

Abstract
Cardiovascular disease is the leading cause of death and disability worldwide with increased oxidative stress and reduced NO bioavailability serving as key risk factors. For decades, elevation in protein abundance and enzymatic activity of xanthine oxidoreductase (XOR) under hypoxic/inflammatory conditions has been associated with organ damage and vascular dysfunction. Recent reports have challenged this dogma by identifying a beneficial function for XOR, under similar hypoxic/acidic conditions, whereby XOR catalyzes the reduction of nitrite (NO2-) to nitric oxide (NO) through poorly defined mechanisms. We previously reported that hydrogen sulfide (H2S/sulfide) confers significant vascular benefit under these same conditions via NO2- mediated mechanisms independent of nitric oxide synthase (NOS). Here we report for the first time the convergence of H2S, XOR, and nitrite to form a concerted triad for NO generation. Specifically, hypoxic endothelial cells show a dose-dependent, sulfide and polysulfide (diallyl trisulfide (DATS)-induced, NOS-independent NO2- reduction to NO that is dependent upon the enzymatic activity of XOR. Interestingly, nitrite reduction to NO was found to be slower and more sustained with DATS compared to H2S. Capacity for sulfide/polysulfide to produce an XOR-dependent impact on NO generation translates to salutary actions in vivo as DATS administration in cystathionine-γ-lyase (CSE) knockout mice significantly improved hindlimb ischemia blood flow post ligation, while the XOR-specific inhibitor, febuxostat (Febx), abrogated this benefit. Moreover, flow-mediated vasodilation (FMD) in CSE knockout mice following administration of DATS resulted in greater than 4-fold enhancement in femoral artery dilation while co-treatment with Febx completely completely abrogated this effect. Together, these results identify XOR as a focal point of convergence between sulfide- and nitrite-mediated signaling, as well as affirm the critical need to reexamine current dogma regarding inhibition of XOR in the context of vascular dysfunction.
Keywords: Sulfide, Nitric oxide, Xanthine oxidase, Nitrite, Polysulfide, Hydrogen sulfide
Graphical abstract
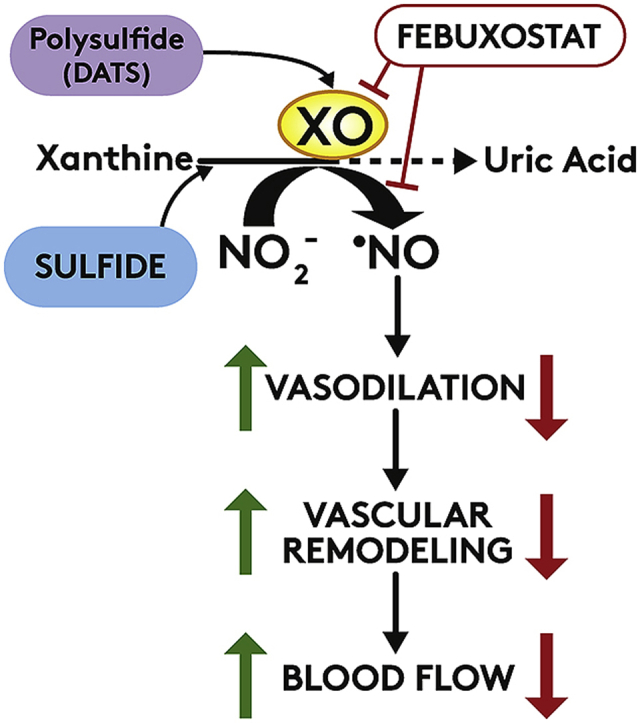
Highlights
-
•
Hydrogen sulfide mediated XOR reduction of nitrite is a major source of NO in hypoxia.
-
•
Hydrogen sulfide inhibits XOR-derived uric acid formation while concomitantly enhancing nitrite reductase activity.
-
•
Hydrogen sulfide/polysulfide acts to influence electron flux between Fe–S clusters and Mo-co domain of XOR.
-
•
Polysulfide increases hypoxic tissue blood flow and NO bioavailability in a XOR dependent manner.
1. Introduction
Cardiovascular disease (CVD), a leading cause of death worldwide, is becoming even more prevalent as modern life amplifies risks [1]. Several factors contribute to increased risk of CVD, including hypertension, obesity, diabetes, and a sedentary lifestyle [2,3]. However, a recently emerging predictor of obesity/diabetes and allied CVD issues is hyperuricemia, which refers to elevated (above 6.0 mg/dL) circulating uric acid (UA) [4,5]. While hyperuricemia is an accepted predictor for poor cardiovascular and metabolic outcomes, its role in causation is yet to be established. Nonetheless, elevation in circulating UA is indicative of enhanced xanthine oxidoreductase (XOR) activity, the sole source of UA in mammals [6]. This is important as increased XOR activity is also associated with cardiovascular disease due to its capacity for oxidant production and subsequent contribution to endothelial dysfunction, and loss of vascular homeostasis [[7], [8], [9]].
Xanthine oxidoreductase refers to both xanthine dehydrogenase (XDH) and the xanthine oxidase (XO) form of the same enzyme. XOR is transcribed from a single gene Xdh, which serves to oxidize hypoxanthine to xanthine, and xanthine to UA while reducing NAD+ to NADH. Under hypoxic/inflammatory conditions, XDH is post-translationally converted to XOR via limited proteolysis (irreversible) or oxidation of critical cysteine residues (reversible) [10]. XOR also oxidizes hypoxanthine and xanthine to UA; yet, its affinity for NAD+ is significantly reduced (9-fold) compared to XDH, and thus XOR utilizes molecular O2 as an electron acceptor resulting in the production O2●- and H2O2 [11]. As such, under hypoxic/inflammatory conditions, XOR is reported to be a significant source of oxidants that contribute to the pathobiology of vascular disease processes [[12], [13], [14], [15], [16], [17]]. In fact, since the discovery of XOR-derived oxidants as central contributors to ischemia/reperfusion injury in 1982, elevation in XOR activity has been dogmatically associated with damage and dysfunction. This deleterious role for XOR has been sustained over the past three decades by numerous reports indicating benefit from XOR inhibition in inflammatory disease processes [13,[18], [19], [20]]. However, this paradigm has been challenged by recent studies describing a novel salutary function for XOR under the same conditions where inhibition would potentially be considered protective. Reports describe a nitrite reductase function, whereby XOR catalyzes the one-electron reduction of NO2- to generate beneficial NO [21,22]. It has been proposed that the micro-environmental conditions requisite for XOR-derived NO generation represent the same hypoxic and acidic conditions that limit or uncouple eNOS activity suggesting XOR as a surrogate source of NO in this milieu [10].
