

Abstract
The investigation of the biodistribution profile of a cell-based medicinal product is a pivotal prerequisite to allow a factual benefit-risk assessment within the non-clinical to clinical translation in product development. Here, a qPCR-based method to determine the amount of human DNA in mouse DNA was validated according to the guidelines of the European Medicines Agency and the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. Furthermore, a preclinical worst-case scenario study was performed in which this method was applied to investigate the biodistribution of 2 × 106 intravenously administered, genetically modified, blood outgrowth endothelial cells from hemophilia A patients after 24 h and 7 days. The validation of the qPCR method demonstrated high accuracy, precision, and linearity for the concentration interval of 1:1 × 103 to 1:1 × 106 human to mouse DNA. The application of this method in the biodistribution study resulted in the detection of human genomes in four out of the eight investigated organs after 24 h. After 7 days, no human DNA was detected in the eight organs analyzed. This biodistribution study provides mandatory data on the toxicokinetic safety profile of an actual candidate cell-based medicinal product. The extensive evaluation of the required validation parameters confirms the applicability of the qPCR method for non-clinical biodistribution studies.
Keywords: Cell-based therapy, gene-based therapy, ATMP, preclinical and non-clinical development, method validation, qPCR, biodistribution, human Alu sequences, blood outgrowth endothelial cells
Graphical Abstract
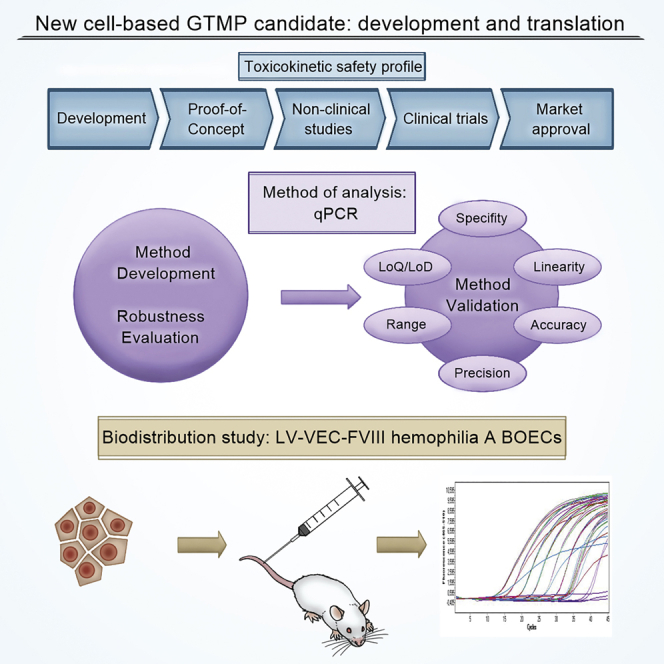
Bittorf and colleagues present the validation of a qPCR-based method to detect human genomic DNA in mouse organs following European and international guidelines. They further introduce a biodistribution study of a medicinal product consisting of lentiviral-transduced BOECs from hemophilic patients to investigate the toxicokinetic safety profile for clinical translation.
Introduction
The mandatory non-clinical study scheme prior to the first administration of a cell-based gene therapy medicinal product (GTMP) to human subjects includes and requires investigation of the biodistribution, comprising mobilization, persistence, and clearance, of the GTMP in a relevant animal model.1,2 Due to the specific characteristics of each different GTMP, the European Medicines Agency (EMA) scientific guidelines recommend selecting the relevant non-clinical studies as well as their design on a case-by-case basis, following a risk-based approach.2,3 Following this approach, a worst-case scenario biodistribution study involving intravenous (i.v.) administration of the GTMP to achieve maximum systemic levels must be taken into account.2 Moreover, for clinical trial approval or marketing authorization, the selection of the methods of analysis applied within the non-clinical study approach has to be justified, and a scientific rationale for the design and the properties of the implemented assays has to be provided to the authorities. Techniques based on nucleic acid amplification to quantify the amount of human genomic DNA (hgDNA) in a heterogeneous sample of murine genomic DNA (mgDNA) are frequently used to evaluate the biodistribution of various cell-based medicinal products4, 5, 6 because of their cost-efficiency, selectivity, and, most notably, their high sensitivity.7,8 However, the performance of a qPCR-based method is influenced by a variety of different parameters that must be taken into account. The choice of primers and probe, the type of thermocycler, DNA polymerase, buffer composition, reaction conditions, and the calculation of quantification cycle (Cq) values all have a strong impact on the specificity, sensitivity, accuracy, and precision of the method.9, 10, 11 Consequently, an appropriate method validation must be performed to ensure that the analytical method is suitable for its intended purpose and that unrestricted interpretability and reliability of the obtained data are achieved. The requirements for an appropriate method validation depend on the intended purpose and differ if the analytical method is applied for early preclinical exploratory studies or for good laboratory practice (GLP)-compliant safety studies. Therefore, different guidelines have been developed by the EMA and the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) defining requirements for method validation.12,13 The European Pharmacopoeia Monograph 2.6.21 also provides specific guidance for the validation of quantitative nucleic acid amplification-based methods.14 The α-satellite DNA on human chromosome 1715 and the human Down syndrome region of chromosome 2116 have been used as PCR targets, resulting in methods with different quantification limits. The selected target for the detection of hgDNA in a mgDNA background must feature a high species specificity to fulfill the sensitivity requirements in accordance with the current state of science and technology.2,17,18 Therefore, the human Alu (hAlu) repeat sequences have been used as a PCR target by several laboratories18, 19, 20 due to the fact that the highly repetitive nature and the species specificity of these sequences allow accurate, specific, and sensitive quantification of hgDNA within mgDNA.21, 22, 23 The developed and validated qPCR method described here is based on the primer and probe sequences published by Creane et al.18 This novel qPCR method and the applied validation approach represent a technological improvement to established research techniques with regard to compliance with regulatory guidelines and requirements. This method of analysis was applied in a biodistribution study simulating a worst-case scenario that involved i.v. administration of human blood outgrowth endothelial cells (BOECs) from hemophilia A (HA) patients in a pre-clinical mouse model. These cells had been transduced with a lentiviral vector carrying the functional human factor VIII (FVIII) gene. The biodistribution study was performed within a project for the development of an advanced therapy medicinal product (ATMP) classified as GTMP for the treatment of severe HA patients who lack coagulation FVIII activity due to mutations or deletions within the FVIII gene.24 It has previously been demonstrated that endothelial cells—and, in particular, liver sinusoidal endothelial cells (LSECs)—are the major FVIII-producing cells;25, 26, 27, 28 thus, endothelial cells could represent an optimal cellular target for HA cell and gene therapy. In particular, BOECs have been described as fully differentiated endothelial cells displaying the typical endothelial cobblestone-like morphology in culture.24,29 They are the progeny of bone-marrow-derived circulating cells, which are putative endothelial progenitor cells and feature a high expansion capacity for in vitro cell culture applications.24,30 The biodistribution of canine or human BOECs from healthy donors has previously been assessed in mice with different methods, such as immunofluorescence microscopy, flow cytometry, and qPCR.31, 32, 33, 34 In addition, BOECs that were genetically modified to produce FVIII, e.g., by lentiviral transduction or plasmid transfection, have been evaluated as a potential treatment for HA patients, including assessment of biodistribution in animal models.32, 33, 34, 35 Here, the biodistribution of an actual and novel candidate cell-based medicinal product consisting of BOECs isolated from hemophilic patients is evaluated. This is a prerequisite for designing and performing the subsequent toxicity studies necessary to ensure patient safety and permit moving the medicinal product toward a first-in-human clinical trial.
