

Abstract
Objective
Dysregulated adipokine profiles contribute to the pathogenesis of diabetic cardiovascular complications. Endothelial cell (EC) dysfunction, a common pathological alteration in cardiovascular disorders, is exaggerated in diabetes. However, it is unclear whether and how dysregulated adipokines may contribute to diabetic EC dysfunction.
Methods and results
Serum C1q/TNF-Related Protein 5 (CTRP5) were determined in control/diabetes patients, and control/diabetic mice (high-fat diet, HFD). We observed for the first time that serum total CTRP5 was increased, high molecular weight (HMW) form was decreased, but the globular form (gCTRP5) was significantly increased in diabetic patients. These pathological alterations were reproduced in diabetic mice. To determine the pathological significance of increased gCTRP5 in diabetes, in vivo, ex vivo and in vitro experiments were performed. Diabetic atherosclerosis and EC dysfunction were significantly attenuated by the in vivo administration of CTRP5 neutralization antibody (CTRP5Ab). EC apoptosis was significantly increased in diabetic EC (isolated from HFD animal aorta) or high glucose high lipid (HGHL) cultured HUVECs. These pathological alterations were further potentiated by gCTRP5 and attenuated by CTRP5Ab. Pathway specific discovery-driven approach revealed that Nox1 expression was one of the signaling molecules commonly activated by HFD, HGHL, and gCTRP5. Treatment with CTRP5Ab reversed HFD-induced Nox1 upregulation. Finally, Nox1siRNA was used to determine the causative role of Nox1 in gCTRP5 induced EC apoptosis in diabetes. Results showed that gCTRP5 activated the mitochondrial apoptotic signal of EC in diabetes, which was blocked by the silencing Nox1 gene.
Conclusion
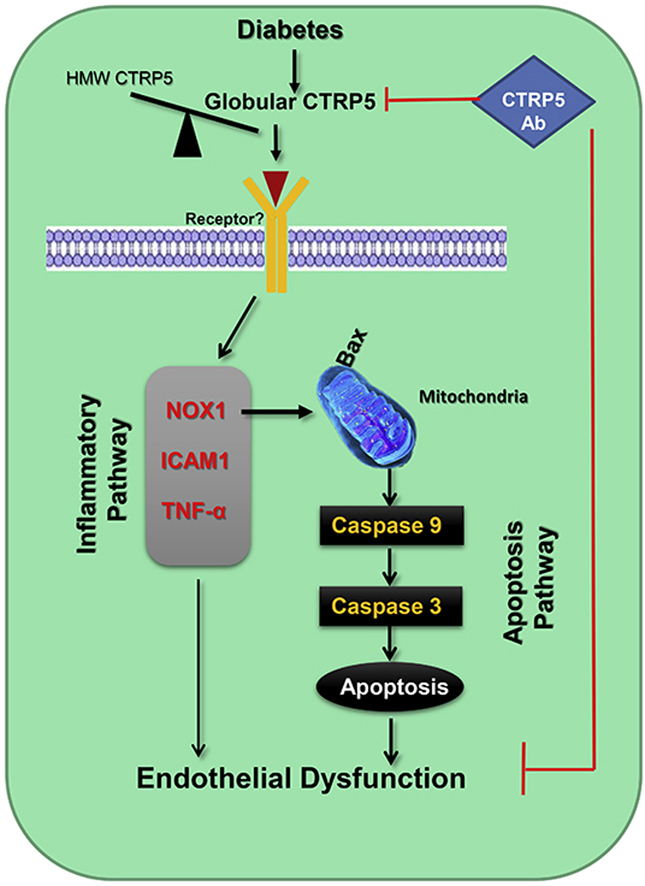
We demonstrated for the first time that gCTRP5 is a novel molecule contributing to diabetic vascular EC dysfunction through Nox1-mediated mitochondrial apoptosis, suggesting that interventions blocking gCTRP5 may protect diabetic EC function, ultimately attenuate diabetic cardiovascular complications.
Keywords: CTRPs, Endothelial dysfunction, Oxidative stress, Diabetes
Graphical abstract
Highlights
-
•
Increased CTRP5 may serve as a marker predicting diabetic macrovascular complications.
-
•
Globular CTRP5 contributes to diabetic vascular endothelial dysfunction.
-
•
Globular CTRP5 upregulates Nox1 levels in diabetic endothelial cells.
-
•
Nox1 mediated mitochondrial apoptosis causes diabetic endothelial dysfunction.
Abbreviations
- EC
endothelial cells
- CTRP5
C1q/TNF-Related Protein 5
- HFD
high-fat diet
- HMW
high molecular weight
- gCTRP5
globular CTRP5
- fCTRP5
full length of CTRP5
- HGHL
high glucose high lipid
- Nox
NADPH oxidase
- HUVEC
human umbilical vein endothelial cells
- DM
diabetes mellitus
- AS
atherosclerosis
- T2DM
type 2 diabetes mellitus
- CAS
carotid atherosclerosis
- CVD
cardiovascular disease
- HbA1c
glycosylated hemoglobin
- FBG
fasting blood glucose
- TC
total cholesterol
- HDL-C
high-density lipoprotein cholesterol
- LDL-C
low-density lipoprotein cholesterol
- TG
triglycerides
- TCH
total cholesterol
- ALT
alanineamino transferease
- AST
aspartate amino transferease
- BMI
Body mass index
- SBP
systolic blood pressure
- DBP
diastolic blood pressure
- LDH
lactate dehydrogenase
- ROS
reactive oxygen species
- AUC
area under the curve
1. Introduction
The prevalence of diabetes mellitus (DM) keeps dramatically rising globally, and it is a major risk factor to cardiovascular disease [1]. Among diabetic vascular complications, atherosclerosis (AS) is a major cause for ischemic heart disease, severe peripheral vascular disease, and stroke [2,3], causing high morbidity and mortality in DM [4,5].
Diabetes is associated with marked fluctuations in the secretory function of adipocytes, and increases the risk of vascular disease development. Dysfunctional adipocytes contribute directly and indirectly to the development of vascular risk factors to vascular disease. Adipokine dysregulations appears to lead to AS in the diabetic setting by influencing vascular endothelial cells (ECs), the first defense barrier and vital defender to the vessel wall [[6], [7], [8]]. However, the causative factor and the underlying mechanism is largely under-investigated.
We and other researchers demonstrated that C1q/tumor necrosis factor-related proteins (CTRPs), a newly discovered secreted protein family, play important role in the remaining homeostasis of vascular system against diabetes and diabetic vasculopathy [[9], [10], [11]]. For Instance, increased CTRP1 promotes endothelial barrier dysfunction under disturbed flow [12]. CTRP9 may exert vasorelaxation and anti-inflammatory effects [13,14]. CTRP3 may serve as a novel biomarker for diabetic retinopathy (DR) preventing inflammatory response [15]. CTRP5 has been identified as a novel metabolic regulating cytokine [[16], [17], [18]]. However, only few CTRP members have been investigated in human studies and results are inconclusive. Due to the complexity of various manifestations CTRPs, the biological function of CTRPs in T2DM, particularly related to accelerated diabetic vascular endothelium dysfunction, is a critical area for research.
Therefore, the objectives of this study were: first, to determine the key CTRPs members in the development of atherosclerosis in diabetes; then to investigate how CTRPs are involved in diabetic vascular endothelium dysfunction in diabetic vasculopathy; and finally, to investigate the responsible underlying mechanisms.
2. Material and methods
2.1. Participants
The human study included 110 subjects aged between 18-80 years-old who were diagnosed in the outpatient or inpatient departments of the Second Affiliated Hospital of Shanxi Medical University between January 2018 and June 2018. It included 40 healthy control subjects (Control group), 35 type 2 diabetes mellitus (T2DM) patients without carotid atherosclerosis (CAS) (T2DM group), and 35 T2DM with carotid atherosclerosis (T2DM + CAS group). All study protocols were approved by the Ethics Committee of the Second Hospital of Shanxi Medical University in accordance with the Declaration of Helsinki. Written informed consents were obtained from all subjects prior to study inclusion. To avoid confounding data, we excluded patients with a history of stroke, type 1 diabetes mellitus (T1DM), valvar heart diseases, severe cardiovascular disease (CVD, cardiac function level at III or IV by New York Heart Association standards), severe hepatic and renal insufficiency, infectious diseases in the past two months, active liver diseases, hemodialysis, malignancy, pregnancy or hyperthyroidism.
