressure, MeOH (20 mL) was added followed by silica gel (5 g). The resulting plug was loaded on to a silica gel column and eluted with CHCl3 followed by 3% MeOH in CHCl3 and then 5% MeOH in CHCl3. Fractions with and Rf = 0.58 (TLC) (CHCl3:CH3OH, 5:1) were pooled and evaporated to afford 16 (1.03 g, yield; 25%). Compound 16 (0.7 mmol) was dissolved in MeOH (10 mL) added 1N NaOH (10 mL) and the mixture was stirred under N2 at room temperature for 10 hours. TLC showed the disappearance of the starting material and one major spot at the origin (CHCl3: CH3OH, 5:1). The reaction mixture was dissolved in water (10 mL), the resulting solution was cooled in an ice bath, and the pH was adjusted to 3–4 with the dropwise addition of 1N HCl. The resulting suspension was frozen in the dry ice-acetone bath, thawed to 4–5 °C in the refrigerator, and filtered. The residue was washed with a small amount of cold water and dried in vacuum using P2O5 to afford the target compound 17 (196 mg, yield 86%) as yellow solid. TLC Rf = 0.19 (5:1 CHCl3:MeOH); mp 154 °C, 1H NMR (DMSO-d6) δ 1.81–1.88 (quin, 2H, CH2CH2CH2NH, J = 7.2 Hz), 2.56–2.59 (t, CH2CH2CH2NH, CH2, J = 7.2 Hz), 3.05–3.09 (m, 2H, CH2CH2CH2NH), 5.91 (s, 1H, C5-CH), 6.18 (bs, 2H, 2-NH2, exch.), 6.54–6.56 (bd, 3H, Ar-CH and NH, J = 8.4 Hz, one proton exch), 7.64–7.66 (d, 2H, CH2, Ar-CH, J = 8.4 Hz), 10.34 (s, 1H, 3-NH, exch.), 10.90 (s, 1H, 7-NH, exch.). Anal. calcd (C16H17N5O3·0.4 HCl): C, 56.20; H, 5.13; N, 20.48. Found: C, 56.39; H, 5.27; N, 20.41.
(4-((3-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)propyl)amino) benzoyl)-L-glutamic acid (4).
To a 250 mL round bottom flask, was added a mixture of compound 17 (100 mg, 0.2 mmol), N-methylmorpholine (0.4 mmol), 2-chloro-4,6-dimethoxy-1,3,5-triazine (0.4 mmol) and anhydrous DMF (7 mL). The resulting mixture was stirred at room temperature under the anhydrous condition for 1.5 hours. N-mehtylmorpholine (0.64 mmol) and L-glutamate diethyl hydrochloride (0.3 mmol) were added in the reaction mixture. The resulting mixture was then stirred at room temperature under the anhydrous condition for 12 hours. After evaporation of the solvent under reduced pressure, MeOH (20 mL) was added followed by silica gel (1 g). The resulting plug was loaded on to a silica gel column and eluted with CHCl3 followed by 3% MeOH in CHCl3 and then with 5% MeOH in CHCl3. Fractions with Rf = 0.45 (CHCl3:CH3OH, 5:1) were pooled and evaporated to afford 18 (96 mg, yield 61%) as solid. Compound 18 (0.15 mmol) was dissolved in MeOH (10 mL) added 1N NaOH (10 mL) and the mixture was stirred under N2 at room temperature for 10 hours. TLC showed the disappearance of the starting material and one major spot at the origin (CHCl3: CH3OH, 5:1). The reaction mixture was dissolved in water (10 mL), the resulting solution was cooled in an ice bath, and the pH was adjusted to 3–4 with dropwise addition of 1N HCl and acetic acid. The resulting suspension was frozen in the dry ice-acetone bath, thawed to 4–5 °C in the refrigerator, and filtered. The residue was washed with a small amount of cold water and dried in vacuum using P2O5 to afford the target compound 4 (58 mg, yield 84%) as a yellow powder. TLC Rf = 0.16 (5:1 CHCl3:MeOH); mp 163 °C; H NMR (DMSO-d6) δ 1.92–1.95 (quin, 2H, CH2CH2CH2NH, J = 7.2 Hz), 1.97–2.09 (m, 2H, β -CH2), 2.32–2.35 (t, 2H, γ-CH2, J = 7.5 Hz), 2.58–2.61 (t, 2H, CH2CH2CH2NH, J = 7.2 Hz), 3.31–3.34 (m, 2H, CH2CH2CH2NH), 4.34–4.37 (m, 1H, α-CH), 5.91–5.91 (d, 1H, Ar-CH, J = 2 Hz), 5.97 (bs, 2H, 2-NH2, exch.), 6.22–6.24 (t, 1H, CH2CH2CH2NH, J = 5.5 Hz, exch.), 6.55–6.56 (d, 2H, Ar-CH, J = 9 Hz), 7.65–7.67 (d, 2H, Ar-CH, J = 9 Hz), 8.09–8.10 (d, 1H, NH, J = 7.5 Hz, exch.), 10.14 (s, 1H, 3-NH, exch.), 10.85 (s, 1H, 7-NH, exch.). Anal. calcd (C21H24N6O6·0.5H2O·0.5CH3COOH): C, 53.33; H, 5.49; N, 16.96. Found: C, 53.27; H, 5.59; N, 16.61.
(4-(N-(3-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)propyl)formamido) benzoyl)-L-glutamic acid (7).