Results
qPCR Method Validation
The general requirements as well as the applicable validation characteristics relevant for the intended use, here the application for early preclinical studies as well as for later GLP-compliant safety studies, are described in the EMA Guideline on Bioanalytical Method Validation12 and the ICH Guideline —Validation of Analytical Procedures: Text and Methodology Q2(R1).13 Although these guidelines should be followed, it is necessary to adapt them to the specific analytical method. The parameters required for the validation of a qPCR-based method are robustness, specificity, linearity, range, accuracy, precision, and quantification limit. In specific cases, the evaluation of both quantification limit and detection limit may be needed.13,14 However, the evaluation of robustness is not included in this article. This topic was addressed during the development phase of the qPCR method, following the recommendations of the aforementioned guidelines. A summary of the method development and evaluation of its robustness can be found in Tables S1–S5 and Figures S1–S7. The acceptance criteria were defined according to both guidelines and the preliminary data obtained during method development and robustness evaluation. Specificity is the ability to unequivocally assess the analyte in the presence of components that may be expected to be present.13 In the study reported here, the analyte is hgDNA contained in a sample matrix comprising different mouse organs and tissues. The specificity of the selected primers and the probe has been demonstrated by Creane et al.18 and confirmed by comparison of the primer sequences with hAlu sequences published in the NCBI GenBank databases, using the BLAST algorithm. Creane et al.18 proved the specific amplification of commercially obtained hgDNA with hAlu primers by analyzing hgDNA diluted in different sample matrices—namely, DNA isolated from mouse, rat, or rabbit organs—and comparing the results with pure samples containing only mouse, rat, or rabbit DNA. In addition, they analyzed six different dilutions of hgDNA diluted in mouse, rat, or rabbit DNA and compared the Cq values to those obtained from equivalent dilutions of hgDNA without the mouse, rat, or rabbit sample matrix. Their analysis of various organs and tissues—namely, thigh, calf, heart, lung, brain, liver, kidney, and spleen—from control mice demonstrated specificity of the selected primers and the probe for different tissues from BALB/c nude mice. Likewise, the analysis of lung, heart, liver, spleen, kidney, gonads, bone with bone marrow, and brain from control NOD/SCID (non-obese diabetic/severe combined immunodeficiency)/gamma(C)(null) mice within this biodistribution study demonstrates specificity for the selected organs and for the mouse model. The numbers of genomes detected in samples isolated from different organs of control mice were compared to those detected in the same concentration of commercially obtained mgDNA (background control), which was isolated from whole blood. The genome concentration in any of the samples from organs of control mice was not statistically significant higher than the concentration of the background control. Moreover, a system-suitability control (SSC) was analyzed. The SSC was prepared by spiking mouse liver samples, consisting of a defined amount of freeze-dried liver powder, from a biodistribution study control mouse with a defined amount of genetically modified HA-BOECs at a ratio of 1,000:1 murine to human haploid genomes, followed by DNA extraction and concentration adjustment. The objective was to demonstrate the suitability of the whole processing chain. Furthermore, the SSC allows an estimation of the recovery that can be achieved after DNA extraction. The mean recovery was 21.94% of the applied genome concentration, as measured by qPCR.
The other validation characteristics—linearity, accuracy, precision, range, and quantification limit—were evaluated simultaneously by applying a validation matrix approach. These parameters were evaluated by analyzing hgDNA diluted in sample matrix (mgDNA) at six different concentrations (reference standards Ref10E1–Ref10E6) in four validation runs using the qPCR method. The reference standard Ref10E1 has a ratio of 1 human genome to 10 murine genomes, the reference standard Ref10E2 has a ratio of 1 to 100, and so forth, up to the maximum ratio of 1 human genome to 1,000,000 murine genomes in Ref10E6 (see Table S6 for a complete description of the calculation and manufacture of reference standards). In addition, pure samples of mgDNA and hgDNA were investigated as a background control and reference standard, respectively, along with Tris-EDTA (TE) buffer as a no-target control and the SSC. Figure 1 shows the different genome concentration values measured for all described reference standards and samples.
Figure 1.

Human Haploid Genome Concentrations for Samples Analyzed during Method Validation, Showing Validation Run 1 as an Example
The concentrations of human haploid genomes measured in 100 ng DNA are indicated as boxplots for five replicates (n = 5) except for the sample mgDNA with four replicates (n = 4). Whiskers represent minimum and maximum values, and the bold horizontal line represents the median. The p value is shown for the determination of the quantification limit (p = 0.0005).
The most critical parameters identified that had an influence on qPCR method performance or within qPCR preparation were the operator performing the experiment and the suitability of the manually prepared reference standards necessary to calculate and provide the calibration curve. Consequently, all validation runs were performed on different days and under different conditions (Table 1). With regard to the data shown in Table S2 and Figure S4, four different operators performed the qPCR method, and four different batches of reference standards for the calibration curves were applied.
Table 1.
Validation Matrix
| Validation Run | qPCR Parameters and Conditions |
|||||
|---|---|---|---|---|---|---|
| Day | Operator | Batch Hydrolysis Probe | Batch Forward/Reverse Primer | Batch Reference Standarda | Batch Mastermix | |
| 1 | 1 | A | 2019-02-12 | 2019-02-12 | 2019-02-13 | 18009400 |
| 2 | 3 | A | 2018-10-24 | 2019-02-12 | 2019-02-13 | 18026520 |
| 3 | 6 | A | 2018-10-24 | 2018-10-16 | 2018-10-15 | 18026520 |
| 4 | 8 | B | 2019-02-12 | 2019-02-12 | 2019-02-13 | 18009400 |
The calibration curves were prepared using two different batches of reference standards. For each validation run, one reference standard batch was used. Each reference standard batch was prepared by a different operator.
The validation characteristics were determined for all reference standards. The linearity of the assay reflects its ability to obtain test results that are directly proportional to the concentration of hgDNA in the sample. Table 2 shows the acceptance criteria and the calculated values for the efficiency of the amplification of the target sequence in percent and the slope and the error of the calibration curve, which are important determinants of linearity. The y-intercept indicates the crossing point for the log concentration of zero and has to be reported.13 Plots of calibration curves for all runs can be found in Figure S8. The acceptance criteria for linearity were fulfilled for all validation runs. The acceptance criteria include the error value, which is the mean squared error of the single data points fit to the regression line, a measure of the accuracy of the quantification results. The error value limit of 0.05 was defined based on the preliminary results presented in Table S2 and Figure S4 and considering the threshold values given in the Roche LightCycler manual. The slope of the calibration curve describes the kinetics of the PCR amplification and indicates how quickly the amount of target nucleic acid can be expected to increase with the amplification cycles; thus, this acceptance criterion also refers to efficiency. The efficiency needs to be stable and in a range of 90%–110% to ensure reproducible results. No acceptance criterion is defined for the y-intercept, as this value only needs to be reported.
Table 2.
Linearity Assessment
| Error | Efficiency (%) | Slope | y-Intercept | |
|---|---|---|---|---|
| Acceptance criterion | ⩽0.05 | 90–110 | −3.100 to 3.580 | not defined |
| Validation run 1 | 0.0190 | 96.80 | −3.486 | 28.70 |
| Validation run 2 | 0.0216 | 96.70 | −3.490 | 28.92 |
| Validation run 3 | 0.0244 | 99.35 | −3.352 | 28.33 |
| Validation run 4 | 0.0109 | 97.40 | −3.454 | 28.15 |
The intra- and inter-assay accuracy and precision are determined to express the closeness of agreement between the value that is accepted as reference value and the value found and the closeness of agreement between a series of measurements obtained from the multiple sampling of the same homogeneous sample, respectively.14 The intra-assay accuracy and precision values were assessed for each validation run individually by analyzing five replicates for each sample. The inter-assay accuracy and precision values were calculated using these five replicates of all samples of validation runs 1–4 obtained under different circumstances, as described earlier. The calculated values and the acceptance criteria for both are reported in Table 3. Accuracy is expressed as percentage of determined value to the nominal value, meaning the nominal genome concentration of the analyte.14 Precision is expressed as the coefficient of variation (CV) in percent. For the reference standard with the lowest concentration of analyte (Ref10E6), the required limit values for accuracy and precision are less strict.12 Therefore, different acceptance criteria for reference standard Ref10E6 were defined. The concentrations and the standard deviations for all samples in all validation runs are reported in Figure S9.
Table 3.
Intra- and Inter-assay Accuracy and Precision
| Acceptance Criteria |
hgDNA |
Ref10E1 |
Ref10E2 |
Ref10E3 |
Ref10E4 |
Ref10E5 |
Ref10E6 |
|
|---|---|---|---|---|---|---|---|---|
| Intra-assay Accuracy and Precision (%)a | ||||||||
| % of Nominal value | accuracy | ±15 | ±15 | ±15 | ±15 | ±15 | ±15 | ±20 |
| CVb | precision | ⩽15 | ⩽15 | ⩽15 | ⩽15 | ⩽15 | ⩽15 | ⩽20 |
| Validation run 1 | accuracy | 80.29 | 115.52 | 114.65 | 99.32 | 99.68 | 92.30 | 99.46 |
| precision | 12.99 | 4.92 | 2.10 | 1.97 | 1.83 | 4.56 | 18.61 | |
| Validation run 2 | accuracy | 70.53c | 113.71 | 115.66 | 99.53 | 102.64 | 88.18 | 107.20 |
| precision | 26.06c | 6.62 | 4.83 | 1.95 | 6.66 | 4.36 | 19.45 | |
| Validation run 3 | accuracy | 76.96 | 129.62 | 115.16 | 101.49 | 96.64 | 89.77 | 95.77 |
| precision | 12.65 | 9.71 | 3.77 | 12.17 | 2.88 | 10.25 | 12.66 | |
| Validation run 4 | accuracy | 92.77d | 109.68c | 109.08 | 105.90 | 102.64 | 91.50 | 101.63 |
| precision | 10.20d | 4.79c | 2.47 | 7.09 | 13.68 | 5.60 | 10.86 | |
| Inter-assay Accuracy and Precision (%)e | ||||||||
| Replicates of all four runs | accuracy | 78.37 | 117.52 | 113.64 | 101.56 | 100.40 | 90.44 | 101.02 |
| precision | 16.82 | 9.29 | 4.00 | 7.16 | 7.73 | 6.34 | 15.28 | |
The intra-assay accuracy and precision values are calculated with five replicates for each single validation run (n = 5).
