

Summary
Parvalbumin (PV) interneuron dysfunction is associated with various brain disorders, including Alzheimer disease (AD). Here, we asked whether early PV neuron hyperexcitability primes the hippocampus for amyloid beta-induced functional impairment. We show that prolonged chemogenetic activation of PV neurons induces long-term hyperexcitability of these cells, disrupts synaptic transmission, and causes spatial memory deficits on the short-term. On the long-term, pyramidal cells also become hyperexcitable, and synaptic transmission and spatial memory are restored. However, under these conditions of increased excitability of both PV and pyramidal cells, a single low-dose injection of amyloid beta directly into the hippocampus significantly impairs PV neuron function, increases pyramidal neuron excitability, and reduces synaptic transmission, resulting in significant spatial memory deficits. Taken together, our data show that an initial hyperexcitable state of PV neurons renders hippocampal function vulnerable to amyloid beta and may contribute to an increased risk for developing AD.
Subject Areas: Neuroscience, Systems Neuroscience, Cellular Neuroscience
Graphical Abstract
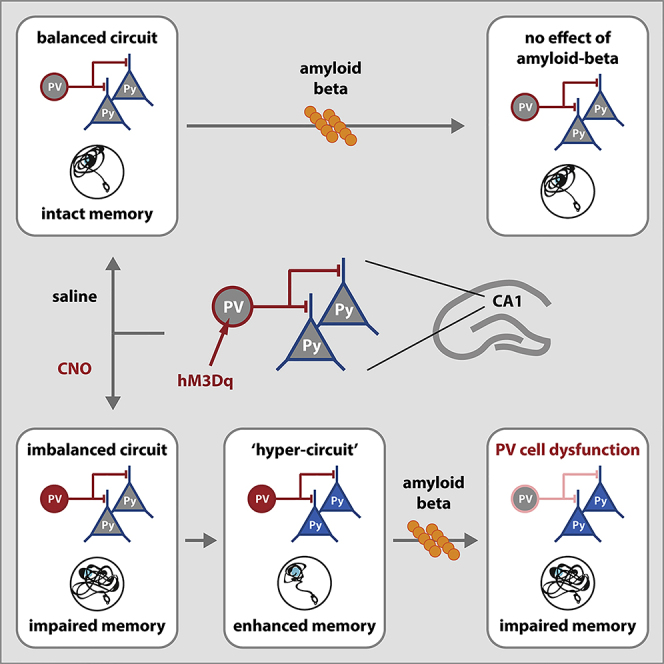
Highlights
-
•
Hyperexcitable hippocampal PV neurons impair spatial memory
-
•
When excitability of pyramidal neurons also increases, spatial memory is restored
-
•
This overall network hyperstate is particularly sensitive to amyloid-beta toxicity
-
•
PV neuron hyperexcitability increases risk for Alzheimer disease
Neuroscience; Systems Neuroscience; Cellular Neuroscience
Introduction
Parvalbumin (PV)-positive interneurons, a major type of inhibitory neurons in the brain, are characterized by both their short action potential duration and their ability to fire at high frequencies (Hu et al., 2014, 2018). PV interneurons have multiple dendrites receiving inputs from diverse afferent pathways (Gulyas et al., 1999; Hioki et al., 2013), as well as numerous perisomatic boutons onto excitatory neurons (Bezaire and Soltesz, 2013; Cornford et al., 2019), together resulting in an integrated feedforward and feedback inhibitory control of both local circuitry and distant neuronal networks (Couey et al., 2013; Espinoza et al., 2018; Pouille and Scanziani, 2001; Sohal et al., 2009; Tukker et al., 2007). Through their role in generating network oscillations (Bartos et al., 2002; Cardin et al., 2009; Gan et al., 2017; Tukker et al., 2007) and their ability to undergo plastic changes upon learning (Donato et al., 2013, 2015), PV interneurons are of crucial importance in memory processes, e.g., in spatial memory consolidation in the hippocampus (Donato et al., 2013; Ognjanovski et al., 2017; Xia et al., 2017). Inhibiting hippocampal PV interneurons during consolidation was found to disrupt context fear memory in mice (Ognjanovski et al., 2017; Xia et al., 2017). Conversely, inhibition of hippocampal PV neurons during Morris water maze (MWM) training was shown to improve spatial memory (Donato et al., 2013). It is also suggested that PV interneurons regulate the precise timing of action potential initiation in pyramidal neuron ensembles encoding hippocampal memories (Agetsuma et al., 2018; Royer et al., 2012). In addition, various studies have proposed that an optimal balance between excitation and inhibition in hippocampal circuits is crucial for successful memory recall (McKay et al., 2013; Oh and Disterhoft, 2015; Ruediger et al., 2011; Stefanelli et al., 2016; Yi et al., 2014). Given the important role of PV neurons in memory formation and recall in hippocampal circuits, it is not surprising that PV cell dysfunction has been linked to several brain diseases that involve memory deficits (Hijazi et al., 2019; Nguyen et al., 2014; Riga et al., 2017).
In Alzheimer disease (AD), a neurodegenerative disease that accounts for the most common cause of dementia (Lane et al., 2018), PV interneuron dysfunction has been recently highlighted (Hijazi et al., 2019; Petrache et al., 2019; Verret et al., 2012). Studies aimed at restoring PV neuron function were successful at reinstating network oscillations and network synchrony and improved cognitive functions in several mouse models of AD (Etter et al., 2019; Hijazi et al., 2019; Iaccarino et al., 2016; Martinez-Losa et al., 2018). PV interneuron impairment and reduced inhibitory transmission are proposed to underlie changes in the excitability of principal networks in AD (Busche et al., 2015; Palop and Mucke, 2010) and may develop over time, including stages of both hyper- (Hijazi et al., 2019; Hollnagel et al., 2019; Kiss et al., 2016; Palop and Mucke, 2010) and hypoexcitability (Martinez-Losa et al., 2018; Petrache et al., 2019; Verret et al., 2012). We previously demonstrated that hippocampal PV interneurons are initially hyperexcitable in a mouse model of AD and that preventing their hyperexcitability early in the disease process is sufficient to rescue spatial memory deficits, avert the loss of inhibitory synaptic transmission, and prevent pyramidal neuron hyperexcitability at later disease stages (Hijazi et al., 2019).
Several studies showed that neuronal hyperexcitability can be caused by amyloid beta (Ab), the peptide responsible for pathological protein aggregation in AD, suggesting that it is a consequence of the disease process rather than a causal factor in the disease. For instance, reducing Ab levels restored neuronal activity and circuit function as well as memory performance, whereas the addition of Ab to brain slices in vitro or to brains in vivo caused significant neuronal hyperactivation (Busche et al., 2015; Hijazi et al., 2019; Keskin et al., 2017; Minkeviciene et al., 2009; Zott et al., 2019). What has not been tested to date is whether neuronal hyperexcitability also increases the risk to develop AD and whether alterations in the excitability of the brain's circuits along with an age-dependent increase in Ab levels could predispose for developing AD. In this study, we tested whether PV hyperexcitability alone could lead to memory deficits and hippocampal circuit dysfunction and importantly, whether such alterations in hippocampal microcircuits predispose for Ab-induced toxicity. Our data show that prolonged chemogenetic activation of PV interneurons in the CA1 of the hippocampus resulted in PV neuron hyperexcitability, memory impairments, and alterations in synaptic transmission similar to what is observed in AD mouse models. On the long term, however, in the absence of Ab, pyramidal cells also became hyperexcitable and synaptic transmission and spatial memory were restored. Importantly, this long-term hyperstate of both PV and pyramidal neurons showed extreme sensitivity to a single low-dose infusion of Ab oligomers, which only under these conditions was sufficient to impair hippocampal spatial memory and network function. Taken together, our data suggest that early hyperexcitable PV neurons can prime neuronal network vulnerability and generate a predisposed state for eliciting AD-like neuronal and behavioral phenotypes.
