

Abstract
Malignant melanoma is the most deadly skin cancer, associated with rising incidence and mortality rates. Most of the patients with melanoma, treated with current targeted therapies, develop a drug resistance, causing tumor relapse. The attainment of a better understanding of novel cancer-promoting molecular mechanisms driving melanoma progression is essential for the development of more effective targeted therapeutic approaches. Recent studies, including the research previously conducted in our laboratory, reported that the histone methyltransferase SETDB1 contributes to melanoma pathogenesis. In this follow-up study, we further elucidated the role of SETDB1 in melanoma, showing that SETDB1 modulated relevant transcriptomic effects in melanoma, in particular, as activator of cancer-related secreted (CRS) factors and as repressor of melanocyte-lineage differentiation (MLD) and metabolic enzymes. Next, we investigated the effects of SETDB1 inhibition via compounds belonging to the mithramycin family, mithramycin A and mithramycin analog (mithralog) EC-8042: melanoma cells showed strong sensitivity to these drugs, which effectively suppressed the expression of SETDB1 and induced changes at the transcriptomic, morphological, and functional level. Moreover, SETDB1 inhibitors enhanced the efficacy of mitogen-activated protein kinase (MAPK) inhibitor-based therapies against melanoma. Taken together, this work highlights the key regulatory role of SETDB1 in melanoma and supports the development of SETDB1-targeting therapeutic strategies for the treatment of melanoma patients.
Graphical Abstract
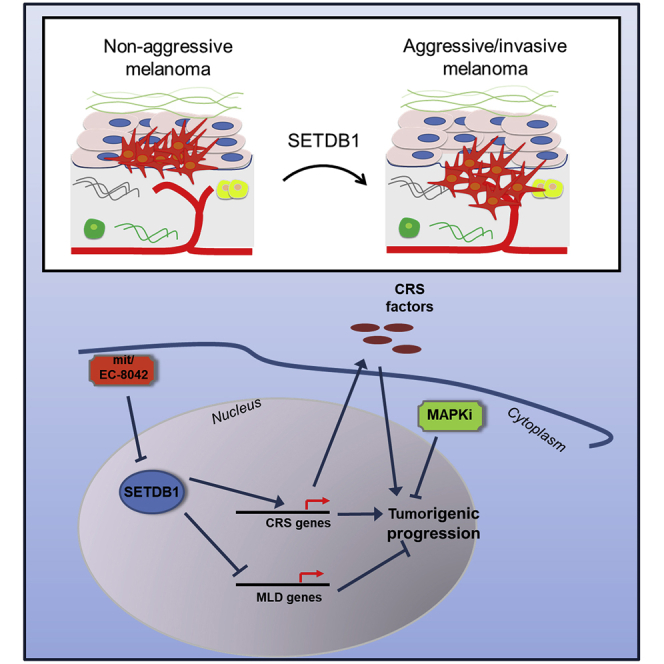
Federico and colleagues investigated the role of SETDB1 in melanoma as master regulator of an oncogenic transcriptional network. Moreover, compounds of the mithramycin family (mithramycin A, EC-8042) strongly impaired SETDB1 levels, leading to antitumor effects. The targeting of SETDB1 represents a promising therapeutic strategy for treating melanoma.
Introduction
Melanoma is one of the most common and aggressive forms of skin cancer. Over the last years, the incidence and mortality rates of malignant melanoma have shown a remarkable increasing trend.1,2 Well-established melanoma treatment options, approved by the US Food and Drug Administration (FDA), include immunotherapies and targeted therapies, used for treating unresectable advanced melanoma, as monotherapy or in combinational treatments.3 Despite the good clinical responses observed in patients with malignant melanoma treated with these therapeutic approaches, poor drug specificity or development of resistance mechanisms occurs in most of the cases.4 Therefore, the definition of innovative therapeutic strategies with improved efficacy against malignant melanoma represents the biggest challenge in this field. Melanoma development and progression are defined by multiple concomitant molecular events leading to the deregulation of cellular mechanisms, such as signal transduction pathways related to cell proliferation and survival. Alterations of key cell signaling pathways (mitogen-activated protein kinase [MAPK], phosphatidylinositol 3-kinase [PI3K], melanocyte inducing transcription factor [MITF], wingless/integrated (WNT)-β-catenin pathways) contribute to the oncogenic potential of melanoma cells.5, 6, 7, 8, 9, 10 The characterizations of novel oncogenic molecular mechanisms driving melanoma tumorigenesis is essential to improve melanoma therapeutic options. Recently, some works described the role of the histone methyltransferase SETDB1 in melanoma, observing an aberrant amplification and/or expression in the melanoma zebrafish model and clinical samples;11, 12, 13 moreover, SETDB1 contributes to melanoma metastases formation in vivo.14 Our previous work13 gave some additional hints about functional effects modulated by SETDB1 and its mechanisms of action during melanomagenesis. With this follow-up study, we could demonstrate that SETDB1 modulated a peculiar oncogenic transcriptional network and that SETDB1-targeting compounds belonging to the mithramycin family (mithramycin A [also referred as “mit”]; demycarosyl-3D-β-D-digitoxosyl-mithramycin SK [DIG-MSK], commonly referred as “EC-8042”) induced a reduction of melanoma cell viability and of cancer cell-specific features.
Results
SETDB1 Overexpression Increases the Expression of Cancer-Related Secreted Factors in Melanoma Cells
Previous work from our group demonstrated that SETDB1 plays a crucial part as the driver of cancer-related features during melanoma progression and that this oncogenic role might be the consequences of the regulation of downstream factors that, in turn, are involved in protumorigenic pathways; moreover, we have also described a regulatory axis between SETDB1 and the glycoprotein thrombospondin 1 (THBS1).13
In this follow-up study, we first aimed to further implement the analysis of our microarray data (GEO: GSE109678) in order to define the functional classes of genes mostly deregulated upon the ectopic overexpression of SETDB1 in melanoma cells. HT 144-SETDB1 overexpressing (OE) versus empty vector (EV) differential expression analysis of quantile-normalized microarray data (empirical Bayes two-group t-test, log2 fold-change [FC] threshold set as > 1, p value [p] 0.05) indicated 152 genes significantly deregulated, including 81 genes upregulated and 71 downregulated in melanoma cells overexpressing SETDB1. A Gene Ontology (GO) enrichment analysis was conducted on upregulated genes (FC > 1) in HT 144-SETDB1 OE cells in comparison to control cells (HT 144 EV). We observed that the most enriched GO terms referred to groups of factors normally released into the extracellular space: keyword “secreted” (number of deregulated genes represented in this GO term [n] = 18, p = 0.000417); GO term: GO: 0005615_extracellular space (n = 19, p = 0.00000277). A more detailed overview of enriched GO terms found related to upregulated SETDB1 target genes is provided in Table S1. This analysis suggested that SETDB1 overexpression induced an upregulation of genes encoding for secreted proteins with a prominent role in cancer progression, including glycoproteins (THBS1), granins (SCG2), metalloproteases (MMP1, MMP3), interleukins (IL-6, IL-8), cytokines (CCL2), and WNT proteins (WNT5A) (Figure 1A). Hereafter, the above-mentioned proteins are referred, in this work, as cancer-related secreted (CRS) factors. The increased levels of genes encoding for CRS factors, following SETDB1 overexpression, were confirmed in HT 144 and C32 melanoma cells (Figures 1B and S1A). Next, to verify whether the transcriptional alteration of CRS factors was followed by changes in melanoma cell secretome, we performed a secreted proteome profiling of HT 144 EV (control) and -SETDB1 OE cells. With the use of a membrane-based antibody array, we could detect the levels of cancer-related proteins present in cell supernatants. Proteome analysis showed that supernatant collected from SETDB1 OE cells tended to have higher levels of the metalloprotease MMP8 and of the cytokines CXCL16 and CCL2 in comparison to cells expressing normal levels of SETDB1. Surprisingly, no changes of THBS1 levels in cell supernatant were detected (Figure 1C). The mechanisms by which tumor cells release factors involved in cancer progression are not yet fully elucidated. In this regard, we focused on the functional role of one putative SETDB1 effector, SCG2, a protein involved in the biosynthesis of secretory granules.15 A previous study correlated the expression of SCG2 with the activation of migratory features in melanoma.16 SCG2 displayed heterogeneous expression in a panel composed of melanoma cell lines and normal human melanocytes (NHMs; Figure S1B). Ectopic overexpression of SCG2 (SCG2 OE) in C32 cells (p < 0.05), characterized by low endogenous SCG2 levels, resulted in the massive accumulation of the SCG2 protein in roundish cytoplasmic structures that resembled the secretory granules (Figures 1D and S1C). This would suggest that the upregulation of SCG2 might be a key event leading to an increased biosynthesis of secretory vesicles, affecting in this way the cell-secretory mechanisms. Finally, we analyzed SCG2 expression in a cohort of clinical specimens obtained from patients with late-stage melanomas, characterized by the presence of metastases (n = 47). In those samples, we could observe a positive concordance between SCG2 and SETDB1 immunohistochemical (IHC) signals (Spearman correlation, r = 0.5928; p < 0.001) (Figure 1E); in addition to that, patients exhibiting moderate to high levels (IHC signal score > 3) of SCG2 also showed a lower survival rate (p = 0.0529) and higher short-term survival (< 12 months survival; p < 0.05) than patients with low SCG2 signals (IHC score ≤ 3), implying a promising prognostic value for SCG2 expression (Figures 1F and S1D).
Figure 1.