Precision is expressed as the coefficient of variation (CV) in percentage.
The intra-assay accuracy and precision values are calculated with five replicates for each single validation run (n = 4).
The intra-assay accuracy and precision values are calculated with five replicates for each single validation run (n = 2).
The inter-assay accuracy and precision values are calculated combining all replicates for every different reference standard of validation runs 1–4.
The results obtained (Tables 2 and 3) lead to the conclusion that suitable precision, accuracy, and linearity can be achieved within the concentration interval of 1:1 × 103 to 1:1 × 106 human genomes diluted in mouse genomes, representing the range of the qPCR method.13,14 Increased concentrations of human genomes, between the interval of 1:1 × 101 to 1:1 × 102 human genomes diluted in mouse genomes (Ref10E1 and Ref10E2), could be detected with acceptable precision. However, the acceptance criterion for accuracy was not quite achieved for reference standard Ref10E2 in validation run 2. Likewise, the acceptance criteria for accuracy and inter-assay precision were not met for Ref10E1 in validation runs 1 and 3. The measurement of undiluted hgDNA failed to achieve both accuracy and precision in almost all validation runs.
The quantification limit is defined as the lowest amount of analyte in a sample or sample matrix that can be quantified with suitable accuracy and precision and expresses the sensitivity of a method.12,14 Applying the qPCR method, the reference standard with the lowest amount of hgDNA that fulfills these acceptance criteria is Ref10E6, which contains 1 human haploid genome within 1 × 106 haploid murine genomes (Table 3). Furthermore, a statistically significant difference in concentration values between the reference standard Ref10E6 and the background control was defined as the acceptance criterion for determining the quantification limit. To test the hypothesis that the reference standard Ref10E6 and the background control sample were associated with statistically significant differences in mean genome concentration, a Welch’s t test was performed, and a significance level of 0.01 was defined. Table 4 shows that a statistically significant difference between the genome concentrations of Ref10E6 and the background control could be calculated for all four validations runs. The Shapiro-Wilk normality test was performed, and Normal Q-Q plots were prepared to prove that the distribution of the data for both samples was sufficiently normal. Additionally, the assumption of the homogeneity of variances was tested and not satisfied via Levene’s F test. The detection limit of an individual analytical procedure is defined as the lowest amount of analyte in a sample that can be detected but not necessarily quantitated as an exact value.13 Based on our preliminary data, only the quantification limit was evaluated.
Table 4.
Quantification Limit Determination
| Acceptance Criterion | Ref10E6a |
mgDNAb |
Welch’s t Test, p ⩽ 0.01 |
||
|---|---|---|---|---|---|
| Conc. | SD | Conc. | SD | ||
| Validation run 1 | 2.75E−02 | 5.12E−03 | 6.39E−03 | 1.13E−03 | t(4.480) = 8.954, p = 0.0005 |
| Validation run 2 | 2.96E−02 | 5.77E−03 | 9.94E−03 | 1.01E−03 | t(4.307) = 7.497, p = 0.001 |
| Validation run 3 | 2.65E−02 | 3.35E−03 | 7.25E−03 | 3.01E−04 | t(4.080) = 12.756, p = 0.0002 |
| Validation run 4 | 2.81E−02 | 3.05E−03 | 1.22E−02 | 2.29E−03 | t(6.994) = 8.893, p = 0.00005 |
The data represent the mean haploid genome concentration (Conc.), meaning human haploid genomes in 100 ng DNA, and the standard deviation (SD) of five replicates (n = 5).
The data represent the mean haploid genome concentration (Conc.), meaning human haploid genomes in 100 ng DNA, and the SD of four replicates (n = 4).
Biodistribution of i.v. Administered Genetically Modified HA-BOECs
Following the recommendations of the EMA guidelines for GTMPs, the biodistribution of genetically modified HA-BOECs in a worst-case scenario was examined (Figure 2). Genetically modified HA-BOECs were characterized using flow cytometry and tested positive for the endothelial surface markers CD31 and VEGFR2. In addition, the BOECs were positive for vascular endothelial (VE)-cadherin and the progenitor and activation marker CD34 but negative for the leukocyte marker CD45.29,36 The results for the characterization of genetically modified HA-BOECs by flow cytometry are shown in Figure S10. Study samples with the same genome concentration as the background control, consisting of mgDNA only, were defined as negative. This means that no human genomes could be amplified in these organs, assuming a quantification limit of 1 human genome in 1,000,000 mouse genomes, as described earlier. Samples of organs and tissues showing a statistically significant lower genome concentration than the background control were also considered negative. In contrast, samples of organs and tissues showing a statistically significant higher genome concentration compared to the background control were defined as positive, meaning that the presence of human genomes was demonstrated. A significance level of 0.05 was defined for samples with concentrations exceeding that of the background control. A Mann-Whitney U test was performed to test the hypothesis that the organs of the two treatment groups and the background control would show statistically significant differences in the mean ranks for genome concentration. The distribution of genome concentration values for tissues and organs from different mice in the treatment groups was not sufficiently normal for the purpose of performing a parametric test. The Mann-Whitney U test indicated that, on average, the genome concentrations found in the organs brain, heart, liver, and lung (mean rank = 8; n = 5) from the 24-h treatment group were significantly higher than the concentration of the background control (mean rank = 3; n = 5), U(n = 10) = 0, p = 0.008. The r value was calculated to determine effect size and was estimated to be 0.83, which is a large effect. For example, the average genome concentration measured in the lungs of five mice after 24 h corresponds to a ratio of 1 BOEC to 2,783.49 mouse cells and a percentage of 0.036%. In contrast, the average genome concentration measured in the livers of five mice after 24 h corresponds to a ratio of 1 BOEC to 412,674.92 mouse cells and a percentage of 0.00024%. No human genomes could be detected in the 24-h treatment group for other organs analyzed such as bone, including bone marrow, kidney, spleen, and gonads (Figure 2A). In the 7-day treatment group, the genome concentration in all organs did not show a statistically significant increase compared to the concentration of the background control. This implies that no signs of biodistribution could be detected after 7 days (Figure 2B).
Figure 2.

Detection of human haploid genomes in eight different mouse organs at two different time points
(A and B) Organs were analyzed 24 h (A) and 7 days (B) after i.v. infusion of genetically modified HA-BOECs into NOD/SCID/gamma(C)(null) mice. The qPCR was performed for quintuplicate samples in one run. The concentrations of human haploid genomes in 100 ng DNA measured in bone, including bone marrow, brain, heart, kidney, liver, lung, spleen, and gonads are indicated as boxplots for five different mice (n = 5). For the organs heart in the 7-day group and gonads in the 24-h group, the boxplot represents four mice (n = 4). Whiskers represent minimum and maximum values, and the bold horizontal line represents the median. Circles represent outliers. The genome concentration measured in samples from brain, heart, liver, and lung after 24 h significantly exceeded the concentration of the background control (Control) mgDNA (∗p = 0.008).
Additionally, a Mann-Whitney U test was performed to test the hypothesis that the organs found to be positive in the 24-h treatment group would show statistically significant differences in the mean ranks for genome concentration compared to the same organs in the 7-day treatment group (Figure 3). The reason for this was to investigate the persistence of genetically modified HA-BOECs after 7 days in different organs. The concentrations found in brain and liver (mean rank = 8; n = 5) after 24 h significantly exceeded the concentrations of genomes detected in these organs after 7 days (mean rank = 3; n = 5), U(n = 10) = 0, p = 0.008, with a large effect size of r = 0.83. Likewise, the concentration measured in the lung in the 24-h treatment group (mean rank = 7; n = 5) was significantly higher than the concentration found in the lung in the 7-day treatment group (mean rank = 2.5; n = 4), U(n = 9) = 0, p = 0.016, with an r value of 0.82, implicating a large effect size. The genome concentrations found in the heart for both time points, 24 h (mean rank = 6.4; n = 5) and 7 days (mean rank = 3.25; n = 5), were not significantly different, U(n = 9) = 3, p = 0.111.
Figure 3.