In addition to NO, another gaseous signaling molecule, hydrogen sulfide (H2S), plays a key regulatory role in various physiological and pathological processes [[23], [24], [25]]. A growing number of reports demonstrate H2S and its enzymatic source, cystathionine γ-lyase (CSE) to be protective in the vasculature [[26], [27], [28], [29], [30]]. Similar to XOR, H2S, and allied sulfide metabolites have been proposed to regulate NO metabolism during hypoxia/inflammation, highlighting novel yet poorly understood biochemical pathways to increase NO bioavailability for promoting ischemic vascular remodeling [21]. Herein, we describe the convergence of H2S/sulfide metabolites, XOR-catalyzed NO2- reduction, and NO signaling where H2S and sulfide metabolites amplify NO production from XOR and NO2- to protect against vascular ischemia.
2. Materials and methods
2.1. Chemicals and reagents
Chemicals and tissue culture reagents were obtained from Sigma unless otherwise noted. Anhydrous sodium sulfide was purchased from Alfa‐Aesar Inc. Anti‐CD31 antibody was from BD Biosciences (San Jose, CA, USA), and anti-α-SMA antibody was obtained from Sigma-Aldrich. Vectashield plus DAPI was from Vector Laboratories. All secondary fluorophore‐labeled antibodies were obtained from Jackson Immunoresearch Inc (West Grove, PA, USA). Human umbilical vein endothelial cells (HUVECs) were from Lifeline Cell Technology, CA, USA.
2.2. Cell culture and treatments
Human umbilical vein endothelial cells (HUVECs) were purchased from Lifeline Cell Technology (Cat. No. FC-0044) and cultured in VascuLife® Basal Medium (Cat. No. LM-0002) supplemented with the appropriate LifeFactors® Kit (No. LL-0003). All cells were grown in tissue culture flasks under normoxic conditions at 37°C, 5% CO2, and 21% O2. Their media was changed every 2–3 days. The HUVECs were starved 16h post-confluence in Endothelial Based Media (EBM) supplemented with 0.5% FBS, nonessential amino acids, Pen Strep, and l-glutamine. Then the HUVECs were incubated in the hypoxic chamber (5% CO2, 37°C, 1% O2) for 4h. HUVECs from passages 2–4 were used in the experiments.
2.3. Animals
Twelve-week-old male CSE knockout (CSE KO) mice were used in this study as reported earlier [27]. Mice were housed at the Louisiana State University Health Sciences Center-Shreveport animal resources facility, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. All animal studies were approved by the LSU Institutional Animal Care and Use Committee (LSU IACUC Protocol# P-08-021) and in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
2.4. Mouse hindlimb ischemia model and treatment routes
Chronic hindlimb ischemia was induced in 12-16-week-old CSE knockout male mice, as we have reported previously [27]. After anesthesia with ketamine (100 mg/kg) and xylazine (10 mg/kg), the left femoral artery was dissected, separated, ligated and excised distal to profunda femoris artery to create the hindlimb ischemia model. Mice were randomly assigned to different experimental groups by one investigator and were treated and evaluated by a second blinded investigator. Polysulfide donor, diallyl trisulfide (DATS; 200 μg/kg) was administered in the retro-orbital capillary plexus with or without the XOR-specific inhibitor, febuxostat (Febx) in drinking water (50 mg/L) during the length of the study.
2.5. Novadaq SPY imaging analysis
The SPY imaging device (Novadaq Technologies) was used to quantify collateral vessel perfusion, as we have previously described [27]. Briefly, a bolus injection of 30 μl ICG (IC-Green, Akorn Pharmaceutical, Lake Forest, IL) was administered retro-orbitally, and angiograms were captured by an array of light-emitting diodes at a wavelength of 806 nm and recorded for 1 min. Angiograms were taken before and post-ligation on days 1, 3, and 7, and percent change in blush rates were calculated as mentioned [27].
2.6. Nitrite reductase activity assay
A Sievers 280i NO analyzer was used to measure H2S mediated NO production as we have reported [21]. Endothelial cells were exposed to 4h of either hypoxia (1% O2) or normoxia (21% O2), each with 5% CO2. Cells were harvested, resuspended at 1 × 106 cells/mL in Hanks’ balanced salt solution (HBSS), and were added to the reaction chamber containing deoxygenated HBSS at pH 7.4. Sodium sulfide (sulfide/H2S) or DATS was injected into the chamber to form a 50 μM solution, and NO production was measured over time. Separate experiments with hypoxic endothelial cells plus nitrite and febuxostat were also performed. Cell free purified protein experiments were performed with purified XO (20 mU), which was then injected into the chamber with 100 μM nitrite. Sodium sulfide (final concentrations of 10 μM or 100 μM) or DATS (final concentrations of 10 μM or 100 μM) was then injected into the chamber and NO production was determined over time. NO production was determined by integrating the emission signal over time calibrated to a standard curve of nitrite (0.1, 0.5, 1, 10, and 100 μM) reduced to NO in sodium iodide/glacial acetic acid. Rate of NO production from either sulfide or DATS was calculated from known NO release derived from standard curve of DETA NONOate. These values were reported as μmols of NO release per minute.
2.7. XOR activity assay
XOR activity was evaluated by measuring uric acid production as described previously [31]. Briefly, purified XO (Sigma Cat# X4500) was added to a 200 μl reaction mixture containing 50mM potassium phosphate buffer (pH 7.4) and 0.15mM xanthine solution (sigma Cat# X-0626) at room temperature. The measurements were recorded at A 290 nm for 5min by a spectrophotometer. XOR activity (ΔA290 nm/minute) was obtained by using the maximum linear absorbance from both the test and blank. XOR activities were calculated by using millimolar extinction coefficient of uric acid at 290 nm using the equation below [31].