To a solution of 4 (110 mg, 0.25 mmol) in 97% formic acid (5 mL) was added acetic anhydride (1 mL), and the reaction mixture was stirred at 25 °C for 3 hours. The solvent was removed under reduced pressure and the residue dissolved in 1 N NaOH at 0 °C. The filtrate was acidified to pH 4 with 0.5 N HCI and stored at 0 °C for 2 hours. The yellow solid was collected by filtration and dried over P2O5 to give 40 mg (yield 35%) of 7. TLC Rf = 0.19 (5:1 CHCl3:MeOH); mp 153 °C; 1H NMR (DMSO-d6) δ 1.92–1.95 (quin, 2H, CH2CH2CH2NCHO, J = 7.2 Hz), 1.97–2.11 (m, 2H, β -CH2), 2.35–2.38 (t, 2H, γ-CH2, J = 7.5 Hz), 2.44–2.47 (t, 2H, CH2CH2CH2NCHO, J = 7.2 Hz), 3.84–3.88 (t, 2H, CH2CH2CH2NCHO, J = 7.5 Hz) 4.38–4.43 (m, 2H, α-CH), 5.85 (s, 1 H, C5-CH), 5.97 (s, 2H, 2-NH2, exch.), 7.45–7.78 (d, 1.72 H, Ar-CH, J = 8.4 Hz, rotamer of formamide), 7.51–7.53 (d, 0.28 H, Ar-CH, J = 8.4 Hz, rotamer of formamide), 7.91–7.95 (d, 2H, Ar-CH, J = 8.4 Hz), 8.64–8.66 (d, 1H, Ar-CONH, J = 7.6 Hz, exch.), 10.23 (bs, H, 3-NH, exch.), 10.93 (s, H, 7-NH, exch.). Anal. calcd (C22H24N6O7·1.5H2O·2.5 HCl): C, 45.25; H, 5.25; N, 13.47. Found: C, 45.56; H, 4.89; N, 13.40.
(4-(N-(3-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)propyl)-2,2,2-trifluoroacetamido)benzoyl)-L-glutamic acid (8).
5 mL trifluoroacetic anhydride was added to a solution of 4 (110 mg, 0.25 mmol) in 10 mL dichloromethane, and the reaction mixture was stirred at 25 °C for 3 hours. The solvent was removed under reduced pressure and the residue was suspended in water. 1N NaOH was added dropwise to the suspension and pH was adjusted to 4. The suspension was stored at 0 °C for 2 hours. The yellow solid was collected by filtration and dried over P2O5 to give 83 mg (yield 76%) of 8. TLC Rf = 0.21 (5:1 CHCl3:MeOH); mp 158 °C; 1H NMR (DMSO-d6) δ 1.87–2.11 (m, 4H, CH2CH2CH2NCOCF3 and β -CH2), 2.33–2.37 (t, 2H, γ-CH2, J = 7.5 Hz), 2.62–2.66 (t, 2H, CH2CH2CH2NCOCF3, J = 7.2 Hz), 3.97–3.99 (t, 2H, CH2CH2CH2NCOCF3, J = 7.2 Hz), 4.38–4.44 (m, 1H, α-CH), 5.92 (s, 1 H, C5-CH), 5.97 (bs, 2H, 2-NH2, exch.), 7.40–7.42 (d, 2 H, Ar-CH, J = 8 Hz), 7.94–7.96 (d, 2H, Ar-CH, J = 8 Hz), 8.76–8.78 (d, 1H, Ar-CONH, J = 8 Hz, exch.), 10.39 (bs, H, 3-NH, exch.), 10.99 (s, H, 7-NH, exch.). Anal. calcd (C23H13N6O7F3·0.5H2O): C, 49.20; H, 4.31; N, 14.97; F, 10.15. Found: C, 49.16; H, 4.52; N, 15.05; F, 9.82.
(4-(N-(3-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)propyl)acetamido)benzoyl)-L-glutamic acid (9).
Compound 4 (110 mg, 0.25 mmol) was added in 5 mL acetic anhydride, and the reaction mixture was stirred under the anhydrous condition at 25 °C for 3 hours. The excess of acetic anhydride was removed under reduced pressure. The residue was suspended in cold water and basified using 1N NaOH. The suspension was then filtered and acidified to pH 4 with 0.5 N HCI and stored at 0 °C for 2 hours. The white solid was collected by filtration and dried over P2O5 to give 57 mg (yield 47%) of 9. TLC Rf = 0.19 (5:1 CHCl3:MeOH); mp 166 °C; 1H NMR (DMSO-d6) δ 1.93–2.14 (m, 4H, CH2CH2CH2NCOCH3, β -CH2), 2.05 (s, 3H, CH2CH2CH2NCOCH3), 2.35–2.39 (t, 2H, γ-CH2, J = 7.5 Hz), 2.64–2.67 (t, 2H, CH2CH2CH2NCOCH3, J = 8 Hz), 3.89–3.92 (t, 2H, CH2CH2CH2NCOCH3, J = 8 Hz), 4.38–4.44 (m, 1H, α-CH), 5.91 (s, 1 H, C5-CH), 5.99 (s, 2H, 2-NH2, exch.), 7.34–7.36 (d, 2H, Ar-CH, J = 8 Hz), 7.93–7.95 (d, 2H, Ar-CH, J = 8 Hz), 8.68–8.70 (d, 1H, Ar-CONH, J = 8 Hz, exch.), 10.16 (bs, H, 3-NH, exch.), 10.83 (s, H, 7-NH, exch.), 12.45 (bs, 2H, 2COOH, exch.). Anal. calcd (C23H26N6O7·0.5 H2O·1.6HCl): C, 49.27; H, 5.21; N, 14.67. Found: C, 48.99; H, 4.99; N, 15.37.
4-(4-(ethoxycarbonyl)phenoxy)butanoic acid (22).