Detailed Comparison of Organs Testing Positive for Human Haploid Genomes
Detailed comparison of the concentration of detected human haploid genomes in 100 ng DNA in the brain, heart, liver, and lung at 24 h and 7 days after i.v. infusion of genetically modified HA-BOECs into NOD/SCID/gamma(C)(null) mice. The qPCR was performed for quintuplicate samples in one run. The concentrations of human haploid genomes measured in the four organs at both time points are indicated as boxplots for five different mice (n = 5). For the heart in the 7-day group, the boxplot represents four mice (n = 4). Whiskers represent minimum and maximum values, and the bold horizontal line represents the median. Circles represent outliers. The genome concentration measured in samples from lung (∗p = 0.016), brain, and liver (∗∗p = 0.008) after 24 h was significantly higher than the genome concentration measured in these organs after 7 days. However, the heart samples (∗∗∗p = 0.111) showed no significant difference.
The suitability of the processes upstream of the analysis of study samples by qPCR with regard to, e.g., cross-contamination was ensured by analyzing all eight organs of the control group mice at both time points to confirm the absence of human genomes. All organs of control mice were analyzed and the resulting data showed that the genome concentration was below or equal to the concentration of the background control with the exception of only one very slightly positive organ; however, this was not statistically significant.
Discussion
The qPCR method used to investigate the biodistribution of cell-based medicinal products must be appropriate to ensure that reliable results are obtained in order to support a foreseen clinical trial application or European marketing authorization request. To achieve this, a well-characterized and validated method of analysis should be used.12 Furthermore, it is important that sufficient information about the applied experimental method is provided to ensure that results can be interpreted in a meaningful way and compared to other experiments.37
The data presented here demonstrate that the concentration of human genomes within mouse genomes can be determined with high accuracy, precision, and linearity between the concentrations of 1:1 × 103 and 1:1 × 106. The method narrowly failed to achieve the required accuracy values for reference standards with higher concentrations of hgDNA. The reference standard containing only hgDNA missed the acceptance criteria for both accuracy and precision, and generally higher standard deviations were observed when analyzing this reference standard. This is probably due to the high initial DNA content present in this reference standard, which might inhibit the qPCR or impede accurate amplification.10 Almost certainly, it can be stated that neither preparation nor stability of the reference standards is an issue, as the more diluted reference standards all fulfill the acceptance criteria for accuracy and precision, even when analyzed after a storage period of 5 months. Each batch of reference standards Ref10E1–Ref10E6 was prepared using the same standardized manufacturing process starting from the undiluted hgDNA. Moreover, the range of the method is appropriate for its intended purpose: concentrations of human genomes within mouse genomes higher than 1:1 × 102 are not expected after the administration of a cell-based product in a mouse model, because lodging of BOECs or their progenitor cells is generally low in healthy organs.38,39 In addition, previously published results have indicated lower concentrations of BOECs in different mouse organs.31 However, the aim was to fully cover the potential quantification range of validation studies in order to unambiguously determine the upper and lower concentration limits at which the qPCR method can detect human genomes in mouse genomes at a suitable level of precision, accuracy, and linearity. In addition, the preliminary results obtained during method development and robustness evaluation already suggested that the undiluted hgDNA reference standard might not meet the acceptance criteria as laid down in Table 3 (see also, in particular, Table S2 and Figure S4). In contrast, the accuracy values for reference standard Ref10E1 (except for validation run 3) and, in particular, for reference standard Ref10E2 were very close to the defined acceptance criteria, and they were assumed to meet the criteria based on the preliminary results and considering the application of a more standardized approach for the method validation. The decision to include the pure hgDNA reference standard was made under the assumption that the linearity assessment should cover the whole quantification range. Consequently, it was a risk-based decision to include all reference standards. Interestingly, when analyzing the SSC, a similar problem to that described by Funakoshi et al.23 was discovered. The concentration of human genomes was only 21.94% of the calculated expected concentration of spiked genomes in the mouse liver. Reasons for this might be loss of genomic DNA during genome extraction, the error and variance that underlie the cell-counting procedure, or the procedure of spiking the liver powder with genetically modified HA-BOECs. This issue will need further investigation.
Assessment of the quantification limit is important, as the sensitivity of the qPCR method must be high enough with respect to the current state of the art.3 However, comparison of the sensitivity of published methods is difficult due to, e.g., differences in how the quantification limit is defined and calculated, missing accuracy and precision values for the reference standard with the lowest concentration, and different techniques for handling and preparation of reference standards for the calibration curves.37 Nevertheless, analyzing and comparing the sensitivity of recently published methods targeting hAlu repeat sequences18,19,23 and other human sequences15,16 indicates that a quantification limit of 1 human genome in 1 × 106 murine genomes could represent the current state of the art. The qPCR method successfully achieved this limit under different experimental conditions in four validation runs, confirming the reliability and robustness of the method. The determination of the detection limit in addition to the quantification limit might increase certainty that no cells remain in the different organs, taking into consideration that this method could also be applied later to investigate a potential leakage of subcutaneously administered BOECs in combination with a medical device. However, there are difficulties that need to be taken into account before calculating the detection limit of a qPCR method according to the ICH Guideline Q2(R1), based, e.g., on the signal-to-noise ratio or based on the positive cutoff point as recommended by the European Pharmacopoeia Monograph 2.6.21.13,14 The estimation of the detection limit in qPCR analysis is complicated by the logarithmic nature of Cq and because Cq is undefined when the template concentration is zero.37 In addition, it has to be considered that hAlu-based qPCR methods probably measure a signal for samples containing only the sample matrix mgDNA. Furthermore, potential background signals of the sample can be derived from an unpredictable non-specific reaction among primers and probes, particularly at high cycle numbers.18,23 Due to these reasons, the quantification limit was determined as described earlier. The extensive evaluation of all required validation parameters following European and international guidelines and the disclosure of the applied qPCR protocol and the respective dataset strengthen the reliability of the results and provide experimental transparency and comparability. This proves that the qPCR method is suitable for its intended purpose, the quantification of hgDNA in mouse organs in a non-clinical setting, in order to support a foreseen clinical trial application or European marketing authorization request. These biodistribution data are mandatory to determine the toxicokinetic safety profile and to move the cell-based medicinal product toward a first-in-human clinical trial.2,12,40 The biodistribution of BOECs has been investigated by different research groups, mainly using mouse models. They applied different methods of analysis, such as cryosection analysis by fluorescence microscopy, flow cytometry, or qPCR.31, 32, 33,41 However, due to the uniqueness and the specific characteristics of each GTMP, with regard to, e.g., the manufacturing process, vector system, administered cell dose, and so forth, reliance on biodistribution data from “similar” products is not sufficient and is unlikely to be accepted by regulatory agencies.1 However, such studies may provide supportive data that facilitate the design of biodistribution studies for other GTMP candidates. Therefore, determination of the biodistribution in several selected organs using the specific GTMP was the next logical step within the preclinical study program. Following relevant scientific EMA guidelines and taking the planned clinical route of administration into account, a worst-case scenario biodistribution study of i.v. administered genetically modified HA-BOECs was performed.2 Following i.v. infusion, the highest concentration of BOECs was found in the lung at the earliest time point of investigation. This confirms the hypothesis that cells initially lodge in the lung after systemic administration.31,33 More precisely, the highest average concentration of genetically modified HA-BOECs was found in the lung after 24 h. This was significantly higher than the concentration of BOECs found in liver, brain, and heart, which had the second highest average, being approximately 63 times lower than that in the lung. No significant differences were found in the concentration of detected genetically modified HA-BOECs in heart, brain, and liver. In contrast to the data published by Milbauer et al.,31 stating that BOECs isolated from healthy human donors could be detected in all investigated organs, no biodistribution of genetically modified HA-BOECs was observed in kidney, spleen, gonads, or bone including bone marrow 24 h and 7 days after i.v. infusion. However, the gradual decrease in cell numbers in almost all investigated tissues observed by Milbauer et al.31 and Somani et al.,34 who investigated different earlier and later time points, should be acknowledged. There are various possibilities based on different processes that could contribute to this gradual decrease in BOECs detected in mouse organs. These processes, in combination with the design of the biodistribution study, are important for evaluating and understanding the toxicokinetic safety profile of BOECs after systemic administration compared to administration in combination with a medical device. The first process is the initial i.v. infusion of the cell suspension into mice followed by the circulation of BOECs in the bloodstream. In particular, the concentration of BOECs circulating in the bloodstream and, consequently, in different organs is expected to be higher at earlier time points after cell infusion, e.g., 24 h, compared