Results
Increased Hippocampal PV Neuron Excitability Induces Spatial Memory Deficits
To selectively increase hippocampal PV neuron activity in vivo in PV-Cre mice, the activating hM3dq receptor was bilaterally expressed in the CA1 area of the hippocampus using Cre-dependent AAV hSyn-DIO-hM3Dq-mCherry (Figures 1A and 1B). On average, 86% of the PV interneurons in the CA1 expressed hM3Dq-mCherry, and 88% mCherry-positive cells expressed PV (Figure 1C). In hippocampal slice preparations of these animals, bath application of clozapine-N-oxide (CNO) at 10 μM resulted in a significant depolarization of mCherry-positive cells and an increase in the number of action potentials at rheobase (Figures 1D–1G), confirming the functionality of the hM3Dq receptor in increasing PV neuron excitability. To validate the effect of CNO in vivo, mice expressing hM3Dq-mCherry in hippocampal PV neurons were injected intraperitoneally (i.p.) with either CNO (2mg/kg) or saline in their home cage and sacrificed 90 min later to quantify expression of cFos, a marker for cellular activation (Flavell and Greenberg, 2008), in the targeted cells. Mice that received CNO injections showed a significant increase in cFos-positive PV neurons compared with saline-injected mice (Figures 1H and 1I).
Figure 1.

Chemogenetic Activation of PV Neurons during MWM Training Induces a Memory Deficit in the Probe Trial
(A) Representative overview (top) and zoom (bottom) images of PV immunostaining (green) and mCherry expression (red) in the CA1 region of the hippocampus of PV-Cre mice injected with AAV hSyn-DIO-hM3Dq-mCherry (blue: DAPI).
(B) Overlay of mCherry labeling and PV staining indicating that mCherry expression is restricted to PV neurons in the CA1 region of the hippocampus. Scale bars: 200 μm (top); 70 μm (bottom).
(C) Bar graphs showing the percentage of PV-positive cells expressing mCherry and of the fraction of mCherry-positive cells expressing PV (n = 4 mice).
(D) Animals were injected with hM3Dq virus and sacrificed 4 weeks later for either slice electrophysiology or immunohistochemistry. Brains were acquired for cFos analysis 90 min following injection of saline or CNO (1mg/kg of body weight, i.p.).
(E) Representative voltage trace of a 10 μM CNO-induced depolarization in an hM3Dq-positive neuron.
(F) Representative voltage responses to 1 s hyperpolarizing or depolarizing current steps from an hM3Dq-expressing PV neuron before and after CNO application indicating an increase in AP firing at rheobase.
(G) Quantification of the increase in AP firing at rheobase before and after CNO (Student's t test: n = 4 cells from 2 mice, p = 0.02).
(H) Example of co-localization of PV+ (magenta), mCherry+ (red), and cFos+ (green) cells in the CA1 region of CNO-injected compared with saline-injected mice. White arrowheads indicate mCherry+/PV+/cFos + cells. Scale bar: 70 μm.
(I) Quantification of percentage cFos+/PV + neurons in the CA1 region of the hippocampus confirms that CNO, but not saline injections, induced cFos immunoreactivity in hM3Dq-expressing PV neurons.
(J) PV-Cre mice were injected with hM3Dq virus or mCherry-control virus and tested in a Morris water maze (MWM) test 4 weeks later. CNO (2 mg/kg) was i.p. injected 30 min prior to each training session.
(K) Spatial learning was assessed measuring the latency to find the hidden platform on four consecutive training days (T1–4). Both PV-Cre mice that expressed hM3Dq and received CNO, as well as mCherry-expressing mice that received CNO injections showed significant learning during training sessions (training two-way repeated measures ANOVA: n = 10 mice per group, F3,54 = 60.62, p < 0.0001).
(L) During the 1-min probe trial, CNO-treated PV-Cre mice expressing hM3Dq spent significantly less time in the target quadrant (TQ) compared with CNO-treated PV-Cre mice expressing mCherry (two-way ANOVA: n = 10 mice per group, F3,68 = 3.16, p = 0.03; post-hoc LSD test: ∗p = 0.012). Compared with chance level (dashed line), CNO-treated PV-Cre mice expressing mCherry spent significantly more time in the target quadrant, whereas CNO-treated PV-Cre expressing hM3Dq did not (Student's t test: p < 0.01). TQ: target quadrant, BL: bottom left quadrant, TR: top right quadrant, and BR: bottom right quadrant. Values are mean ± SEM.
Next, we confirmed that increasing PV neuron excitability during MWM training impairs spatial memory performance as reported previously (Donato et al., 2013). To that end, PV-Cre mice were injected with either hSyn-DIO-hM3Dq-mCherry- or the control hSyn-DIO-mCherry-expressing virus in the hippocampal CA1 region. Thirty minutes prior to each training session, mice received CNO (2 mg/kg) (Figure 1J). There was no significant difference between groups in the latency to find the platform during the four training days (Figure 1K); however during the probe test, hM3Dq-expressing mice that had received CNO spent significantly less time in the target quadrant compared with control mCherry-expressing mice and relative to chance level (Figure 1L). These findings demonstrate that increasing PV interneuron activity during learning impairs hippocampal spatial memory.
Prolonged Activation of Hippocampal PV Neurons Induces Spatial Memory Deficits, PV Hyperexcitability, and Network Imbalance
Next, we aimed to test whether prolonged chemogenetic activation of PV neurons leads to hippocampal circuit alterations and memory deficits in the absence of acute effects of CNO. To that end, mice expressing hM3Dq virus in hippocampal PV interneurons received daily injections of CNO (1 mg/kg) or saline for a period of 3 weeks and were subjected to an MWM test 48 h after the last injection (Figure 2A). CNO- and saline-injected mice showed a similar decrease in the latency to find the platform with training (Figure 2B). However, in the probe trial, CNO-injected mice displayed a clear spatial memory deficit and explored the target quadrant at chance level, significantly less than saline-injected control mice (Figure 2C).
Figure 2.

Prolonged Activation of Hippocampal PV Interneurons Causes Spatial Memory Deficits, PV Neuron Hyperexcitability, and Network Imbalance on the Short Term
(A) PV-Cre mice were injected with hM3Dq virus at 8–10 weeks of age. After 4 weeks, CNO (1 mg/kg) or saline were i.p. injected daily for a period of 3 weeks. An MWM test was performed 48 h after discontinuation of CNO injections. Electrophysiological recordings were performed following the MWM test.
(B) Spatial learning was assessed measuring the latency to find the hidden platform on four consecutive training days (T1–4). Both PV-Cre mice that expressed hM3Dq and had received CNO injections or saline injections showed significant learning during training (training two-way repeated measures ANOVA: n = 11/12 mice per group, F3,60 = 40.50, p < 0.0001).
(C) During the 1-min probe trial, CNO-injected PV-Cre mice expressing hM3Dq spent significantly less time in the target quadrant (TQ) compared with PV-Cre mice receiving control saline injections (two-way ANOVA: n = 11/12 mice per group, F3,84 = 4.12, p = 0.009; post-hoc LSD test: ∗∗p = 0.002). Compared with chance level (dashed line), saline-injected PV-Cre mice spent significantly more time in the target quadrant, whereas CNO-injected PV-Cre mice did not (Student's t test: p < 0.01).
(D) PV interneuron resting membrane potential was not different between CNO-injected or saline-injected PV-Cre mice expressing hM3Dq (Student's t test: n = 18/24 cells from 5 mice per group, p = 0.514).
(E) PV interneuron input resistance was significantly increased in CNO-injected compared with saline-injected PV-Cre mice expressing hM3Dq (Student's t test: n = 18/24 cells from 5 mice per group, ∗p = 0.018).
(F) Voltage responses to 1 s hyperpolarizing or depolarizing current steps from a PV interneuron in saline-injected PV-Cre (gray) and CNO-injected PV-Cre (blue) mice.
(G) Average action potential (AP) frequency in response to 0–250 pA depolarizing current steps illustrating a significant increase in PV interneuron excitability in CNO-injected PV-Cre mice compared with saline-injected PV-Cre mice (group x current two-way repeated measures ANOVA: n = 18/24 cells from 5 mice per group, F1,30 = 7.13, p = 0.012).