Units/mg protein = (ΔA/min × 1000)/(1.22 × 104 × mg mL-1 reaction mixture); where, the molar absorption coefficient of uric acid is 1.22 × 104 M-1cm-1.
2.8. Flow mediated dilation (FMD)
Vascular function was determined by mouse FMD with modifications from the previously reported method [32]. Experimental cohorts of Control (PBS), Febx (50 g/L in drinking water), DATS (200 μg/kg retro-orbital) or Febx + DATS, were treated for 3 days, and subjected to FMD. Briefly, mice were anesthetized with isoflurane and fur was removed from the hindlimbs. The animals were then placed on a warmed ultrasound table equipped with ECG. A vascular occluder (5 mm diameter, Harvard Apparatus) was placed around the proximal hindlimb to induce transient occlusion of the vessels of the distal hindlimb as an ischemic trigger. The Doppler ultrasound probe (VEVO 3100, VisualSonics) was manually aligned over the femoral artery, distal to the occluder, to take baseline recordings of the blood vessel for diameter (M mode) and mean velocity (PW mode). The vascular occluder was inflated manually with an air-filled syringe for 1-min, and deflated. Measurements of diameter and blood flow velocity were recorded for 180s at 30s intervals. The recorded loops were analyzed by Vevo LAB analysis software.
2.9. Statistical analysis
Data were reported as mean ± standard error of the mean (SEM) for all groups. Statistical analysis was performed with GraphPad Prism using Student’s t-test, one-way ANOVA and two-way ANOVA with Tukey post-hoc test, Mann–Whitney or Kruskal–Wallis analysis of variance with Dunn's multiple-comparison tests. A p-value of <0.05 was considered to be statistically significant.
3. Results
3.1. Sulfide mediates in vitro reduction of NO2- to NO
XOR-catalyzed, nitrite-dependent NO release from HUVECs was assessed. When normoxic or hypoxic HUVECs were placed in the NOA reaction chamber, addition of increasing concentrations of nitrite (final concentrations of 25, 50 and 100 μM) (Fig. 1A&C) showed no response, indicating no change in NO production. However, addition of same concentrations of sodium sulfide (sulfide/Na2S) induced significant NO production, especially in hypoxic cells (Fig. 1B&D). Duration of hypoxia treatment was also assessed for impact on abundance of NO generated upon reaction with sulfide. Total NO generated by hypoxic HUVECs increased with increasing doses of sulfide (final concentrations of 25 and 50 μM) at increasing times of hypoxia (0, 3, 4 and 5h) (Fig. 1E). While time in hypoxia elevated NO generation, the relationship was not linear; thus, a hypoxic time of 4h (unless specified) was chosen for the remaining cell experiments. Generation of NO from hypoxic (4h) HUVECs increased with increasing doses of sulfide (final concentrations of 25, 50 and 100 μM) compared to corresponding normoxic controls (Fig. 1F). The presence of sulfide or nitrite alone did not result in NO production (Supplementary Fig. 1).
Fig. 1.

Sulfide induces NO release from hypoxic endothelial cells. HUVECs (1 × 106 cells) were assessed for sulfide-mediated NO generation using enhanced chemiluminescence. Buffer was exposed to 25, 50, 100 μM of nitrite (A) or Sulfide (B) in the NOA reaction chamber to establish a normoxic cell baseline; or under hypoxia nitrite (C) or Sulfide (D). Quantitation of NO generation from cells exposed to 25 and 50 μM of Sulfide at various times of hypoxia (0h, 3h, 4h and 5h) (E) NO generation from cells exposed with 25, 50 and 100 μM of Sulfide at 4h normoxia or hypoxia (F). n = 6; *p = 0.000002, 0.000060 and 0.000183 respectively for panel E; *p<0.005 for panel F.
3.2. Polysulfide is more potent than sulfide in stimulating NO2--dependent NO generation by XOR
To explore characteristics of sulfide dependent nitrite reduction to NO by XOR, we compared different sulfide donors Na2S (sulfide) versus DATS polysulfide. A significant increase in NO formation by hypoxic HUVECs was observed with addition of either 100 μM of Sulfide (Fig. 2A) or DATS (Fig. 2B) in the NOA reaction chamber. However, release of NO via sulfide addition was immediate, compared to slower and more sustained NO release with DATS treatment. To determine contributions of nitric oxide synthases (NOS) and investigate the role of XOR as an important source of NO generation, hypoxic (4h) HUVECs were exposed to the global NOS inhibitor l-NAME (200 μM) or Febx (10 nM) (Fig. 2C–F). The presence of l-NAME did not alter observed levels of NO release (Fig. 2C&D); however, Febx completely abrogated NO generation verifying XOR dependence (Fig. 2E&F). The average rate of NO production was quantified from sulfide (Fig. 2G) and DATS (Fig. 2H). To further confirm the role of sulfide(s) in XOR-catalyzed nitrite reduction, purified XO (0.02 U) and NaNO2- (100 μM) were added to the reaction followed by sulfide or DATS (Fig. 3). A significant increase in NO formation was observed with either 100 μM sulfide or 100 μM DATS (Fig. 3A). However, addition of DATS produced a 4-fold greater amount of NO than sulfide; yet, at a slower and more sustained rate (Fig. 3B&C).
Fig. 2.

NO generation is independent of NOS. Exposure of hypoxic cells (HUVECs; 4h) to Sulfide (100 μM) (A) or DATS (50 μM) (B). Exposure of hypoxic cells (HUVECs) to Sulfide (100 μM) and l-NAME (200 μM) (C) or DATS (100 μM) and l-NAME (200 μM) (D). Exposure of hypoxic cells (HUVECs) to Sulfide (100 μM) and Febx (10 nM) (E) or DATS (100 μM) and Febx (10 nM) (F). Average rate of NO production, quantitation from A-F for sulfide (G), *p = 0.001, and DATS (H), *p = 0.0005, where n = 6.