to later time points, e.g., 7 days.32,33 The amount of hgDNA measured in mouse organs after 24 h might be higher than after 7 days, due to the fact that all mouse organs are harvested and frozen directly while still containing blood with a probably higher number of circulating BOECs. Second, there is the possibility that a proportion of the circulating BOECs or those engrafted or migrated into mouse tissues are dying and being cleared during this time and, therefore, cannot be detected by qPCR.31,34 The degradation of dead cells by caspases and DNases starts almost immediately upon phagocytosis. Therefore, reliable quantification of hgDNA isolated from dead cells is unlikely, and preliminary data from other research groups suggest that hAlu-based qPCR amplifies DNA mainly from living cells.18,21 It is expected that the recipient natural killer cells or macrophages will deplete the living BOEC population, and the extent of this will depend on the level of the immunological dysfunction of the mouse model used.31,42 NOD/SCID/gamma(C)(null) mice are considered to be a suitable mouse model, as they feature high engraftment levels of human cells due to multiple immunological dysfunctions, e.g., incompetence of T, B, and natural killer cells.43 A third possibility is the migration of circulating BOECs or BOECs engrafted in different tissues. In general, endothelial cell migration is essential for angiogenesis, but, depending on the situation, intercellular signals, and environmental cues, endothelial cells can also migrate during vasculogenesis and in a damaged vessel to restore vessel integrity.44 BOECs might migrate to different tissues during the duration of 7 days; as a result, they might not be detected by qPCR in the harvested organs. Consequently, endothelial cell migration might influence the amount of human BOECs measured in different mouse organs. However, the impact of these processes might differ depending on the route of administration. The application of BOECs into a medical device should result in a lower concentration of BOECs in the bloodstream and, consequently, in different organs compared to the i.v. infusion of BOECs as a worst-case scenario. Interestingly, the third highest average genome concentration was found in the brain. This raises the question of whether human BOECs can be incorporated into the blood-brain barrier or actually cross this barrier.45 Modified cells or cells of a different origin, especially those of the macrophage lineage, might have the ability to cross the blood-brain barrier.46 It is possible that the harvesting procedure plays a role here, considering that parts of the blood-brain barrier might also be harvested and contain incorporated human cells or cell fragments. Moreover, in contrast to qPCR-based methods, no or only an extremely small amount of specifically labeled BOECs could be found in the brain by, e.g., fluorescence microscopy or detection of radioactivity.7,38,41 The utilization of human in vitro blood-brain barrier models or the histological assessment of different brain segments could help to answer these questions.47 The use of histological analysis to confirm qPCR results for the investigated organs would be an advantageous approach, as qPCR can only detect DNA, whereas histological analysis can detect cells and also evaluate them, e.g., for viability.22 In comparison to other research groups, no biodistribution of BOECs could be detected in bone marrow samples.31,33,41 However, unlike in other studies, the bone marrow was not separated from the bone but analyzed as one sample. Following EMA guidelines for rodent animal models, the organ-harvesting protocol required the removal of the femur, including bone marrow.48 As endothelial progenitor cells originate from the bone marrow24 and BOECs have been detected in bone marrow samples by several different research groups applying different analytical methods,31, 32, 33,41 it is likely that readministered BOECs can be found in pure bone marrow after i.v. administration. The measurement of a combined sample instead of a pure bone marrow sample could be the reason why no human genomes were detected, as the levels may have fallen below the quantification limit.
In conclusion, the biodistribution of a cell-based medicinal product must always be investigated on a case-by-case basis for each specific product. Here, the application of state-of-the-art and validated methods is important to ensure the adequacy and reliability of the generated data. In this article, the validation and application of a highly sensitive, precise, and accurate qPCR-based method for the evaluation of hgDNA in mouse organs is presented. The validation approach—including defined specifications for accuracy, precision, linearity, range, and quantification limit—follows prevailing European and international guidelines, thus making this a method of choice for required GLP-compliant non-clinical animal studies. The next step will be to determine the biodistribution after applying cells in the foreseen clinical route of administration. Subsequently, the evaluation and comparison of these data with the results presented here for the worst-case scenario biodistribution study of i.v. administered genetically modified HA-BOECs will follow. This will provide the opportunity to further evaluate the toxicokinetic safety profile of this cell-based medicinal product.
Materials and Methods
Blood Sampling, BOEC Isolation, Transduction, Culture Conditions, Characterization, and i.v. Infusion
Blood sampling from adult severe HA patients was performed at the hospital A.O.U Città della Salute e della Scienza, Turin, Italy, followed by standardized transportation of the blood at room temperature to the Department of Health Sciences, Università del Piemonte Orientale ‘‘A. Avogadro’’, Novara, Italy. Blood sampling from adult severe hemophilia A patients was approved by the ethics committee “Comitato Etico Interaziendale A.O.U. Maggiore della Carità, ASL BI, ASL NO, ASL VCO” (Protocol 810/CE, study no. CE 125/17). Isolation of human HA-BOECs was performed applying the protocol previously outlined by Ormiston et al.36 with a minor modification. An earlier cell passaging step 7 days after initial isolation of the peripheral blood mononuclear cells was introduced to reduce expansion time and increase the final cell yield, as described previously.49 Afterward, HA-BOECs were transduced using a lentiviral vector carrying the B-domain-deleted form of human FVIII under the VE-cadherin promoter (LV-VEC.FVIII) at an MOI (multiplicity of infection) of 20. This led to the generation of genetically modified HA-BOECs. BOECs were shipped on dry ice to the University Hospital Würzburg, Würzburg, Germany, in a temperature-controlled transport process for further expansion. Afterward genetically modified HA-BOECs were characterized using flow cytometry for the markers CD31, VEGFR2, VE-cadherin, CD34, and CD45 (Figure S10). The cells were frozen using Cryo-SFM Medium (PromoCell, Heidelberg, Germany) as soon as they reached a population doubling level of 32 to 36, equivalent to about 10 passages with an associated population doubling time of approximately 2 days. The cells were only applied if they passed a defined quality control program, including tests, e.g., for lentiviral integrated copy numbers and absence of microbiological, endotoxin, and mycoplasma contaminations. The same transport process was applied for the shipment of the expanded and genetically modified HA-BOECs back to the Department of Health Sciences, Università del Piemonte Orientale. There, the cryopreserved cells were thawed and cultured for recovery until they reached about 90% confluency. The adherent cell monolayer was washed with Dulbecco’s Phosphate Buffered Saline (DPBS) (Sigma-Aldrich, Munich, Germany) and subsequently detached using TrypLE Select Enzyme (1×), no phenol red (GIBCO by Life Technologies, Darmstadt, Germany). Cell counting was performed using the Neubauer improved counting chamber, and cell suspensions of 2 × 106 genetically modified HA-BOECs were prepared from the same cell suspension for each mouse individually. The cells were centrifuged at 190 × g, washed with DPBS-, and centrifuged again at 190 × g. Pelleted cells, comprising 2 × 106 genetically modified HA-BOECs, were resuspended in 100 μL 0.9% saline (Industria Farmaceutica Galenica Senese, Monteroni D’Arbia, Siena, Italy). Finally, the resuspended cells were i.v. infused into each mouse using a 1-mL syringe (Norm-Ject, Henke Sass Wolf, Tuttlingen, Germany) with a 27G needle (AGANI needle, Terumo Italia, Rome, Italy).
Mice and Animal Husbandry
Eight- to 10-week-old NOD/SCID/gamma(C)(null) mice (Charles River Laboratories Italia, Calco, Italy) were used for the biodistribution study. Mice were kept under specific pathogen-free conditions, and animal care was performed at the Department of Health Sciences, Università del Piemonte Orientale, Novara, Italy. Mice were kept in separated standard polypropylene cages for treatment and control groups, and water and diet were provided ad libitum. Animal studies were approved by the Animal Care and Use Committee of the Università del Piemonte Orientale “A. Avogadro,” Novara, Italy, and by the Italian Ministry of Health (project no. DB064.5), confirming that all experiments conformed to regulatory standards.
Organ and Tissue Harvesting
Mice were sacrificed, and harvesting of organs was performed at the Department of Health Sciences, Università del Piemonte Orientale, Novara, Italy, 24 h or 7 days post-i.v. infusion of genetically modified HA-BOECs. The wearing of disposable protective clothing, overshoes, protective gown, cap, face mask, and gloves and the compliance with sanitary regulations, e.g., disinfection of gloves before entering the animal facilities, were verified. Surgical instruments, scissors, forceps, and scalpels were cleaned and sterilized by heating up to 180°C for 3 h in an oven. Before starting organ harvesting, the entire work surfaces and surgical instruments were intensively cleaned with DNA AWAY solution (Fisher Scientific Molecular BioProducts, Schwerte, Germany), according to the manufacturer’s instructions. Likewise, after successful harvesting of all the organs from one mouse, the work surfaces and all surgical instruments were again cleaned with DNA AWAY solution to avoid and minimize any potential cross-contamination. Mice were sacrificed by cervical dislocation, and the harvesting of organs and tissues was performed consecutively, starting with the organs from control mice, in the following order: lung, heart, liver, spleen, kidney, gonads, bone with bone marrow, and brain. Whole organs were harvested for all mice. For sampling bone including bone marrow, the femur was harvested and separated from skeletal muscle. All harvested organs and tissues were transferred into Cryo.s 2 mL Cryo Tubes (Greiner Bio-One, Frickenhausen, Germany) and immediately stored on dry ice for intermediate storage. Afterward, organs were stored at −80°C before shipping in a temperature-monitored transport process on dry ice to the facilities of the University Hospital Würzburg, where −80°C storage was maintained until further processing.