Overexpression of SETDB1 in Melanoma Cell Lines Resulted in the Activation of Protumorigenic-Secreted Factors
(A) Histogram plot showing microarray expression data (shown as log2 fold change) of genes encoding for protumorigenic-secreted factors (THBS1, SCG2, MMP1, MMP3, IL-6, IL-8, CCL2, and WNT5A) in HT 144-SETDB1 OE cells compared to EV cells. (B) Quantitative real-time PCR analysis of the CRS factor-encoding genes SCG2, MMP3, IL8, CCL2, and WNT5A expression 5. (C) Secretome analysis of HT 144 EV and -SETDB1 OE cell supernatant. Right panel: blots showing the signals of 55 different secreted proteins detected in the supernatant of cultured cells; signals of MMP8 (1), CXCL16 (2), CCL2 (3), and THBS1 (4) are highlighted. Left panel: quantification of mean pixel density related to MMP8, CXCL16, CCL2, and THBS1 signals. Number or replicates: 3. (D) SCG2 immunofluorescence detection in C32 EV (control) and -SCG2 OE cell lines. SCG2-overexpressing cells showed a strong SCG2 signal accumulation at granule structures (indicated by arrows). DAPI stained the nuclei. Scale bars, 20 μm. (E) Tissue microarray analysis (TMA) of a cohort of melanoma patients showing that SETDB1 expression positively correlated with SCG2 expression in melanoma metastases tissue samples. SETDB1 and SCG2 correlation plot is shown. Number of analyzed patients’ samples (n), Spearman correlation coefficient (r), and p value are reported. (F) Kaplan-Meier survival analysis of patients with melanoma metastases, classified according to intratumoral SCG2 expression (IHC overall score).
Taken together, high levels of SETDB1 promoted the expression of several pro-tumorigenic factors which are either secreted or involved in the organization of the secretory machinery.
SETDB1 Negatively Affects the Expression of the Melanocytic-Lineage Differentiation Markers
Differentially gene-expression data from HT 144-SETDB1 OE versus EV also included downregulated genes. Gene-set functional annotations of SETDB1-induced downregulated genes indicated as particularly enriched the terms related to differentiation mechanisms of melanocytes, like melanin biosynthesis (GO: 0042438; n = 5, p = 0.000000103), melanosome formation (GO: 0033162; n = 3, p = 0.000594), and developmental pigmentation (GO: 0048066; n = 2, p = 0.024749764). Moreover, some metabolic terms, such as “negative regulation of lipid biosynthetic process” (GO: 0051055; n = 2, p = 0.017740531), were found enriched as well. A more detailed overview of enriched GO terms related to the downregulated SETDB1-target genes is provided in Table S2. Melanoma-malignant transformation is defined by the acquisition of a more undifferentiated status, which is the consequences of the loss of melanocytic-lineage differentiation (hereafter labeled as “MLD”) factors, such as dopachrome tautomerase (DCT), tyrosinase-related protein 1 (TYRP1), tyrosinase (TYR), and premelanosome (PMEL);17,18 our microarray data showed a strong impairment of the expression of MLD genes in melanoma cells carrying SETDB1 overexpression in comparison with control cells. Moreover, SETDB1 OE melanoma cells exhibited the downregulation of several metabolic enzymes with a tumor-suppressor role or poorly expressed in melanoma and other cancer types, including apolipoprotein E (APOE), QPRT, and PDK419, 20, 21 (Figure 2A). Quantitative PCR showed a clear and significant decrease of DCT (MLD marker) and APOE (metabolic marker) expression; looking at the protein levels by western blot, C32 cells exhibited decreased levels of both proteins in the SETDB1 OE condition, whereas HT 144-SETDB1 OE showed only a slight reduction (Figures 2B, 2C, S2A, and S2B). The transcriptional alterations observed in SETDB1 OE melanoma cells resembled a cell model recently established by our group, consisting of human-induced pluripotent stem cell-derived melanoblasts (hIPSC-Mb), an intermediate differentiation stage of melanocytes that has been shown to share several features with melanoma tumor cells.22 We matched the hIPSC-Mb gene signature, obtained from the comparison to the transcriptome of totally differentiated melanocytes (NHMs), with the deregulated genes found in HT 144 -SETDB1 OE cells (versus EV), and we observed that the 2 cell conditions had 31 upregulated and 20 downregulated genes in common (Figure 2D). Notably, the expression of genes encoding for MLD markers and for the metabolic factor APOE was lower in melanoblasts than in differentiated melanocytes (Table 1); this observation was confirmed by quantitative PCR (Figure 2E). The obtained data indicated that SETDB1 interfered with the expression of MLD and metabolic enzymes, recapitulating a melanoblast-like gene signature.
Figure 2.

Increasing Levels of SETDB1 Reduced the Expression of Melanocytic and Metabolic Markers
(A) Log2 fold-change expression of genes encoding for melanocytic-lineage differentiation (MLD) markers (DCT, TYRP1, TYR, and PMEL; blue bars) and for metabolic factors (APOE, QPRT, and PDK4; orange bars) observed in HT 144-SETDB1 OE cells in comparison with EV cells. (B) Validation of DCT and APOE downregulation in HT 144-SETDB1 OE cells. Number or replicates: 3–4. (C) DCT and APOE western blots of C32-SETDB1 OE and control (EV). GAPDH was used as a loading control. (D) Venn diagrams representing the number of common upregulated (left panel) and downregulated (right) genes obtained by comparing the gene-expression signature of HT 144 -SETDB1 OE cells (blue) to the transcriptional profile of hIPSC-Mb cells (red). (E) mRNA expression analysis of MLD markers (DCT, TYRP1, TYR, and PMEL) and of APOE in hIPSC-Mb versus NHM cells. Number or replicates: 3.
Table 1.
List of Genes Found Deregulated Both in “HT 144-SETDB1 OE versus EV” and “hIPSC-Mb versus NHM” Gene-Expression Analysis, with Relative Fold Change
| Upregulated Genes in Common |
Downregulated Genes in Common |
||||
|---|---|---|---|---|---|
| Gene Symbol | FC HT 144-SETDB1 OE versus EV | FC hIPSC-Mb versus NHM | Gene Symbol | FC HT 144-SETDB1 OE versus EV | FC hIPSC-Mb versus NHM |
| ANKRD1 | 1.02 | 5.09 | APOE | −1.95 | −2.21 |
| AXL | 1.09 | 3.07 | CAPN3 | −1.26 | −5.46 |
| C12orf75 | 1.10 | 2.74 | CEACAM1 | −1.13 | −2.34 |
| CCL2 | 1.23 | 2.66 | CYGB | −1.04 | −3.00 |
| CD24 | 1.17 | 5.66 | DCT | −2.30 | −7.08 |
| DCBLD2 | 1.24 | 2.55 | FCRLA | −1.15 | −5.15 |
| DKK1 | 2.52 | 3.04 | HES6 | −1.27 | −3.89 |
| EFNB2 | 1.28 | 2.65 | IGSF11 | −1.16 | −4.57 |
| ERRFI1 | 1.75 | 5.07 | ISG20 | −1.16 | −2.55 |
| FLNB | 1.22 | 3.43 | LOC641738 | −1.04 | −3.37 |
| IL6 | 1.84 | 3.26 | LZTS1 | −1.00 | −2.90 |
| IL8 | 2.46 | 5.57 | NBL1 | −1.25 | −2.20 |
| KRT81 | 1.81 | 2.08 | PIR | −1.04 | −3.60 |
| LYPD1 | 1.08 | 2.72 | PLTP | −1.07 | −2.27 |
| MMP1 | 1.35 | 3.31 | PMEL | −1.82 | −6.45 |
| NPTX2 | 1.16 | 2.76 | TBC1D7 | −1.04 | −3.30 |
| NRP1 | 1.90 | 2.50 | TSPAN10 | −1.25 | −6.27 |
| NT5E | 1.26 | 3.81 | TUBB4A | −1.46 | −3.28 |
| PMEPA1 | 1.07 | 2.56 | TYR | −1.42 | −7.31 |
| PPAP2B | 1.01 | 2.30 | TYRP1 | −1.51 | −7.23 |
| PTGS2 | 1.32 | 2.18 | |||
| RND3 | 1.67 | 2.86 | |||
| SCG2 | 1.10 | 2.51 | |||
| SCG5 | 1.45 | 4.33 | |||
| SLC25A24 | 1.16 | 3.02 | |||
| SMAGP | 1.42 | 2.16 | |||
| TGM2 | 1.03 | 2.58 | |||
| THBS1 | 2.31 | 5.23 | |||
| TNFRSF12A | 1.54 | 3.35 | |||
| TPM1 | 1.98 | 5.24 | |||
| WNT5A | 1.40 | 4.53 | |||
mit Treatment Impairs SETDB1 Expression and Melanoma Cell Viability
Our work addressed the crucial contribution of SETDB1 in melanoma progression as a driver of regulatory mechanisms leading to the acquisition of protumorigenic properties. Next, we investigated the effects caused by the inhibition of SETDB1 in melanoma cells using a drug-based approach. Although a selective SETDB1 inhibitor has not been identified yet, several compounds have been shown to reduce SETDB1 expression levels, including common antitumor drugs (paclitaxel),23 unspecific histone methyltransferases (DZnep Chemical Abstracts Service [CAS] 935693-62-2),13,24 and mit.25, 26, 27 Within the scope of our study, we chose mit (CAS 18378-89-7) to interfere with SETDB1 expression. mit is an antitumor antibiotic actually adopted for clinical studies (phase 2) for the treatment of a broad range of malignancies (ClinicalTrials.gov: NCT01624090).
mit represses SETDB1 thanks to its DNA-binding properties, able to block the interactions between SETDB1 regulatory sites and the transcription factor SP-1.28 We tested the capacity of mit to interfere with the expression of SETDB1 in melanoma-stimulating melanoma cells with high SETDB1 endogenous levels (A375 and SK-HI-SETDB1) to increase mit doses for 24 h. SETDB1 expression after 24 h was drastically diminished in mit-exposed melanoma cells in a dose-dependent fashion (Figures 3A and 3B). The gradual impairment of SETDB1 levels was accompanied by cytotoxic effects: melanoma cells treated with increasing mit concentrations at different time points (24, 48, and 72 h) showed a strong reduction of their viability (mean half-maximal inhibitory concentration [IC50] values for A375: 1.04 μM after 24 h of treatment, 34.33 nM after 48 h of observation, 15.5 nM upon 72 h treatment; IC50 SK-HI-SETDB1: 912.9, 43.72, and 29.06 nM after 24, 48, and 72 h, respectively) (Figures S3A–S3F). Whereas SETDB1-positive melanoma cells displayed high sensitivity to mit treatment, no particular cytotoxic effects were detected after 24 h treatment on nontumor cells (human dermal fibroblasts), as well as on melanoma cells carrying SETDB1 knockdown (KD; Figure 3C), suggesting that cytotoxic effects modulated by mit on melanoma cells are dependent on SETDB1 expression.