To 100 mL rbf was added a mixture of compound 20 (1.66 g, 10 mmol), cesium carbonate (3.26 g, 10 mmol), TBAI (3.70 g, 10 mmol) and anhydrous DMF (20 mL). Compound 19 (2.23 g, 10 mmol) was added dropwise to the mixture. The reaction mixture was then stirred at room temperature for 3 hours. Ethyl acetate was added into the reaction mixture. The combined mixture was washed with two portions of water. After evaporation of the solvent under reduced pressure MeOH (20 mL) was added followed by silica gel (1 g). The resulting plug was loaded on to a silica gel column and eluted with 1:10 (ethyl acetate: hexane). Fractions with and Rf = 0.64 (hexane:ethyl acetate 1:1) were pooled and evaporated to afford tert-butyl esters (1.47 g, yield; 47%). Trifluoroacetic acid was then added into the tert-butyl esters and mixture was stirred at room temperature for 30 min. Excess of trifluoroacetic acid was evaporated and MeOH (20 mL) was added followed by silica gel (1 g). The resulting plug was loaded on to a silica gel column and eluted with 1:10 (ethyl acetate: hexane). Fractions with and Rf = 0.45 (TLC) (Hexane: ethylacetate, 1:1) were pooled and evaporated to afford 22 (1 g, yield 83%) as white solid. TLC Rf = 0.45 (1:1 hexane:EtOAc); mp 112 °C; 1H NMR (CDCl3) δ 2.13–2.20 (quin, 2H, CH2CH2CH2O, J = 7.2 Hz), 2.60–2.64 (t, 2H, CH2CH2CH2O, J = 7.2 Hz), 3.90 (s, 3H, OCH3), 4.08–4.11 (t, 2H, CH2CH2CH2O, J = 7.2 Hz), 6.91–6.93 (d, 2H, Ar-CH, J = 8.8 Hz), 7.99–8.01 (d, 2H, Ar-CH, J = 8.8 Hz).
4-((4-(ethoxycarbonyl)phenyl)thio)butanoic acid (23).
Carboxylic acid 23 was synthesized from compound 21 using the synthetic procedure as for carboxylic acid 22 in 74% yield as white solid over two-step. TLC Rf = 0.52 (1:1 hexane:EtOAc); mp 76 °C; 1H NMR (CDCl3) δ 1.39–1.43 (t, 2H, OCH2CH3, J = 7.2 Hz), 2.03–2.06 (quin, 2H, CH2CH2CH2S, J = 7.2 Hz), 2.53–2.60 (t, 2H, CH2CH2CH2O, J = 7.2 Hz), 3.07–3.10 (t, 2H, CH2CH2CH2S, J = 7.2 Hz), 4.36–4.41 (q, 2H, OCH2CH3, J = 7.2 Hz), 7.33–7.35 (d, 2H, Ar-CH, J = 8.4 Hz ), 7.95–7.97 (d, 2H, CH2, Ar-CH, J = 8.4 Hz).
4-(3-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)propoxy)benzoic acid (32).
To 22 (2.52 g, 10 mmol) in a 250 mL flask was added oxalyl chloride (5.14 g, 60 mmol) and anhydrous CH2Cl2 (20 mL). The resulting solution was refluxed for 1 hour and then cooled to room temperature. After the solvent was evaporated under reduced pressure, the residue was dissolved in 20 mL of Et2O. The resulting solution was added dropwise to an ice-cooled diazomethane (generated in situ from 15 g of Diazald® by using Aldrich Mini Diazald® apparatus) in an ice bath over 10 min. The resulting mixture was allowed to stand for 30 min and then stirred for an additional 1 hour. To this solution was added 48% HBr (20 mL). The resulting mixture was refluxed for 1.5 hours. After the mixture was cooled to room temperature, the organic layer was separated, and the aqueous layer was extracted with Et2O (2 × 200 mL). The combined organic layer and Et2O extract was washed with two portions of 10% Na2CO3 solution and dried over Na2SO4. Evaporation of the solvent afforded 28 in 94% crude yield. To a suspension of 2,6-diaminopyrimidin-4-one (1.26 g, 10 mmol) in anhydrous DMF (25 mL) was added 28 (9.4 mmol). The resulting mixture was stirred under N2 at room temperature for 3 days. After evaporation of the solvent under reduced pressure, MeOH (20 mL) was added followed by silica gel (5 g). The resulting plug was loaded on to a silica gel column and eluted with CHCl3 followed by 3% MeOH in CHCl3 and then 5% MeOH in CHCl3. Fractions with and Rf = 0.48 (TLC) (CHCl3: CH3OH, 5:1) were pooled and evaporated to afford 30 (1 g, yield 31%). Compound 30 (1 g, 2.92 mmol) was dissolved in MeOH (10 mL) added 1N NaOH (10 mL) and the mixture was stirred under N2 at room temperature for 10 hours. TLC showed the disappearance of the starting material and one major spot at the origin (CHCl3: MeOH 5:1). The reaction mixture was dissolved in water (10 mL), the resulting solution was cooled in an ice bath, and the pH was adjusted to 3–4 with the dropwise addition of 1N HCl. The resulting suspension was frozen in the dry ice-acetone bath, thawed to 4–5 °C in the refrigerator, and filtered. The residue was washed with a small amount of cold water and dried in vacuum using P2O5 to afford the compound 32 (638 mg, yield 66%) as white solid. TLC Rf = 0.15 (5:1 CHCl3:MeOH); mp 155 °C; 1H NMR (DMSO-d6) δ 2.02–2.07 (quin, 2H, CH2CH2CH2O, J = 7.2 Hz), 2.63–2.69 (t, 2H, CH2CH2CH2O, J = 7.2 Hz), 4.03–4.06 (t, 2H, CH2CH2CH2O, J = 7.2 Hz), 5.95 (s, 1H, C5-CH), 6.60 (bs, 2H, 2-NH2, exch.), 6.99–7.02 (d, 2H, Ar-CH, J = 8.8 Hz), 7.86–7.89 (d, 2H, Ar-CH, J = 8.8 Hz), 10.67 (s, 1H, 3-NH, exch.), 11.18 (s, 1H, 7-NH, exch.).
4-((3-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)propyl)thio)benzoic acid (33).
Carboxylic acid 33 was synthesized from compound 23 using the synthetic procedure as for carboxylic acid 32 from compound 22 in 22% yield as white solid over five steps. TLC Rf = 0.22 (1:1 hexane:EtOAc); mp 143 °C; 1H NMR (DMSO-d6) δ 1.89–1.96 (quin, 2H, CH2CH2CH2S, J = 7.2 Hz), 2.64–2.67 (t, 2H, CH2CH2CH2S, J = 7.2 Hz), 3.03–3.07 (t, 2H, CH2CH2CH2S, J = 7.2 Hz), 5.95 (s, 1H, C5-CH), 6.02 (bs, 2H, 2-NH2, exch.), 7.34–7.36 (d, 2H, Ar-CH, J = 8.8 Hz), 7.82–7.84 (d, 2H, Ar-CH, J = 8.8 Hz), 10.47 (s, 1H, 3-NH, exch.), 11.06 (s, 1H, 7-NH, exch.).