DNA Extraction, Concentration Measurement, and Concentration Adjustment
Organ processing and DNA extraction were performed consecutively, but organ samples were not stored for longer than 6 months. Mice organs were freeze-dried followed by mechanical homogenization using liquid nitrogen and a pestle to obtain a fine dry powder. Genomic DNA extraction was performed using the protocol of the DNeasy Blood & Tissue Kit (“Purification of Total DNA from Animal Tissues (Spin-Column Protocol)” in the DNeasy Blood & Tissue Handbook; QIAGEN, Hilden, Germany). The DNeasy Blood & Tissue Kits with batch numbers 157035017 and 160026736 were used for DNA extraction. Additionally, an RNA lysis step was implemented using 4 μL of 100 mg/mL RNase A (QIAGEN, Hilden, Germany) within step two of the protocol, according to the manufacturer’s instructions. The quantity of isolated genomic DNA was measured with a NanoQuant Plate (Tecan, Männedorf, Switzerland) in combination with an Infinite 200 plate reader (Tecan, Männedorf, Switzerland). The manufacturer’s instructions for measurement of DNA concentration in small-volume samples of nucleic acids in absorbance mode were followed, and the respective software i-control, v.1.11. for Infinite readers was used. Measurements to prevent cross-contamination included the treatment of all work surfaces and equipment with DNA AWAY solution prior to use, and the use of certified DNA- and DNase-free consumables only, a dedicated set of pipettes, and protective clothing. The DNA concentration of samples was adjusted by dilution with TE buffer solution (pH 8.0) (Sigma-Aldrich, Munich, Germany) to the desired concentration of 50 ng/μL following storage at 2°C–8°C before qPCR analysis.
Oligonucleotides, qPCR Conditions, and Analysis
The sequences of the primers and hydrolysis probe have been described previously.18 The primers were as follows: forward: 5′-TGGTGGCTCTCTCCTGTAAT-3′ (hAlu fwd); and reverse: 5′-GATCTCGGCTCACTGCAA-3′ (hAlu rev). The hydrolysis probe (hAlu probe) was labeled with carboxyfluorescein (FAM) at the 5′ end and the Eclipse quencher (Eurofins Genomics, Ebersberg, Germany) attached to the minor groove binder molecule at the 3′ end and comprised the following sequence: 5′-TGAGGCAGGAGAATCGCTTGAACC-3′. Primers and probe were synthesized by Eurofins Genomics (Ebersberg, Germany). Primers were purified using HPSF, a trademark of Eurofins Genomics. HPSF stands for High-Purity, Salt-Free and is a column-based purification. The hydrolysis probe was purified by high performance liquid chromatography (HPLC). The qPCR reaction mix included the LightCycler 480 Probes Master Mix (Roche Diagnostics, Mannheim, Germany) at 1× concentration, 400 nM hAlu fwd and hAlu rev primer, 50 nM hAlu probe, PCR-grade water (Roche Diagnostics, Mannheim, Germany), and 100 ng genomic DNA in a total volume of 20 μL for each well. The LightCycler 480 Probes Master Mix is a ready-to-use hot-start PCR mix and contains FastStart Taq DNA Polymerase, reaction buffer, deoxyribonucleotide triphosphate (dNTP) mix with deoxyuridine triphosphate (dUTP) instead of deoxythymidine triphosphate (dTTP), and 6.4 mM MgCl2. No further additives such as DMSO were used in the qPCR protocol. The validation as well as the entire analyses for the biodistribution study were performed on a LightCycler 480 System II (Roche Diagnostics, Mannheim, Germany). The qPCR program consisted of 1 preincubation step of 50°C for 2 min; 1 initial denaturation step of 95°C for 5 min, followed by 45 cycles of 95°C for 10 s and 60°C for 30 s for annealing and amplification; and 1 cycle of 40°C for 10 s for cooling. The applied qPCR analysis program was the LightCycler 480 software, v.1.5.1.62, and the detection format was the Monocolor Hydrolysis Probe. The Absolute Quantification/Second Derivative Maximum analysis method was used to determine Cq and concentration values. Further settings were: color compensation off, “high confidence” mode, and selected channel 465-510. The advantage of this analysis method is that it requires little user input; e.g., the baseline settings and the threshold concentration are defined automatically by the instrument. The method takes the shape of the calibration curve into account when calculating the DNA concentration of a sample. Therefore, an error value is calculated for the calibration curve instead of the correlation coefficient. The correlation coefficient describes the correlation of data points with a monotonic decreasing or increasing function. The error describes the deviation of data points from a fitted function, independent of its shape, decrease, or increase. The use of only DNA- and DNase-free consumables was verified in the respective manufacturing protocols. All consumables and reagents, including the DNA LoBind Tubes (1.5 mL; Eppendorf, Hamburg, Germany), LightCycler 480 Multiwell Plate 96, LightCycler 480 Sealing Foil, LightCycler 480 Probes Master (all Roche Diagnostics, Mannheim, Germany), and Tris-EDTA buffer solution (TE-buffer) were certified free from DNA and DNase contaminations. Measurements to prevent cross-contamination also included the treatment of all work surfaces and equipment with DNA AWAY solution prior to use as well as the use of a dedicated set of pipettes and protective clothing. The reference standards used to generate and calculate the calibration curve (hgDNA, Ref10E1–Ref10E6) were prepared using commercially available characterized hgDNA (EMD Millipore, Hayward, CA, USA) as reference material and performing six 10-fold serial-dilution steps of hgDNA into commercially available characterized mgDNA (EMD Millipore Corporation, Hayward, CA, USA). The source of the hgDNA reference material is human whole blood, and the preparation was free of contaminating deoxyribonucleases. The purity of the hgDNA solution was determined by measuring the A260/A280 ratio with acceptance criteria between 1.70 and 1.95. More than 90% of the hgDNA was shown to be larger than 100 kilobase pairs (kbp) in size, and no inhibition of restriction enzyme activity was observed. The accuracy of the concentration was ±10%. In addition, the applied methods are outlined in the certificate of analysis provided by the manufacturer. Four different batches of reference material were used to prepare the reference standards: two for method development and robustness evaluation and two for method validation. The concentration calculation for these reference standards was based on the concentrations of human genomes within mouse genomes for each standard, and the complete calculation can be found in Table S6. The DNA concentrations of the reference standards, background control, and SSC were adjusted to the desired concentration of 50 ng/μL, and they were stored at 2°C–8°C until qPCR analysis.
Biodistribution Study Protocol, Method of Analysis, and Data Evaluation
The objective of the biodistribution study was to determine whether 2 × 106 i.v. administered genetically modified BOECs from severe HA patients biodistribute and persist in the lung, heart, liver, spleen, kidney, gonads, bone with bone marrow, or brain after 24 h and 7 days. It was performed as a worst-case scenario biodistribution study, and the BOECs were infused i.v. to achieve maximum systemic levels. In contrast, the actual clinical route of administration consists of subcutaneous application of BOECs in combination with a medical device. For this reason, less biodistribution is expected for the clinical route of administration. However, due to the possibility of a damaged medical device or a potential leakage of BOECs from the medical device into the bloodstream of patients, the investigation of biodistribution for both routes of administrations, starting with i.v. application, were considered to be an important part of the toxicokinetic safety profile. As the probability of distribution of BOECs into the bloodstream of patients, as described earlier, is expected to be low, the number of animals used for the worst-case scenario biodistribution study was kept to a minimum. Mice were randomly allocated to study groups, and animal care, cell infusion, and organ harvesting were performed by independent operators. The treatment and handling of animals were the same across study groups and control groups. Mice in the treatment groups were infused with a single dose of 2 × 106 genetically modified HA-BOECs (n = 5). The mice in the control groups received only PBS (n = 5). The cell dose of 2 × 106 genetically modified HA-BOECs was the same as the dose used in the proof-of-concept studies applying the clinical route of administration. Animals from the control and the treatment groups were kept in different cages for 24 h (n = 2) or for 1 week (n = 3) until organ harvesting. The harvesting of organs and tissues was performed consecutively, starting with the organs of control mice to minimize cross-contamination. Eight different organs—i.e., bone including bone marrow, brain, heart, kidney, liver, lung, spleen, and gonads—were harvested, followed by extraction of genomic DNA from all organs, DNA quantification, and concentration adjustment of samples for qPCR analysis. These steps were performed sequentially, meaning that the next step was only started if all samples were processed completely. The operators performing genomic DNA extraction, DNA quantification, and concentration adjustment were different and independent from the operators performing qPCR experiments and data evaluation. The same qPCR method and protocol validated as described earlier was applied for the analysis of the biodistribution samples. The only exception was the integration of an external calibration curve for the determination of the concentration of human genomes in samples. The reference standards prepared for the generation of the external calibration curve were examined in three independent qPCR runs before application. If the acceptance criteria for all three independent runs were met, the external calibration curve of one of these qPCR runs was integrated into the qPCR protocol and applied for the evaluation of all biodistribution samples. The general requirements for the application of an external calibration curve are that the qPCR amplification is highly reproducible, reaction conditions are constant, and the detection format and analysis mode are the same as for the experiment from which the calibration curve was imported. Furthermore, it is important to include and define a reference standard that falls within the range of the imported external calibration curve and to introduce control samples that were quantified in a previous experiment to compare the calculated values. The application of an external calibration curve increases the comparability of results, as all sample concentrations from different experiments are calculated based on the same underlying calibration curve. The identification and exclusion of outliers was defined beforehand. Criteria were the observation of a deviating amplification curve, caused, e.g., by a bubble, or an uncertain Cq or concentration value indicated by the LightCycler 480 Software. Furthermore, for the experiment performed to provide the calibration curve, a concentration value for the reference standards (hgDNA and Ref10E1–Ref10E6), mgDNA, or SSC that differed by more than 50% from the median concentration of five replicates was excluded. Finally, all samples and replicates were excluded for which a handling error was documented previously. For the biodistribution samples, concentration values determined with a Cq value of 40 were excluded, as a Cq of 40 was set as the threshold value for the LightCycler 480 System II.