(H and I) Pyramidal neuron resting membrane potential (H) and input resistance (I) were unaltered in CNO-injected PV-Cre mice compared with saline-injected PV-Cre controls (Student's t test: n = 29/25 cells from 5 mice per group, p = 0.437 and p = 0.534).
(J) Voltage responses to 1 s hyperpolarizing or depolarizing current steps from a pyramidal neuron in a saline-injected PV-Cre mouse (gray) or a CNO-injected PV-Cre mouse (blue).
(K) AP frequency in response to 0–250 pA depolarizing current steps illustrating no significant change in pyramidal neuron excitability in CNO-injected PV-Cre mice compared with saline-injected PV-Cre controls (group x current two-way repeated measures ANOVA: n = 29/25 cells from 5 mice per group, F1,38 = 0.02, p = 0. 882).
(L) Example traces of spontaneous inhibitory postsynaptic currents (sIPSC) recorded from hippocampal pyramidal neurons in saline-injected PV-Cre mouse (gray) or a CNO-injected PV-Cre mouse (blue).
(M) Decreased sIPSC frequency in CNO-injected PV-Cre mice compared with saline-injected controls (Mann-Whitney test: n = 14/12 cells from 4 mice per group, ∗∗∗p = 0.000).
(N) No alterations were observed in the amplitudes of sIPSCs (Mann-Whitney test: n = 14/12 cells from 4 mice per group, p = 0.526).
(O) Example traces of spontaneous excitatory postsynaptic currents (sEPSC) recorded from hippocampal pyramidal neurons in saline-injected PV-Cre mouse (gray) or a CNO-injected PV-Cre mouse (blue).
(P and Q) No significant alterations were observed in the frequency (P) or in the amplitude (Q) of sEPSCs in CNO-injected PV-Cre mice compared with saline-injected PV-Cre controls (Mann-Whitney test: n = 14/12 cells from 4 mice per group, p = 0.595 and p = 0.347). Values are mean ± SEM.
We then asked whether PV neuron excitability had changed due to prolonged chemogenetic activation. Intrinsic properties of PV interneurons were assessed using whole-cell patch clamp recording from mCherry-expressing cells. An increase in the excitability of PV interneurons in the CNO-injected group compared with the saline group was confirmed (Figures 2D–2G; Table S1). Specifically, PV interneurons of CNO-injected mice had no alterations in resting membrane potential (Figure 2D) but did show a significantly larger input resistance compared with saline-injected mice (Figure 2E). Importantly, PV neurons from CNO-injected mice also presented a significant decrease in rheobase, a significant increase in action potential firing frequency with increasing current injections, and a significantly smaller action potential half-width (Figures 2F and 2G; Table S1), indicating hyperexcitability. Interestingly, no changes in the excitability of pyramidal neurons was observed at this time point (Figures 2H–2K, Table S1).
To explore how hyperexcitable PV neurons affect synaptic transmission in the hippocampus, we recorded spontaneous inhibitory postsynaptic currents (sIPSCs) and excitatory postsynaptic currents (sEPSCs) from hippocampal pyramidal neurons after discontinuation of CNO treatment. Surprisingly, a significant decrease was observed in the frequency of sIPSCs received by CA1 pyramidal neurons in CNO-injected mice compared with saline controls (Figures 2L and 2M), whereas sIPSC amplitudes were not affected (Figure 2N), suggesting a decrease in the number of inhibitory synaptic inputs. No differences were found in the frequency or amplitudes of sEPSCs recorded at this time point (Figures 2O–2Q). Taken together, these data show that prolonged activation of hippocampal PV neurons induces spatial memory deficits, accompanied with hyperexcitability of PV neurons and a decrease in inhibitory input in the hippocampus.
Spatial Memory Deficits after Prolonged Activation of Hippocampal PV Neurons Are Transient
Next, we examined whether memory deficits caused by prolonged PV neuron activation are long-lasting. Accordingly, we tested mice 8 weeks after the last CNO or saline injections (Figure 3A). Both CNO- and saline-injected mice showed no learning deficits during the MWM training (Figure 3B), and in the probe test both groups spent significantly more time in the target quadrant compared with chance level (Figure 3C), indicating intact spatial memory. Interestingly, CNO-treated mice showed a significantly higher preference for the target quadrant than saline-injected mice, suggesting that, in contrast to an earlier impairment in spatial memory shortly after CNO treatment, prolonged activation of PV neurons results in an improvement of spatial memory on the long term.
Figure 3.

Prolonged Chemogenetic Activation of Hippocampal PV Interneurons Induces a Hyperexcitable Hippocampal Network but Does Not Impair Spatial Memory on the Long Term
(A) PV-Cre mice were injected with hM3Dq virus at 8–10 weeks of age. After 4 weeks, CNO (1 mg/kg) or saline were i.p. injected daily for a period of 3 weeks. Behavioral testing and electrophysiological recordings were performed 8 weeks after discontinuation of CNO injections.
(B) Spatial learning was assessed measuring the latency to find the hidden platform on four consecutive training days (T1–4). PV-Cre mice that both expressed hM3Dq and had received CNO injections or saline injections showed significant learning during training (training two-way repeated measures ANOVA: n = 10 mice per group, F2,27 = 43.65, p < 0.001).
(C) During the one-minute probe trial, CNO-injected PV-Cre mice expressing hM3Dq spent significantly more time in the target quadrant (TQ) compared with PV-Cre mice receiving control saline injections (two-way ANOVA: n = 10 mice per group, F3,72 = 4.42, p = 0.006, post-hoc LSD test: ∗∗p = 0.009). Compared with chance level (dashed line), both saline-injected PV-Cre mice and CNO-injected PV-Cre mice spent significantly more time in the target quadrant (Student's t test: p < 0.01 for saline and p < 0.001 for CNO).
(D) PV interneuron resting membrane potential was not different between CNO-injected or saline-injected PV-Cre mice expressing hM3Dq (Student's t test: n = 14/20 cells from 4 mice per group, p = 0.776).
(E) PV interneuron input resistance was significantly increased in CNO-injected compared with saline-injected PV-Cre mice expressing hM3Dq (Student's t test: n = 14/20 cells from 4 mice per group, ∗∗∗p = 0.000).
(F) Voltage responses to 1 s hyperpolarizing or depolarizing current steps from a PV interneuron in saline-injected PV-Cre (gray) and CNO-injected PV-Cre (blue) mice.
(G) Average action potential (AP) frequency in response to 0–250 pA depolarizing current steps illustrating a significant increase in PV interneuron excitability in CNO-injected PV-Cre mice compared with saline-injected PV-Cre mice (group x current two-way repeated measures ANOVA: n = 14/20 cells from 4 mice per group, F1,42 = 4.32, ∗p = 0.044).
(H) Pyramidal neuron resting membrane potential was unaltered in CNO-injected PV-Cre mice compared with saline-injected PV-Cre controls (Student's t test: n = 25/24 cells from 5 mice per group, p = 0.123).
(I) Pyramidal neuron input resistance was significantly increased in CNO-injected compared with saline-injected PV-Cre mice expressing hM3Dq (Student's t test: n = 25/24 cells from 5 mice per group, ∗p = 0.031).
(J) Voltage responses to 1 s hyperpolarizing or depolarizing current steps from a pyramidal neuron in a saline-injected PV-Cre mouse (gray) or a CNO-injected PV-Cre mouse (blue).
(K) AP frequency in response to 0–250 pA depolarizing current steps illustrating a significant increase in pyramidal neuron excitability in CNO-injected PV-Cre mice compared with saline-injected PV-Cre controls (group x current two-way repeated measures ANOVA: n = 25/24 cells from 5 mice per group, F1,21 = 7.74, ∗p = 0.011).
(L) Example traces of spontaneous inhibitory postsynaptic currents (sIPSC) recorded from hippocampal pyramidal neurons in saline-injected PV-Cre mouse (gray) or a CNO-injected PV-Cre mouse (blue).
(M and N) No significant alterations were observed in the frequency (M) or in the amplitude (N) of sIPSCs in CNO-injected PV-Cre mice compared with saline-injected PV-Cre controls (Mann-Whitney test: n = 11/14 cells from 4 mice per group, p = 0.727 and p = 0.267).