Fig. 3.

Both Sulfide and DATS induce XO-catalyzed NO2-reduction to NO. Purified XO (20 mU) in HBSS was added to nitrite (100 μM) followed by sulfide (100 μM Sulfide) or DATS (100 μM) (A). Data in A are representative profiles of NO generation from the NOA reaction chamber. Total NO produced (nmol) by sulfide and DATS quantified from panel A (B). Average rate of NO formation induced by sulfide and DATS is presented (C). n = 4.
3.3. Sulfide and xanthine regulate XOR-catalyzed reduction of NO2- to NO in a concentration-dependent manner
Although NO production from XOR and NO2- is enhanced with greater concentrations of sulfide, XOR-catalyzed xanthine oxidation to UA is initially increased and then reduced as sulfide concentration increases, (Fig. 4A). The presence of xanthine (100 μM) did not affect DATS (1 μM)-mediated XOR reduction of NO2-; however, the same concentration of xanthine significantly increased sulfide (1 μM)-induced NO formation from XOR and NO2- (Fig. 4B). Similar results were observed with greater concentrations of DATS or sulfide (10 or 100 μM).
Fig. 4.

Dose-dependent effects of Sulfide and DATS on xanthine oxidation and NO2-reduction. Purified XO (20 mU, dissolved in HBSS) was exposed to Sulfide (0–50 μM), and both XOR-dependent oxidation of xanthine to UA (λ = 295 nm) and XOR-dependent NO2- reduction to NO were assessed (A). Total NOx was determined in hypoxic HUVECs treated with or without xanthine (100 μM) and Sulfide or DATS (1–100 μM) (B). Average rate of NO formation induced by 10 μM or 100 μM sulfide (C) and DATS in the presence or absence of Zinc acetate and/or DTPA (D). For panel A, *p<0.0001, #p<0.05, n = 4, compared to group with 0 μM of Sulfide; for B-D *p = 0.006, **p = 0.0014, ***p = 0.0170; #p = 0.04; n = 4.
3.4. The influence of XOR metal centers on sulfide/polysulfide-stimulated, XOR-catalyzed reduction of nitrite to NO
Diethylenetriamine pentaacetate (DTPA) was used to assess the impact of Fe/S clusters in XOR. DTPA has a high affinity for divalent cations such as Fe and was thus used to determine if blockade (by Fe chelation) of the two Fe/S centers in XOR affects sulfide/polysulfide-mediated stimulation of XOR-derived NO from NO2-. The presence of DTPA (100 μM) significantly diminished NO generation stimulated by either 10 μM sulfide (Fig. 4C) or 10 μM DATS (Fig. 4D) indicating the Fe/S centers are requisite for sulfide/polysulfide-mediated effects. Likewise, the presence of zinc acetate diminished sulfide/polysulfide-mediated effects suggesting the Mo-co is also required for this process to be operative (Fig. 4C&D).
3.5. The effects of electron flux on XOR reduction of nitrite
Electron flux between the FAD and the Mo-co domains of XOR is illustrated in Fig. 5A. Dichlorophenolindophenol (DCPIP) and diphenyleneiodonium (DPI) were used to evaluate electron flux near the Mo-co and the FAD, respectfully. DCPIP can intercept electrons between the Mo-co and the first Fe/S center whereas DPI is a global flavin inhibitor. Both DCPIP and DPI inhibited XOR-catalyzed NO formation from NO2- stimulated by xanthine whereas DPI did not alter DATS-mediated XOR-dependent NO generation (Fig. 5B and C). Moreover, and interestingly, the presence of DATS does not result in the reduction of the FAD to FADH2 (Fig. 5D), whereas oxidation of XOR is significantly reduced upon dithionite addition (Fig. 5D).
Fig. 5.

Electron flux between the Mo-co and proximal Fe/S center is requisite for Sulfide and DATS-mediated effects. Schematic of electron flux in XOR reduction of nitrite (A). Purified XO (20 mU, dissolved in HBSS) plus NO2- (100 μM) were exposed to either DPI and/or DCPIP (after/before addition of DATS) and NO release was assessed (B) and quantified (C). The capacity for DATS to reduce the FAD of XOR was determined by following alterations in absorbance (λ = 450 nm) of purified XO in KPi pH 7.4 (D). n = 4; *p<0.001.
3.6. Sulfide-mediated stimulation of XOR-catalyzed reduction of NO2-in vivo regulates vascular function
To examine the impact of sulfide-mediated stimulation of XOR-derived NO generation in vivo, a murine chronic ischemia model of FAL was used. Administration of DATS (0.5 mg/kg twice per day) significantly restored ischemic hindlimb blood flow by seven days post ligation (Fig. 6A&B). Conversely, treatment with the polysulfide donor DATS and the XOR inhibitor Febx did not significantly alter the blood flow in ischemic hindlimbs compared to the Febx group (Fig. 6B). We measured the formation of mature vessel density in CSE KO mice by dual staining with anti-CD31 and anti α-SMA antibodies, and DAPI counterstaining. Representative images of ischemic muscle tissues from CSE KO mice (PBS, DATS, Febx alone and DATS + Febx) showing vessel densities in Supplementary Fig. 2A. Angiogenic and arteriogenesis indices were significantly increased in DATS-treated CSE KO ischemic tissue compared to PBS or Febx alone controls (Supplementary Fig. 2B&C). Additionally, co-treatment of Febx with DATS significantly reduced the ischemic angiogenesis index compared to DATS alone treatment. We next examined the effects of Febx with DATS in the FAL model to understand the role of XOR on NO availability. Fig. 6C&D report plasma, ischemic, and non-ischemic tissue levels of NOx (NO2- + NO3-) in CSE knockout mice treated with DATS, Febx, or both (DATS + Febx). Febx significantly inhibited both DATS-induced ischemic angiogenesis and NOx bioavailability indicating XOR is important for DATS-induced NO formation.
Fig. 6.