2.2. Carotid ultrasonography
Carotid intima-middle thickness (cIMT) was defined as the distance between the two parallel echogenic lines on the far wall of the artery in the longitudinal plane image [19]. Carotid atherosclerotic plaques were defined as a protrusion into the lumen, adding 50% to the thickness of the surrounding intima-media or maximal thickness of 1.5 mm in the carotid bifurcation or along the carotid arterial tree. We diagnosed carotid atherosclerosis (CAS) in the presence of cIMT thickening (cIMT≥1.0 mm) or atheromatous plaque presence.
2.3. Biochemical measurements
We collected patient blood samples after overnight fasting. We measured glycosylated hemoglobin (HbA1c), fasting blood glucose (FBG), total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), triglycerides (TG), total cholesterol (TCH), alanineamino transferease (ALT) and aspartate amino transferease (AST) with standard laboratory techniques on a Hitachi 912 Analyzer (Roche Diagnostics, Germany). Body mass index (BMI), systolic blood pressure (SBP), and diastolic blood pressure (DBP) were measured. Insulin was assessed by commercially available ELISA kit. We used the homeostasis model assessment of insulin resistance (HOMA-IR) to evaluate insulin function. The calculation formula is fasting blood glucose (mg/dL) × fasting blood insulin (μU/mL) /405.
2.4. Enzyme-linked immunosorbent assay (ELISA)
Serum CTRP5 levels were determined by commercial ELISA kit (Cat #SK00594-06, Aviscera Bioscience) per manufacturer's instructions. The level of TNF-α and IL-6 in cell culture supernatant were determined by commercial ELISA kit (Cat #EK0525, #EK0410, Boster Biological Technology) per manufacturer's instructions.
2.5. Type 2 diabetic mice model
All experiments of this study were performed in adherence to the NIH Guidelines on the Use of Laboratory Animals and approved by the Thomas Jefferson University and Shanxi Medical University Committee on Animal Care. Apoe knockout mice (male Apoe-/- mice, 8–10 weeks old) were utilized in this study. The mice were either fed the normal diet (ND), or high-fat diet (HFD) (60% kcal fat, 20% kcal protein, 20% kcal carbohydrate, Cat #D12492; Research Diets) for 12 weeks to induce type 2 diabetes, characterized by abnormal glucose (FBG >12 mmol/L) and insulin resistance. To detect glucose tolerance, mice were intraperitoneally injected with d-glucose (1.5 g/kg) after overnight fasting (12 h), and blood glucose was measured before injection (0 min) and after injection at 15, 30, 60 and 120 min. To assess insulin tolerance, a single dose of Novolin R regular insulin (Novo Nordisk) (0.5 units/kg) was intraperitoneally administered to the mice after fasting for 4 h, and the blood glucose was measured at 0,15, 30, 60, and 120 min.
In order to investigate the function of CTRP5, CTRP5 neutralization antibody (CTRP5Ab, 0.5 μg/g LifeSpan BioSciences) was applied to ND or HFD mice.
2.6. Arterial ring preparation and determination of vasorelaxation
Mouse thoratic aortic vessel segments were harvested and cultured in the Dulbecco's modification of eagle's medium (DMEM)containing normal glucose/normal lipid in the presence and absence of globular CTRP5/CTRP5 (gCTRP5/CTRP5, Aviscera Bioscience). After culturing at 37 °C, 5% CO2 ambient incubator for 24 h, segments were suspended upon stainless steel hooks, and placed in Krebs buffer solution (NaCl 118, KCl 4.75, CaCl2·2H2O 2.54, KH2PO4 1.19, MgSO4·7H2O 1.19, NaHCO3 25, and glucose 10.0 mM) that is constantly permeated with 95% O2 and 5% CO2. Segments were first stretched to generate 2.5 mN force, followed by 0.5 mN increments every 15 min until achieving 4 mN total force. After the vessel segments were balanced and prepared for analysis, norepinephrine (NE, 10−7-10−4M) was administrated. Endothelium-dependent vasorelaxation was determined by measuring the cumulative response to acetylcholine (Ach, 10−7-10−4M). The endothelium-independent vasorelaxation was determined by administrated NaNO2 (10−4M) [13].
2.7. Atherosclerotic lesion analysis
The entire aorta was isolated from ND or HFD mice with or without treatment and fixed in 4% paraformaldehyde overnight. Staining was performed with Sudan IV (Sigma) according to the previous study [20]. Briefly, after fixation, the vessels were stained with Sudan IV dye for 3 min twice. Non-specific staining was then performed with 80% ethanol for 2.5 min. The images were collected by Olympus DP72 with Desense Pro software to analyze the area of red plaques.
2.8. Cell cultures
Human umbilical vein endothelial cells (HUVECs, Cell Applications) were randomized to receive corresponding intervention after reaching 80% confluence. The normal glucose/normal lipid contains 5.5 mM d-glucose in DMEM medium, and the high glucose/high lipids (HGHL) contains 25 mM d-glucose and 200 μM palmitates in DMEM medium [21]. Both culture medium comprises 10% fetal bovine serum (FBS) and premixed endothelial cell growth supplements (Cell Applications) per manufacturer's instructions. The cells were used in 3-4 passages in all experiments.
Primary mouse aortic endothelial cells obtained from ND, HFD and HFD + CTRP5Ab group mice as described in a previous study [22]. Briefly, the aorta was harvested followed by isolating endothelial cells with collagenase type II solution (2 mg/ml). The endothelial cells were collected by centrifugation and suspended in the culture medium. When the cells reached 80% confluence, they were collected and analyzed for the following assay.
2.9. Real-time quantitative PCR
Total RNA from cells or tissues was extracted via a TRIzol reagent (Invitrogen, Carlsbad, CA) per manufacturer instruction. Briefly, cDNA was prepared from 1 μg total RNA using SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific). PCR was then performed using the SYBR-Green Master mix (Thermo Fisher Scientific) in a Quantstudio 5 Real-Time PCR machine (Applied Biosystems). Other primers were purchased from Integrated DNA Technologies (Supplements Table 1). 18S ribosomal RNA served as an internal control. PCR array panels (RT2 Profiler PCR Arrays, Qiagen) was performed per manufacturer's instructions. Data were normalized by the standard comparative cycle threshold (CT) method.
Table 1.
General characteristics of study subjects.
| Variable | Control (n = 40) | T2DM (n = 35) | T2DM + CAS (n = 35) | P |
|---|---|---|---|---|
| Sex (M/F) | 19/21 | 19/16 | 19/16 | 0.791 |
| Smoker (Y/N) | 6/34 | 9/26 | 13/22 | 0.090 |
| Drinker (Y/N) | 5/35 | 7/28 | 8/27 | 0.482 |
| Age (year) | 50.73 ± 11.18 | 50.57 ± 9.89 | 55.69 ± 10.56*# | 0.072 |
| BMI (kg/m2) | 23.29 ± 2.44 | 23.95 ± 3.44 | 25.20 ± 3.17** | <0.001 |
| SBP (mmHg) | 118.28 ± 10.61 | 128.80 ± 12.03** | 138.57 ± 14.83**## | <0.001 |
| DBP (mmHg) | 73.50 ± 10.00 | 78.00 ± 10.00 | 71.00 ± 12.00 | 0.415 |
| FBG (mmol/L) | 4.80 ± 0.48 | 7.77 ± 2.29** | 7.44 ± 3.01** | <0.001 |
| Insulin (μU/ml) | 7.75 ± 2.08 | 10.40 ± 3.60** | 10.20 ± 3.20** | <0.001 |
| HOMA-IR | 1.66 ± 0.47 | 3.55 ± 1.91** | 3.56 ± 1.58** | <0.001 |
| HbA1c (%) | 4.90 ± 0.58 | 8.00 ± 3.10** | 7.90 ± 2.10** | <0.001 |
| TG (mmol/L) | 1.46 ± 0.94 | 1.48 ± 1.42 | 2.01 ± 1.65* | 0.012 |
| TC (mmol/L) | 4.17 ± 1.19 | 4.94 ± 1.22** | 4.93 ± 1.47* | 0.004 |
| LDL-C (mmol/L) | 2.08 ± 0.59 | 2.64 ± 1.12* | 2.89 ± 1.03** | <0.001 |
| HDL-C (mmol/L) | 1.40 ± 0.29 | 1.16 ± 0.29** | 1.04 ± 0.22** | <0.001 |
| ALT (U/L) | 13.25 ± 6.75 | 24.30 ± 23.6** | 17.40 ± 11.90* | <0.001 |
| AST (U/L) | 18.80 ± 5.68 | 22.10 ± 11.00 | 20.20 ± 6.60 | 0.066 |
| ESR (mm/h) | 13.00 ± 10.00 | 14.00 ± 8.00 | 11.00 ± 11.00 | 0.879 |
| hsCRP (mg/L) | 1.95 ± 1.60 | 2.31 ± 2.60 | 2.71 ± 3.64 | 0.201 |
| CTRP5 (ng/ml) | 100.21 ± 37.94 | 168.48 ± 39.25** | 230.93 ± 34.97**## | <0.001 |
*P < 0.05, **P < 0.01 Comparison with Control.