(O) Example traces of spontaneous excitatory postsynaptic currents (sEPSC) recorded from hippocampal pyramidal neurons in saline-injected PV-Cre mouse (gray) or a CNO-injected PV-Cre mouse (blue).
(P and Q) No significant alterations were observed in the frequency (P) or in the amplitude (Q) of sEPSCs in CNO-injected PV-Cre mice compared with saline-injected PV-Cre controls (Mann-Whitney test: n = 11/14 cells from 4 mice per group, p = 0.195 and p = 0.193). Values are mean ± SEM.
We then investigated whether the observed changes in memory performance over time was paralleled by a rescue of PV neuron excitability and hippocampal synaptic transmission. Surprisingly, PV interneurons were still hyperexcitable in CNO-injected mice 8 weeks after the treatment (Figures 3D–3G, Table S2). There was no difference in the resting membrane potential of PV neurons (Figure 3D), and they showed a significantly larger input resistance compared with saline-injected mice (Figure 3E). Notably, PV neurons from CNO-injected mice presented a significant decrease in rheobase (Table S2) and a significant increase in action potential firing frequency with increasing current injections (Figures 3F and 3G), demonstrating that PV neurons were still hyperexcitable at this time point. Interestingly, we also observed an increase in pyramidal neuron excitability in CNO-injected mice (Figures 3H–3K, Table S2). Resting membrane potential of pyramidal neurons was not changed (Figure 3H), but there was a significant increase in their input resistance in CNO-injected mice compared with saline-injected control mice (Figure 3I). Pyramidal neurons from CNO-injected mice also showed a significant decrease in rheobase, a significant increase in action potential firing frequency with increasing current injections, and a decrease in action potential amplitudes (Figures 3J and 3K; Table S2). Synaptic transmission onto pyramidal neurons was unaltered at this time point (Figures 3L–3Q), as no significant differences in frequency or amplitudes of either sIPSCs (Figures 3L–3N) or sEPSCs (Figures 3O–3Q) were observed. Together, these data suggest that an initial hyperexcitability of PV neurons induces, on the long-term, a new, functional hyperstate marked by an overall increase in excitability of both PV neurons and pyramidal cells, restoration of synaptic transmission, and an improvement of spatial memory.
Increasing Hippocampal PV Neuron Excitability Creates a Vulnerable Network State that Is Sensitive to Amyloid Beta
Our data so far demonstrated that memory deficits due to PV neuron hyperexcitability alone are not permanent, likely because hippocampal pyramidal neurons show a compensatory increase in excitability in an attempt to restore E/I balance. We then tested whether and how Ab oligomers would affect this state of the network. To answer this question, 0.3 μL of a 0.5 nM solution of Ab oligomers (0.15 femtomole Ab), were infused bilaterally in the CA1 region of the hippocampus of CNO-injected mice 1–2 weeks before the MWM test. In control mice that had previously received saline injections instead of CNO, this low concentration of Ab had no effect on spatial memory, PV and pyramidal neuron intrinsic properties, or synaptic transmission in the hippocampus (Figure S1, Table S3). In contrast, this single low dose of Ab in the hippocampus of CNO-injected mice was sufficient to cause a significant spatial memory deficit (Figures 4A–4C). Although there was no apparent difference during training (Figure 4B), Ab-infused mice showed impaired memory compared with PBS-infused control mice in the probe trial (Figure 4C).
Figure 4.

Increasing Hippocampal PV Neuron Excitability Creates a Vulnerable Network State that Is Sensitive to Ab
(A) PV-Cre mice were injected with hM3Dq virus at 8–10 weeks of age. After 4 weeks, CNO (1 mg/kg) or saline were i.p. injected daily for a period of 3 weeks. Bilateral infusion of 0.15 fmole of Ab or PBS control in the CA1 region of the hippocampus of PV-Cre mice was completed 6.5 weeks after discontinuation of CNO injections. An MWM test was performed 1.5 week after Ab infusions. Electrophysiological recordings were performed following the MWM test.
(B) Spatial learning was assessed measuring the latency to find the hidden platform on four consecutive training days (T1–4). Both Ab-infused and PBS-infused PV-Cre mice that expressed hM3Dq and had received CNO injections showed significant learning during training (training two-way repeated measures ANOVA: n = 17/13 mice per group, F3,72 = 47.65, p < 0.000).
(C) During the one-minute probe trial, Ab-infused PV-Cre mice spent significantly less time in the target quadrant (TQ) compared with PBS-infused PV-Cre mice (two-way ANOVA: n = 17/13 mice per group, F3,112 = 48.98, p = 0.000, post-hoc LSD test: ∗∗∗p = 0.000). Compared with chance level (dashed line), PBS-infused PV-Cre mice spent significantly more time in the target quadrant, whereas Ab-infused PV-Cre did not (Student's t test: p < 0.001 for CNO and p = 0.059 for CNO + Ab).
(D) PV interneuron resting membrane potential was significantly increased in Ab-infused PV-Cre mice compared with PBS-infused PV-Cre mice (Student's t test: n = 16/15 cells from 4 mice per group, ∗∗p = 0.004).
(E) There was no difference in PV interneuron input resistance between Ab-infused and PBS-infused PV-Cre mice that expressed hM3Dq and had received CNO injections (Student's t test: n = 16/15 cells from 4 mice per group, p = 0.740).
(F) Voltage responses to 1 s hyperpolarizing or depolarizing current steps from a PV interneuron in PBS-infused (blue) and Ab-infused (red) PV-Cre mice that expressed hM3Dq and had received CNO injections.
(G) Average action potential (AP) frequency in response to 0–250 pA depolarizing current steps illustrating a significant decrease in PV interneuron firing frequency in Ab-infused compared with PBS-infused PV-Cre mice expressing hM3Dq and had received CNO injections (group x current two-way repeated measures ANOVA: n = 16/15 cells from 4 mice per group, F1,30 = 5.76, ∗p = 0.023).
(H) Pyramidal neuron resting membrane potential was unaltered in Ab-infused compared with PBS-infused PV-Cre mice expressing hM3Dq and had received CNO injections (Student's t test: n = 14/15 cells from 4 mice per group, p = 0.300).
(I) Pyramidal neuron input resistance was not changed in Ab-infused compared with PBS-infused PV-Cre mice that expressed hM3Dq and had received CNO injections (Student's t test: n = 14/15 cells from 4 mice per group, p = 0.070).
(J) Voltage responses to 1 s hyperpolarizing or depolarizing current steps from a pyramidal neuron in PBS-infused (blue) and Ab-infused (red) PV-Cre mice that expressed hM3Dq and had received CNO injections.
(K) AP frequency in response to 0–250 pA depolarizing current steps illustrating a significant increase in pyramidal neuron excitability in Ab-infused compared with PBS-infused PV-Cre mice that expressed hM3Dq and had received CNO injections starting at 150 pA (group x current two-way repeated measures ANOVA: n = 14/15 cells from 4 mice per group, F1,30 = 1.25, ∗p = 0.02).
(L) Example traces of spontaneous inhibitory postsynaptic currents (sIPSC) recorded from hippocampal pyramidal neurons in PBS-infused (blue) or Ab-infused (red) PV-Cre mice that expressed hM3Dq and had received CNO injections.
(M and N) No significant alterations were observed in the frequency (M) or in the amplitude (N) of sIPSCs in Ab-infused compared with PBS-infused PV-Cre mice that expressed hM3Dq and had received CNO injections (Mann-Whitney test: n = 14/16 cells from 5 mice per group, p = 0.983 and p = 0.822).
(O) Example traces of spontaneous excitatory postsynaptic currents (sEPSC) recorded from hippocampal pyramidal neurons in PBS-infused (blue) or Ab-infused (red) PV-Cre mice that expressed hM3Dq and had received CNO injections.
(P) Decreased sEPSC frequency in Ab-infused compared with PBS-infused PV-Cre mice that expressed hM3Dq and had received CNO injections (Mann-Whitney test: n = 18/13 cells from 5 mice per group, ∗∗∗p = 0.000).