Figure 3.

Mithramycin A (mit) Treatment Impaired SETDB1 Levels and Melanoma Cell Viability
(A) qPCR analysis of SETDB1 expression in A375 and SK-HI-SETDB1 cells exposed to DMSO (control) and to increasing doses of mit for 24 h. SETDB1 transcript levels decreased with an increasing concentration of mit. One-way ANOVA test was applied; p value < 0.0001. Number or replicates: 4. (B) Western blot detection of SETDB1 and GAPDH in A375 and SK-HI-SETDB1 cells, indicating a mit dose-dependent reduction of SETDB1 protein levels. (C) Cell viability assay upon treatment with different mit doses for 24 h of human dermal fibroblasts (red) and of A375 (blue) and SK-HI-SETDB1 (green) melanoma cell lines, either carrying a nontargeting (“NT”; continuous lines) or a SETDB1 short hairpin RNA (shRNA) (“SETDB1 KD”; segmented lines) vector. (D) 100× light microscope acquisitions of A375 and SK-HI-SETDB1 cells exposed either to DMSO or 300 nM mit for 24 h.
We could establish that the maximum concentration of mit, leading to a strong SETDB1 downregulation, together with an impact on melanoma cell viability after 24 h, was 300 nM; no further reduction of SETDB1 expression and cell viability was observed with higher mit dosages (data not shown). 24 h exposure to 300 nM mit was then established as the standard mit treatment for all of the subsequent mit-drug response experiments. Melanoma cells treated for 24 h with 300 nM mit also exhibited drastic morphological alterations, appearing bigger in size, with larger nuclei, with a reduction of the cell-cytoplasm ratio, and for SK-HI-SETDB1 cells, less and smaller dendritic structures (Figure 3D). Our data indicated the powerful effects mediated by mit on SETDB1 expression and on melanoma cell viability and morphology.
mit-Treated Melanoma Cells Exhibit Altered Tumorigenic Properties
We wanted next to evaluate whether SETDB1 inhibition resulted in a functional regression of melanoma cells. We previously demonstrated that SETDB1 drives important protumorigenic features, such as cell migratory and invasive behavior.13 We performed an in vitro functional characterization of A375 and SK-HI-SETDB1 melanoma cells upon mit treatment. Compared to DMSO-treated cells, cells exposed to 300 nM mit for 24 h exhibited significantly slower migratory and invasive behaviors (Figures 4A and 4B). To further assess the invasive capacities of the highly invasive A375 melanoma cells, we established a two-dimensional (2D) invasion assay: tumor cells were cocultured with dermal fibroblasts, interspaced by a short gap. Both cell types, each of them labeled with a different fluorescent dye, would migrate toward each other until the interspaced gap was filled, and the two cell layers started to interact. At this point, DMSO or mit treatments were added, and after 24 h, the invasive behavior of tumor cells was evaluated by estimating the amount of A375 cells scattered across the fibroblast layer (Figure 4C). Interestingly, we observed that DMSO-treated cells massively occupied the fibroblast regions, whereas treatment with mithramycin led to a strong reduction of A375-invading cells (Figure 4D). Putting these observations together, we could speculate that SETDB1 inhibition by mit impacted on melanoma cell functionality, leading to a less-aggressive behavior and to a functional regression.
Figure 4.

Assessment of Melanoma Cell Migratory and Invasive Properties after Mithramycin A (mit) Treatment
(A) Quantification of the migration capacity of melanoma cells treated with DMSO or mit. Left panel: representative images of scratch assays with A375 cells; open-gap area is highlighted. Right side: quantification of open-gap area on scratched A375 and SK-HI-SETDB1 cell layers after 24 h stimulation. (B) Transwell assay showing a strong reduction of invasion rate of mit-treated melanoma cells. Left side: microscopic images of invading fluorescent cells after DMSO or mit treatment. Right side: quantification of the invasion rate for A375 and SK-HI-SETDB1 cells following mit exposure, measured as the relative fluorescence units (RFUs) released from DMSO- and mit-treated invading cells. (C) Schematic overview of the 2D invasion system. Cells used for this experiment were previously labeled with different fluorochromes (TOMATO for red fluorescence; GFP for green fluorescence). (D) Fluorescent images of 2D invasion assays. GFP-A375 and TOMATO-fibroblast cells were cocultured and subsequently exposed to 300 nM mit (or DMSO). After 24 h, the amount of tumor cells invading the fibroblast layer was evaluated. A375 cells showed a massive invasive behavior impaired by mit treatment.
mit Reverts the Regulatory Effects Modulated by SETDB1 on Its Downstream Effectors
To further determine the consequences of mit-mediated SETDB1 inhibition, we investigated the variations in the regulatory network of SETDB1. We tested the expression levels of genes encoding for CRS, MLD, and metabolic markers in A375 and SK-HI-SETDB1 cells, exposed for 24 h either to DMSO as vehicle control or 300 nM mit. A375 mit-treated cells showed a concomitant repression of CRS protein-encoding genes (THBS1, SCG2, MMP1, MMP3, IL8, CCL2, and WNT5A) and the upregulation of MLD (DCT, TYRP1, and PMEL) and metabolic (APOE) genes (Figure 5A). Similarly, the inhibition of SETDB1 in SK-HI-SETDB1 also induced the significant downregulation of CRS markers (THBS1, SCG2, MMP1, MMP3, and WNT5A) and the upregulation of the APOE gene (Figure S4A), whereas no relative differences were detected relative to MLD gene expression (data not shown). The mit-based SETDB1-silencing approach was useful to further study the impact of SETDB1 on the transcriptional regulation of its downstream target genes. We focused, in particular, on the SETDB1-mediated deregulation of the DCT gene—a reporter plasmid containing the melanocyte-specific cis elements of the DCT promoter29—which placed in front of a GFP open reading frame (ORF; “DCT promoter-GFP” vector), was generated and inserted in A375 cells, characterized by high SETDB1 and very low DCT endogenous expression. Transfected cells emitted no or only a faint green fluorescence signal; upon mit treatment, however, cells exhibited a general increase of the GFP signal, indicating the activation of the DCT promoter and in particular, of the promoter region responsive to melanocyte-differentiation signals, following SETDB1 inhibition (Figure S4B). Protein analysis of A375 DMSO- or mit-treated cells confirmed the increase of THBS1 (CRS marker) and the reduction of DCT (MLD marker) and APOE (metabolic marker) expression (Figure 5B). Furthermore, supernatant of A375 cells exposed to mithramycin exhibited lower levels of oncogenic-related secreted proteins (MMP8, IL-8, CXCL16, CCL2) than DMSO-treated cells; again, secreted THBS1 amounts resulted unchanged (Figure 5C). Finally, mit-mediated SETDB1 inhibition led to a strong abrogation of SCG2 expression and cytoplasmic accumulation (Figure 5D). Taken together, mit induced profound changes in SETDB1-mediated molecular signature and protein activation in melanoma; these events might contribute to the drastic shift from an aggressive tumor cell-like behavior toward a less-aggressive and more differentiated phenotype also observed at the functional level.
Figure 5.

Mithramycin A (mit) Treatment Abrogated the SETDB1-Centered Regulatory Mechanisms
(A) qPCR analysis of a panel of CRS, MLD, and metabolic genes in A375 cells treated either with DMSO or with 300 nM mit. Number or replicates: 3–5. (B) Western blot for THBS1, DCT, APOE, and GAPDH detection in the whole-cell lysate of A375 cells treated with either DMSO or mit. (C) Secretome profiling blots and quantification plot of A375 cells cultured for 24 h in the presence or absence of 300 nM mit. The plot displays the reduction of MMP8, IL-8, CXCL16, and CCL2 secretion levels from cells exposed to mit compared with the ones treated with DMSO. THBS1 levels resulted unaffected. Number or replicates: 3. (D) SCG2 immunofluorescence analysis of DMSO- and mit-A375-treated cells. Scale bars, 20 μm.
Combinatorial Treatment of Melanoma Cells with mit and BRAF/MEK Inhibitors Increases the Efficacy of Targeted Therapies
Next, we aimed to define the effects of combinational treatments, consisting of stimulating A375 and SK-HI-SETDB1 (BRAF-mutated melanoma cell lines) with mit, in combination with MAPK inhibitors, well-established targeted therapy compounds—vemurafenib (BRAF inhibitor; FDA approved in 2011) and trametinib (MEK inhibitor; approved in 2013) —used to treat BRAF-mutated melanoma patients. We treated A375 and SK-HI-SETDB1 cells for 24 h with increasing concentrations of mit, alone or in combination with sublethal doses of either vemurafenib (3 μM; “mit+vem”) or trametinib (3 μM; “mit+tra”). Melanoma cells treated with combinatorial treatments showed decreased cell viability in comparison to mit-alone-treated cells (Figure 6A). Therefore, A375 and SK-HI-SETDB1 functional properties were assessed upon mono- and combinatorial treatment. Vemurafenib- or trametinib-treated A375 and SK-HI-SETDB1 melanoma cells exhibited a modest impairment of migration and invasive rate compared to DMSO-treated cells, whereas combinatorial treatments with mit induced stronger anti-migratory and anti-invasive effects (Figures 6B and 6C). These data suggested that targeted therapies with MAPK inhibitors (vemurafenib and trametinib), in combination with mit, showed an increased efficacy on melanoma cells.
Figure 6.

Treatment with Mithramycin A (mit) in Combination with BRAF/MEK Inhibitors Contributed to Melanoma Cell Death and to an Impairment of Tumor Cell Features
(A) Top panels: cell viability assay of A375 and SK-HI-SETDB1 melanoma cells treated with increasing doses of mit alone or in combination with either sublethal concentrations of vemurafenib (3 μM; mit+vem) or trametinib (3 μM; mit+tra). Cells showed a greater sensibility to the combined therapies after 24 h. Viability of DMSO-treated (control) cells was set as 100%. Bottom panels: boxplots showing A375 and SK-HI-SETDB1 mit, mit+vem, and mit+tra IC50 (nM) after 24 h exposures. (B) Migration scratch assay of A375 and SK-HI-SETDB1 melanoma cells treated according to the established drug-treatment conditions: DMSO, vemurafenib (vem), mit+vem, trametinib (tra), mit+tra. At the endpoint, the percentages of the open-gap area were defined. (C) Invasion assay showing the invasive capacities of A375 and SK-HI-SETDB1 melanoma cells exposed to DMSO, vem, mit+vem, tra, or mit+tra for 24 h.