#P < 0.05.
##P < 0.01 Comparison with T2DM.
Data are mean ± SD. BMI, body mass index; SBP, systolic blood pressure; DBP, diastolic blood pressure; FBG, fasting blood-glucose; HOMA-IR, homeostasis model insulin resistance index; HbA1c, glycated hemoglobin A1c; TG, riglycerides; TC, total cholesterol; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; ALT, alanine aminotransferase; AST, aspartic aminotransferase; ESR, erythrocyte sedimentation rate; hsCRP, highsensitive C-reactive protein; CTRP5, C1q/TNF-related protein 5.
2.10. Western blot analysis
Western blot assay was conducted as described in the previous study [13]. Briefly, cells or tissues were harvested and 50 μg total proteins per sample were separated by gel electrophoresis, transferred to a poly-vinylidene fluoride membrane. The membrane was incubated overnight with primary antibodies (Supplements Table 2) at 4 °C, followed by secondary HRP-conjugated antibody (anti-mouse or anti-rabbit antibody, Cell Signaling) at room temperature for 2 h, visualized by enhanced chemiluminescence (Thermo Fisher Scientific) and captured images on ChemiDoc MP imaging system (Bio-rad). The results of Western blot were quantified by densitometry (Image Lab).
Table 2.
Correlation between T2DM + CAS and laboratory characteristics Univariate logistics regression analysis.
| Variable | OR | OR 95%CI | P-value |
|---|---|---|---|
| Sex (M/F) | 1 | 0.390–2.561 | 1 |
| Age (year) | 1.051 | 1.001–1.104 | 0.045 |
| Smoker (Y/N) | 1.707 | 0.614–4.744 | 0.305 |
| Drinker (Y/N) | 1.185 | 0.378–3.720 | 0.771 |
| BMI (kg/m2) | 1.197 | 1.009–1.420 | 0.039 |
| SBP (mmHg) | 1.056 | 1.015–1.098 | 0.006 |
| DBP (mmHg) | 0.973 | 0.923–1.026 | 0.314 |
| FBG (mmol/L) | 1.012 | 0.842–1.215 | 0.902 |
| Insulin (μU/ml) | 0.889 | 0.709–1.115 | 0.309 |
| HOMA-IR | 0.968 | 0.713–1.314 | 0.834 |
| HbA1c (%) | 0.881 | 0.661–1.174 | 0.387 |
| TG (mmol/L) | 1.218 | 0.850–1.745 | 0.282 |
| TC (mmol/L) | 0.900 | 0.558–1.452 | 0.666 |
| LDL-C (mmol/L) | 1.466 | 0.755–2.847 | 0.259 |
| HDL-C (mmol/L) | 0.159 | 0.023–1.091 | 0.061 |
| ALT (U/L) | 0.965 | 0.930–1.001 | 0.060 |
| AST (U/L) | 0.954 | 0.904–1.008 | 0.092 |
| ESR (mm/h) | 1.014 | 0.960–1.070 | 0.619 |
| CRP (mg/L) | 1.014 | 0.882–1.165 | 0.843 |
| CTRP5 (ng/ml) | 1.048 | 1.026–1.071 | <0.001 |
Values were determined by using univariate logistics regression analysis. BMI, body mass index; SBP, systolic blood pressure; DBP, diastolic blood pressure; FBG, fasting blood-glucose; HOMA-IR, homeostasis model insulin resistance index; HbA1c, glycated hemoglobin A1c; TG, riglycerides; TC, total cholesterol; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; ALT, alanine aminotransferase; AST, aspartic aminotransferase; ESR, erythrocyte sedimentation rate; hsCRP, highsensitive C-reactive protein; CTRP5, C1q/TNF-related protein 5.
Mouse and human serum were used for Non-reducing Western blotting. Serum samples were run on 4–20% gradient gel without β-mercaptoethanol and SDS additives.
2.11. Cell viability assay and lactate dehydrogenase (LDH) assay
Cell viability was determined using the MTT [3-(4,5-dimethythiazol-2-yl) 2,5-diphenyltetrazolium bromide] assay per the previous study [23]. In brief, HUVECs were exposed to vehicle, HGHL, and CTRP5 (gCTRP5, full-length CTRP5 [fCTRP5]) (Aviscera Bioscience) at different concentrations with/without the present of HGHL for 24 h. Then, the 0.5 mg/ml MTT was applied and the absorbance at 570 nm was measured by SpectraMax M5 microplate reader (Molecular Devices).
HUVECs were exposed to vehicle, gCTRP5, or HGHL with or without gCTRP5 for 24 h. The cells were harvested and cell medium was collected for LDH assay as performed previously [23]. Briefly, at the end of the observation period, conditioned media, and the cells were collected. LDH activity was determined using pre-prepared reaction mixture solution. The absorbance of samples at 490 nm was measured by the SpectraMax M5 microplate reader (Molecular Devices). The percentage of LDH release was calculated as follows: (A-B)/(C-B) × 100, where A is LDH activity in conditioned media, B is LDH activity in culture media (without cells), and C is LDH activity in cell lysates.
2.12. Detection of reactive oxygen species (ROS) production
The ROS production was assessed by CellROX Deep Red oxidative stress indicator (Invitrogen), which is a fluorogenic dye specifically targeted to ROS in live cells. After treatment, HUVEC were stained with ROS sensor (5 μM) and Hoechst 33342 Solution (1 μg/mL, Thermo Scientific) for nuclei. Then, the cells were washed with PBS. The images were acquired via an Olympus BX51 Fluorescence Microscope and analyzed by Image J software (NIH).
2.13. TUNEL staining
EC apoptosis was determined by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining via Roche In Situ Cell Death Detection Kit (Sigma) per manufacturer's protocol. The nuclei were stained with DAPI. The index of apoptosis was determined by the number of TUNEL positive nuclei/the total nuclei in the field. The images were acquired by Olympus BX51 Fluorescence Microscope, and analyzed by Image J (NIH) software.
2.14. Small interfering RNA transfection
siRNA duplex oligonucleotides were designed for Nox1 target sequences, silencing Nox1 gene expression. Nox1-siRNA and non-target control siRNA were purchased from Santa Cruz. When HUVEC reached 80% confluence, cells were transfected with siRNA via Attractene Transfection Reagent (Qiagen) per manufacturer protocol (final siRNA concentration 100 nM). Cells were then challenged by different interventions in experiments.
2.15. Statistical analysis
Data with normal distribution are expressed as mean ± SD. Skewed distributed variables are expressed with median ± interquartile ranges (IQR). For continuous variables, normal distribution was evaluated by the Shapiro-Wilk test. The unpaired Student t-test was performed for analysis of differences between two groups. For multiple groups, one-way ANOVA was carried out. For non-normal distribution data, differences between groups were tested by the Mann-Whitney U test. The differences in the distribution of categorical variables were evaluated by Chi-square test. The effects of different variables were calculated using univariate logistics regression analysis. We built a multivariate model among the significant variables noted by univariate analysis to determine potential markers for T2DM with CAS. All statistical analyses were performed with SPSS 25 or Graph Pad Prism 8. P values < 0.05 were considered significant.
3. Results
3.1. Serum circulatory CTRP5 may serve as an independent predictor in T2DM combined with CAS
A total of 110 subjects were enrolled in this study. Demographic data, baseline clinical and biochemical characteristics are presented in Table 1. Statistical analysis showed that the levels of CTRP5, SBP, FBG, insulin, HOMA-IR, HbA1c, TC, LDL-C, ALT were increased, and the levels of HDL-C were decreased in the T2DM group compared to the control group. In the T2DM combined with CAS group, CTRP5 level was further increased compared to the T2DM group without CAS (Table 1), which was confirmed by Western blot assay (Fig 1A–1D). Univariate logistics regression analysis indicated that CAS combined with T2DM was positively correlated with age, BMI, SBP and CTRP5 (Table 2).