(Q) No alterations were observed in the amplitudes of sEPSCs (Mann-Whitney test: n = 18/13 cells from 5 mice per group, p = 0.370). Values are mean ± SEM.
We then investigated the effects of Ab infusion in CNO-treated mice on neuronal intrinsic properties and synaptic transmission. The resting membrane potential of PV neurons in mice that received Ab was significantly higher than in the PBS controls (Figure 4D, Table S4). Input resistance was not altered, yet PV neurons from mice that received Ab infusions fired less action potentials with increasing current injections compared with PBS controls (Figures 4E–4G). Pyramidal neurons on the other hand showed a tendency toward increased excitability after Ab infusion (Table S4). Resting membrane potential and input resistance of pyramidal neurons were not altered (Figures 4H and 4I); however, pyramidal neurons in Ab-infused mice showed a significant increase in action potential firing frequency with increasing current injections (Figures 4J and 4K) and a significantly smaller action potential half-width (Table S4) compared with PBS controls. Finally, synaptic transmission onto pyramidal neurons after Ab infusion was measured. There were no differences in frequency or amplitude of recorded sIPSCs (Figures 4L–4N), but a significant decrease in sEPSCs frequency (Figures 4O and 4P), not amplitude (Figure 4Q), was observed. Together, these data suggest that prolonged activation of hippocampal PV neurons induced a hippocampal circuit state that is sensitive to disruption by Ab oligomers. In this state, Ab impairs PV neuron function, increases pyramidal neuron excitability, and disrupts synaptic transmission, resulting in a spatial memory deficit.
Discussion
PV neurons are inhibitory neurons that support synchronous firing of excitatory neurons, generate gamma oscillations, and control basic microcircuit functions as well as complex brain network computations (Bartos et al., 2002; Hu et al., 2014). Yet, due to their high energy-demanding fast-spiking properties (Kann, 2016), PV neurons are also vulnerable to various stressors, and their dysfunction has been associated with several brain diseases (Marin, 2012), including AD (Palop and Mucke, 2016). We previously demonstrated, in an amyloidosis mouse model of AD, that PV neuron dysfunction may be initiated by PV neuron hyperexcitability, and that early memory deficits in these mice are rescued when PV neuron excitability is restored early (Hijazi et al., 2019). In the present study we show that PV neuron hyperexcitability alone can cause memory impairments and that inducing PV hyperexcitability is sufficient to create a vulnerable hippocampal circuit onto which disease-related stressors can act. Specifically, prolonged chemogenetic activation of hippocampal PV neurons can effectively induce PV neuron hyperexcitability, a phenotype that is still consistent at least 8 weeks after the end of the CNO treatment. Moreover, this sustained PV neuron hyperexcitability state comes with an increase in the excitability of pyramidal neurons, resulting in a hyperstate of the hippocampal circuitry that is vulnerable to Ab-induced impairment of both neuronal network function and spatial memory.
A Pharmacogenetic Model of PV Neuron Hyperexcitability
In APP/PS1 mice, initial hyperexcitability of PV neurons at 3–4 months of age is followed by a decrease in PV neuron function and inhibitory transmission at 5–6 months of age (Hijazi et al., 2019; Kiss et al., 2016). A similar decrease in PV neuron function has been observed in several other mouse models of AD (Nava-Mesa et al., 2014; Petrache et al., 2019; Verret et al., 2012). In such genetic models, however, increased Ab levels and neuronal dysfunction develop in parallel, and their separate contributions to disease pathogenesis or mutual interdependencies cannot easily be disentangled. We therefore established a model in which hyperexcitability of PV neurons is induced in the absence of Ab. This allowed us first to answer the question whether an early impairment of inhibitory transmission would affect spatial memory on the long term and second, whether the subsequently induced state of the hippocampal network would be prone to disruptive effects of Ab. Following three weeks of PV neuron activation, mice showed clear spatial memory deficits in the MWM test. Electrophysiological recordings confirmed that PV neurons were hyperexcitable at this time point, whereas pyramidal neuron intrinsic properties were not affected. Surprisingly, inhibitory synaptic inputs onto pyramidal neurons were reduced, as indicated by a significant decrease in sIPSCs frequencies. This may suggest a compensatory mechanism that prevents aberrant overinhibition of pyramidal neurons by hyperexcitable PV neurons. Interestingly, in APP/PS1 mice, PV neuron hyperexcitability was accompanied with an increase in sIPSCs frequency (Hijazi et al., 2019), highlighting a different effect of early, genetically driven exposure to Ab in modulating synaptic compensatory mechanisms. Clearly, the interaction between PV cell hyperexcitability and a normal, age-dependent increase in Ab levels needs further investigation.
Dynamic Adaptations in Hippocampal Network Properties after Inducing PV Hyperexcitability
Interestingly, PV neuron hyperexcitability alone did not result in memory impairment, but memory enhancement 8 weeks after discontinuation of the CNO treatment. When we investigated the underlying cell physiological changes, PV neurons were still hyperexcitable, and not hypoactive as observed in AD mouse models, demonstrating that PV neuron hyperexcitability alone cannot explain spatial memory deficits in AD mice. Interestingly, we found that pyramidal neuron excitability was also increased, which is likely a compensatory activity to restore the balance of the inhibitory and excitatory tone in the hippocampus. This is further confirmed by the fact that we found no differences in excitatory or inhibitory synaptic transmission onto pyramidal neurons at this point in time. This new hyperstate after 8 weeks, showing an overall increase in excitability of both inhibitory and excitatory neurons, may thus have developed to maintain E/I balance. It may also explain why spatial memory is enhanced, as it has been shown previously that increasing the excitability neurons promotes their allocation to memory engrams (Han et al., 2007; Lisman et al., 2018; McKay et al., 2013; Rao-Ruiz et al., 2019). Correlating levels of neuronal activation during recall to actual memory performance would help to elucidate this further. Behavioral recovery from a PV neuron hyperexcitability-induced memory impairment is in itself an interesting observation, and further investigation into the underlying molecular, cellular, and network mechanisms could reveal brain resilience mechanisms that are relevant for the etiology of AD. In vivo recordings of oscillatory network activity throughout such an intervention might additionally help provide an important link between the cellular changes reported in this study and the behavioral output.
PV Neuron Hyperexcitability Creates a Vulnerable Network State that Is Sensitive to Amyloid Beta
We next hypothesized that, although the homeostatic adaptations in response to PV cell hyperexcitability may be beneficial under healthy conditions in order to compensate for neuron-specific alterations, they may increase sensitivity to disease-associated cellular stressors. In AD, this stressor is likely the gradual increase in soluble Ab levels. Indeed, our data show that a low concentration of Ab in vivo is able to significantly impair memory performance in mice that had received CNO injections to increase PV neuron excitability, while having no effect in saline-injected mice. On a cellular level, Ab affected the resting membrane potential of PV neurons in CNO-injected and, not in saline-injected, mice, an aberrant phenotype that is also observed in various mouse models of AD (Hijazi et al., 2019; Verret et al., 2012). Ab also reduced the firing frequency of previously activated PV neurons, a characteristic of crucial importance for healthy circuit functioning (Bartos et al., 2002; Cardin et al., 2009; Tukker et al., 2007; Vida et al., 2006). Moreover, Ab increased pyramidal neuron excitability and caused a major reduction in excitatory synaptic transmission in the hippocampus, similar to what is reported in AD mouse models (Hijazi et al., 2019; Nava-Mesa et al., 2014; Styr and Slutsky, 2018). Thus, soluble Ab, at low concentrations, causes the dysfunction of vulnerable PV neurons and of associated vulnerable hippocampal circuitry.