In vitro effects of DATS on XOR-derived NO are operative in vivo. Representative images demonstrating restoration of ischemic limb blood flow from CSE KO mice treated with DATS, Febx, or DATS + Febx (A) and quantification thereof (B). Total NO levels of Plasma (C) and ischemic and non-ischemic tissue of skeletal muscle (D) Vessel dilation (% change) in femoral artery at 30, 60 and 300 s in PBS, DATS, Febx + DATS or Febx alone. n = 4 and * = p<0.0001, # = p<0.005, ‡p = 0.007, & p = 0.0086.
Lastly, we assessed the role of XOR on DATS dependent vascular reactivity with Febx treatment using a non-invasive flow mediated vasodilation (FMD) model (similar to human assessment) in CSE KO mice [33]. In this FMD model, transient hindlimb ischemia (1 min) with a pressure cuff in CSE KO mice did not induce femoral artery vasodilation upon reperfusion (Fig. 6E). Notably, this was corrected by DATS with an increase in FMD (8% dilation) compared to PBS treatment. However, Febx treatment completely blunted DATS mediated femoral artery dilation in the FMD model. Together, these results demonstrate that Febx inhibits DATS-induced increases in NO bioavailability, the ability of DATS to restore flow-mediated vasodilation and reduces blood flow restoration in ischemic hindlimbs.
4. Discussion
Xanthine oxidoreductase (XOR) is a ubiquitously expressed molybdoflavin enzyme that catalyzes the terminal two steps of purine catabolism in primates. Upregulation of XOR under hypoxic/inflammatory conditions has been associated with deleterious outcomes for decades, especially in the vascular compartment where XOR-derived oxidants and UA are thought to mediate vascular dysfunction and end organ damage [17,34]. Hypoxia inducible factor-1α (HIF-1α) has been implicated in upregulation genes implicated in regulation of metabolism and angiogenesis, including Xdh [35,36]. However, the mechanisms of oxygen-mediated effects on XO-XDH has been elusive. Recent reports have identified a salutary function for XOR in this same hypoxic/inflammatory milieu as a source of NO [10,21,37,38]. It has been convincingly demonstrated that XOR (both XO and XDH) can catalyze the reduction of NO2- to NO under hypoxic/acidic conditions similar to those that favor NOS uncoupling and thus diminish NOS-catalyzed NO production. We and others have also established that sulfide demonstrates significant vascular benefit/protection under these same hypoxic conditions via NO2--mediated mechanisms independent of NOS [21,27,39].
Previous work from our group and others has reported that hypoxia/ischemia significantly enhances endothelial XOR mRNA and protein expression, and activity [7,21,35,40]. In the present work, we used hypoxic cells (HUVECs), to observe a dose-dependent, sulfide-induced, NOS independent NO2- reduction to NO (Figs. 1 and 2). Interestingly, we also found the polysulfide, DATS to mediate NO generation from NO2- (Fig. 3) in a manner that was more sustained than with sulfide. Furthermore, both sulfide and DATS-mediated NO generation from NO2- was dependent upon the enzymatic activity of XOR (Fig. 4A). These in vitro data mechanistically elucidate our previous observations of febuxostat-mediated inhibition of H2S-induced blood flow recovery in a murine model of chronic ischemia [21].
The driving molecular effect of sulfide/polysulfide on the capacity of XOR to reduce NO2- must be reduction of XOR as the oxidized enzyme is incapable of catalyzing reduction reactions. For example, reduction of NO2- occurs at the Mo-co domain, when the valence of the Mo is either Mo(V) or Mo(IV) [41]. It has also been reported that hypo/xanthine (directly reduces Mo-co) and NADH (reduces Mo-co via retrograde electron flow from FAD) can be used to achieve the reduction of the Mo-co and subsequent NO generation in the presence of NO2-. In the experiments described in Fig. 1, Fig. 2, Fig. 3, there was no XOR reducing substrate (hypo/xanthine or NADH) present and thus the required reduction (Mo(IV)) or partial reduction (Mo(V)) of the enzyme must be provided by sulfide or DATS. If this is the case, then the presence of sulfide should be manifest in alteration of XOR-catalyzed oxidation of xanthine to UA as this two-electron oxidation process is frustrated if the enzyme is partially (Mo(V)) or fully (Mo(IV)) reduced. Indeed, Sulfide (1–50 μM), diminishes UA formation from xanthine while concomitantly enhancing NO formation (Fig. 4A). However, this same phenomenon was not realized with DATS as there was no difference in NO generation in the presence or absence of xanthine (Fig. 4B). This may indicate that DATS is capable of greater XOR reduction (e.g. greater Mo(IV)) and thus inhibits the reaction with xanthine and thus, xanthine has no observable impact on subsequent NO generation from NO2-. Likewise, this may explain why there is only a partial effect of xanthine in the presence of sulfide if indeed sulfide only solicits a partial reduction of the Mo-co to Mo(V); a valence state capable of reducing NO2- to NO [41]. However, it does not readily explain why lower levels of sulfide (0.05 μM, Fig. 4A) enhance XOR-catalyzed oxidation of xanthine to UA. We postulate that these levels of sulfide may induce subtle turnover of XOR sufficient to consume all residual O2; the presence of which inhibits XOR-catalyzed NO2- reduction. While these hypotheses are plausible, only extensive low-temperature electron paramagnetic resonance (EPR) experiments will be able to provide evidence of their validity.