Fig. 1.

Serum CTRP5 profile was altered in T2DM. A Human serum CTRP5 monomer and gCTRP5 were detected by Western blot. B Human serum HMW CTRP5 was detected by non-reducing Western blot. C Quantification of Western blot from human serum CTRP5 monomer and gCTRP5. D Quantification of non-reducing Western blot from human serum HMW CTRP5. n = 5. **p < 0.01 vs. Control. E ROC curve analysis revealed that CTRP5 distinguishes patients with and without T2DM + CAS. F Mouse serum CTRP5 monomer and gCTRP5 were detected by Western blot. G Mouse serum HMW CTRP5 was detected by non-reducing Western blot. H Quantification of Western blot for mouse serum CTRP5 monomer and gCTRP5. I Quantification of non-reducing Western blot for mouse serum HMW CTRP5. n = 10. **p < 0.01 vs. ND. J CTRP5 distribution was analyzed by Real-time quantitative PCR. Results were expressed as mean ± SD. n = 3. **p < 0.01 vs. CTRP5, C1q/TNF-Related Protein 5; gCTRP5, globular CTRP5. T2DM, type 2 diabetes; HMW, high molecule weight; CON, Control; ND, normal diet; HFD, high-fat diet.
To reveal the relationship between CTRP5 and T2DM with CAS, we built a multivariate model among the significant variables noted by univariate analysis to determine a potential marker for T2DM with CAS. Notably, results showed that CTRP5 was an independent predictor for CAS in T2DM (hazard ratio, 1.053; 95% CI, 1.026–1.080; p < 0.001) (Table 3). In addition, the ROC curve indicated that CTRP5 may serve as a diagnostic marker for CAS combined with T2DM as the area under the curve (AUC) was 0.885 (95% CI, 0.806–0.964; p < 0.001) (Table 4, Fig. 1E).
Table 3.
Multivariate logistics regression analysis.
| Variable | OR | OR 95%CI | P-value |
|---|---|---|---|
| Age (year) | 1.025 | 0.955–1.100 | 0.499 |
| SBP (mmHg) | 1.047 | 0.986–1.113 | 0.135 |
| BMI (kg/m2) | 0.846 | 0.627–1.140 | 0.272 |
| CTRP5 (ng/ml) | 1.053 | 1.026–1.080 | <0.001 |
Values were determined by using multivariate logistics regression analysis.
CTRP5, C1q/TNF-related protein 5.
Table 4.
ROC curves, for T2DM + CAS diagnosis, by circulating CTRP5 level.
| Variable | AUC | SE | P-value | 95%CI |
|---|---|---|---|---|
| CTRP5 (ng/ml) | 0.885 | 0.040 | <0.001 | 0.806–0.964 |
CTRP5, C1q/TNF-related protein 5.
3.2. Globular CTRP5 (gCTRP5) increased in diabetic condition
CTRP5 monomer forms complex aggregates that circulate in the body as high, medium, and low-molecular-weight forms [24]. To further clarify the alteration of CTRP5's profile in the setting of diabetes, human serum samples were analyzed with reducing/non-reducing Western blotting assay. The results demonstrated an increased total CTRP5 (Fig. 1A, up bands and Fig. 1C), a decreased high molecular weight (HMW) (Fig. 1B, 1D) but a significantly increased monomer and globular form of CTRP5 (Fig. 1A, down bands) in T2DM compared with the control group.
To further verify these findings in animals, type 2 diabetic mice models were established. The characteristics of HFD mice demonstrated an effective diabetic model with increased body weight, abnormal glucose tolerance and insulin resistance (Supplementary Figs. 1A–1D). Consistent with the patient's analysis, the CTRP5's profile in animal serum showed a similar pattern as that of human serum (Fig. 1F–1I).
Quantitative real-time PCR analysis was explored to clarify CTRP5 tissue distribution. The results demonstrated that although CTRP5 was widely expressed in multiple tissues, the level of CTRP5 in visceral adipose tissue, perivascular adipose tissue and aorta were ranked on the top three most significantly changed in HFD mice compared with the ND mice, suggesting that CTRP5 may function in the physiological and pathological process of vascular system. Interestingly, the level of CTRP5 in the lung, heart, and spleen was down-regulated in HFD mice compared with the ND mice (Fig. 1J).
3.3. CTRP5 antibody attenuated diabetic vascular injury and gCTRP5 promoted endothelial dysfunction
Next, to explore CTRP5 biological function in the setting of diabetes and to determine whether it induces diabetic endothelial dysfunction, HFD mice were subjected to CTRP5 neutralization antibody (CTRP5Ab) administration. Atherosclerotic plaques were assessed. The results showed that the area of atherosclerotic plaque in the aorta of Apoe-/- mice increased significantly after HFD 12 weeks, while CTRP5Ab treated group markedly reduced the lesions (Fig. 2A, 2B).
Fig. 2.

CTRP5 antibody attenuated vascular injury and gCTRP5 induced diabetic endothelial dysfunction. A Representative figures of the atherosclerotic lesion. CTRP5Ab treatment reduced atherosclerosis lesion area compared to HFD with vehicle. B Bar graphs represented the analysis results of the lesion area. **p < 0.01 vs. ND. ##p < 0.01 vs. HFD. n = 10. C The Aortic segment relaxation was significantly restored in the HFD + CTRP5Ab group compared to the segments from the HFD group with vehicle. D Analysis of relaxation of the aortic segment after Acetylcholine (ACh) was introduced in ND, HFD, and HFD + ACh groups. E gCTRP5 (1 μg/ml) induced endothelial dysfunction with/without HGHL (high glucose: 25.5 mmol/L; high palmitate: 200 μmol/L), which was evaluated by vessel ring assay. F Analysis of vessel ring relaxation of the aortic segment. G gCTRP5 (1 μg/ml) did not cause smooth muscle dysfunction, as the response to acidified NaNO2, an endothelium-independent vasodilator was unchanged. H Analysis of vessel ring relaxation of the aortic segment. **p < 0.01, ##p < 0.01 vs. respective control. In aortic vascular rings experiments, each mouse aorta generated 2-3 rings, n = 8 rings/group. Results are expressed as mean ± SD. ND, normal diet; HFD, high-fat diet; ACh, Acetylcholine; Ab, antibody; ND, normal diet; HFD, high-fat diet.
Then, to investigate the role of CTRP5 in endothelial dysfunction, the aortic segments from ND, HFD, and CTRP5Ab treated HFD mice were analyzed. The vasorelaxation impairment was pronounced in HFD mice compared to ND mice. Notably, the delivery of CTRP5Ab manifestly ameliorated the decline of vasorelaxation caused by HFD (Fig. 2C, 2D).
To identify which form of CTRP5 (monomer CTRP5 or gCTRP5) was responsible for the endothelial dysfunction, isolated aortic segments from ND Apoe-/- mice were incubated in vehicle or gCTRP5/CTRP5 with or without HGHL for 24 h ex vivo. Administrating acetylcholine (an endothelium-dependent vasodilator) resulted in concentration-dependent vasorelaxation in ND mice, while the vasorelaxation was significantly blunted in HGHL treated vessels. Moreover, relaxation was further suppressed by gCTRP5 addition (Fig. 2E, 2F). Importantly, vasorelaxation response to acifified NaNO2, an endothelium-independent NO donor, was not impaired in HGHL-incubated aortic rings with/without gCTRP9 (Fig. 2G, 2H). Surprisingly, monomer CTRP5 did not show any additional suppressive effect on HGHL-induced endothelial dysfunction (data not shown). Taken together, these results demonstrated that gCTRP9 exacerbates HGHL-induced endothelial dysfunction without affecting smooth muscle function, suggesting that elevated gCTRP9 in diabetic individuals may contribute to endothelial dysfunction.
3.4. gCTRP5 aggravated vascular endothelial apoptosis in T2DM
To identify the mechanism responsible for gCTRP5-inducedendothelial dysfunction, we designed three serial experiments to evaluate the effect of gCTRP5 on the function of endothelial cells. First, to obtain direct evidence, HUVECs were incubated for 24 h with various doses of gCTRP5 in the presence or absence of HGHL.Then cell viability was evaluated by MTT cell proliferation assay, and cell toxic assay was assessed by LDH measurement. The vitality of HUVECs was reduced by HGHL in a dose-dependent manner (Supplementary Fig. 2). Of note, the viability of EC was significantly reduced by 1 μg/mL gCTRP5, and it was further reduced in the presence of HGHL. However, it showed no difference between the treatments of 5 μg/mL gCTRP5 and 10 μg/mL gCTRP5 (Fig. 3A). Interestingly, the treatment of the full length of CTRP5 (fCTRP5, 10 μg/mL) did not aggravate the decrease of EC vitality induced by HGHL (Fig. 3B). Consistently, LDH results showed that gCTRP5 (1 μg/mL) markedly induced the cytotoxicity of endothelial cells, especially when co-treated with HGHL (Fig. 3C).