(4-(3-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)propoxy)benzoyl)-L-glutamic acid (5).
To a 250 mL rbf, was added a mixture of compound 32 (80 mg, 0.24 mmol), N-methylmorpholine (0.48 mmol), 2-chloro-4,6-dimethoxy-1,3,5-triazine (0.48 mmol) and anhydrous DMF (10 mL). The resulting mixture was stirred at room temperature under the anhydrous condition for 1.5 hours. N-mehtylmorpholine (0.48 mmol) and L-glutamate di-tert-butyl hydrochloride (0.36 mmol) were added in reaction mixture. The resulting mixture was then stirred at room temperature under the anhydrous condition for 12 hours. After evaporation of the solvent under reduced pressure, MeOH (20 mL) was added followed by silica gel (1 g). The resulting plug was loaded on to a silica gel column and eluted with CHCl3 followed by 1% MeOH in CHCl3. Fractions with Rf = 0.52 (CHCl3:CH3OH, 5:1) were pooled and evaporated to afford 34 (56 mg, yield 45%) as solid. Compound 34 (56 mg, 0.11 mmol) was dissolved in MeOH (10 mL) added 1N NaOH (10 mL) and the mixture was stirred under N2 at room temperature for 10 hours. TLC showed the disappearance of the starting material and one major spot at the origin (CHCl3: MeOH 5:1). The reaction mixture was dissolved in water (10 mL), the resulting solution was cooled in an ice bath, and the pH was adjusted to 3–4 with the dropwise addition of 1N HCl. The resulting suspension was frozen in the dry ice-acetone bath, thawed to 4–5 °C in the refrigerator, and filtered. The residue was washed with a small amount of cold water and dried in vacuum using P2O5 to afford the target compound (5) (34 mg, yield 68%) as white powder. TLC Rf = 0.12 (5:1 CHCl3:MeOH); mp 173 °C; 1H NMR (DMSO-d6) δ 1.91–2.09 (m, 4H, CH2CH2CH2O and β -CH2), 2.33–2.37 (t, 2H, γ-CH2, J = 7.6 Hz), 2.45–2.68 (t, 2H, CH2CH2CH2O, J = 7.6 Hz), 4.03–4.06 (t, 2H, CH2CH2CH2O, J = 7.6 Hz), 4.35–4.40 (m, 1H, α-CH), 5.90 (s, 1H, C5-CH), 5.99 (bs, 2H, 2-NH2, exch.), 6.99–7.02 (d, 2H, CH2, Ar-CH, J = 8.8 Hz), 7.84–7.87 (d, 2H, Ar-CH, J = 8.8 Hz), 8.44–8.46 (d, 1H, Ar-CONH, J = 7.5 Hz, exch.), 10.16 (s, 1H, 3-NH, exch.), 10.89 (s, 1H, 7-NH, exch.). Anal. calcd (C21H23N5O7 2 H2O): C, 51.11; H, 5.51; N, 14.19: Found: C, 50.94; H, 5.45; N, 14.00.
(4-((3-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-6-yl)propyl)thio)benzoyl)-L-glutamic acid (6).
Target compound 6 was synthesized from compound 33 using the synthetic procedure as for target compound 5 from compound 32 in 38% yield as buff color solid over two steps. TLC Rf = 0.14 (5:1 CHCl3:MeOH); mp 171 °C; 1H NMR (DMSO-d6) δ 1.95–2.13 (m, 4H, CH2CH2CH2S and β -CH2), 2.33 –2.37 (t, 2H, γ-CH2, J = 7.2 Hz), 2.62–2.66 (t, 2H, CH2CH2CH2S, J = 7.2 Hz), 3.02–3.06 (t, 2H, CH2CH2CH2S, J = 7.2 Hz), 4.38–4.39 (m, 1H, α-CH), 5.89 (s, 1H, C5-CH), 5.99 (bs, 2H, 2-NH2, exch.), 7.35–7.37 (d, 2H, Ar-CH, J = 8.4 Hz), 7.80–7.82 (d, 2H, Ar-CH, J = 8.4 Hz), 8.57–8.59 (d, 1H, Ar-CONH, J = 7.5 Hz, exch.), 10.16 (s, 1H, 3-NH, exch.), 10.88 (s, 1H, 7-NH, exch.). Anal. calcd (C21H23N5O6S·0.5 H2O): C, 52.27; H, 5.01; N, 14.51; S, 6.65. Found: C, 52.08; H, 4.86; N, 14.51; S, 6.47.
6.2. Molecular Modeling and Computational Studies
All the compounds were docked on to the X-ray crystal structures of human FRα (PDB 5IZQ, 3.60 Å)35, 37, 46, FRβ45 (PDB 4KN2, 2.6 Å), and GARFTase (PDB 5J9F, 2.1 Å)35, 37 to analyze the potential binding modes, binding energies, and favorable or unfavorable interactions. The energy-minimized crystallized ligand 335 and PMX were redocked with an RMSD (root-mean-square deviation) of 0.89 and 0.91 for the best-scored pose, thus validating the docking process. The crystal structure PDBs were obtained from the protein database. All docking procedures were performed using various modules of Schrödinger Maestro suite (Schrödinger, LLC, New York, NY, 2019).44 The polypeptide structures of FRα, FRβ and GARFTase were optimized and prepared for docking using the Maestro Protein Preparation Wizard to assess bond order and missing hydrogens, followed by energy minimization using the OPLS3e force field. Gaps in the protein structures were not corrected as they were far from the active site. The Maestro induced-fit Grid Generation module was then used to define a 15 × 15 × 15 Å grid from the center of all the ligands. Ligands used in the computational docking study were built using the Maestro 2D Build module. The Maestro LigPrep module was then used to generate conformers of each compound subjected to energy minimization using the OPLS3e force field protocol. The resulting compounds were docked into the prepared FRα, FRβ, and GARFTase structures using the Maestro Induced Fit Docking. Induced Fit Docking was performed with standard precision with flexible ligand sampling. A total of 20 initial poses were generated for each compound. Based on the pose score, the top 4 poses were selected and subjected to energy minimization using the OPLS3e force field. Finally, the top 2 poses per compound were generated and ranked according to Glide score, which is an approximation of binding energy defined by receptor-ligand complex energies. The top pose was analyzed and presented in the Biological Evaluation and Discussion. Docking scores are listed in table 2S (supporting information).