Statistics
The statistical analysis was performed using the statistics software IBM SPSS Statistics v.23. The quantification limit within the validation was determined with a Welch’s t test comparing the means of the reference standard with the lowest DNA concentration and the background control sample. A significance level of 0.01 was defined. Statistical comparison of the biodistribution of tail-vein-infused genetically modified HA-BOECs in several mouse organs, measured as the concentration of human genomes, after 24 h and 7 days was carried out using a Mann-Whitney U test. The Pearson r correlation was calculated to evaluate effect size. The significance level was defined as 0.05. A Shapiro-Wilk normality test was performed previously, and Normal Q-Q plots were prepared to determine whether a parametric or non-parametric statistical test was mandatory. Additionally, the assumption of the homogeneity of variances was tested via Levene’s F test before the parametric test was performed.
Author Contributions
P.B. conducted the research, analyzed and interpreted the data, and was responsible for drafting the manuscript. T.B., S.M., and C.O. conducted part of the research and analyzed and helped interpret the data. A.F., M.Z., H.W., and J.B. were involved in study design, interpretation of data, and preparation of the manuscript. O.P. and R.S. were involved in interpretation of data and preparation of the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by HemAcure, having received funding from the European Union’s Horizon 2020 Research and Innovation Programme under grant agreement no. 667421. This publication was supported by the Open Access Publication Fund of the University of Würzburg. P.B. was supported by a grant from the German Excellence Initiative to the Graduate School of Life Sciences, University of Würzburg, Würzburg, Germany. Work was conducted in Würzburg, Bavaria, Germany and Novara, Italy.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.05.029.
Supplemental Information
References
- 1.European Medicines Agency . EMEA/CHMP/GTWP/125459/2006; 2008. Guideline on the non-clinical studies required before first clinical use of gene therapy medicinal products.https://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003942.pdf [Google Scholar]
- 2.European Medicines Agency . EMA/CAT/80183/2014; 2018. Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products.https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-non-clinical-clinical-aspects-gene-therapy-medicinal-products_en.pdf [Google Scholar]
- 3.European Medicines Agency . EMA/CAT/GTWP/671639/2008 Rev. 1; 2019. Guideline on quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells.https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-quality-non-clinical-clinical-aspects-medicinal-products-containing-genetically_en.pdf [Google Scholar]
- 4.Sensebé L., Fleury-Cappellesso S. Biodistribution of mesenchymal stem/stromal cells in a preclinical setting. Stem Cells Int. 2013;2013:678063. doi: 10.1155/2013/678063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shim G., Lee S., Han J., Kim G., Jin H., Miao W., Yi T.G., Cho Y.K., Song S.U., Oh Y.K. Pharmacokinetics and in vivo fate of intra-articularly transplanted human bone marrow-derived clonal mesenchymal stem cells. Stem Cells Dev. 2015;24:1124–1132. doi: 10.1089/scd.2014.0240. [DOI] [PubMed] [Google Scholar]
- 6.Zscharnack M., Krause C., Aust G., Thümmler C., Peinemann F., Keller T., Smink J.J., Holland H., Somerson J.S., Knauer J. Preclinical good laboratory practice-compliant safety study to evaluate biodistribution and tumorigenicity of a cartilage advanced therapy medicinal product (ATMP) J. Transl. Med. 2015;13:160. doi: 10.1186/s12967-015-0517-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reyes B., Coca M.I., Codinach M., López-Lucas M.D., Del Mazo-Barbara A., Caminal M., Oliver-Vila I., Cabañas V., Lope-Piedrafita S., García-López J. Assessment of biodistribution using mesenchymal stromal cells: Algorithm for study design and challenges in detection methodologies. Cytotherapy. 2017;19:1060–1069. doi: 10.1016/j.jcyt.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Salvadori M., Cesari N., Murgia A., Puccini P., Riccardi B., Dominici M. Dissecting the Pharmacodynamics and Pharmacokinetics of MSCs to Overcome Limitations in Their Clinical Translation. Mol. Ther. Methods Clin. Dev. 2019;14:1–15. doi: 10.1016/j.omtm.2019.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bustin S., Huggett J. qPCR primer design revisited. Biomol Detect. Quantif. 2017;14:19–28. doi: 10.1016/j.bdq.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hedman J., Rådström P. Overcoming inhibition in real-time diagnostic PCR. Methods Mol. Biol. 2013;943:17–48. doi: 10.1007/978-1-60327-353-4_2. [DOI] [PubMed] [Google Scholar]
- 11.Rodríguez A., Rodríguez M., Córdoba J.J., Andrade M.J. Design of primers and probes for quantitative real-time PCR methods. Methods Mol. Biol. 2015;1275:31–56. doi: 10.1007/978-1-4939-2365-6_3. [DOI] [PubMed] [Google Scholar]
- 12.European Medicines Agency . EMEA/CHMP/EWP/192217/2009 Rev. 1 Corr. 2∗∗; 2011. Guideline on bioanalytical method validation.https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf [Google Scholar]
- 13.ICH Expert Working Group (2005). ICH Harmonised Tripartite Guideline: Validation of Analytical Procedures: Text and Methodology Q2(R1). https://database.ich.org/sites/default/files/Q2_R1__Guideline.pdf.