Fig. 3.

gCTRP5 aggravated diabetic vascular EC apoptosis. A HUVEC viability was evaluated by MTT assay. HUVEC were either treated with vehicle or different doses of gCTRP5 (0–5 μg /mL) in the presence or absence of HGHL. n = 8. **p < 0.01, *p < 0.05 vs. vehicle. #p < 0.05, ##p < 0.01 vs. HGHL. B HUVEC viability was evaluated with MTT assay after the cells were treated with vehicle, HGHL or HGHL combined with gCTRP5 (1 μg/mL) or fCTRP5 (10 μg/mL). n = 8. **p < 0.01 vs. vehicle. ##p < 0.01 vs. HGHL. C HUVEC cytotoxicity was determined by LDH assay after the cells were incubated with vehicle or gCTRP5 (1 μg/mL) with or without HGHL. n = 8. **p < 0.01 vs. vehicle. ##p < 0.01 vs. HGHL. D Aortic EC's apoptosis was determined by TUNEL staining. Aortic endothelial cells were isolated from ND, HFD and HFD + CTRP5Ab mice. Green, apoptosis positive cells. Blue, DAPI stained nuclei. E The number of TUNEL positive nuclei/the total nuclei. **p < 0.01 vs. vehicle. ##p < 0.01 vs. HGHL. F Western blot was performed to analyze protein expression of cleaved caspase 3 and caspase 3 in aortic ECs from ND, HFD and HFD + CTRP5Ab group mice. G Quantification of the Western blot results. n = 10. **p < 0.01 vs. ND. ##p < 0.01 vs. HFD. H Western blot was performed to analyze protein expression of cleaved caspase 3 and caspase 3 in HUVECs after incubated in vehicle or gCTRP5 (1 μg/mL) with or without HGHL for 24 h. I Quantification of the western blot results. Data were expressed as mean ± SD. n = 5. **p < 0.01, *p < 0.05 vs. vehicle. ##p < 0.01 vs. HGHL. gCTRP5, globular CTRP5; fCTRP5, full length CTRP5; HGHL, high glucose and high lipid; HUVEC, human umbilical vein cells; ND, normal diet; HFD, high-fat diet.
Next, to evaluate if gCTRP5 aggravates the apoptosis of vascular EC in T2DM, the TUNEL assay was explored on endothelial cells isolated from ND and HFD mice with or without CTRP5Ab treatment. The TUNEL positive cells were significantly increased in aortic EC isolated from HFD mice than ND mice, and CTRP5Ab significantly relieved the increased apoptotic level in treated HFD animal group (Fig. 3D, 3E). Thirdly, the apoptotic signal alteration was assessed ex vivo. The cleaved caspase 3 were markedly increased in aortic endothelial cells isolated from HFD mice than the cells from ND mice, and this increase was markedly alleviated in CTRP5Ab treated mice group (Fig. 3F, 3G). Additionally, cleaved caspase 3 was markedly increased after HUVECs were challenged for 24 h by HGHL, and the increase was exaggerated in the presence of gCTRP5 (Fig. 3H, 3I). Collectively, gCTRP5 contributes to the enhanced EC apoptosis in the setting of diabetes.
3.5. gCTRP5 upregulated the inflammatory signals including Nox1 in diabetic vascular EC
We next investigated the underlying molecular mechanism of gCTRP5 induced endothelial dysfunction. PCR array panels including oxidative stress signals, an inflammation panel, and endoplasmic reticulum stress (ERS) panel were assessed to screen the genes responsible for gCTRP5-exacerbated HUVECs dysfunction. Among the screened genes, the inflammation and oxidative stress related genes were ranked on the top (Fig. 4A). The most significantly changed gene after gCTRP5 treatment was oxidative stress related gene Nox1 (Fig. 4B and 4C), which is a subtype of NADPH oxidase (NOX) and is the main source of ROS production in the vascular system exposed to high glucose [25]. Hence, the role of gCTRP5 in the regulation of Nox1 was further explored both ex vivo and in vitro. Nox1 expression was significantly increased in HFD mouse aortic ECs compared to that of ND mice, and this increase was clearly relieved in CTRP5Ab induced group (Fig. 4F, 4G). Meanwhile, we found that the Nox1 expression was significantly increased after HUVECs were challenged for 24 h with HGHL, and gCTRP5 further elevated the Nox1 level when co-treatment with HGHL (Fig. 4H, 4I). Additionally, the inflammation-related gene TNF-α and IL-6 presented the similar upregulatory pattern when exposed to gCTRP5 and HGHL (Fig. 4A) and so did the level of TNF-α and IL-6 in the supernatants (Fig. 4D, 4E). Hence, gCTRP5 stimulation promoted oxidative stress and inflammatory response in the setting of diabetes; furthermore, the CTRP5 neutral antibody reduced the amplitude of inflammatory responses as demonstrated by the experiments described above.
Fig. 4.

gCTRP5 upregulated Nox1 expression in vascular EC in T2DM. A Gene array assay of endothelial cell biology in HUVEC which were treated with HGHL or gCTRP5 (1 μg/mL) with HGHL condition for 24 h n = 3. **p < 0.01, *p < 0.05 vs HGHL. B Quantitative Real time PCR verified mRNA expression of target genes. n = 6. ##p < 0.01 vs. HGHL. C Quantitative Real time PCR verified mRNA expression of Nox1 genes. n = 6. **p < 0.01, *p < 0.05 vs. vehicle. ##p < 0.01 vs. HGHL. D TNF-α level in cell culture supernatant was detected by ELISA. E IL-6 level in cell culture supernatant was detected by ELISA. n = 5. **p < 0.01, *p < 0.05 vs. vehicle. ##p < 0.01, #p < 0.05 vs. HGHL. F Western blot analysis of Nox1 protein expression in aortic ECs from ND, HFD and HFD + CTRP5Ab mice. G Quantification of the western blot results. n = 6. **p < 0.01, *p < 0.05 vs. ND. ##p < 0.01 vs. HFD. H Western blot analysis of Nox1 protein expression in HUVEC after incubation with gCTRP5 (1 μg/mL) with or without HGHL for 24 h. I Quantification of the Western blot results. n = 5. **p < 0.01, *p < 0.05 vs. vehicle. ##p < 0.01 vs. HGHL. Data were expressed as mean ± SD. gCTRP5, globular CTRP5; HGHL, high glucose and high lipid; HUVEC, human umbilical vein cells; ND, normal diet; HFD, high-fat diet.
3.6. gCTRP5 exacerbated apoptosis of EC in T2DM via Nox1/mitochondrial pathway
Since gCTRP5 may regulate the ROS generation and since mitochondria not only regulates the production of ATP and ROS but also plays an important role in cell apoptosis [26,27], we hypothesized that gCTRP5 induced endothelial dysfunction may be related to the mitochondrial apoptotic pathway. The intensity of fluorescence indicated that gCTRP5 significantly aggravated ROS generation when co-incubated with HGHL (Fig. 5A, 5B). Next, we assessed the gCTRP5's role in apoptotic signals in vivo and in vitro. The caspase 9 activation and Bcl2/Bax level were evaluated, which indicates whether the mitochondrial apoptotic pathway was involved. Compared with the ND group, the caspase 9 activation and Bax level of aortic EC isolated from HFD mice were significantly augmented, while Bcl2 expression was markedly suppressed. These pathologic alterations were partially reversed in CTRP5Ab administrated mice (Fig. 5C–5F). However, the evaluation on caspase 8 (Cytosolic apoptosis signal) did not show the statistical difference. Meanwhile, gCTRP5 significantly increased the cleaved caspase 9 and Bax level in HGHL-cultued HUVECs, and supressed Bcl2 level (Fig. 5G–5J). These above results suggested that gCTRP5 aggravated HGHL-induced endothelial apoptosis through the oxidative stress-induced, mitochondrial-mediated cell death pathway.
Fig. 5.