6.3. Cell lines and assays of antitumor drug activities
The engineered CHO sublines including RFC-, PCFT-, and FRα-null MTXRIIOuaR2–4 (R2) and RFC- (PC43–10), PCFT- (R2/PCFT4), FRα- (RT16), and FRβ- (D4) expressing CHO sublines were previously described.33, 42, 49–51, 53 The CHO cells were grown in α-minimal essential medium (MEM) supplemented with 10% bovine calf serum (Invitrogen, Carlsbad, CA), L-glutamine (2 mM), penicillin (1000 U/mL), and streptomycin (1000 μg/mL) at 37 °C with 5% CO2. R2 transfected cells (PC43–10, RT16, D4, R2/PCFT4) were cultured in complete α-MEM media plus G418 (1 mg/mL). Prior to the cell proliferation assays (see below), RT16 and D4 cells were cultured in complete folate-free RPMI 1640 (without added folate), plus 10% dialyzed fetal bovine serum (FBS) (Sigma-Aldrich) and penicillin/streptomycin for 3 days. Human KB carcinoma cells were purchased from ATCC (Manassas, VA) continuously maintained in complete folate-free (FF) RPMI with 10% fetal bovine serum and 1% penicillin-streptomycin and L-glutamine.
For growth inhibition studies, cells (CHO, KB) were plated in 96 well dishes (~2000 cells/well, total volume of 200 μL) and treated with a range of drug concentrations (0–1000 nM) in complete folate-free RPMI 1640 medium with 10% dialyzed FBS, supplemented with 2 nM (KB, RT16, D4 CHO cells) or 25 nM (PC43–10, R2/PCFT4) leucovorin, as described.33–35, 38–42, 51, 54 To confirm FR-mediated drug uptake, 200 nM folic acid was added to parallel incubations for KB, RT16 and D4 cells. After 96 h, viable cells were assayed with Cell-Titer Blue™ reagent (Promega, Madison, WI), and fluorescence was measured with a fluorescence plate reader. Fluorescence measurements were used for calculations of IC50 values, corresponding to the drug concentrations at which cells showed 50% loss of proliferation.
To confirm the targeted pathway or enzyme, in vitro growth inhibition of KB tumor cells was measured with complete folate- and glycine-free RPMI 1640 supplemented with 10% dialyzed fetal bovine serum in the presence of thymidine (10 μM), adenosine (60 μM) and/or glycine (130 μM). 33–35, 38–42, 51, 54 For de novo purine biosynthesis inhibitors, additional protection experiments used AICA (320 μM) to distinguish inhibitory effects at GARFTase from those at AICARFTase. 33–35, 38–42, 51, 54
Supplementary Material
Acknowledgments
This work was supported in part by grants from the National Institutes of Health R01 CA53535 (LHM and ZH), R01 CA125153 (AG), R01 CA152316 (LHM and AG), and R01 CA166711 (AG, LHM and CED), the Eunice and Milton Ring Endowed Chair for Cancer Research (LHM), and the Duquesne University Adrian Van Kaam Chair in Scholarly Excellence (AG). AD was supported by T32 CA009531 (LHM) and F30 CA228221 (AD).
Abbreviations Used
- AICA
5-Aminoimidazole-4-carboxamide
- AICARFTase
5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase
- CHO
Chinese hamster ovary
- FBS
fetal bovine serum
- FDA
Food and Drug Administration
- FF
folate free
- FR
folate receptor
- 10-CHOTHF
N10-formyl tetrahydrofolate
- GAR
glycinamide ribonucleotide
- GARFTase
glycinamide ribonucleotide formyltransferase
- GPI
glycosyl-phosphatidylinositol
- IC50
50 percent inhibitory concentration
- IUPAC
Chemical names follow International Union of Pure and Applied Chemistry
- LCV
leucovorin
- MHz
megahertz
- MTX
methotrexate
- MEM
minimal essential media
- NMR
nuclear magnetic resonance spectra
- NSCLC
non-small cell lung cancer
- NCHO
N- formyl
- N-COCF3
N-trifluoroacetyl
- N-COCH3
- ppm
parts per million
- PMX
pemetrexed
- PCFT
proton-coupled folate transporter
- RTX
Raltitrexed
- RFC
reduced folate carrier
- RMSD
root-mean-square deviation
- RPMI
Roswell Park Memorial Institute
- TLC
thin layer chromatography
- UV
ultraviolet
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Statistical Analysis
Descriptive statistical tests (e.g., t-tests) were conducted using GraphPad 6.0 software (La Jolla, CA).
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors declare no competing financial interest.