Whereas the Mo-co is the recognized site of NO2- reduction, electron donation to the enzyme and subsequent electron flux to the Mo-co domain can be achieved via reduction of the FAD or Fe/S centers. To establish the impact of sulfide/polysulfide on electron flux in XOR, experiments were designed to either inhibit the capacity of the Fe/S centers (Fe chelation via DTPA) to transfer electrons or the Mo-co (zinc acetate) to accept electrons. As seen in Fig. 5, both inhibition of the Fe/S centers and the Mo-co results in diminution of XOR-catalyzed NO2- reduction indicating these two redox centers of enzyme are requisite for sulfide/polysulfide-mediated effects. Likewise, experiments were designed to determine if an operational FAD is required for sulfide/polysulfide-mediated stimulation of NO generation (Fig. 5C). The presence of DPI, a global Flavin inhibitor, frustrated xanthine-driven NO generation from XOR and NO2- as seen previously [42]; however, blockade of the FAD did not impact DATS-induced NO generation by XOR suggesting the FAD is not required for DATS-mediated effects. This observation is validated by experiments demonstrating incapacity of DATS to reduce the FAD to FADH2 (Fig. 5D). Importantly, oxidized XOR was reduced by dithionite confirming Fe–S centers were responsive. Furthermore, experiments whereby electrons are “stolen” from the enzyme between the Mo-co and the first adjacent Fe/S cluster confers loss of NO2- reductase activity indicating electron flux between this Fe/S center and the Mo-co is requisite for sulfide/polysulfide-stimulated XOR-derived NO generation from NO2-. Again, while these experiments are indicative of required/contributory electron transfer, low-temperature EPR will be required for definitive confirmation.
While the mechanistic details defining sulfide/polysulfide-driven, XOR-dependent NO generation are crucial to understanding the micro-environmental conditions where this process may be operational, the capacity for sulfide or DATS to produce an XOR-dependent impact on NO generation that translates to salutary actions in vivo is both unique and biologically important. As we have previously reported, DATS significantly improved blood flow in a chronic ischemia model of FAL, whereas the presence of Febx abrogated this benefit suggesting XOR activity is required to mediate salutary actions associated with DATS in this setting. Although we observed a significant increase in angiogenesis with DATS (Supplementary Fig. 2), Febx co-treatment did significantly inhibit DATS-mediated angiogenesis, indicating a beneficial role of XOR for NO-mediated angiogenesis. Further, XOR inhibition also blunted vasoreactivity as Febx inhibited DATS-mediated vasodilation, as assessed by FMD (Fig. 6E). Additionally, DATS-treated mice demonstrate elevated NOx in their plasma and skeletal muscle which is diminished by co-administration of Febx confirming an important role for XOR. These in vivo results affirm the critical need for reexamination of dogma regarding inhibition of XOR in the context of vascular dysfunction. For example, analysis of data derived from the recent CARES study revealed that febuxostat increased the risk for all-cause mortality in hyperuricemic patients with preexisting cardiovascular conditions when compared to allopurinol [43]. If this conclusion is indeed valid, then a potential contributor to enhanced risk may be the superior efficacy of febuxostat for inhibiting XOR bound to the glycocalyx of the vascular endothelium, a common occurrence in patients with cardiovascular disease [44]. In conjunction with our current findings, it is possible that XOR may provide some pathophysiological benefit via reduction of NO2- to NO, and that irreversible inhibition of this pathway via Febx would be detrimental. Thus, alternative approaches that blunt XOR uric acid formation while increasing nitrite reduction to NO may be more therapeutically ideal.
In summary, we report a sulfide/polysulfide-induced XOR-dependent reduction of NO2- to NO. This process requires neither the commonly XOR-associated reducing substrates hypo/xanthine or NADH nor the reduction of the FAD cofactor whereas it does involve electron flux between the Mo-co and proximal Fe/S center. Administration of a polysulfide (DATS) to mice with experimental hindlimb ischemia elicits improved blood perfusion in CSE/H2S deficient mice, in an XOR-dependent manner suggesting in vitro results reported herein are operative in vivo. Moreover, vasodilation mediated by DATS was completely inhibited upon simultaneous treatment with Febx, indicating that sulfide mediated vascular function is XOR-dependent (Fig. 6D). Together, these results serve to identify XOR as a focal point of convergence between sulfide- and nitrite-mediated signaling in the vasculature.
Declaration of competing interest
C.G.K. has provisional patents regarding nitric oxide/nitrite and hydrogen sulfide chemistry and is a co-founder of Innolyzer LLC. X.S. has intellectual property regarding hydrogen sulfide chemistry.
Acknowledgements
This work was supported by an Institutional Development Award (IDeA) from the National Institutes of General Medical Sciences of the National Institutes of Health under grant number P20GM121307 to C.G.K. and AG043376-02S1, GM109098 and AHA (19TPA34850089) to E.E.K.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101447.
Appendix A. Supplementary data
Supplementary Fig. 1. Baseline nitric oxide analyzer profiles. Buffer was exposed to increasing concentrations (25, 50, 100 μM) of nitrite (A) or Sulfide (B) in the NOA reaction chamber to establish a cell-free baseline.
Supplementary Fig. 2. DATS therapy induces blood flow and collateral formation in CSE KO mice. (A) Representative photomicrographs of CD31/α-SMA counterstained with DAPI in ischemic skeletal muscle sections of PBS, DATS, Febx alone or DATS + Febx treated CSE KO mice at day 7 post-ligation. Quantitative image analysis of ischemic muscle showing angiogenesis index (CD31/DAPI) (B) and arteriogenesis index (α-SMA/DAPI) (C). Significance indicators are shown within the figure. n = 5 per cohort.