Mithralog EC-8042 Effectively Induces the Inhibition of SETDB1 Expression and Anti-Tumorigenic Effects in Melanoma
In this study, we provided evidence that mit abolished SETDB1 expression in melanoma cells, reverting its oncogenic potential. In the past years, a renovated interest in adopting mit for clinical purposes has been reported;30,31 however, the application of mit for targeted therapy approaches against cancer is today still limited by lack of efficacy and the presence of side effects. The development of mithramycin analogs (mithralogs) with improved anticancer properties and reduced toxicity represents a novel and promising alternative; in particular, it has been previously shown that the mithralog DIG-MSK (EC-8042) induced strong antitumor effects with less toxicity.32 For this reason, we included in this work an evaluation of the effects produced by stimulation of melanoma cells with the EC-8042 compound. Similarly to what was observed with mit, melanoma cells exposed to increasing doses of EC-8042 (10 nM–10 μM range) showed reduced levels of SETDB1 (Figures 7A and S5A); moreover, EC-8042 treatment resulted in impaired melanoma cell viability (mean IC50 values for A375: 2.75 μM after 24 h of EC-8042 treatment, 203 nM after 48 h, and 67.3 nM upon 72 h treatment; IC50 SK-HI-SETDB1: 5.84 μM, 161.7 nM, and 72.21 nM after 24, 48, and 72 h, respectively) (Figure S5B). Conversely, SETDB1 KD melanoma cells and fibroblasts exhibited no particular sensitivity to EC-8042 treatment (Figure 7B). Combinatorial treatments, including EC-8042 and MAPK inhibitors (vemurafenib/trametinib), led to augmented A375 melanoma cell death (Figure 7C). Next, to test whether EC-8042-mediated SETDB1 inhibition resulted in the loss of SETDB1-mediated tumorigenic effects, we performed in vitro functional assays of melanoma cells treated with 1 μM EC-8042 for 24 h. A375 and SK-HI-SETDB1 cells showed slower migratory and invasive capacities upon EC-8042 exposure in comparison with DMSO-treated cells (Figures 7D and 7E). Finally, we evaluated the expression of SETDB1 downstream targets in melanoma cells following EC-8042 treatment; in a similar way to mit-treated cells, A375 exposed to 1 μM EC-8042 for 24 h displayed lower expression of CRS genes (THBS1, SCG2, MMP1, MMP3, IL8, CCL2, and WNT5A), together with higher expression of MLD (DCT and PMEL) and metabolic (APOE) markers compared to control cells. We could also observe the downregulation of THBS1, SCG2, MMP1, MMP3, and WNT5A and the upregulation of APOE in EC-8042-treated SK-HI-SETDB1 cells (Figures 7F, 7G, S5C, and S5D). To conclude, we demonstrated that EC-8042 effectively targeted SETDB1 expression and showed a strong anti-tumorigenic effect on melanoma cells, representing a valuable and promising therapeutic alternative for SETDB1 targeting in melanoma.
Figure 7.

Mithralog EC-8042 Targeted SETDB1 in Melanoma Cells
(A) Western blot detection of SETDB1 in A375 and SK-HI-SETDB1 cells, showing an EC-8042 dose-dependent SETDB1 inhibition. (B) Cell viability assay of dermal fibroblasts, A375 NT, A375 SETDB1 KD, SK-HI-SETDB1 NT, and SK-HI-SETDB1 SETDB1 KD cells upon EC-8042 stimulation for 24 h. (C) Boxplot representing the A375 IC50 relative to mono (EC-8042 [“EC”]) and combinatorial (EC-8042 + vemurafenib [“EC+vem”]; EC-8042 + trametinib [“EC+tra”]) treatments after 24 h. (D) Migration and (E) Invasion functional assays performed on A375 and SK-HI-SETDB1 cells following exposure to 1 μM EC-8042 for 24 h. (F) qPCR analysis of a panel of CRS, MLD, and metabolic genes in A375 cells treated either with DMSO or with 1 μM EC-8042. Number or replicates: 3–4. (G) THBS1, DCT, APOE, and GAPDH immunodetection in A375 cells treated with either DMSO or EC-8042.
Discussion
Our previous work indicated that SETDB1 induces transcriptomic alterations in melanoma. Functional annotation revealed that many genes upregulated in SETDB1 OE cell lines encoded for protumorigenic factors. Differential expression analysis indicated that SETDB1 upregulation increased the expression of secreted onco-proteins. CRS factors are strongly expressed and drive important tumorigenic features in melanoma; they included proteins belonging to thrombospondin (THBS1),33,34 granin (SCG2),16 matrix metalloproteases (MMP1, MMP3),35 ILs (IL-6, IL-8),36,37 chemokines (CCL2),38 and WNT (WNT5A)39 protein families. We also observed that upregulation of SETDB1 resulted in an increase of melanoma cell release of several secreted proteins, such as MMP8, CXCL16, and CCL2. During tumor development, changes in the tumor cell secretome occur frequently and for this reason, represent a hallmark of carcinogenesis.40 Contrary to our expectations, the level of secreted THBS1 was not affected by SETDB1 overexpression. Due to its multimodular structure, THBS1 is involved in different intra- and extracellular signaling pathways;41,42 in light of this, we speculated that SETDB1 may affect only the intracellular role of THBS1 in melanoma cells. Secretory mechanisms during tumorigenesis are still poorly elucidated. A broad range of cell types release secreted factors via a tight regulation of secretory granule exocytosis mechanisms;43 SCG2 is involved in the biosynthetic processes of secretory granules. Here, we proved that SCG2 protein is mostly located at the perinuclear region, assembled in vesicle-like intracellular structures. Hence, upregulation of SCG2 may be linked to an increase of secretory granule biogenesis, representing a key event for the activation of the secretory machinery and for the release of protumorigenic-secreted factors. Our data also suggested that SCG2 expression has a potential prognostic value, since it is particularly expressed in clinical samples related to patients with melanoma metastases and with low survival rate.
Functional annotation analysis of SETDB1 OE-melanoma cell transcriptome also indicated several downregulated genes that could be classified into two different classes: MLD markers and metabolic enzymes. MLD markers found downregulated following overexpression of SETDB1 in melanoma cells included DCT, TYRP1, TYR, and PMEL. These proteins modulate development and functions of melanocytes.44,45 In melanoma, the role of melanocytic markers is controversial: whereas several studies report a loss of DCT, TYR, and TYRP1 during melanoma progression,46,47 another work shows that melanoma cells can express these markers.48 The expression of differentiation markers positively correlates with a favorable clinical outcome in patients with metastatic melanoma.49 Reduced levels of MLD markers in SETDB1 OE cells suggested that SETDB1-positive cell populations might possess a less-differentiated phenotype. The metabolic factor APOE was also silenced upon SETDB1 induction. APOE mediates anti-tumorigenic effects inducing melanoma regression.19 Therefore, our findings indicated that SETDB1 drives the activation of a secreted protein associated with tumorigenesis and the repression of pro-differentiation/anti-tumorigenic factors. Mechanistically, given its multiple subcellular location,50 SETDB1 might be involved in multiple regulatory mechanisms, as the results of interaction with chromatin regulators, transcription factors, or other interacting partners indicate. It has been recently shown that SETDB1 is able to interact and form complexes with nonhistone substrates, such as P53 and Akt,27,51,52 driving cancer cell growth and oncogenic features. Akt activation, in particular, is involved in the repression of differentiation mechanisms, such as melanin production and melanosome formation, modulating the expression of tyrosinase and tyrosinase-related proteins.53, 54, 55 For this reason, Akt might play a critical role in modulating SETDB1-driven repression of a differentiated phenotype in melanoma.
This bivalent gene-expression signature closely resembled the expression profiling of hIPSC-Mb, which is recently described as a model to investigate molecular features that often occur during melanoma pathogenesis.22 The analysis of the common deregulated genes observed in melanoma cells following SETDB1 overexpression and in hIPSC-Mb (undifferentiated lineage), in comparison to NHMs (differentiated lineage), reported genes belonging to CRS, MLD, and metabolic classes. Since positive-SETDB1 melanoma cell lines are characterized by a more aggressive behavior,12,13 we could speculate that this features reflected the melanoblast-like gene signature.
Given the prominent role of SETDB1 in melanoma progression, a therapeutic strategy able to efficiently target SETDB1 might represent a promising option for melanoma treatment. The antitumor antibiotic mit abrogates the expression of SETDB1 by modulating SP-1 protein activity at the SETDB1 promoter.25 mit modulates antitumor effects in several malignancies, including melanoma;28,56, 57, 58, 59, 60 the compound was used in a phase II clinical trial for the treatment of lung cancer, esophageal cancer, breast cancer, mesothelioma, and gastrointestinal tumors (ClinicalTrials.gov: NCT01624090). Lung cancer cells treated with mit exhibit impaired proliferation and decreased levels of SETDB1; interestingly, these effects were observed only in SETDB1-positive cancer cells.61 A recent work conducted by Guo et al.27 showed that mit impaired SETDB1 levels in different cell lines, including A375 melanoma cells, resulting in strong in vitro and in vivo antitumor effects. Here, we evaluated the effects of mit treatment on viability of melanoma cells with high endogenous levels of SETDB1, observing that they were particularly sensitive to mit treatment, whereas nontumor cells, like dermal fibroblasts, as well as SETDB1-KD melanoma cells, showed an unaltered viability rate upon mit exposure. Furthermore, mit treatment reduced SETDB1 expression in melanoma cells in a dose-dependent fashion. These findings recapitulated the inhibitory effects induced by mit in lung cancer cells. mit treatment also induced drastic morphological changes of melanoma cells, exhibiting larger nuclei and less-pronounced membrane protrusions. Morphological alterations are predictive of tumor cell behavior;62 melanoma cells with ectopic SETDB1 overexpression show an elongated, spindle-shaped morphology,13 which is often observed in aggressive tumor cells, whereas mit treatment reverted this morphological feature.