Amyloid Load and PV Cell Hyperexcitability as Dual Risk Factors in AD
Since the introduction of the amyloid cascade hypothesis, it is becoming increasingly clear that risk factors other than Ab need to be considered to explain the complex etiology of AD (e.g., Herrup, 2015). From previous studies it is known that Ab can cause PV neuron hyperexcitability (Hijazi et al., 2019) but also that PV neurons contribute significantly to Ab production and Ab load (Iaccarino et al., 2016; Rice et al., 2020). This suggests a model in which increasing Ab concentrations increase PV neuron excitability, whereas hyperexcitable PV neurons in turn increase Ab release and Ab load. Both Ab load and PV hyperexcitability can thus be considered risk factors in AD, and together they can trigger a detrimental cycle of pathological protein aggregation, neuronal circuit dysfunction, and cognitive decline. It has also been shown, however, that Ab can induce pyramidal neuron hyperexcitability (Busche et al., 2012; Zott et al., 2019) and that other interneurons also become dysfunctional in AD (Schmid et al., 2016). The question thus remains whether Ab toxicity shows any bias toward specific neuronal types and whether the data we obtained are the result of direct or indirect effects of Ab on PV neurons. Interestingly, previous studies have proposed that PV neurons are resistant to neurodegeneration, as the number of PV neurons was found unchanged in postmortem brain tissue of AD patients (Ferrer et al., 1991; Hof et al., 1991; Morawski et al., 2012). Although other studies have challenged these findings (Brady and Mufson, 1997; Sampson et al., 1997; Sanchez-Mejias et al., 2020; Takahashi et al., 2010; Verret et al., 2012), our data do not necessarily contradict either of these views and merely show that PV neurons become functionally impaired in early AD stages (Hijazi et al., 2019), irrespective of how their viability changes on the long term. More research investigating different types of neurons and their specific vulnerability to Ab toxicity during disease progression is needed to better understand the complexity of circuit dysfunction in AD. However, given the fact that PV interneurons seem to be affected in multiple brain disorders, they remain an attractive target for identifying novel disease mechanisms and interventions.
Limitations of the Study
To test whether hyperexcitable PV interneurons are a predisposing risk factor for AD, we created a model where PV cells were made hyperexcitable in the absence of Ab and then tested the impact of Ab injection on the hippocampal network. It is important to note that Ab injections are artificial and do not mimic AD pathology in vivo. Ideally, these experiments would be repeated in a mouse model of AD where PV neuron hyperexcitability can be induced before Ab levels start to increase. However, at this moment the artificial separation in time of PV neuron hyperexcitability and the presence of Ab allowed us to clearly demonstrate that hippocampal network vulnerability alone can increase risk for Ab toxicity. Also, although our study centered on the effects of Ab, AD is also characterized by the accumulation of hyperphosphorylated Tau protein. In a recent study, it was shown that Tau suppresses neuronal activity and that this tau-mediated suppression dominates over Ab-induced hyperactivity (Busche et al., 2019). The combined and time-dependent effects of Ab and Tau in relation to PV neuron excitability clearly need further investigation in the future. Finally, although our study focused on CA1 networks, the effects of changes in PV neuron excitability in the rest of the hippocampus remain unknown. Various studies have highlighted the importance of CA2 and CA3 PV neuron plasticity in learning and memory (Dominguez et al., 2019; Donato et al., 2013; Nasrallah et al., 2019), and these specific networks are also impaired in AD (Blanken et al., 2017; Padurariu et al., 2012; Suthana et al., 2010; Yassa et al., 2010). It would therefore be interesting for future studies to explore how alterations in PV neuron activity in different subregions of the hippocampus affect learning and memory and how these circuits are altered in response to hyperexcitable PV neurons. Despite these limitations, our study clearly demonstrates that PV neuron hyperexcitability increases the vulnerability of hippocampal circuitry to Ab-induced toxicity and impairs spatial memory accordingly.
Resource Availability
Lead Contact
Ronald E. van Kesteren (ronald.van.kesteren@vu.nl).
Materials Availability
No new unique materials or reagents were generated in this study.
Data and Code Availability
All electrophysiological data are available in the Supplemental Information. No new unique code or software were generated in this study.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
S.H. received funding from the EU-FP7-PEOPLE program (CognitionNet; grant 607508). R.E.v.K. received funding from the Dutch Alzheimer Association (Alzheimer Nederland; grant WE.03-2017-03) and from the Netherlands Organisation for Health Research and Development (ZonMw; grant 91218018).
Author Contributions
S.H., R.E.v.K., and A.B.S. designed the experiments; T.S.H. assisted in electrophysiological experiments and data analysis. H.D.M. supervised electrophysiological experiments. R.v.L assisted with stereotaxic surgeries and perfusions. S.H. performed all behavioral and electrophysiological experiments; S.H., R.E.v.K., H.D.M., and A.B.S. wrote the manuscript.
Declaration of Interests
The authors declare no conflict of interest.
Published: July 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101271.
Supplemental Information
References
- Agetsuma M., Hamm J.P., Tao K., Fujisawa S., Yuste R. Parvalbumin-positive interneurons regulate neuronal ensembles in visual cortex. Cereb. Cortex. 2018;28:1831–1845. doi: 10.1093/cercor/bhx169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos M., Vida I., Frotscher M., Meyer A., Monyer H., Geiger J.R., Jonas P. Fast synaptic inhibition promotes synchronized gamma oscillations in hippocampal interneuron networks. Proc. Natl. Acad. Sci. U S A. 2002;99:13222–13227. doi: 10.1073/pnas.192233099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezaire M.J., Soltesz I. Quantitative assessment of CA1 local circuits: knowledge base for interneuron-pyramidal cell connectivity. Hippocampus. 2013;23:751–785. doi: 10.1002/hipo.22141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanken A.E., Hurtz S., Zarow C., Biado K., Honarpisheh H., Somme J., Brook J., Tung S., Kraft E., Lo D. Associations between hippocampal morphometry and neuropathologic markers of Alzheimer's disease using 7 T MRI. Neuroimage Clin. 2017;15:56–61. doi: 10.1016/j.nicl.2017.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady D.R., Mufson E.J. Parvalbumin-immunoreactive neurons in the hippocampal formation of Alzheimer's diseased brain. Neuroscience. 1997;80:1113–1125. doi: 10.1016/s0306-4522(97)00068-7. [DOI] [PubMed] [Google Scholar]
- Busche M.A., Chen X., Henning H.A., Reichwald J., Staufenbiel M., Sakmann B., Konnerth A. Critical role of soluble amyloid-beta for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U S A. 2012;109:8740–8745. doi: 10.1073/pnas.1206171109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche M.A., Kekus M., Adelsberger H., Noda T., Forstl H., Nelken I., Konnerth A. Rescue of long-range circuit dysfunction in Alzheimer's disease models. Nat. Neurosci. 2015;18:1623–1630. doi: 10.1038/nn.4137. [DOI] [PubMed] [Google Scholar]
- Busche M.A., Wegmann S., Dujardin S., Commins C., Schiantarelli J., Klickstein N., Kamath T.V., Carlson G.A., Nelken I., Hyman B.T. Tau impairs neural circuits, dominating amyloid-beta effects, in Alzheimer models in vivo. Nat. Neurosci. 2019;22:57–64. doi: 10.1038/s41593-018-0289-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin J.A., Carlen M., Meletis K., Knoblich U., Zhang F., Deisseroth K., Tsai L.H., Moore C.I. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornford J.H., Mercier M.S., Leite M., Magloire V., Hausser M., Kullmann D.M. Dendritic NMDA receptors in parvalbumin neurons enable strong and stable neuronal assemblies. Elife. 2019;8:e49872. doi: 10.7554/eLife.49872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couey J.J., Witoelar A., Zhang S.J., Zheng K., Ye J., Dunn B., Czajkowski R., Moser M.B., Moser E.I., Roudi Y. Recurrent inhibitory circuitry as a mechanism for grid formation. Nat. Neurosci. 2013;16:318–324. doi: 10.1038/nn.3310. [DOI] [PubMed] [Google Scholar]