gCTRP5 aggravated diabetic vascular EC apoptosis via the mitochondrial pathway. A Representative fluorescent images showed mitochondrial ROS productions in HUVEC which were incubated with gCTRP5 (1 μg/mL) with or without HGHL. B Bar graph of quantification for Fluorescence (FL) density. n = 5. **p < 0.01 vs. vehicle. ##p < 0.01 vs. HGHL. C Western blot analysis of protein expression of Bax, Bcl2, cleaved caspase 9 and caspase 9 in aortic endothelial cells isolated from ND, HFD, and HFD + CTRP5Ab mice. D, E and F Quantification of the Western blot results. n = 6–10. **p < 0.01 vs. ND. ##p < 0.01, #p < 0.05 vs. HFD. G Western blot presented protein expression of cleaved Bax, Bcl2, cleaved caspase 9 and caspase 9 in HUVECs after the treatment of gCTRP5 (1 μg/mL) with or without HGHL for 24 h. H, I and J Quantification of the Western blot results. n = 5. **p < 0.01, *p < 0.05 vs. vehicle. ##p < 0.01 vs. HGHL. Data are expressed as mean ± SD. gCTRP5, globular CTRP5; ROS, reactive oxygen species; HGHL, high glucose and high lipid; HUVEC, human umbilical vein cells; ND, normal diet; HFD, high-fat diet.
To investigate the causative role of Nox1 in gCTRP5 induced mitochondrial apoptosis in endothelial cells, Nox1 specific small interfering RNA (Nox1 siRNA) and negative control siRNA (NCsiRNA) was utilized. As depicted in Fig. 6A–6F, after successfully knocked down Nox1 by siRNA (Fig. 6B) in HUVEC, gCTRP5 (1 μg/mL) failed to facilitate the HGHL-induced increase in Bax, cleaved caspase 9, and cleaved caspase 3 level. It also abolished gCTRP5-induced Bcl2 reduction. In addition, Bax expression levels and caspase 9 and caspase 3 activation declined in the Nox1 knockdown group compared to the HGHL group with control siRNA (HGHL + NCsiRNA), and the Bcl2 expression was preserved (Fig. 6C–6F). Consistently, mitochondrial ROS specific staining demonstrated that Nox1 deficiency alleviated gCTRP5-aggravated mitochondrial apoptosis in HGHL cultured EC (Fig. 6G, 6H). Taken together, these results indicate that gCTRP5 exacerbates EC apoptosis in T2DM via the Nox1/mitochondrial cell death pathway.
Fig. 6.

gCTRP5 disrupted mitochondrial signals via augmented Nox1 level in EC. A Western blot examined the expression of Nox1, Bax, Bcl2 proteins and the activity of caspase3, caspase9 in HUVEC which was treated with HGHL or HGHL + gCTRP5 after NCsiRNA or Nox1siRNA transfection. B, C, D, E and F Quantification of Western blot results. n = 5. **p < 0.01 vs. HGHL + NCsiRNA. ##p < 0.01 vs. HGHL + gCTRP5+NCsiRNA. G Representative fluorescent images exhibited mitochondrial ROS productions in HUVEC which treated with HGHL + gCTRP5 for 24 h after transfection of NCsiRNA or Nox1siRNA. H Fluorescence (FL) density quantification for ROS generation. n = 5. ##p < 0.01 vs. HGHL + gCTRP5+NCsiRNA. Data were expressed as mean ± SD. gCTRP5, globular CTRP5; ROS, reactive oxygen species; HGHL, high glucose and high lipid; HUVEC, human umbilical vein cells; NCsiRNA, negative control siRNA; Nox1siRNA, NADPH oxidase1 specific siRNA.
4. Discussion
In the current study, we first report that increased circulating CTRP5 may serve as a novel diagnostic marker and an independent predictor for diabetes with CAS. We also report that the increased globular form of CTRP5 contributing to accelerated endothelial dysfunction in the setting of diabetes. Additionally, we found that gCTRP5 contributes to endothelial cell apoptosis by promoting an oxidative stress induced and Nox-1 mediated mitochondrial cell death pathway. and.CAS is the pathological basis of diabetic macrovascular complications and it can evaluate the severity of macroangiopathy in T2DM. Hence, identification of a predictor of diabetic macrovascular complications (such as CAS) is greatly in need. CTRP5 was identified by multivariate logistics regression analysis followed by the ROC curve assay. The results indicated that CTRP5 may serve as a predictor for diabetic macrovascular complications in T2DM. In addition to our report, accumulated data showed that increased CTRP5 can predict inflammatory response in COPD and metabolic syndrome, and is associated with in-stent restenosis [[28], [29], [30]].
APN exists in multiple oligomers: trimers, hexamers, and HMW, each with distinct biological activities [31]. Globular APN (gAPN), known as the product of full-length adiponectin (fAPN) cleaved by leukocyte elastase secreted from activated monocytes and/or neutrophils, has also been found in human plasma [32,33]. Like APN, CTRP5 proteins form trimers as their basic structural units and further assemble into higher-order oligomeric complexes [24]. However, what and why the forms of CTRP5 in diabetes change, and whether there exist functional globular fragments need to be clarified. In the current study, we first demonstrated that total CTRP5 was increased in T2DM patients serum, HMW form was decreased, but full-length CTRP5 and gCTRP5 were significantly increased. Consistent with the clinical results, the animal experiments suggested that the cleaved CTRP5 may play a role in the development of CAS.
Moreover, both LDH and MTT results showed that gCTRP5, not fCTRP5, reduced EC viability, strongly suggesting that gCTRP5 may be a pathologic response to the setting of T2DM and may play a central role in it.
Several studies have suggested that members of CTRPs may be associated with endothelial function [[13], [14], [15]] with unclear mechanisms. Our data from ex vivo and in vivo experiments demonstrated that gCTRP5 resulted in direct endothelial dysfunction and aggravated HGHL-induced endothelial dysfunction. Besides hyperglycemia, free fatty acid levels have been found to be notably increased in diabetic patients and animals, of which palmitic acid is the predominant component. Hence, HG alone could not mimic the diabetic microenvironment completely, especially in the case of T2DM. Therefore, combination of HG and 200 μM palmitates were used to mimic T2DM [21]. In addition, CTRP5Ab effectively alleviates AS plaque formation in Apoe-/- HFD mice and ameliorates mitochondrial apoptotic signals, suggesting circulating gCTRP5 may be one of the causes of diabetic macrovascular complications in T2DM.
EC monolayers provides a barrier by constituting the luminal surface of all blood vessels [34]. Endothelial cell apoptosis is responsible for endothelial dysfunction that results in endothelial barrier destruction. The emerging evidence showed that mitochondrial dysfunction are the key in the development of endothelial injury and diabetic vasculopathy [35]. Our second discovery was that HFD-induced caspase 3 activation was alleviated by CTRP5Ab, revealing that CTRP5 contributes to endothelial apoptosis in T2DM. We selected 200 μM as the high palmitate concentration in our in vitro experiments based on the lipid range of clinical hyperlipidemia. We further confirmed that gCTRP5 aggravates HGHL-induced EC apoptosis, indicating gCTRP5 is one of the mechanisms responsible for increased susceptibility to vascular disease in diabetic individual than normal people.
Finally, we demonstrated that Nox 1 is the key molecule to promote gCTRP5 associated EC injury. We screened multiples genes to identify potential mechanisms responsible for gCTRP5 regulatory pathways in endothelial cells. Nox1, a subtype of NADPH oxidase (NOX) draw our attention. Excess production of ROS plays a major role in diabetes-accelerated AS [36,37]. Nox has been implicated as the major source of ROS generation in the vessel in response to diabetes [25]. Among all Nox isoforms (Nox1, Nox2, Nox4, and Nox5) involved in the vasculature [[38], [39], [40]], we found that gCTRP5 significantly upregulated Nox1expression. These findings were confirmed in in vivo and ex vivo diabetic models. CTRP5Ab ameliorated Nox1 protein increase in HFD. gCTRP5 could aggravate HGHL-induced upregulation of Nox1 protein expression in vitro. Hence, we showed that Nox1 is involved in gCTRP5-induced endothelial apoptosis in T2DM. Given mitochondrial dysfunction is one of the major causes of vascular injury in T2DM [35,[41], [42], [43]], we investigated the regulatory effect of gCTRP5 on mitochondria. Our data showed that CTRP5Ab administration alleviated caspase 9 overactivation and restored Bcl2/Bax expression in HFD. These results indicate that gCTRP5 leads to EC apoptosis through the mitochondrial pathway in the diabetic setting.