Melanoma cells treated with mit exhibited impaired migratory and invasive capacity. Similar effects have been described in glioma, salivary adenoid cystic carcinoma, and lung cancer exposed to mit.63, 64, 65 Melanoma cell motility was further investigated by exploiting an in vitro 2D invasion model. This system aimed at recreating the interactions found in a solid-tumor microenvironment between tumor cells and stromal cells (fibroblasts), which sustain tumor cell growth, malignant transformation, and drug-resistance mechanisms.66 mit exposure, however, severely limited the interactions and the functionality of melanoma cells that were in direct contact with fibroblasts, failing to migrate and invade the fibroblast layer. mit-mediated SETDB1 inhibition impacted on transcriptional and secretory mechanism melanoma cells, resulting in reduced levels of CRS factor expression and secretion, together with the upregulation of MLD and metabolic enzymes. Again, SETDB1 deregulation defined a bivalent gene-expression pattern, reflecting melanoma cell properties and behavior.
Next, we assessed the anti-tumorigenic effects of mit in combinatorial treatments, together with compounds able to specifically target and inhibit mutated protein variants of the MAPK pathway, found frequently deregulated in melanoma. BRAF (vemurafenib) and MEK (trametinib) inhibitors achieve remarkable clinical responses, inducing a strong tumor regression in melanoma patients; however, these approaches are limited by acquired mechanisms of drug resistance.4 A promising strategy, aiming at overcoming resistance, would be the definition of novel combinatorial/multi-targeted therapeutic treatments. Our findings indicated that mit, combined with vemurafenib or trametinib, cooperatively enhanced melanoma cell death and anti-migratory and anti-invasive effects, whereas treatments with single MAPK inhibitors were not sufficient to impair melanoma cell motility. Hence, we support a combinatorial and multi-target treatment of melanoma cells based on concomitant SETDB1 and MAPK pathway inhibition as a powerful and effective therapeutic option.
Lastly, we included a novel promising therapeutic agent in our study. Despite showing strong antitumor effects, the application of mit for cancer treatment is still limited because of its side effects. This brought interest in the development of mithralogs with improved properties and lower toxicity. DIG-MSK (EC-8042) is a biosynthetic mithralog able to interfere with the Sp-1 transcriptional process.67 EC-8042 showed strong antitumor activity in vitro and in vivo, together with reduced toxicity, than mit in vivo.32 Therefore, the EC-8042 molecule achieved beneficial effects in preclinical studies for the treatment of several malignancies, including triple-negative breast cancer, ovarian cancer, sarcoma, head and neck cancer, and prostate cancer.68, 69, 70, 71, 72, 73 For all of these reasons, we investigated the effects of EC-8042 in melanoma. Here, we showed that EC-8042 suppressed the expression of SETDB1; melanoma cells were highly sensitive to EC-8042 and in a similar way, as observed following mit treatment, exerted antitumor effects related to SETDB1 mechanisms of action and augmented the action of MAPK inhibitors. Together, these data suggested that EC-8042 might be considered as a valuable option, ideally in relation to SETDB1-abrogative effects and in combination with MAPK inhibitors, for the establishment of novel treatments used in the therapy of melanoma.
In conclusion, our findings support the central role of SETDB1 as a key regulator of molecular events leading melanoma progression and as a promising candidate target for treating advanced melanoma. The development of novel therapeutic strategies aiming at selectively targeting SETDB1 expression and functions may result in significant clinical benefits for melanoma treatment.
Materials and Methods
Cell Lines and Compounds
Melanoma cell lines used in this study were purchased from ATCC and the Leibnitz Institute Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ). The SK-HI-SETDB1 cell line is a subclone generated by the parental SKMEL28 cell line, as previously described,13 characterized by high mean SETDB1 amplification. Identity of melanoma cells was authenticated by a cell-line authentication test. Cells were routinely tested for mycoplasma contamination with the VenorGeM Classic mycoplasma detection kit (Minerva Biolabs). Melanoma cells, NHMs, and dermal fibroblasts (isolated from a healthy patient’s foreskin) were cultured in mouse embryonic fibroblast (MEF) medium, composed of DMEM medium (Life Technologies), 10% heat-inactivated fetal calf serum (FCS; Biochrom), 0.1 mM β-mercaptoethanol (Life Technologies), 1% nonessential amino acids (Sigma-Aldrich), and 1% penicillin/streptomycin (Sigma-Aldrich), and stably kept at 37°C and 5% CO2 in a humidified incubator. Drug-based assays were performed, exposing cultured cells to DMSO-dissolved mit (BioTrend), EC-8042 (under development at EntreChem SL), vemurafenib (Selleckchem), or trametinib (Selleckchem), at defined concentrations (final DMSO concentration in cell culture medium < 0.1%).
Plasmids and Lentiviral Cell Transduction
SETDB1 OE and control (EV) constructs were generated, as previously described.13 Constitutive mammalian vectors containing fluorescent reporter genes (GFP, TOMATO) were used for 2D invasion assay. The DCT promoter-GFP reporter construct (derived from Addgene; #17448) contained the ORF of GFP under the control of a partial DCT promoter sequence. Competent DH5α bacterial cells (Sigma-Aldrich) were used for transformation. A high quantity of pure plasmid DNA was obtained with the EndoFree Plasmid Maxi Kit (QIAGEN), and the DNA plasmid sequence was confirmed by DNA sequencing (LGC Genomics). Lentiviruses containing DNA plasmids were produced in human HEK293T cells and then used for cell transduction in the presence of 2–8 μg/mL polybrene (Sigma-Aldrich) to improve the transduction efficiency. Transduced cells were selected with antibiotic (1–3 μg/mL puromycin, Carl Roth; or 10–15 μg/mL blasticidin, Sigma-Aldrich), accordingly, with the used lentivirus.
Real-Time PCR
Total RNA from cultured cells was isolated using the RNeasy Mini Kit (QIAGEN), following the manufacturer’s instructions. cDNA was generated by reverse transcription of 500 ng of total RNA using the Revert Aid First Strand cDNA synthesis kit (Thermo Fisher Scientific), according to the manufacturer’s protocol. Gene expression was assessed by a real-time PCR reaction using the SYBR Green method. Briefly, cDNA was mixed with SYBR Green Master Mix (Applied Biosystems, Life Technologies); each PCR reaction was loaded in triplicates onto a 96-well plate and run on a 7500 Real-Time PCR System device (Applied Biosystems, Life Technologies). Primers were designed using PrimerBlast or obtained from a Primer Bank database;74 primer efficiency was determined by amplification of the target from serial fold dilutions. The full list of primer pairs used in this study is provided in Table 2. 18s ribosomal RNA (18s) expression was used as an endogenous control for all of the experiments;75 expression results were analyzed using either ΔCt value (for determining the endogenous SCG2 expression in a panel of melanoma cell lines) or ΔΔCt value (for all of the remaining experiments) methods.76 qPCR data were analyzed using 7500 Software, version 2 0.5 (Applied Biosystems).
Table 2.
List of the Primers Used in This Work
| Amplification Target | Forward Sequence, 5′–3′ | Reverse Sequence, 5′–3′ |
|---|---|---|
| h18S | GAGGATGAGGTGGAACGTGT | TCTTCAGTCGCTCCAGGTCT |
| hSETDB1 | CATCCAGGGCAGTGACTAATTG | CGGAGCTTCTGGTCTTTTGG |
| hTHBS1 | GCCATCCGCACTAACTACATT | TCCGTTGTGATAGCATAGGGG |
| hDCT | CCACAGTTCTGACGCTGACA | ACAAGCAAGCAAAGCGGAAA |
| hSCG2 | CCAGGTCACTGGGGAGTCTGCT | TGAGCATCAACAATGCCA |
| hTYRP1 | AAACTTTGGAGAGGGAAAATCT | CACAGGCAATATCCATTGTTG |
| hAPOE | GTTGCTGGTCACATTCCTGG | GCAGGTAATCCCAAAAGCGAC |
| hMMP1 | AAAATTACACGCCAGATTTGCC | GGTGTGACATTACTCCAGAGTTG |
| hMMP3 | CGGTTCCGCCTGTCTCAAG | CGCCAAAAGTGCCTGTCTT |
| hCCL2 | CCTTCATTCCCCAAGGGCTC | GGTTTGCTTGTCCAGGTGGT |
| hWNT5A | ATTCTTGGTGGTCGCTAGGTA | CGCCTTCTCCGATGTACTGC |
| hIL8 | TTTTGCCAAGGAGTGCTAAAGA | AACCCTCTGCACCCAGTTTTC |
| hPMEL | AGGTGCCTTTCTCCGTGAG | AGCTTCAGCCAGATAGCCACT |
| hTYR | GCACAGATGAGTACATGG | TGGGGTTCTGGATTTGTC |
Western Blot
Cultured cells were harvested and then lysed in radioimmunoprecipitation assay (RIPA) buffer (4 M NaCl, 1% IGEPAL, 10% sodium deoxycholate, 10% SDS, 1 M Tris, pH 8). Lysates’ protein concentrations were assessed with the Pierce bicinchoninic acid (BCA) Protein Assay Kit (Thermo Fisher Scientific), following the manufacturer’s instructions. About 15–50 μg of whole-cell lysates was loaded and run on NuPAGE Novex 4%–12% Bis-Tris protein gels (Thermo Fisher Scientific). Proteins were then transferred onto polyvinylidene fluoride (PVDF) membranes (Merck Millipore). Membranes were first incubated for 1 h at room temperature (RT) with a blocking solution (3% BSA in Tris-buffered saline [TBS]) to avoid any unspecific antibody-protein binding and then probed with anti-SETDB1 (Bio-Rad; VMA00243KT), anti-THBS1 (Thermo Fischer Scientific; MA5-13398), anti-DCT (Santa Cruz; sc-74439), anti-APOE (Santa Cruz; sc-390925), or anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Cell Signaling Technology; 2118S) antibodies, diluted in the same blocking buffer during the overnight incubation at 4°C on a shaker. The following day, membranes were washed three times in 1× TBS-Tween buffer and then incubated with the designated horseradish peroxidase (HRP)-conjugated secondary antibody (anti-mouse immunoglobulin G [IgG] HRP-linked antibody, Cell Signaling Technology, 7076S; anti-rabbit IgG HRP-linked antibody, Cell Signaling Technology, 7074S) for 1 h at 4°C. Afterward, membranes were shortly exposed to Luminata Forte Western HRP Substrate (Merck Millipore) and developed with the ChemiDoc Touch Imaging System (Bio-Rad). Acquired images were analyzed by ImageJ software (NIH) and Image Lab (Bio-Rad).