- Dominguez S., Rey C.C., Therreau L., Fanton A., Massotte D., Verret L., Piskorowski R.A., Chevaleyre V. Maturation of PNN and ErbB4 signaling in area CA2 during adolescence underlies the emergence of PV interneuron plasticity and social memory. Cell Rep. 2019;29:1099–1112.e4. doi: 10.1016/j.celrep.2019.09.044. [DOI] [PubMed] [Google Scholar]
- Donato F., Rompani S.B., Caroni P. Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature. 2013;504:272–276. doi: 10.1038/nature12866. [DOI] [PubMed] [Google Scholar]
- Donato F., Chowdhury A., Lahr M., Caroni P. Early- and late-born parvalbumin basket cell subpopulations exhibiting distinct regulation and roles in learning. Neuron. 2015;85:770–786. doi: 10.1016/j.neuron.2015.01.011. [DOI] [PubMed] [Google Scholar]
- Espinoza C., Guzman S.J., Zhang X., Jonas P. Parvalbumin(+) interneurons obey unique connectivity rules and establish a powerful lateral-inhibition microcircuit in dentate gyrus. Nat. Commun. 2018;9:4605. doi: 10.1038/s41467-018-06899-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etter G., van der Veldt S., Manseau F., Zarrinkoub I., Trillaud-Doppia E., Williams S. Optogenetic gamma stimulation rescues memory impairments in an Alzheimer's disease mouse model. Nat. Commun. 2019;10:5322. doi: 10.1038/s41467-019-13260-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I., Soriano E., Tunon T., Fonseca M., Guionnet N. Parvalbumin immunoreactive neurons in normal human temporal neocortex and in patients with Alzheimer's disease. J. Neurol. Sci. 1991;106:135–141. doi: 10.1016/0022-510x(91)90250-b. [DOI] [PubMed] [Google Scholar]
- Flavell S.W., Greenberg M.E. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu. Rev. Neurosci. 2008;31:563–590. doi: 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan J., Weng S.M., Pernia-Andrade A.J., Csicsvari J., Jonas P. Phase-locked inhibition, but not excitation, underlies hippocampal ripple oscillations in awake mice in vivo. Neuron. 2017;93:308–314. doi: 10.1016/j.neuron.2016.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas A.I., Megias M., Emri Z., Freund T.F. Total number and ratio of excitatory and inhibitory synapses converging onto single interneurons of different types in the CA1 area of the rat hippocampus. J. Neurosci. 1999;19:10082–10097. doi: 10.1523/JNEUROSCI.19-22-10082.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J.H., Kushner S.A., Yiu A.P., Cole C.J., Matynia A., Brown R.A., Neve R.L., Guzowski J.F., Silva A.J., Josselyn S.A. Neuronal competition and selection during memory formation. Science. 2007;316:457–460. doi: 10.1126/science.1139438. [DOI] [PubMed] [Google Scholar]
- Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015;18:794–799. doi: 10.1038/nn.4017. [DOI] [PubMed] [Google Scholar]
- Hijazi S., Heistek T.S., Scheltens P., Neumann U., Shimshek D.R., Mansvelder H.D., Smit A.B., van Kesteren R.E. Early restoration of parvalbumin interneuron activity prevents memory loss and network hyperexcitability in a mouse model of Alzheimer’s disease. Mol. Psychiatry. In press. 2019 doi: 10.1038/s41380-019-0483-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hioki H., Okamoto S., Konno M., Kameda H., Sohn J., Kuramoto E., Fujiyama F., Kaneko T. Cell type-specific inhibitory inputs to dendritic and somatic compartments of parvalbumin-expressing neocortical interneuron. J. Neurosci. 2013;33:544–555. doi: 10.1523/JNEUROSCI.2255-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof P.R., Cox K., Young W.G., Celio M.R., Rogers J., Morrison J.H. Parvalbumin-immunoreactive neurons in the neocortex are resistant to degeneration in Alzheimer's disease. J. Neuropathol. Exp. Neurol. 1991;50:451–462. doi: 10.1097/00005072-199107000-00006. [DOI] [PubMed] [Google Scholar]
- Hollnagel J.O., Elzoheiry S., Gorgas K., Kins S., Beretta C.A., Kirsch J., Kuhse J., Kann O., Kiss E. Early alterations in hippocampal perisomatic GABAergic synapses and network oscillations in a mouse model of Alzheimer's disease amyloidosis. PLoS One. 2019;14:e0209228. doi: 10.1371/journal.pone.0209228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H., Gan J., Jonas P. Interneurons. Fast-spiking, parvalbumin(+) GABAergic interneurons: from cellular design to microcircuit function. Science. 2014;345:1255263. doi: 10.1126/science.1255263. [DOI] [PubMed] [Google Scholar]
- Hu H., Roth F.C., Vandael D., Jonas P. Complementary tuning of Na(+) and K(+) channel gating underlies fast and energy-efficient action potentials in GABAergic interneuron axons. Neuron. 2018;98:156–165.e6. doi: 10.1016/j.neuron.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino H.F., Singer A.C., Martorell A.J., Rudenko A., Gao F., Gillingham T.Z., Mathys H., Seo J., Kritskiy O., Abdurrob F. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature. 2016;540:230–235. doi: 10.1038/nature20587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann O. The interneuron energy hypothesis: implications for brain disease. Neurobiol. Dis. 2016;90:75–85. doi: 10.1016/j.nbd.2015.08.005. [DOI] [PubMed] [Google Scholar]
- Keskin A.D., Kekus M., Adelsberger H., Neumann U., Shimshek D.R., Song B., Zott B., Peng T., Forstl H., Staufenbiel M. BACE inhibition-dependent repair of Alzheimer's pathophysiology. Proc. Natl. Acad. Sci. U S A. 2017;114:8631–8636. doi: 10.1073/pnas.1708106114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss E., Gorgas K., Schlicksupp A., Gross D., Kins S., Kirsch J., Kuhse J. Biphasic alteration of the inhibitory synapse scaffold protein gephyrin in early and late stages of an Alzheimer disease model. Am. J. Pathol. 2016;186:2279–2291. doi: 10.1016/j.ajpath.2016.05.013. [DOI] [PubMed] [Google Scholar]
- Lane C.A., Hardy J., Schott J.M. Alzheimer's disease. Eur. J. Neurol. 2018;25:59–70. doi: 10.1111/ene.13439. [DOI] [PubMed] [Google Scholar]
- Lisman J., Cooper K., Sehgal M., Silva A.J. Memory formation depends on both synapse-specific modifications of synaptic strength and cell-specific increases in excitability. Nat. Neurosci. 2018;21:309–314. doi: 10.1038/s41593-018-0076-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin O. Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 2012;13:107–120. doi: 10.1038/nrn3155. [DOI] [PubMed] [Google Scholar]
- Martinez-Losa M., Tracy T.E., Ma K., Verret L., Clemente-Perez A., Khan A.S., Cobos I., Ho K., Gan L., Mucke L. Nav1.1-overexpressing interneuron transplants restore brain rhythms and cognition in a mouse model of Alzheimer's disease. Neuron. 2018;98:75–89.e5. doi: 10.1016/j.neuron.2018.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay B.M., Oh M.M., Disterhoft J.F. Learning increases intrinsic excitability of hippocampal interneurons. J. Neurosci. 2013;33:5499–5506. doi: 10.1523/JNEUROSCI.4068-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkeviciene R., Rheims S., Dobszay M.B., Zilberter M., Hartikainen J., Fulop L., Penke B., Zilberter Y., Harkany T., Pitkanen A. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J. Neurosci. 2009;29:3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawski M., Bruckner G., Jager C., Seeger G., Matthews R.T., Arendt T. Involvement of perineuronal and perisynaptic extracellular matrix in Alzheimer's disease neuropathology. Brain Pathol. 2012;22:547–561. doi: 10.1111/j.1750-3639.2011.00557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrallah K., Therreau L., Robert V., Huang A.J.Y., McHugh T.J., Piskorowski R.A., Chevaleyre V. Routing hippocampal information flow through parvalbumin interneuron plasticity in area CA2. Cell Rep. 2019;27:86–98.e3. doi: 10.1016/j.celrep.2019.03.014. [DOI] [PubMed] [Google Scholar]