The following is the Supplementary data to this article:
References
- 1.Kochanek K.D.X.J., Murphy S.L., Miniño A.M., Kung H.C. Deaths: final data for 2009. Natl. Vital Stat. Rep. 2011;60(3) [PubMed] [Google Scholar]
- 2.Centers for Disease C Prevention. Million hearts: strategies to reduce the prevalence of leading cardiovascular disease risk factors--United States, 2011. MMWR Morb. Mortal. Wkly. Rep. 2011;60(36):1248–1251. [PubMed] [Google Scholar]
- 3.Ritchey M.D., Wall H.K., Gillespie C., George M.G., Jamal A., Division for Heart D., Stroke Prevention C.D.C. Million hearts: prevalence of leading cardiovascular disease risk factors--United States, 2005-2012. MMWR Morb. Mortal. Wkly. Rep. 2014;63(21):462–467. [PMC free article] [PubMed] [Google Scholar]
- 4.Niskanen L.K., Laaksonen D.E., Nyyssonen K., Alfthan G., Lakka H.M., Lakka T.A., Salonen J.T. Uric acid level as a risk factor for cardiovascular and all-cause mortality in middle-aged men: a prospective cohort study. Arch. Intern. Med. 2004;164(14):1546–1551. doi: 10.1001/archinte.164.14.1546. [DOI] [PubMed] [Google Scholar]
- 5.Skak-Nielsen H., Torp-Pedersen C., Finer N., Caterson I.D., Van Gaal L., James W.P., Maggioni A.P., Sharma A.M., Coutinho W., Andersson C. Uric acid as a risk factor for cardiovascular disease and mortality in overweight/obese individuals. PloS One. 2013;8(3) doi: 10.1371/journal.pone.0059121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Becker B.F. Towards the physiological function of uric acid. Free Radic. Biol. Med. 1993;14(6):615–631. doi: 10.1016/0891-5849(93)90143-i. [DOI] [PubMed] [Google Scholar]
- 7.Kelley E.E., Hock T., Khoo N.K., Richardson G.R., Johnson K.K., Powell P.C., Giles G.I., Agarwal A., Lancaster J.R., Jr., Tarpey M.M. Moderate hypoxia induces xanthine oxidoreductase activity in arterial endothelial cells. Free Radic. Biol. Med. 2006;40(6):952–959. doi: 10.1016/j.freeradbiomed.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 8.Berry C.E., Hare J.M. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J. Physiol. 2004;555(Pt 3):589–606. doi: 10.1113/jphysiol.2003.055913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Battelli M.G., Polito L., Bolognesi A. Xanthine oxidoreductase in atherosclerosis pathogenesis: not only oxidative stress. Atherosclerosis. 2014;237(2):562–567. doi: 10.1016/j.atherosclerosis.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Kelley E.E. A new paradigm for XOR-catalyzed reactive species generation in the endothelium. Pharmacol. Rep. 2015;67(4):669–674. doi: 10.1016/j.pharep.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelley E.E., Khoo N.K., Hundley N.J., Malik U.Z., Freeman B.A., Tarpey M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic. Biol. Med. 2010;48(4):493–498. doi: 10.1016/j.freeradbiomed.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hosoya T., Ohno I., Nomura S., Hisatome I., Uchida S., Fujimori S., Yamamoto T., Hara S. Effects of topiroxostat on the serum urate levels and urinary albumin excretion in hyperuricemic stage 3 chronic kidney disease patients with or without gout. Clin. Exp. Nephrol. 2014;18(6):876–884. doi: 10.1007/s10157-014-0935-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feoli A.M., Macagnan F.E., Piovesan C.H., Bodanese L.C., Siqueira I.R. Xanthine oxidase activity is associated with risk factors for cardiovascular disease and inflammatory and oxidative status markers in metabolic syndrome: effects of a single exercise session. Oxid Med Cell Longev. 2014;2014:587083. doi: 10.1155/2014/587083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tam H.K., Kelly A.S., Metzig A.M., Steinberger J., Johnson L.A. Xanthine oxidase and cardiovascular risk in obese children. Child. Obes. 2014;10(2):175–180. doi: 10.1089/chi.2013.0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gokce Cokal B., Yurtdas M., Keskin Guler S., Gunes H.N., Atac Ucar C., Aytac B., Durak Z.E., Yoldas T.K., Durak I., Cubukcu H.C. Serum glutathione peroxidase, xanthine oxidase, and superoxide dismutase activities and malondialdehyde levels in patients with Parkinson's disease. Neurol. Sci. 2017;38(3):425–431. doi: 10.1007/s10072-016-2782-8. [DOI] [PubMed] [Google Scholar]
- 16.Battelli M.G., Polito L., Bortolotti M., Bolognesi A. Xanthine oxidoreductase-derived reactive species: physiological and pathological effects. Oxid Med Cell Longev. 2016;2016:3527579. doi: 10.1155/2016/3527579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmidt H.M., Kelley E.E., Straub A.C. The impact of xanthine oxidase (XO) on hemolytic diseases. Redox Biol. 2019;21:101072. doi: 10.1016/j.redox.2018.101072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El-Bassossy H.M., Watson M.L. Xanthine oxidase inhibition alleviates the cardiac complications of insulin resistance: effect on low grade inflammation and the angiotensin system. J. Transl. Med. 2015;13:82. doi: 10.1186/s12967-015-0445-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saban-Ruiz J., Alonso-Pacho A., Fabregate-Fuente M., de la Puerta Gonzalez-Quevedo C. Xanthine oxidase inhibitor febuxostat as a novel agent postulated to act against vascular inflammation. Antiinflamm Antiallergy Agents Med Chem. 2013;12(1):94–99. doi: 10.2174/1871523011312010011. [DOI] [PubMed] [Google Scholar]
- 20.Pacher P., Nivorozhkin A., Szabo C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol. Rev. 2006;58(1):87–114. doi: 10.1124/pr.58.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bir S.C., Kolluru G.K., McCarthy P., Shen X., Pardue S., Pattillo C.B., Kevil C.G. Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia-inducible factor-1alpha and vascular endothelial growth factor-dependent angiogenesis. J Am Heart Assoc. 2012;1(5) doi: 10.1161/JAHA.112.004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H., Cui H., Kundu T.K., Alzawahra W., Zweier J.L. Nitric oxide production from nitrite occurs primarily in tissues not in the blood: critical role of xanthine oxidase and aldehyde oxidase. J. Biol. Chem. 2008;283(26):17855–17863. doi: 10.1074/jbc.M801785200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang R. Signaling pathways for the vascular effects of hydrogen sulfide. Curr. Opin. Nephrol. Hypertens. 2011;20(2):107–112. doi: 10.1097/MNH.0b013e3283430651. [DOI] [PubMed] [Google Scholar]