Tissue Microarray (TMA) Analysis
Tissues obtained from patients with melanoma metastases were prepared and assembled in TMA blocks, as previously described,77 and stained with anti-SETDB1 or anti-SCG2 (GeneTex; GTX116446) antibodies. Next, TMAs were scanned by the National Centre for Tumour Diseases (NCT)-Gewebebank facility, Pathology Unit (Heidelberg, Germany). Acquired images were scored by two independent observers; overall scores for each sample were based on the immunohistochemistry score system (score range: 0–12).77 Each patient included in this study released a valid informed consent, in accordance with the ethical vote 2010-318N-MA (Heidelberg University, Germany).
Light and Fluorescent Microscopy
To evaluate morphological alterations of cultured cells upon treatment with the SETDB1 inhibitor, cells were seeded into 6-well plates and treated the following day; after an additional 24 h, cells were observed under a DM LS Light Microscope (Leica). Melanoma cells previously transduced with the DCT promoter-GFP vector were treated in the same way; fluorescent images were acquired with an Eclipse Ti fluorescence microscope (Nikon).
Immunofluorescence
Around 3 × 104 cells were seeded in 8-chamber culture slides (Falcon). After 48 h (or 24 h after treatments), cells were fixed in 4% paraformaldehyde (PFA) for 5 min on ice and an additional 10 min at RT and then permeabilized with 0.1% Triton X-100 for 10 min. Next, cells were preincubated for 2 h with blocking solution (3% BSA) before anti-SCG2 primary antibody incubation overnight at 4°C. The following day, cells were washed and incubated with goat anti-rabbit IgG heavy + light chains (H+L) Alexa Fluor 488 (Abcam; ab150077)-conjugated secondary antibodies for 2 h at RT in the dark. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Roche) for 15 min. Slide mounting was performed with fluorescence mounting medium (Dako). Images were acquired and analyzed by Nikon Imaging Software (NIS)-Elements software (Nikon).
ELISA Proteome Profiler
1 × 106 cells were seeded in T-75 flasks and cultured for 48 h (or 24 h after treatment). Then, cell supernatants were centrifuged to remove particulates; 1 mL of each cell culture supernatant was collected and tested with the Proteome Profiler Human Angiogenesis Array Kit (R&D Systems), following the manufacturer’s instructions. Protein signals were quantified by HLImage++ image analysis software (Western Vision) and ImageJ (NIH).
Transcriptional Profiling Analysis
A functional (GO) annotation of differentially expressed genes was conducted using the Database for Annotation, Visualization and Integrated Discovery (DAVID) tool (https://david.ncifcrf.gov) on gene-expression data from our previous work (HT 144-SETDB1 OE versus EV microarray data13). Transcriptional profiling data of hIPSC-Mb versus NHM were obtained from published work.22
Cell Viability Assay
About 2.5–5 × 103 cells were seeded in 96-well plates and treated the following day with mit or EC-8042, alone or in combinatorial treatments with MAPK inhibitors (vemurafenib or trametinib) at different concentration ranges, depending on the exposure time. After 24, 48, or 72 h, Alamar Blue (Invitrogen) was added to cultured cells’ medium (10% final concentration), and following 4 h incubation, fluorescence emitted by viable cells was read at the excitation wavelength of 560 nm and emission of 590 nm and measured with an Infinite 200 spectrophotometer (Tecan).
Migration Assay
Cells were seeded in culture 2-well inserts (Ibidi) at a density of 3.5 × 104 cells/field. 24 h later, cells were exposed to defined drug treatments and to 1 μg/mL aphidicolin (Sigma-Aldrich), a cell-proliferation inhibitor. Subsequently, inserts were removed, and gap closing, an indicator of cell migration rate, was monitored until the 24-h treatment endpoint. Acquired images were analyzed with TScratch software (CSElab).
Transwell Invasion Assay and 2D Invasion System
Cell invasive properties were tested with the Tumor Invasion System (Corning), following the manufacturer’s guidelines, with some modifications: about 2.5 × 104 cells were cultured in each top chamber, diluted in 0.5% FCS-MEF medium, whereas 20% FCS-MEF medium was added to the lower chamber to stimulate cell invasion. 4 h after seeding, cells were treated, and the following day, invading cells were stained with 4 μg/mL calcein-acetoxymethyl (AM; Corning) in PBS solution for 1 h. Then, images were acquired and fluorescence read with a microplate reader at the excitation of 494 nm and emission of 517 nm. Cell invasive rate was quantified as relative fluorescence units (RFUs). Tumor cell invasive properties were also evaluated by establishing a 2D invasion system.78 Culture 2-well inserts were filled with about 3 × 104 melanoma cells on one side and 3 × 104 fibroblasts on the other. The following day, inserts were removed to allow the two cell types (both labeled with a different fluorochrome) to interact. Once the gap was fully closed, cells were exposed to inhibitor treatment. Fluorescent images were acquired before and after treatment to define the amount of tumor cells invading the fibroblast layer upon drug exposure.
Statistical Analysis
Experiments were run at least in triplicates. Data are represented as mean ± SEM and analyzed using Prism 5.0 software (GraphPad). Statistical two-tailed Student’s t test was applied to compare two conditions, whereas one-way ANOVA statistical test was used to compare multiple conditions. Spearman correlation defined the association of two parameters, and Kaplan-Meier method was used for survival analysis. Statistical significance is indicated with the p value scale (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; “ns” refers to p > 0.5).
Author Contributions
A.F. designed and carried out most of the experiments and wrote the manuscript. T.S. performed additional experiments. L.L, D.N., and V.U. contributed to the analysis and interpretation of the results. V.U., F.M., and L.-E.N. contributed to the implementation of the research. J.U. supervised the project. All authors discussed the results and edited the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Sayran Arif-Said and Jennifer Dworacek for excellent technical assistance. We thank Elias Orouji, Laura Hüser, Karol Granados Blanco, and Sun Qian, who provided scientific insights and expertise during the course of this research. We thank the NCT-Gewebebank facility, Pathology Unit, University of Heidelberg, for the TMA slide-scanning service. This work is part of the doctoral theses of Aniello Federico and Tamara Steinfass. The study was funded by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation; project number 25933240/RTG 2099).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.06.001.
Supplemental Information
References
- 1.Leiter U., Eigentler T., Garbe C. Epidemiology of skin cancer. Adv. Exp. Med. Biol. 2014;810:120–140. doi: 10.1007/978-1-4939-0437-2_7. [DOI] [PubMed] [Google Scholar]
- 2.Whiteman D.C., Green A.C., Olsen C.M. The Growing Burden of Invasive Melanoma: Projections of Incidence Rates and Numbers of New Cases in Six Susceptible Populations through 2031. J. Invest. Dermatol. 2016;136:1161–1171. doi: 10.1016/j.jid.2016.01.035. [DOI] [PubMed] [Google Scholar]
- 3.Gazzé G. Combination therapy for metastatic melanoma: a pharmacist’s role, drug interactions & complementary alternative therapies. Melanoma Manag. 2018;5:MMT07. doi: 10.2217/mmt-2017-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalal B.S., Upadhya D., Pai V.R. Chemotherapy Resistance Mechanisms in Advanced Skin Cancer. Oncol. Rev. 2017;11:326. doi: 10.4081/oncol.2017.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ascierto P.A., Kirkwood J.M., Grob J.J., Simeone E., Grimaldi A.M., Maio M., Palmieri G., Testori A., Marincola F.M., Mozzillo N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012;10:85. doi: 10.1186/1479-5876-10-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jakob J.A., Bassett R.L., Jr., Ng C.S., Curry J.L., Joseph R.W., Alvarado G.C., Rohlfs M.L., Richard J., Gershenwald J.E., Kim K.B. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118:4014–4023. doi: 10.1002/cncr.26724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee J.H., Choi J.W., Kim Y.S. Frequencies of BRAF and NRAS mutations are different in histological types and sites of origin of cutaneous melanoma: a meta-analysis. Br. J. Dermatol. 2011;164:776–784. doi: 10.1111/j.1365-2133.2010.10185.x. [DOI] [PubMed] [Google Scholar]
- 8.Aguissa-Touré A.H., Li G. Genetic alterations of PTEN in human melanoma. Cell. Mol. Life Sci. 2012;69:1475–1491. doi: 10.1007/s00018-011-0878-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wellbrock C., Arozarena I. Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment Cell Melanoma Res. 2015;28:390–406. doi: 10.1111/pcmr.12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sinnberg T., Menzel M., Ewerth D., Sauer B., Schwarz M., Schaller M., Garbe C., Schittek B. β-Catenin signaling increases during melanoma progression and promotes tumor cell survival and chemoresistance. PLoS ONE. 2011;6:e23429. doi: 10.1371/journal.pone.0023429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ceol C.J., Houvras Y., Jane-Valbuena J., Bilodeau S., Orlando D.A., Battisti V., Fritsch L., Lin W.M., Hollmann T.J., Ferré F. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471:513–517. doi: 10.1038/nature09806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miura S., Maesawa C., Shibazaki M., Yasuhira S., Kasai S., Tsunoda K., Maeda F., Takahashi K., Akasaka T., Masuda T. Immunohistochemistry for histone h3 lysine 9 methyltransferase and demethylase proteins in human melanomas. Am. J. Dermatopathol. 2014;36:211–216. doi: 10.1097/DAD.0b013e3182964e02. [DOI] [PubMed] [Google Scholar]