REFERENCES
- 1.Stokstad ELR, Historical perspective on key advances in the biochemistry and physiology of folates Picciano MF , Stokstad ELR And Gregory JF Iii (Ed ) Contemporary Issues in Clinical Nutrition , Vol 1990, 13 Folic Acid Metabolism in Health And Disease; 196th National Meeting, Los Angele (Ed), New York, New York, Usa; Chichester, England, Uk Illus: 1–22. [Google Scholar]
- 2.Shuvalov O; Petukhov A; Daks A; Fedorova O; Vasileva E; Barlev NA, One-carbon metabolism and nucleotide biosynthesis as attractive targets for anticancer therapy. Oncotarget 2017, 8 (14), 23955–23977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hou Z; Matherly LH, Biology of the major facilitative folate transporters SLC19A1 and SLC46A1. Curr. Top. Membr 2014, 73 (Exchangers), 175–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matherly LH; Hou Z; Deng Y, Human reduced folate carrier: translation of basic biology to cancer etiology and therapy. Cancer metastasis reviews 2007, 26 (1), 111–28. [DOI] [PubMed] [Google Scholar]
- 5.Elnakat H; Ratnam M, Distribution, functionality and gene regulation of folate receptor isoforms: implications in targeted therapy. Adv Drug Deliv Rev 2004, 56 (8), 1067–84. [DOI] [PubMed] [Google Scholar]
- 6.Matherly LH; Hou Z; Gangjee A, The promise and challenges of exploiting the proton-coupled folate transporter for selective therapeutic targeting of cancer. Cancer chemotherapy and pharmacology 2018, 81 (1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao R; Goldman ID, The molecular identity and characterization of a Proton-coupled Folate Transporter--PCFT; biological ramifications and impact on the activity of pemetrexed. Cancer metastasis reviews 2007, 26 (1), 129–39. [DOI] [PubMed] [Google Scholar]
- 8.Desmoulin SK; Hou Z; Gangjee A; Matherly LH, The human proton-coupled folate transporter: Biology and therapeutic applications to cancer. Cancer Biol Ther 2012, 13 (14), 1355–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qiu A; Jansen M; Sakaris A; Min SH; Chattopadhyay S; Tsai E; Sandoval C; Zhao R; Akabas MH; Goldman ID, Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 2006, 127 (5), 917–28. [DOI] [PubMed] [Google Scholar]
- 10.Inoue K; Nakai Y; Ueda S; Kamigaso S; Ohta K.-y.; Hatakeyama M; Hayashi Y; Otagiri M; Yuasa H, Functional characterization of PCFT/HCP1 as the molecular entity of the carrier-mediated intestinal folate transport system in the rat model. American Journal of Physiology-Gastrointestinal and Liver Physiology 2008, 294 (3), G660–G668. [DOI] [PubMed] [Google Scholar]
- 11.Chabner BA; Allegra CJ, Cancer chemotherapy and biotherapy principles and practice: Antifolates. 5 ed.; Wolters Kluwer : Lippincott Williams & Wilkins: Philadelphia; Baltimore; New York, 2011; p 109–138. [Google Scholar]
- 12.Visentin M; Zhao R; Goldman ID, The antifolates. Hematol Oncol Clin North Am 2012, 26 (3), 629–48, ix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wessels JA; Huizinga TW; Guchelaar HJ, Recent insights in the pharmacological actions of methotrexate in the treatment of rheumatoid arthritis. Rheumatology (Oxford) 2008, 47 (3), 249–55. [DOI] [PubMed] [Google Scholar]
- 14.Chattopadhyay S; Moran RG; Goldman ID, Pemetrexed: biochemical and cellular pharmacology, mechanisms, and clinical applications. Molecular cancer therapeutics 2007, 6 (2), 404–17. [DOI] [PubMed] [Google Scholar]
- 15.Gonen N; Assaraf YG, Antifolates in cancer therapy: structure, activity and mechanisms of drug resistance. Drug Resist Updat 2012, 15 (4), 183–210. [DOI] [PubMed] [Google Scholar]
- 16.Gibbs DD; Theti DS; Wood N; Green M; Raynaud F; Valenti M; Forster MD; Mitchell F; Bavetsias V; Henderson E; Jackman AL, BGC 945, a novel tumor-selective thymidylate synthase inhibitor targeted to alpha-folate receptor-overexpressing tumors. Cancer Res 2005, 65 (24), 11721–8. [DOI] [PubMed] [Google Scholar]
- 17.Vergote IB; Marth C; Coleman RL, Role of the folate receptor in ovarian cancer treatment: evidence, mechanism, and clinical implications. Cancer metastasis reviews 2015, 34 (1), 41–52. [DOI] [PubMed] [Google Scholar]
- 18.Xia W; Low PS, Folate-targeted therapies for cancer. Journal of medicinal chemistry 2010, 53 (19), 6811–24. [DOI] [PubMed] [Google Scholar]
- 19.Parker N; Turk MJ; Westrick E; Lewis JD; Low PS; Leamon CP, Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Analytical biochemistry 2005, 338 (2), 284–93. [DOI] [PubMed] [Google Scholar]
- 20.Puig-Kroger A; Sierra-Filardi E; Dominguez-Soto A; Samaniego R; Corcuera MT; Gomez-Aguado F; Ratnam M; Sanchez-Mateos P; Corbi AL, Folate receptor beta is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer Res 2009, 69 (24), 9395–403. [DOI] [PubMed] [Google Scholar]
- 21.Shen J; Hu Y; Putt KS; Singhal S; Han H; Visscher DW; Murphy LM; Low PS, Assessment of folate receptor alpha and beta expression in selection of lung and pancreatic cancer patients for receptor targeted therapies. Oncotarget 2018, 9 (4), 4485–4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weitman SD; Weinberg AG; Coney LR; Zurawski VR; Jennings DS; Kamen BA, Cellular Localization of the Folate Receptor: Potential Role in Drug Toxicity and Folate Homeostasis. Cancer Research 1992, 52 (23), 6708–6711. [PubMed] [Google Scholar]