- 14.European Pharmacopoiea Commission, Council of Europe European Directorate for the Quality of Medicines (EDQM) European Pharmacopoeia. 9th ed. Council of Europe; 2008. Nucleic acid amplification techniques; pp. 214–219. [Google Scholar]
- 15.Becker M., Nitsche A., Neumann C., Aumann J., Junghahn I., Fichtner I. Sensitive PCR method for the detection and real-time quantification of human cells in xenotransplantation systems. Br. J. Cancer. 2002;87:1328–1335. doi: 10.1038/sj.bjc.6600573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song P., Xie Z., Guo L., Wang C., Xie W., Wu Y. Human genome-specific real-time PCR method for sensitive detection and reproducible quantitation of human cells in mice. Stem Cell Rev. Rep. 2012;8:1155–1162. doi: 10.1007/s12015-012-9406-3. [DOI] [PubMed] [Google Scholar]
- 17.U.S. Department of Health and Human Services, Food and Drug Administration . 2013. Guidance for Industry: Preclinical Assessment of Investigational Cellular and Gene Therapy Products (FDA-2012-D-1038)https://www.fda.gov/media/87564/download [Google Scholar]
- 18.Creane M., Howard L., O’Brien T., Coleman C.M. Biodistribution and retention of locally administered human mesenchymal stromal cells: Quantitative polymerase chain reaction-based detection of human DNA in murine organs. Cytotherapy. 2017;19:384–394. doi: 10.1016/j.jcyt.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 19.McBride C., Gaupp D., Phinney D.G. Quantifying levels of transplanted murine and human mesenchymal stem cells in vivo by real-time PCR. Cytotherapy. 2003;5:7–18. doi: 10.1080/14653240310000038. [DOI] [PubMed] [Google Scholar]
- 20.Ramot Y., Meiron M., Toren A., Steiner M., Nyska A. Safety and biodistribution profile of placental-derived mesenchymal stromal cells (PLX-PAD) following intramuscular delivery. Toxicol. Pathol. 2009;37:606–616. doi: 10.1177/0192623309338383. [DOI] [PubMed] [Google Scholar]
- 21.Schneider T., Osl F., Friess T., Stockinger H., Scheuer W.V. Quantification of human Alu sequences by real-time PCR--an improved method to measure therapeutic efficacy of anti-metastatic drugs in human xenotransplants. Clin. Exp. Metastasis. 2002;19:571–582. doi: 10.1023/a:1020992411420. [DOI] [PubMed] [Google Scholar]
- 22.Prigent J., Herrero A., Ambroise J., Smets F., Deblandre G.A., Sokal E.M. Human Progenitor Cell Quantification After Xenotransplantation in Rat and Mouse Models by a Sensitive qPCR Assay. Cell Transplant. 2015;24:1639–1652. doi: 10.3727/096368914X681955. [DOI] [PubMed] [Google Scholar]
- 23.Funakoshi K., Bagheri M., Zhou M., Suzuki R., Abe H., Akashi H. Highly sensitive and specific Alu-based quantification of human cells among rodent cells. Sci. Rep. 2017;7:13202. doi: 10.1038/s41598-017-13402-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hebbel R.P. Blood endothelial cells: utility from ambiguity. J. Clin. Invest. 2017;127:1613–1615. doi: 10.1172/JCI93649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Do H., Healey J.F., Waller E.K., Lollar P. Expression of factor VIII by murine liver sinusoidal endothelial cells. J. Biol. Chem. 1999;274:19587–19592. doi: 10.1074/jbc.274.28.19587. [DOI] [PubMed] [Google Scholar]
- 26.Follenzi A., Benten D., Novikoff P., Faulkner L., Raut S., Gupta S. Transplanted endothelial cells repopulate the liver endothelium and correct the phenotype of hemophilia A mice. J. Clin. Invest. 2008;118:935–945. doi: 10.1172/JCI32748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumaran V., Benten D., Follenzi A., Joseph B., Sarkar R., Gupta S. Transplantation of endothelial cells corrects the phenotype in hemophilia A mice. J. Thromb. Haemost. 2005;3:2022–2031. doi: 10.1111/j.1538-7836.2005.01508.x. [DOI] [PubMed] [Google Scholar]
- 28.Zanolini D., Merlin S., Feola M., Ranaldo G., Amoruso A., Gaidano G., Zaffaroni M., Ferrero A., Brunelleschi S., Valente G. Extrahepatic sources of factor VIII potentially contribute to the coagulation cascade correcting the bleeding phenotype of mice with hemophilia A. Haematologica. 2015;100:881–892. doi: 10.3324/haematol.2014.123117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin-Ramirez J., Hofman M., van den Biggelaar M., Hebbel R.P., Voorberg J. Establishment of outgrowth endothelial cells from peripheral blood. Nat. Protoc. 2012;7:1709–1715. doi: 10.1038/nprot.2012.093. [DOI] [PubMed] [Google Scholar]
- 30.Lin Y., Weisdorf D.J., Solovey A., Hebbel R.P. Origins of circulating endothelial cells and endothelial outgrowth from blood. J. Clin. Invest. 2000;105:71–77. doi: 10.1172/JCI8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Milbauer L.C., Enenstein J.A., Roney M., Solovey A., Bodempudi V., Nichols T.C., Hebbel R.P. Blood outgrowth endothelial cell migration and trapping in vivo: a window into gene therapy. Transl. Res. 2009;153:179–189. doi: 10.1016/j.trsl.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin Y., Chang L., Solovey A., Healey J.F., Lollar P., Hebbel R.P. Use of blood outgrowth endothelial cells for gene therapy for hemophilia A. Blood. 2002;99:457–462. doi: 10.1182/blood.v99.2.457. [DOI] [PubMed] [Google Scholar]
- 33.Matsui H., Shibata M., Brown B., Labelle A., Hegadorn C., Andrews C., Hebbel R.P., Galipeau J., Hough C., Lillicrap D. Ex vivo gene therapy for hemophilia A that enhances safe delivery and sustained in vivo factor VIII expression from lentivirally engineered endothelial progenitors. Stem Cells. 2007;25:2660–2669. doi: 10.1634/stemcells.2006-0699. [DOI] [PubMed] [Google Scholar]
- 34.Somani A., Nguyen J., Milbauer L.C., Solovey A., Sajja S., Hebbel R.P. The establishment of murine blood outgrowth endothelial cells and observations relevant to gene therapy. Transl. Res. 2007;150:30–39. doi: 10.1016/j.trsl.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 35.Ozelo M.C., Vidal B., Brown C., Notley C., Hegadorn C., Webster S., Harpell L., Ahlin J., Winterborn A., Handforth J. Omental implantation of BOECs in hemophilia dogs results in circulating FVIII antigen and a complex immune response. Blood. 2014;123:4045–4053. doi: 10.1182/blood-2013-12-545780. [DOI] [PubMed] [Google Scholar]
- 36.Ormiston M.L., Toshner M.R., Kiskin F.N., Huang C.J., Groves E., Morrell N.W., Rana A.A. Generation and Culture of Blood Outgrowth Endothelial Cells from Human Peripheral Blood. J. Vis. Exp. 2015;(106):e53384. doi: 10.3791/53384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bustin S.A., Benes V., Garson J.A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M.W., Shipley G.L. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 38.Wei J., Jarmy G., Genuneit J., Debatin K.M., Beltinger C. Human blood late outgrowth endothelial cells for gene therapy of cancer: determinants of efficacy. Gene Ther. 2007;14:344–356. doi: 10.1038/sj.gt.3302860. [DOI] [PubMed] [Google Scholar]
- 39.Asahara T., Murohara T., Sullivan A., Silver M., van der Zee R., Li T., Witzenbichler B., Schatteman G., Isner J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 40.Silva Lima B., Videira M.A. Toxicology and Biodistribution: The Clinical Value of Animal Biodistribution Studies. Mol. Ther. Methods Clin. Dev. 2018;8:183–197. doi: 10.1016/j.omtm.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smits P.A., Kleppe L.S., Witt T.A., Mueske C.S., Vile R.G., Simari R.D. Distribution of circulation-derived endothelial progenitors following systemic delivery. Endothelium. 2007;14:1–5. doi: 10.1080/10623320601177254. [DOI] [PubMed] [Google Scholar]
- 42.Yoshino H., Ueda T., Kawahata M., Kobayashi K., Ebihara Y., Manabe A., Tanaka R., Ito M., Asano S., Nakahata T., Tsuji K. Natural killer cell depletion by anti-asialo GM1 antiserum treatment enhances human hematopoietic stem cell engraftment in NOD/Shi-scid mice. Bone Marrow Transplant. 2000;26:1211–1216. doi: 10.1038/sj.bmt.1702702. [DOI] [PubMed] [Google Scholar]
- 43.Ito M., Hiramatsu H., Kobayashi K., Suzue K., Kawahata M., Hioki K., Ueyama Y., Koyanagi Y., Sugamura K., Tsuji K. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100:3175–3182. doi: 10.1182/blood-2001-12-0207. [DOI] [PubMed] [Google Scholar]
- 44.Michaelis U.R. Mechanisms of endothelial cell migration. Cell. Mol. Life Sci. 2014;71:4131–4148. doi: 10.1007/s00018-014-1678-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geng J., Wang L., Qu M., Song Y., Lin X., Chen Y., Mamtilahun M., Chen S., Zhang Z., Wang Y., Yang G.Y. Endothelial progenitor cells transplantation attenuated blood-brain barrier damage after ischemia in diabetic mice via HIF-1α. Stem Cell Res. Ther. 2017;8:163. doi: 10.1186/s13287-017-0605-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ellison S.M., Liao A., Wood S., Taylor J., Youshani A.S., Rowlston S., Parker H., Armant M., Biffi A., Chan L. Pre-clinical Safety and Efficacy of Lentiviral Vector-Mediated Ex Vivo Stem Cell Gene Therapy for the Treatment of Mucopolysaccharidosis IIIA. Mol. Ther. Methods Clin. Dev. 2019;13:399–413. doi: 10.1016/j.omtm.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Appelt-Menzel A., Cubukova A., Günther K., Edenhofer F., Piontek J., Krause G., Stüber T., Walles H., Neuhaus W., Metzger M. Establishment of a Human Blood-Brain Barrier Co-culture Model Mimicking the Neurovascular Unit Using Induced Pluri- and Multipotent Stem Cells. Stem Cell Reports. 2017;8:894–906. doi: 10.1016/j.stemcr.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.European Medicines Agency (2010). Guideline on repeated dose toxicity (CPMP/SWP/1042/99 Rev 1 Corr∗). https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-repeated-dose-toxicity-revision-1_en.pdf.
- 49.Kolbe M., Dohle E., Katerla D., Kirkpatrick C.J., Fuchs S. Enrichment of outgrowth endothelial cells in high and low colony-forming cultures from peripheral blood progenitors. Tissue Eng. Part C Methods. 2010;16:877–886. doi: 10.1089/ten.tec.2009.0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.