- 13.Orouji E., Federico A., Larribère L., Novak D., Lipka D.B., Assenov Y., Sachindra S., Hüser L., Granados K., Gebhardt C. Histone methyltransferase SETDB1 contributes to melanoma tumorigenesis and serves as a new potential therapeutic target. Int. J. Cancer. 2019;145:3462–3477. doi: 10.1002/ijc.32432. [DOI] [PubMed] [Google Scholar]
- 14.Shi X., Tasdogan A., Huang F., Hu Z., Morrison S.J., DeBerardinis R.J. The abundance of metabolites related to protein methylation correlates with the metastatic capacity of human melanoma xenografts. Sci. Adv. 2017;3:eaao5268. doi: 10.1126/sciadv.aao5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerdes H.H., Rosa P., Phillips E., Baeuerle P.A., Frank R., Argos P., Huttner W.B. The primary structure of human secretogranin II, a widespread tyrosine-sulfated secretory granule protein that exhibits low pH- and calcium-induced aggregation. J. Biol. Chem. 1989;264:12009–12015. [PubMed] [Google Scholar]
- 16.Peitsch W.K., Doerflinger Y., Fischer-Colbrie R., Huck V., Bauer A.T., Utikal J., Goerdt S., Schneider S.W. Desmoglein 2 depletion leads to increased migration and upregulation of the chemoattractant secretoneurin in melanoma cells. PLoS ONE. 2014;9:e89491. doi: 10.1371/journal.pone.0089491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lekmine F., Chang C.K., Sethakorn N., Das Gupta T.K., Salti G.I. Role of microphthalmia transcription factor (Mitf) in melanoma differentiation. Biochem. Biophys. Res. Commun. 2007;354:830–835. doi: 10.1016/j.bbrc.2007.01.075. [DOI] [PubMed] [Google Scholar]
- 18.Pinner S., Jordan P., Sharrock K., Bazley L., Collinson L., Marais R., Bonvin E., Goding C., Sahai E. Intravital imaging reveals transient changes in pigment production and Brn2 expression during metastatic melanoma dissemination. Cancer Res. 2009;69:7969–7977. doi: 10.1158/0008-5472.CAN-09-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pencheva N., Tran H., Buss C., Huh D., Drobnjak M., Busam K., Tavazoie S.F. Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell. 2012;151:1068–1082. doi: 10.1016/j.cell.2012.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riker A.I., Enkemann S.A., Fodstad O., Liu S., Ren S., Morris C., Xi Y., Howell P., Metge B., Samant R.S. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med. Genomics. 2008;1:13. doi: 10.1186/1755-8794-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun Y., Daemen A., Hatzivassiliou G., Arnott D., Wilson C., Zhuang G., Gao M., Liu P., Boudreau A., Johnson L., Settleman J. Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells. Cancer Metab. 2014;2:20. doi: 10.1186/2049-3002-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larribère L., Kuphal S., Sachpekidis C., Sachindra, Hüser L., Bosserhoff A., Utikal J. Targeted Therapy-Resistant Melanoma Cells Acquire Transcriptomic Similarities with Human Melanoblasts. Cancers (Basel) 2018;10:451. doi: 10.3390/cancers10110451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noh H.J., Kim K.A., Kim K.C. p53 down-regulates SETDB1 gene expression during paclitaxel induced-cell death. Biochem. Biophys. Res. Commun. 2014;446:43–48. doi: 10.1016/j.bbrc.2014.02.053. [DOI] [PubMed] [Google Scholar]
- 24.Lee J.K., Kim K.C. DZNep, inhibitor of S-adenosylhomocysteine hydrolase, down-regulates expression of SETDB1 H3K9me3 HMTase in human lung cancer cells. Biochem. Biophys. Res. Commun. 2013;438:647–652. doi: 10.1016/j.bbrc.2013.07.128. [DOI] [PubMed] [Google Scholar]
- 25.Ryu H., Lee J., Hagerty S.W., Soh B.Y., McAlpin S.E., Cormier K.A., Smith K.M., Ferrante R.J. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. USA. 2006;103:19176–19181. doi: 10.1073/pnas.0606373103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karanth A.V., Maniswami R.R., Prashanth S., Govindaraj H., Padmavathy R., Jegatheesan S.K., Mullangi R., Rajagopal S. Emerging role of SETDB1 as a therapeutic target. Expert Opin. Ther. Targets. 2017;21:319–331. doi: 10.1080/14728222.2017.1279604. [DOI] [PubMed] [Google Scholar]
- 27.Guo J., Dai X., Laurent B., Zheng N., Gan W., Zhang J., Guo A., Yuan M., Liu P., Asara J.M. AKT methylation by SETDB1 promotes AKT kinase activity and oncogenic functions. Nat. Cell Biol. 2019;21:226–237. doi: 10.1038/s41556-018-0261-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi E.S., Nam J.S., Jung J.Y., Cho N.P., Cho S.D. Modulation of specificity protein 1 by mithramycin A as a novel therapeutic strategy for cervical cancer. Sci. Rep. 2014;4:7162. doi: 10.1038/srep07162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yokoyama K., Yasumoto K., Suzuki H., Shibahara S. Cloning of the human DOPAchrome tautomerase/tyrosinase-related protein 2 gene and identification of two regulatory regions required for its pigment cell-specific expression. J. Biol. Chem. 1994;269:27080–27087. [PubMed] [Google Scholar]
- 30.Jia Z., Zhang J., Wei D., Wang L., Yuan P., Le X., Li Q., Yao J., Xie K. Molecular basis of the synergistic antiangiogenic activity of bevacizumab and mithramycin A. Cancer Res. 2007;67:4878–4885. doi: 10.1158/0008-5472.CAN-06-3494. [DOI] [PubMed] [Google Scholar]
- 31.Jia Z., Gao Y., Wang L., Li Q., Zhang J., Le X., Wei D., Yao J.C., Chang D.Z., Huang S., Xie K. Combined treatment of pancreatic cancer with mithramycin A and tolfenamic acid promotes Sp1 degradation and synergistic antitumor activity. Cancer Res. 2010;70:1111–1119. doi: 10.1158/0008-5472.CAN-09-3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Núñez L.E., Nybo S.E., González-Sabín J., Pérez M., Menéndez N., Braña A.F., Shaaban K.A., He M., Morís F., Salas J.A. A novel mithramycin analogue with high antitumor activity and less toxicity generated by combinatorial biosynthesis. J. Med. Chem. 2012;55:5813–5825. doi: 10.1021/jm300234t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jayachandran A., Anaka M., Prithviraj P., Hudson C., McKeown S.J., Lo P.H., Vella L.J., Goding C.R., Cebon J., Behren A. Thrombospondin 1 promotes an aggressive phenotype through epithelial-to-mesenchymal transition in human melanoma. Oncotarget. 2014;5:5782–5797. doi: 10.18632/oncotarget.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Borsotti P., Ghilardi C., Ostano P., Silini A., Dossi R., Pinessi D., Foglieni C., Scatolini M., Lacal P.M., Ferrari R. Thrombospondin-1 is part of a Slug-independent motility and metastatic program in cutaneous melanoma, in association with VEGFR-1 and FGF-2. Pigment Cell Melanoma Res. 2015;28:73–81. doi: 10.1111/pcmr.12319. [DOI] [PubMed] [Google Scholar]
- 35.Moro N., Mauch C., Zigrino P. Metalloproteinases in melanoma. Eur. J. Cell Biol. 2014;93:23–29. doi: 10.1016/j.ejcb.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 36.Hoejberg L., Bastholt L., Schmidt H. Interleukin-6 and melanoma. Melanoma Res. 2012;22:327–333. doi: 10.1097/CMR.0b013e3283543d72. [DOI] [PubMed] [Google Scholar]
- 37.Singh R.K., Varney M.L. IL-8 expression in malignant melanoma: implications in growth and metastasis. Histol. Histopathol. 2000;15:843–849. doi: 10.14670/HH-15.843. [DOI] [PubMed] [Google Scholar]
- 38.Payne A.S., Cornelius L.A. The role of chemokines in melanoma tumor growth and metastasis. J. Invest. Dermatol. 2002;118:915–922. doi: 10.1046/j.1523-1747.2002.01725.x. [DOI] [PubMed] [Google Scholar]
- 39.Weeraratna A.T., Jiang Y., Hostetter G., Rosenblatt K., Duray P., Bittner M., Trent J.M. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell. 2002;1:279–288. doi: 10.1016/s1535-6108(02)00045-4. [DOI] [PubMed] [Google Scholar]
- 40.Paltridge J.L., Belle L., Khew-Goodall Y. The secretome in cancer progression. Biochim. Biophys. Acta. 2013;1834:2233–2241. doi: 10.1016/j.bbapap.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 41.Sid B., Sartelet H., Bellon G., El Btaouri H., Rath G., Delorme N., Haye B., Martiny L. Thrombospondin 1: a multifunctional protein implicated in the regulation of tumor growth. Crit. Rev. Oncol. Hematol. 2004;49:245–258. doi: 10.1016/j.critrevonc.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 42.Resovi A., Pinessi D., Chiorino G., Taraboletti G. Current understanding of the thrombospondin-1 interactome. Matrix Biol. 2014;37:83–91. doi: 10.1016/j.matbio.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 43.Burgoyne R.D., Morgan A. Secretory granule exocytosis. Physiol. Rev. 2003;83:581–632. doi: 10.1152/physrev.00031.2002. [DOI] [PubMed] [Google Scholar]