- 23.Wibowo AS; Singh M; Reeder KM; Carter JJ; Kovach AR; Meng W; Ratnam M; Zhang F; Dann CE, Structures of human folate receptors reveal biological trafficking states and diversity in folate and antifolate recognition. Proceedings of the National Academy of Sciences 2013, 110 (38), 15180–15188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen J; Putt KS; Visscher DW; Murphy L; Cohen C; Singhal S; Sandusky G; Feng Y; Dimitrov DS; Low PS, Assessment of folate receptor-beta expression in human neoplastic tissues. Oncotarget 2015, 6 (16), 14700–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ross JF; Wang H; Behm FG; Mathew P; Wu M; Booth R; Ratnam M, Folate receptor type beta is a neutrophilic lineage marker and is differentially expressed in myeloid leukemia. Cancer 1999, 85 (2), 348–57. [DOI] [PubMed] [Google Scholar]
- 26.Hu Y; Wang B; Shen J; Low SA; Putt KS; Niessen HWM; Matteson EL; Murphy L; Ruppert C; Jansen G; Oliver SJ; Feng Y; Dimitrov DS; Nickerson-Nutter C; Low PS, Depletion of activated macrophages with a folate receptor-beta-specific antibody improves symptoms in mouse models of rheumatoid arthritis. Arthritis research & therapy 2019, 21 (1), 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakashima-Matsushita N; Homma T; Yu S; Matsuda T; Sunahara N; Nakamura T; Tsukano M; Ratnam M; Matsuyama T, Selective expression of folate receptor beta and its possible role in methotrexate transport in synovial macrophages from patients with rheumatoid arthritis. Arthritis and rheumatism 1999, 42 (8), 1609–16. [DOI] [PubMed] [Google Scholar]
- 28.Wu D; Zhang P; Ma J; Xu J; Yang L; Xu W; Que H; Chen M; Xu H, Serum biomarker panels for the diagnosis of gastric cancer. Cancer medicine 2019, 8 (4), 1576–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Y; Unno K; Hyjek E; Liu H; Zimmerman T; Karmakar S; Putt KS; Shen J; Low PS; Wickrema A, Expression of functional folate receptors in multiple myeloma. Leukemia & lymphoma 2018, 59 (12), 2982–2989. [DOI] [PubMed] [Google Scholar]
- 30.Kurkjian C, LoRusso P, Sankhala KK, Birrer MJ, Kirby M, Ladd S, Hawes S,Running KL, O’Leary JJ, Moore KN, A phase I, first-in-human studyto evaluate the safety, pharmacokinetics (PK), and pharmacodynamics (PD) ofIMGN853 in patients (Pts) with epithelial ovarian cancer (EOC) and other FOLR1-positive solid tumors. J. Clin. Oncol 2013, 31 (15 Suppl.), 2573. [Google Scholar]
- 31.Reddy JA; Dorton R; Bloomfield A; Nelson M; Dircksen C; Vetzel M; Kleindl P; Santhapuram H; Vlahov IR; Leamon CP, Pre-clinical evaluation of EC1456, a folate-tubulysin anti-cancer therapeutic. Scientific Reports 2018, 8 (1), 8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banerji U, Garces AHI, Michalarea V, Ruddle R, Raynaud FI Riisnaes R Rodrigues DN, Tunariu N, Porter JC, Ward SE, Parmar M, Turner AJ, Seeramreddi S, Hall E, Dean EJ, Basu B, George A, Kaye SB, Banerjee SN, De Bono JS , An investigator-initiated phase I study of ONX-0801, a first-in-class alpha folate receptor targeted, small molecule thymidylate synthase inhibitor in solid tumors. Journal of Clinical Oncology 2017, 35 (15 suppl), 2503. [Google Scholar]
- 33.Desmoulin SK; Wang Y; Wu J; Stout M; Hou Z; Fulterer A; Chang MH; Romero MF; Cherian C; Gangjee A; Matherly LH, Targeting the proton-coupled folate transporter for selective delivery of 6-substituted pyrrolo[2,3-d]pyrimidine antifolate inhibitors of de novo purine biosynthesis in the chemotherapy of solid tumors. Molecular Pharmacology 2010, 78 (4), 577–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Golani LK; George C; Zhao S; Raghavan S; Orr S; Wallace A; Wilson MR; Hou ZJ; Matherly LH; Gangjee A, Structure-Activity Profiles of Novel 6-Substituted Pyrrolo[2,3-d]pyrimidine Thienoyl Antifolates with Modified Amino Acids for Cellular Uptake by Folate Receptors alpha and beta and the Proton-Coupled Folate Transporter. Journal of medicinal chemistry 2014, 57 (19), 8152–8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Golani LK; Wallace-Povirk A; Deis SM; Wong J; Ke J; Gu X; Raghavan S; Wilson MR; Li X; Polin L; de Waal PW; White K; Kushner J; O’Connor C; Hou Z; Xu HE; Melcher K; Dann CE 3rd; Matherly LH; Gangjee A, Tumor Targeting with Novel 6-Substituted Pyrrolo [2,3-d] Pyrimidine Antifolates with Heteroatom Bridge Substitutions via Cellular Uptake by Folate Receptor alpha and the Proton-Coupled Folate Transporter and Inhibition of de Novo Purine Nucleotide Biosynthesis. Journal of medicinal chemistry 2016, 59 (17), 7856–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kugel Desmoulin S; Wang L; Hales E; Polin L; White K; Kushner J; Stout M; Hou Z; Cherian C; Gangjee A; Matherly LH, Therapeutic Targeting of a Novel 6-Substituted Pyrrolo [2,3-d]pyrimidine Thienoyl Antifolate to Human Solid Tumors Based on Selective Uptake by the Proton-Coupled Folate Transporter. Molecular Pharmacology 2011, 80 (6), 1096–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ravindra M; Wilson MR; Tong N; O’Connor C; Karim M; Polin L; Wallace-Povirk A; White K; Kushner J; Hou ZJ; Matherly LH; Gangjee A, Fluorine -Substituted Pyrrolo[2,3-d]Pyrimidine Analogues with Tumor Targeting via Cellular Uptake by Folate Receptor alpha and the Proton-Coupled Folate Transporter and Inhibition of de Novo Purine Nucleotide Biosynthesis. Journal of medicinal chemistry 2018, 61 (9), 4228–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L; Cherian C; Desmoulin SK; Mitchell-Ryan S; Hou ZJ; Matherly LH; Gangjee A, Synthesis and Biological Activity of 