- Nava-Mesa M.O., Jimenez-Diaz L., Yajeya J., Navarro-Lopez J.D. GABAergic neurotransmission and new strategies of neuromodulation to compensate synaptic dysfunction in early stages of Alzheimer's disease. Front. Cell. Neurosci. 2014;8:167. doi: 10.3389/fncel.2014.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen R., Morrissey M.D., Mahadevan V., Cajanding J.D., Woodin M.A., Yeomans J.S., Takehara-Nishiuchi K., Kim J.C. Parvalbumin and GAD65 interneuron inhibition in the ventral hippocampus induces distinct behavioral deficits relevant to schizophrenia. J. Neurosci. 2014;34:14948–14960. doi: 10.1523/JNEUROSCI.2204-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ognjanovski N., Schaeffer S., Wu J., Mofakham S., Maruyama D., Zochowski M., Aton S.J. Parvalbumin-expressing interneurons coordinate hippocampal network dynamics required for memory consolidation. Nat. Commun. 2017;8:15039. doi: 10.1038/ncomms15039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh M.M., Disterhoft J.F. Increased excitability of both principal neurons and interneurons during associative learning. Neuroscientist. 2015;21:372–384. doi: 10.1177/1073858414537382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padurariu M., Ciobica A., Mavroudis I., Fotiou D., Baloyannis S. Hippocampal neuronal loss in the CA1 and CA3 areas of Alzheimer's disease patients. Psychiatr. Danub. 2012;24:152–158. [PubMed] [Google Scholar]
- Palop J.J., Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat. Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop J.J., Mucke L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2016;17:777–792. doi: 10.1038/nrn.2016.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrache A.L., Rajulawalla A., Shi A., Wetzel A., Saito T., Saido T.C., Harvey K., Ali A.B. Aberrant excitatory-inhibitory synaptic mechanisms in entorhinal cortex microcircuits during the pathogenesis of Alzheimer's disease. Cereb. Cortex. 2019;29:1834–1850. doi: 10.1093/cercor/bhz016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouille F., Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- Rao-Ruiz P., Yu J., Kushner S.A., Josselyn S.A. Neuronal competition: microcircuit mechanisms define the sparsity of the engram. Curr. Opin. Neurobiol. 2019;54:163–170. doi: 10.1016/j.conb.2018.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice H.C., Marcassa G., Chrysidou I., Horre K., Young-Pearse T.L., Muller U.C., Saito T., Saido T.C., Vassar R., de Wit J. Contribution of GABAergic interneurons to amyloid-beta plaque pathology in an APP knock-in mouse model. Mol. Neurodegener. 2020;15:3. doi: 10.1186/s13024-019-0356-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riga D., Kramvis I., Koskinen M.K., van Bokhoven P., van der Harst J.E., Heistek T.S., Jaap Timmerman A., van Nierop P., van der Schors R.C., Pieneman A.W. Hippocampal extracellular matrix alterations contribute to cognitive impairment associated with a chronic depressive-like state in rats. Sci. Transl. Med. 2017;9:eaai8753. doi: 10.1126/scitranslmed.aai8753. [DOI] [PubMed] [Google Scholar]
- Royer S., Zemelman B.V., Losonczy A., Kim J., Chance F., Magee J.C., Buzsaki G. Control of timing, rate and bursts of hippocampal place cells by dendritic and somatic inhibition. Nat. Neurosci. 2012;15:769–775. doi: 10.1038/nn.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruediger S., Vittori C., Bednarek E., Genoud C., Strata P., Sacchetti B., Caroni P. Learning-related feedforward inhibitory connectivity growth required for memory precision. Nature. 2011;473:514–518. doi: 10.1038/nature09946. [DOI] [PubMed] [Google Scholar]
- Sampson V.L., Morrison J.H., Vickers J.C. The cellular basis for the relative resistance of parvalbumin and calretinin immunoreactive neocortical neurons to the pathology of Alzheimer's disease. Exp. Neurol. 1997;145:295–302. doi: 10.1006/exnr.1997.6433. [DOI] [PubMed] [Google Scholar]
- Sanchez-Mejias E., Nunez-Diaz C., Sanchez-Varo R., Gomez-Arboledas A., Garcia-Leon J.A., Fernandez-Valenzuela J.J., Mejias-Ortega M., Trujillo-Estrada L., Baglietto-Vargas D., Moreno-Gonzalez I. Distinct disease-sensitive GABAergic neurons in the perirhinal cortex of Alzheimer's mice and patients. Brain Pathol. 2020;30:345–363. doi: 10.1111/bpa.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid L.C., Mittag M., Poll S., Steffen J., Wagner J., Geis H.R., Schwarz I., Schmidt B., Schwarz M.K., Remy S. Dysfunction of somatostatin-positive interneurons associated with memory deficits in an Alzheimer's disease model. Neuron. 2016;92:114–125. doi: 10.1016/j.neuron.2016.08.034. [DOI] [PubMed] [Google Scholar]
- Sohal V.S., Zhang F., Yizhar O., Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanelli T., Bertollini C., Luscher C., Muller D., Mendez P. Hippocampal somatostatin interneurons control the size of neuronal memory ensembles. Neuron. 2016;89:1074–1085. doi: 10.1016/j.neuron.2016.01.024. [DOI] [PubMed] [Google Scholar]
- Styr B., Slutsky I. Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer's disease. Nat. Neurosci. 2018;21:463–473. doi: 10.1038/s41593-018-0080-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthana N.A., Krupa A., Donix M., Burggren A., Ekstrom A.D., Jones M., Ercoli L.M., Miller K.J., Siddarth P., Small G.W. Reduced hippocampal CA2, CA3, and dentate gyrus activity in asymptomatic people at genetic risk for Alzheimer's disease. Neuroimage. 2010;53:1077–1084. doi: 10.1016/j.neuroimage.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H., Brasnjevic I., Rutten B.P., Van Der Kolk N., Perl D.P., Bouras C., Steinbusch H.W., Schmitz C., Hof P.R., Dickstein D.L. Hippocampal interneuron loss in an APP/PS1 double mutant mouse and in Alzheimer's disease. Brain Struct. Funct. 2010;214:145–160. doi: 10.1007/s00429-010-0242-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukker J.J., Fuentealba P., Hartwich K., Somogyi P., Klausberger T. Cell type-specific tuning of hippocampal interneuron firing during gamma oscillations in vivo. J. Neurosci. 2007;27:8184–8189. doi: 10.1523/JNEUROSCI.1685-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L., Mann E.O., Hang G.B., Barth A.M., Cobos I., Ho K., Devidze N., Masliah E., Kreitzer A.C., Mody I. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012;149:708–721. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vida I., Bartos M., Jonas P. Shunting inhibition improves robustness of gamma oscillations in hippocampal interneuron networks by homogenizing firing rates. Neuron. 2006;49:107–117. doi: 10.1016/j.neuron.2005.11.036. [DOI] [PubMed] [Google Scholar]
- Xia F., Richards B.A., Tran M.M., Josselyn S.A., Takehara-Nishiuchi K., Frankland P.W. Parvalbumin-positive interneurons mediate neocortical-hippocampal interactions that are necessary for memory consolidation. Elife. 2017;6:e27868. doi: 10.7554/eLife.27868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassa M.A., Stark S.M., Bakker A., Albert M.S., Gallagher M., Stark C.E. High-resolution structural and functional MRI of hippocampal CA3 and dentate gyrus in patients with amnestic Mild Cognitive Impairment. Neuroimage. 2010;51:1242–1252. doi: 10.1016/j.neuroimage.2010.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F., Ball J., Stoll K.E., Satpute V.C., Mitchell S.M., Pauli J.L., Holloway B.B., Johnston A.D., Nathanson N.M., Deisseroth K. Direct excitation of parvalbumin-positive interneurons by M1 muscarinic acetylcholine receptors: roles in cellular excitability, inhibitory transmission and cognition. J. Physiol. 2014;592:3463–3494. doi: 10.1113/jphysiol.2014.275453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zott B., Simon M.M., Hong W., Unger F., Chen-Engerer H.J., Frosch M.P., Sakmann B., Walsh D.M., Konnerth A. A vicious cycle of beta amyloid-dependent neuronal hyperactivation. Science. 2019;365:559–565. doi: 10.1126/science.aay0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All electrophysiological data are available in the Supplemental Information. No new unique code or software were generated in this study.