- 44.Guyonneau L., Murisier F., Rossier A., Moulin A., Beermann F. Melanocytes and pigmentation are affected in dopachrome tautomerase knockout mice. Mol. Cell. Biol. 2004;24:3396–3403. doi: 10.1128/MCB.24.8.3396-3403.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raposo G., Marks M.S. Melanosomes--dark organelles enlighten endosomal membrane transport. Nat. Rev. Mol. Cell Biol. 2007;8:786–797. doi: 10.1038/nrm2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lenggenhager D., Curioni-Fontecedro A., Storz M., Shakhova O., Sommer L., Widmer D.S., Seifert B., Moch H., Dummer R., Mihic-Probst D. An Aggressive Hypoxia Related Subpopulation of Melanoma Cells is TRP-2 Negative. Transl. Oncol. 2014;7:206–212. doi: 10.1016/j.tranon.2014.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fang D., Tsuji Y., Setaluri V. Selective down-regulation of tyrosinase family gene TYRP1 by inhibition of the activity of melanocyte transcription factor, MITF. Nucleic Acids Res. 2002;30:3096–3106. doi: 10.1093/nar/gkf424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pak B.J., Lee J., Thai B.L., Fuchs S.Y., Shaked Y., Ronai Z., Kerbel R.S., Ben-David Y. Radiation resistance of human melanoma analysed by retroviral insertional mutagenesis reveals a possible role for dopachrome tautomerase. Oncogene. 2004;23:30–38. doi: 10.1038/sj.onc.1207007. [DOI] [PubMed] [Google Scholar]
- 49.Takeuchi H., Kuo C., Morton D.L., Wang H.J., Hoon D.S. Expression of differentiation melanoma-associated antigen genes is associated with favorable disease outcome in advanced-stage melanomas. Cancer Res. 2003;63:441–448. [PubMed] [Google Scholar]
- 50.Tachibana K., Gotoh E., Kawamata N., Ishimoto K., Uchihara Y., Iwanari H., Sugiyama A., Kawamura T., Mochizuki Y., Tanaka T. Analysis of the subcellular localization of the human histone methyltransferase SETDB1. Biochem. Biophys. Res. Commun. 2015;465:725–731. doi: 10.1016/j.bbrc.2015.08.065. [DOI] [PubMed] [Google Scholar]
- 51.Fei Q., Shang K., Zhang J., Chuai S., Kong D., Zhou T., Fu S., Liang Y., Li C., Chen Z. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat. Commun. 2015;6:8651. doi: 10.1038/ncomms9651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang G., Long J., Gao Y., Zhang W., Han F., Xu C., Sun L., Yang S.C., Lan J., Hou Z. SETDB1-mediated methylation of Akt promotes its K63-linked ubiquitination and activation leading to tumorigenesis. Nat. Cell Biol. 2019;21:214–225. doi: 10.1038/s41556-018-0266-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oka M., Nagai H., Ando H., Fukunaga M., Matsumura M., Araki K., Ogawa W., Miki T., Sakaue M., Tsukamoto K. Regulation of melanogenesis through phosphatidylinositol 3-kinase-Akt pathway in human G361 melanoma cells. J. Invest. Dermatol. 2000;115:699–703. doi: 10.1046/j.1523-1747.2000.00095.x. [DOI] [PubMed] [Google Scholar]
- 54.Chae J.K., Subedi L., Jeong M., Park Y.U., Kim C.Y., Kim H., Kim S.Y. Gomisin N Inhibits Melanogenesis through Regulating the PI3K/Akt and MAPK/ERK Signaling Pathways in Melanocytes. Int. J. Mol. Sci. 2017;18:471. doi: 10.3390/ijms18020471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou S., Sakamoto K. Pyruvic acid/ethyl pyruvate inhibits melanogenesis in B16F10 melanoma cells through PI3K/AKT, GSK3β, and ROS-ERK signaling pathways. Genes Cells. 2019;24:60–69. doi: 10.1111/gtc.12654. [DOI] [PubMed] [Google Scholar]
- 56.Ream N.W., Perlia C.P., Wolter J., Taylor S.G., 3rd Mithramycin therapy in disseminated germinal testicular cancer. JAMA. 1968;204:1030–1036. [PubMed] [Google Scholar]
- 57.Dutcher J.P., Coletti D., Paietta E., Wiernik P.H. A pilot study of alpha-interferon and plicamycin for accelerated phase of chronic myeloid leukemia. Leuk. Res. 1997;21:375–380. doi: 10.1016/s0145-2126(96)00108-7. [DOI] [PubMed] [Google Scholar]
- 58.Liu R., Zhi X., Zhou Z., Zhang H., Yang R., Zou T., Chen C. Mithramycin A suppresses basal triple-negative breast cancer cell survival partially via down-regulating Krüppel-like factor 5 transcription by Sp1. Sci. Rep. 2018;8:1138. doi: 10.1038/s41598-018-19489-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quarni W., Dutta R., Green R., Katiri S., Patel B., Mohapatra S.S., Mohapatra S. Mithramycin A Inhibits Colorectal Cancer Growth by Targeting Cancer Stem Cells. Sci. Rep. 2019;9:15202. doi: 10.1038/s41598-019-50917-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sachrajda I., Ratajewski M. Mithramycin A suppresses expression of the human melanoma-associated gene ABCB8. Mol. Genet. Genomics. 2011;285:57–65. doi: 10.1007/s00438-010-0586-8. [DOI] [PubMed] [Google Scholar]
- 61.Rodriguez-Paredes M., Martinez de Paz A., Simó-Riudalbas L., Sayols S., Moutinho C., Moran S., Villanueva A., Vázquez-Cedeira M., Lazo P.A., Carneiro F. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene. 2014;33:2807–2813. doi: 10.1038/onc.2013.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Friedl P., Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147:992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 63.Seznec J., Silkenstedt B., Naumann U. Therapeutic effects of the Sp1 inhibitor mithramycin A in glioblastoma. J. Neurooncol. 2011;101:365–377. doi: 10.1007/s11060-010-0266-x. [DOI] [PubMed] [Google Scholar]
- 64.Li J., Gao H., Meng L., Yin L. Mithramycin inhibits epithelial-to-mesenchymal transition and invasion by downregulating SP1 and SNAI1 in salivary adenoid cystic carcinoma. Tumour Biol. 2017;39 doi: 10.1177/1010428317708697. 1010428317708697. [DOI] [PubMed] [Google Scholar]
- 65.Lin R.K., Hsu C.H., Wang Y.C. Mithramycin A inhibits DNA methyltransferase and metastasis potential of lung cancer cells. Anticancer Drugs. 2007;18:1157–1164. doi: 10.1097/CAD.0b013e3282a215e9. [DOI] [PubMed] [Google Scholar]
- 66.Flach E.H., Rebecca V.W., Herlyn M., Smalley K.S., Anderson A.R. Fibroblasts contribute to melanoma tumor growth and drug resistance. Mol. Pharm. 2011;8:2039–2049. doi: 10.1021/mp200421k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fernández-Guizán A., Mansilla S., Barceló F., Vizcaíno C., Núñez L.E., Morís F., González S., Portugal J. The activity of a novel mithramycin analog is related to its binding to DNA, cellular accumulation, and inhibition of Sp1-driven gene transcription. Chem. Biol. Interact. 2014;219:123–132. doi: 10.1016/j.cbi.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 68.Pandiella A., Morís F., Ocaña A., Núñez L.E., Montero J.C. Antitumoral activity of the mithralog EC-8042 in triple negative breast cancer linked to cell cycle arrest in G2. Oncotarget. 2015;6:32856–32867. doi: 10.18632/oncotarget.5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fernández-Guizán A., López-Soto A., Acebes-Huerta A., Huergo-Zapico L., Villa-Álvarez M., Núñez L.E., Morís F., Gonzalez S. Pleiotropic Anti-Angiogenic and Anti-Oncogenic Activities of the Novel Mithralog Demycarosyl-3D-ß-D-Digitoxosyl-Mithramycin SK (EC-8042) PLoS ONE. 2015;10:e0140786. doi: 10.1371/journal.pone.0140786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vizcaíno C., Núñez L.E., Morís F., Portugal J. Genome-wide modulation of gene transcription in ovarian carcinoma cells by a new mithramycin analogue. PLoS ONE. 2014;9:e104687. doi: 10.1371/journal.pone.0104687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tornin J., Martinez-Cruzado L., Santos L., Rodriguez A., Núñez L.E., Oro P., Hermosilla M.A., Allonca E., Fernández-García M.T., Astudillo A. Inhibition of SP1 by the mithramycin analog EC-8042 efficiently targets tumor initiating cells in sarcoma. Oncotarget. 2016;7:30935–30950. doi: 10.18632/oncotarget.8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hermida-Prado F., Villaronga M.A., Granda-Díaz R., Del-Río-Ibisate N., Santos L., Hermosilla M.A., Oro P., Allonca E., Agorreta J., Garmendia I. The SRC Inhibitor Dasatinib Induces Stem Cell-Like Properties in Head and Neck Cancer Cells that are Effectively Counteracted by the Mithralog EC-8042. J. Clin. Med. 2019;8:1157. doi: 10.3390/jcm8081157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shinde D., Albino D., Zoma M., Mutti A., Mapelli S.N., Civenni G., Kokanovic A., Merulla J., Perez-Escuredo J., Costales P. Transcriptional Reprogramming and Inhibition of Tumor-propagating Stem-like Cells by EC-8042 in ERG-positive Prostate Cancer. Eur. Urol. Oncol. 2019;2:415–424. doi: 10.1016/j.euo.2018.08.024. [DOI] [PubMed] [Google Scholar]
- 74.Spandidos A., Wang X., Wang H., Dragnev S., Thurber T., Seed B. A comprehensive collection of experimentally validated primers for Polymerase Chain Reaction quantitation of murine transcript abundance. BMC Genomics. 2008;9:633. doi: 10.1186/1471-2164-9-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bas A., Forsberg G., Hammarström S., Hammarström M.L. Utility of the housekeeping genes 18S rRNA, beta-actin and glyceraldehyde-3-phosphate-dehydrogenase for normalization in real-time quantitative reverse transcriptase-polymerase chain reaction analysis of gene expression in human T lymphocytes. Scand. J. Immunol. 2004;59:566–573. doi: 10.1111/j.0300-9475.2004.01440.x. [DOI] [PubMed] [Google Scholar]
- 76.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 77.Wagner N.B., Weide B., Reith M., Tarnanidis K., Kehrel C., Lichtenberger R., Pflugfelder A., Herpel E., Eubel J., Ikenberg K. Diminished levels of the soluble form of RAGE are related to poor survival in malignant melanoma. Int. J. Cancer. 2015;137:2607–2617. doi: 10.1002/ijc.29619. [DOI] [PubMed] [Google Scholar]
- 78.Nnetu K.D., Knorr M., Käs J., Zink M. The impact of jamming on boundaries of collectively moving weak-interacting cells. New J. Phys. 2012;14:115012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.