Our study has several limitations. Firstly, the gCTRP5 quantification was based on Western blot assay because gCTRP5 ELISA assay is not currently available. Although Western is sensitive (with detective sensitivity lower than 10 ng) it is not the best quantitative measurement. Secondly, the local concentration of gCTRP5 (perivascular area) remains completely unknown, although it is likely higher than that in circulation. Hence, more research is needed to identify the proper dose of gCTRP5. Thirdly, we utilized male mice in our study because female mice have superior resistance to macrovascular atherosclerosis than males in response to high fat diet due to the enhanced angiogenesis [44]. Moreover, male mice have higher susceptibility than female mice in cardiovascular disease [45]. Fourthly, the clinical population was Asia Han only and the sample size is relatively small. Finally, the mechanism responsible for gCTRP5 selective signaling (as full CTRP5 had no significant effect) remains to be identified.
5. Conclusions
In summary, our findings provide supporting evidence that gCTRP5 contributes to vascular endothelial cell apoptosis by Nox1-mediated mitochondrial dysfunction in T2DM. Elevated gCTRP5 levels play a causative role in diabetic macrovascular endothelium dysfunction in T2DM. Targeting gCTRP5 and its related signaling is a promising therapeutic avenue restoring endothelial functional endothelial cells and mitigating the diabetic macrovascular complication in diabetes. In these connection, we have demonstrated that CTRP5 may serve a valuable role as a biomarker screening for diabetic CAS complications.
Declaration of competing interest
The authors declare no competing interests, financial or otherwise.
Acknowledgments
This work was supported by the following grants: American Diabetes Association [1-17-IBS-297]; W.W. Smith Charitable Trust; Natural Science Foundation of China [81670278, 81670313]; Shanxi “1331 Project” Key Subjects Construction (1331KSC); Key Laboratory of Cellular Physiology (Shanxi Medical University).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101476.
Contributor Information
Jimin Cao, Email: caojimin@126.com.
Yajing Wang, Email: yajing.wang@jefferson.edu.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Cho N.H., Shaw J.E., Karuranga S., Huang Y., da Rocha Fernandes J.D., Ohlrogge A.W., Malanda B. IDF Diabetes Atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018;138:271–281. doi: 10.1016/j.diabres.2018.02.023. [DOI] [PubMed] [Google Scholar]
- 2.Giacco F., Brownlee M. Oxidative stress and diabetic complications. Circ. Res. 2010;107(9):1058–1070. doi: 10.1161/circresaha.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paneni F., Beckman J.A., Creager M.A., Cosentino F. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur. Heart J. 2013;34(31):2436–2443. doi: 10.1093/eurheartj/eht149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Narayan K.M. Type 2 diabetes: why we are winning the battle but losing the war? 2015 Kelly West award lecture. Diabetes Care. 2016;39(5):653–663. doi: 10.2337/dc16-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu J., Zou M.H. Molecular insights and therapeutic targets for diabetic endothelial dysfunction. Circulation. 2009;120(13):1266–1286. doi: 10.1161/CIRCULATIONAHA.108.835223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schalkwijk C.G., Stehouwer C.D. Vascular complications in diabetes mellitus: the role of endothelial dysfunction. Clin. Sci. (Lond.) 2005;109(2):143–159. doi: 10.1042/CS20050025. [DOI] [PubMed] [Google Scholar]
- 7.Gimbrone M.A., Jr., Garcia-Cardena G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc. Pathol. 2013;22(1):9–15. doi: 10.1016/j.carpath.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang H., Dellsperger K.C., Zhang C. The link between metabolic abnormalities and endothelial dysfunction in type 2 diabetes: an update. Basic Res. Cardiol. 2012;107(1):237. doi: 10.1007/s00395-011-0237-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kacso I.M., Potra A.R., Bondor C.I., Moldovan D., Rusu C., Patiu I.M., Racasan S., Orasan R., Vladutiu D., Spanu C., Rusu A., Nita C., Moldovan R., Ghigolea B., Kacso G. Adiponectin predicts cardiovascular events in diabetes dialysis patients. Clin. Biochem. 2015;48(13-14):860–865. doi: 10.1016/j.clinbiochem.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y., Wang X., Lau W.B., Yuan Y., Booth D., Li J.J., Scalia R., Preston K., Gao E., Koch W., Ma X.L. Adiponectin inhibits tumor necrosis factor-alpha-induced vascular inflammatory response via caveolin-mediated ceramidase recruitment and activation. Circ. Res. 2014;114(5):792–805. doi: 10.1161/CIRCRESAHA.114.302439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu G.Z., Liang B., Lau W.B., Wang Y., Zhao J., Li R., Wang X., Yuan Y., Lopez B.L., Christopher T.A., Xiao C., Ma X.L., Wang Y. High glucose/High Lipids impair vascular adiponectin function via inhibition of caveolin-1/AdipoR1 signalsome formation. Free Radic. Biol. Med. 2015;89:473–485. doi: 10.1016/j.freeradbiomed.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Z.H., Li C., Chen J.W., Shen Y., Gao J., Shen W.F., Zhang R.Y., Wang X.Q., Lu L. C1q/TNF-related protein 1 promotes endothelial barrier dysfunction under disturbed flow. Biochem. Biophys. Res. Commun. 2017;490(2):580–586. doi: 10.1016/j.bbrc.2017.06.081. [DOI] [PubMed] [Google Scholar]
- 13.Zheng Q., Yuan Y., Yi W., Lau W.B., Wang Y., Wang X., Sun Y., Lopez B.L., Christopher T.A., Peterson J.M., Wong G.W., Yu S., Yi D., Ma X.L. C1q/TNF-related proteins, a family of novel adipokines, induce vascular relaxation through the adiponectin receptor-1/AMPK/eNOS/nitric oxide signaling pathway. Arterioscler. Thromb. Vasc. Biol. 2011;31(11):2616–2623. doi: 10.1161/ATVBAHA.111.231050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jung C.H., Lee M.J., Kang Y.M., Lee Y.L., Seol S.M., Yoon H.K., Kang S.W., Lee W.J., Park J.Y. C1q/TNF-related protein-9 inhibits cytokine-induced vascular inflammation and leukocyte adhesiveness via AMP-activated protein kinase activation in endothelial cells. Mol. Cell. Endocrinol. 2016;419:235–243. doi: 10.1016/j.mce.2015.10.023. [DOI] [PubMed] [Google Scholar]
- 15.Yan Z., Zhao J., Gan L., Zhang Y., Guo R., Cao X., Lau W.B., Ma X., Wang Y. CTRP3 is a novel biomarker for diabetic retinopathy and inhibits HGHL-induced VCAM-1 expression in an AMPK-dependent manner. PloS One. 2017;12(6) doi: 10.1371/journal.pone.0178253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmid A., Kopp A., Aslanidis C., Wabitsch M., Muller M., Schaffler A. Regulation and function of C1Q/TNF-related protein-5 (CTRP-5) in the context of adipocyte biology. Exp. Clin. Endocrinol. Diabetes. 2013;121(5):310–317. doi: 10.1055/s-0032-1333299. [DOI] [PubMed] [Google Scholar]
- 17.Lei X., Rodriguez S., Petersen P.S., Seldin M.M., Bowman C.E., Wolfgang M.J., Wong G.W. Loss of CTRP5 improves insulin action and hepatic steatosis. Am. J. Physiol. Endocrinol. Metab. 2016;310(11):E1036–E1052. doi: 10.1152/ajpendo.00010.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li C., Chen J.W., Liu Z.H., Shen Y., Ding F.H., Gu G., Liu J., Qiu J.P., Gao J., Zhang R.Y., Shen W.F., Wang X.Q., Lu L. CTRP5 promotes transcytosis and oxidative modification of low-density lipoprotein and the development of atherosclerosis. Atherosclerosis. 2018;278:197–209. doi: 10.1016/j.atherosclerosis.2018.09.037. [DOI] [PubMed] [Google Scholar]
- 19.Chambless L.E., Folsom A.R., Clegg L.X., Sharrett A.R., Shahar E., Nieto F.J., Rosamond W.D., Evans G. Carotid wall thickness is predictive of incident clinical stroke: the Atherosclerosis Risk in Communities (ARIC) study. Am. J. Epidemiol. 2000;151(5):478–487. doi: 10.1093/oxfordjournals.aje.a010233. [DOI] [PubMed] [Google Scholar]