- 24.Kolluru G.K., Shen X., Kevil C.G. A tale of two gases: NO and H2S, foes or friends for life? Redox Biol. 2013;1(1):313–318. doi: 10.1016/j.redox.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kolluru G.K., Shen X., Bir S.C., Kevil C.G. Hydrogen sulfide chemical biology: pathophysiological roles and detection. Nitric Oxide. 2013;35:5–20. doi: 10.1016/j.niox.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al-Magableh M.R., Kemp-Harper B.K., Hart J.L. Hydrogen sulfide treatment reduces blood pressure and oxidative stress in angiotensin II-induced hypertensive mice. Hypertens. Res. 2015;38(1):13–20. doi: 10.1038/hr.2014.125. [DOI] [PubMed] [Google Scholar]
- 27.Kolluru G.K., Bir S.C., Yuan S., Shen X., Pardue S., Wang R., Kevil C.G. Cystathionine gamma-lyase regulates arteriogenesis through NO-dependent monocyte recruitment. Cardiovasc. Res. 2015;107(4):590–600. doi: 10.1093/cvr/cvv198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altaany Z., Moccia F., Munaron L., Mancardi D., Wang R. Hydrogen sulfide and endothelial dysfunction: relationship with nitric oxide. Curr. Med. Chem. 2014 doi: 10.2174/0929867321666140706142930. [DOI] [PubMed] [Google Scholar]
- 29.Jang H., Oh M.Y., Kim Y.J., Choi I.Y., Yang H.S., Ryu W.S., Lee S.H., Yoon B.W. Hydrogen sulfide treatment induces angiogenesis after cerebral ischemia. J. Neurosci. Res. 2014;92(11):1520–1528. doi: 10.1002/jnr.23427. [DOI] [PubMed] [Google Scholar]
- 30.Rajpal S., Katikaneni P., Deshotels M., Pardue S., Glawe J., Shen X., Akkus N., Modi K., Bhandari R., Dominic P., others Total sulfane sulfur bioavailability reflects ethnic and gender disparities in cardiovascular disease. Redox Biol. 2018;15:480–489. doi: 10.1016/j.redox.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bergmeyer H.-U. In: Methods of Enzymatic Analysis. Bergmeyer H.-U., editor. Elsevier; 1974. p. 682. [Google Scholar]
- 32.Schuler D., Sansone R., Freudenberger T., Rodriguez-Mateos A., Weber G., Momma T.Y., Goy C., Altschmied J., Haendeler J., Fischer J.W., others Measurement of endothelium-dependent vasodilation in mice--brief report. Arterioscler. Thromb. Vasc. Biol. 2014;34(12):2651–2657. doi: 10.1161/ATVBAHA.114.304699. [DOI] [PubMed] [Google Scholar]
- 33.Henning C., Branopolski A., Schuler D., Dimitroulis D., Huelsemann P., Nicolaus C., Sansone R., Ludolf Postma J., Eberhard D., Le Noble F., others Requirement of beta1 integrin for endothelium-dependent vasodilation and collateral formation in hindlimb ischemia. Sci. Rep. 2019;9(1):16931. doi: 10.1038/s41598-019-53137-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernandez M.L., Stupar D., Croll T., Leavesley D., Upton Z. Xanthine oxidoreductase: a novel therapeutic target for the treatment of chronic wounds? Adv Wound Care (New Rochelle) 2018;7(3):95–104. doi: 10.1089/wound.2016.0724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Terada L.S., Piermattei D., Shibao G.N., McManaman J.L., Wright R.M. Hypoxia regulates xanthine dehydrogenase activity at pre- and posttranslational levels. Arch. Biochem. Biophys. 1997;348(1):163–168. doi: 10.1006/abbi.1997.0367. [DOI] [PubMed] [Google Scholar]
- 36.Zimna A., Kurpisz M. Hypoxia-inducible factor-1 in physiological and pathophysiological angiogenesis: applications and therapies. BioMed Res. Int. 2015;2015:549412. doi: 10.1155/2015/549412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cantu-Medellin N., Kelley E.E. Xanthine oxidoreductase-catalyzed reduction of nitrite to nitric oxide: insights regarding where, when and how. Nitric Oxide. 2013;34:19–26. doi: 10.1016/j.niox.2013.02.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weidert E.R., Schoenborn S.O., Cantu-Medellin N., Choughule K.V., Jones J.P., Kelley E.E. Inhibition of xanthine oxidase by the aldehyde oxidase inhibitor raloxifene: implications for identifying molybdopterin nitrite reductases. Nitric Oxide. 2014;37:41–45. doi: 10.1016/j.niox.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan S., Kevil C.G. Nitric oxide and hydrogen sulfide regulation of ischemic vascular remodeling. Microcirculation. 2016;23(2):134–145. doi: 10.1111/micc.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hassoun P.M., Yu F.S., Shedd A.L., Zulueta J.J., Thannickal V.J., Lanzillo J.J., Fanburg B.L. Regulation of endothelial cell xanthine dehydrogenase xanthine oxidase gene expression by oxygen tension. Am. J. Physiol. 1994;266(2 Pt 1):L163–L171. doi: 10.1152/ajplung.1994.266.2.L163. [DOI] [PubMed] [Google Scholar]
- 41.Cantu-Medellin N., Kelley E.E. Xanthine oxidoreductase-catalyzed reactive species generation: a process in critical need of reevaluation. Redox Biol. 2013;1(1):353–358. doi: 10.1016/j.redox.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelley E.E., Batthyany C.I., Hundley N.J., Woodcock S.R., Bonacci G., Del Rio J.M., Schopfer F.J., Lancaster J.R., Jr., Freeman B.A., Tarpey M.M. Nitro-oleic acid, a novel and irreversible inhibitor of xanthine oxidoreductase. J. Biol. Chem. 2008;283(52):36176–36184. doi: 10.1074/jbc.M802402200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.White W.B., Saag K.G., Becker M.A., Borer J.S., Gorelick P.B., Whelton A., Hunt B., Castillo M., Gunawardhana L., Investigators C. Cardiovascular safety of febuxostat or allopurinol in patients with gout. N. Engl. J. Med. 2018;378(13):1200–1210. doi: 10.1056/NEJMoa1710895. [DOI] [PubMed] [Google Scholar]
- 44.Malik U.Z., Hundley N.J., Romero G., Radi R., Freeman B.A., Tarpey M.M., Kelley E.E. Febuxostat inhibition of endothelial-bound XO: implications for targeting vascular ROS production. Free Radic. Biol. Med. 2011;51(1):179–184. doi: 10.1016/j.freeradbiomed.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.