6-Substituted Pyrrolo[2,3-d]pyrimidine Thienoyl Regioisomers as Inhibitors of de Novo Purine Biosynthesis with Selectivity for Cellular Uptake by High Affinity Folate Receptors and the Proton-Coupled Folate Transporter over the Reduced Folate Carrier. Journal of medicinal chemistry 2012, 55 (4), 1758–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L; Cherian C; Desmoulin SK; Polin L; Deng YJ; Wu JM; Hou ZJ; White K; Kushner J; Matherly LH; Gangjee A, Synthesis and Antitumor Activity of a Novel Series of 6-Substituted Pyrrolo[2,3-d]pyrimidine Thienoyl Antifolate Inhibitors of Purine Biosynthesis with Selectivity for High Affinity Folate Receptors and the Proton-Coupled Folate Transporter over the Reduced Folate Carrier for Cellular Entry. Journal of medicinal chemistry 2010, 53 (3), 1306–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L; Wallace A; Raghavan S; Deis SM; Wilson MR; Yang S; Polin L; White K; Kushner J; Orr S; George C; O’Connor C; Hou ZJ; Mitchell-Ryan S; Dann CE; Matherly LH; Gangjee A, 6-Substituted Pyrrolo[2,3-d]pyrimidine Thienoyl Regioisomers as Targeted Antifolates for Folate Receptor alpha and the Proton-Coupled Folate Transporter in Human Tumors. Journal of medicinal chemistry 2015, 58 (17), 6938–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y; Cherian C; Orr S; Mitchell-Ryan S; Hou Z; Raghavan S; Matherly LH; Gangjee A, Tumor-targeting with novel non-benzoyl 6-substituted straight chain pyrrolo[2,3-d]pyrimidine antifolates via cellular uptake by folate receptor alpha and inhibition of de novo purine nucleotide biosynthesis. Journal of medicinal chemistry 2013, 56 (21), 8684–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deng YJ; Wang YQ; Cherian C; Hou ZJ; Buck SA; Matherly LH; Gangjee A, Synthesis and discovery of high affinity folate receptor-specific glycinamide ribonucleotide formyltransferase inhibitors with antitumor activity. Journal of medicinal chemistry 2008, 51 (16), 5052–5063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meanwell NA, Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. Journal of medicinal chemistry 2011, 54 (8), 2529–2591. [DOI] [PubMed] [Google Scholar]
- 44.Maestro, Schrödinger Release 2016–4; Schrödinger, LLC: New York, NY, 2016. [Google Scholar]
- 45.Wibowo AS; Singh M; Reeder KM; Carter JJ; Kovach AR; Meng WY; Ratnam M; Zhang FM; Dann CE, Structures of human folate receptors reveal biological trafficking states and diversity in folate and antifolate recognition. P Natl Acad Sci USA 2013, 110 (38), 15180–15188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen C; Ke JY; Zhou XE; Yi W; Brunzelle JS; Li J; Yong EL; Xu HE; Melcher K, Structural basis for molecular recognition of folic acid by folate receptors. Nature 2013, 500 (7463), 486-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chackal S; Houssin R; Henichart JP, An efficient synthesis of the new benzo[c]pyrido[2,3,4-kl]acridine skeleton. J Org Chem 2002, 67 (10), 3502–3505. [DOI] [PubMed] [Google Scholar]
- 48.Smart BP; Pan YH; Weeks AK; Bollinger JG; Bahnson BJ; Gelb MH, Inhibition of the complete set of mammalian secreted phospholipases A(2) by indole analogues: a structure-guided study. Bioorgan Med Chem 2004, 12 (7), 1737–1749. [DOI] [PubMed] [Google Scholar]
- 49.Flintoff WF; Davidson SV; Siminovitch L, Isolation and partial characterization of three methotrexate-resistant phenotypes from Chinese hamster ovary cells. Somatic Cell Genet 1976, 2 (3), 245–61. [DOI] [PubMed] [Google Scholar]
- 50.Wong SC; Proefke SA; Bhushan A; Matherly LH, Isolation of Human Cdnas That Restore Methotrexate Sensitivity and Reduced Folate Carrier Activity in Methotrexate Transport-Defective Chinese-Hamster Ovary Cells. J Biol Chem 1995, 270 (29), 17468–17475. [DOI] [PubMed] [Google Scholar]
- 51.Deng YJ; Zhou XL; Desmoulin SK; Wu JM; Cherian C; Hou ZJ; Matherly LH; Gangjee A, Synthesis and Biological Activity of a Novel Series of 6-Substituted Thieno[2,3-d]pyrimidine Antifolate Inhibitors of Purine Biosynthesis with Selectivity for High Affinity Folate Receptors over the Reduced Folate Carrier and Proton-Coupled Folate Transporter for Cellular Entry. Journal of medicinal chemistry 2009, 52 (9), 2940–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dekhne AS; Shah K; Ducker GS; Katinas JM; Wong-Roushar J; Nayeen MJ; Doshi A; Ning C; Bao X; Fruhauf J; Liu J; Wallace-Povirk A; O’Connor C; Dzinic SH; White K; Kushner J; Kim S; Huttemann M; Polin L; Rabinowitz JD; Li J; Hou Z; Dann CE 3rd; Gangjee A; Matherly LH, Novel Pyrrolo[3,2- d]pyrimidine Compounds Target Mitochondrial and Cytosolic One-carbon Metabolism with Broad-spectrum Antitumor Efficacy. Mol Cancer Ther 2019, 18 (10), 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Flintoff WF; Nagainis CR, Transport of methotrexate in Chinese hamster ovary cells: a mutant defective in methotrexate uptake and cell binding. Arch Biochem Biophys 1983, 223 (2), 433–40. [DOI] [PubMed] [Google Scholar]
- 54.Golani LK; George C; Zhao S; Raghavan S; Orr S; Wallace A; Wilson MR; Hou Z; Matherly LH; Gangjee A, Structure-activity profiles of novel 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolates with modified amino acids for cellular uptake by folate receptors alpha and beta and the proton-coupled folate transporter. Journal of medicinal chemistry 2014, 57, 8152–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.