- 20.Wang S., Zhang C., Zhang M., Liang B., Zhu H., Lee J., Viollet B., Xia L., Zhang Y., Zou M.H. Activation of AMP-activated protein kinase alpha2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. Nat. Med. 2012;18(6):902–910. doi: 10.1038/nm.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chokpaisarn J., Urao N., Voravuthikunchai S.P., Koh T.J. Quercus infectoria inhibits Set7/NF-kappaB inflammatory pathway in macrophages exposed to a diabetic environment. Cytokine. 2017;94:29–36. doi: 10.1016/j.cyto.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J.M., Chen A.F., Zhang K. Isolation and primary culture of mouse aortic endothelial cells. JoVE. 2016;118 doi: 10.3791/52965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y., Lau W.B., Gao E., Tao L., Yuan Y., Li R., Wang X., Koch W.J., Ma X.L. Cardiomyocyte-derived adiponectin is biologically active in protecting against myocardial ischemia-reperfusion injury. Am. J. Physiol. Endocrinol. Metab. 2010;298(3):E663–E670. doi: 10.1152/ajpendo.00663.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong G.W., Krawczyk S.A., Kitidis-Mitrokostas C., Revett T., Gimeno R., Lodish H.F. Molecular, biochemical and functional characterizations of C1q/TNF family members: adipose-tissue-selective expression patterns, regulation by PPAR-gamma agonist, cysteine-mediated oligomerizations, combinatorial associations and metabolic functions. Biochem. J. 2008;416(2):161–177. doi: 10.1042/BJ20081240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christ M., Bauersachs J., Liebetrau C., Heck M., Gunther A., Wehling M. Glucose increases endothelial-dependent superoxide formation in coronary arteries by NAD(P)H oxidase activation: attenuation by the 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor atorvastatin. Diabetes. 2002;51(8):2648–2652. doi: 10.2337/diabetes.51.8.2648. [DOI] [PubMed] [Google Scholar]
- 26.Li X., Fang P., Li Y., Kuo Y.M., Andrews A.J., Nanayakkara G., Johnson C., Fu H., Shan H., Du F., Hoffman N.E., Yu D., Eguchi S., Madesh M., Koch W.J., Sun J., Jiang X., Wang H., Yang X. Mitochondrial reactive oxygen species mediate lysophosphatidylcholine-induced endothelial cell activation. Arterioscler. Thromb. Vasc. Biol. 2016;36(6):1090–1100. doi: 10.1161/ATVBAHA.115.306964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Placido A.I., Pereira C.M.F., Correira S.C., Carvalho C., Oliveira C.R., Moreira P.I. Phosphatase 2A inhibition affects endoplasmic reticulum and mitochondria homeostasis via cytoskeletal alterations in brain endothelial cells. Mol. Neurobiol. 2017;54(1):154–168. doi: 10.1007/s12035-015-9640-1. [DOI] [PubMed] [Google Scholar]
- 28.Jiang F., Yang M., Zhao X., Liu R., Yang G., Liu D., Liu H., Zheng H., Zhu Z., Li L. C1q/TNF-Related Protein5 (CTRP5) as a biomarker to predict metabolic syndrome and each of its components. Int. J. Endocrinol. 2018;2018:7201473. doi: 10.1155/2018/7201473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li D., Wu Y., Tian P., Zhang X., Wang H., Wang T., Ying B., Wang L., Shen Y., Wen F. Adipokine CTRP-5 as a potential novel inflammatory biomarker in chronic obstructive pulmonary disease. Medicine (Baltim.) 2015;94(36) doi: 10.1097/MD.0000000000001503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen Y., Li C., Zhang R.Y., Zhang Q., Shen W.F., Ding F.H., Lu L. Association of increased serum CTRP5 levels with in-stent restenosis after coronary drug-eluting stent implantation: CTRP5 promoting inflammation, migration and proliferation in vascular smooth muscle cells. Int. J. Cardiol. 2017;228:129–136. doi: 10.1016/j.ijcard.2016.11.034. [DOI] [PubMed] [Google Scholar]
- 31.Tsao T.S., Murrey H.E., Hug C., Lee D.H., Lodish H.F. Oligomerization state-dependent activation of NF-kappa B signaling pathway by adipocyte complement-related protein of 30 kDa (Acrp30) J. Biol. Chem. 2002;277(33):29359–29362. doi: 10.1074/jbc.C200312200. [DOI] [PubMed] [Google Scholar]
- 32.Fruebis J., Tsao T.S., Javorschi S., Ebbets-Reed D., Erickson M.R., Yen F.T., Bihain B.E., Lodish H.F. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc. Natl. Acad. Sci. U. S. A. 2001;98(4):2005–2010. doi: 10.1073/pnas.041591798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waki H., Yamauchi T., Kamon J., Kita S., Ito Y., Hada Y., Uchida S., Tsuchida A., Takekawa S., Kadowaki T. Generation of globular fragment of adiponectin by leukocyte elastase secreted by monocytic cell line THP-1. Endocrinology. 2005;146(2):790–796. doi: 10.1210/en.2004-1096. [DOI] [PubMed] [Google Scholar]
- 34.Nakashima Y., Wight T.N., Sueishi K. Early atherosclerosis in humans: role of diffuse intimal thickening and extracellular matrix proteoglycans. Cardiovasc. Res. 2008;79(1):14–23. doi: 10.1093/cvr/cvn099. [DOI] [PubMed] [Google Scholar]
- 35.Qin R., Zhang L., Lin D., Xiao F., Guo L. Sirt1 inhibits HG-induced endothelial injury: role of Mff-based mitochondrial fission and Factin homeostasis-mediated cellular migration. Int. J. Mol. Med. 2019;44(1):89–102. doi: 10.3892/ijmm.2019.4185. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Brandes R.P., Weissmann N., Schroder K. NADPH oxidases in cardiovascular disease. Free Radic. Biol. Med. 2010;49(5):687–706. doi: 10.1016/j.freeradbiomed.2010.04.030. [DOI] [PubMed] [Google Scholar]
- 37.Armitage M.E., La M., Schmidt H.H., Wingler K. Diagnosis and individual treatment of cardiovascular diseases: targeting vascular oxidative stress. Expet Rev. Clin. Pharmacol. 2010;3(5):639–648. doi: 10.1586/ecp.10.40. [DOI] [PubMed] [Google Scholar]
- 38.Gray S.P., Di Marco E., Okabe J., Szyndralewiez C., Heitz F., Montezano A.C., de Haan J.B., Koulis C., El-Osta A., Andrews K.L., Chin-Dusting J.P., Touyz R.M., Wingler K., Cooper M.E., Schmidt H.H., Jandeleit-Dahm K.A. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation. 2013;127(18):1888–1902. doi: 10.1161/CIRCULATIONAHA.112.132159. [DOI] [PubMed] [Google Scholar]
- 39.Chen F., Qian L.H., Deng B., Liu Z.M., Zhao Y., Le Y.Y. Resveratrol protects vascular endothelial cells from high glucose-induced apoptosis through inhibition of NADPH oxidase activation-driven oxidative stress. CNS Neurosci. Ther. 2013;19(9):675–681. doi: 10.1111/cns.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lassegue B., Griendling K.K. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2010;30(4):653–661. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tait S.W., Green D.R. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010;11(9):621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 42.Tan C., Dlugosz P.J., Peng J., Zhang Z., Lapolla S.M., Plafker S.M., Andrews D.W., Lin J. Auto-activation of the apoptosis protein Bax increases mitochondrial membrane permeability and is inhibited by Bcl-2. J. Biol. Chem. 2006;281(21):14764–14775. doi: 10.1074/jbc.M602374200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Z., Mo X., Gong Q., Pan Q., Yang X., Cai W., Li C., Ma J.X., He Y., Gao G. Critical effect of VEGF in the process of endothelial cell apoptosis induced by high glucose. Apoptosis. 2008;13(11):1331–1343. doi: 10.1007/s10495-008-0257-y. [DOI] [PubMed] [Google Scholar]
- 44.Rudnicki M., Abdifarkosh G., Rezvan O., Nwadozi E., Roudier E., Haas T.L. Female mice have higher angiogenesis in perigonadal adipose tissue than males in response to high-fat diet. Front. Physiol. 2018;9:1452. doi: 10.3389/fphys.2018.01452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patten R.D. Models of gender differences in cardiovascular disease. Drug Discov. Today Dis. Model. 2007;4(4):227–232. doi: 10.1016/j.ddmod.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.