

ABSTRACT
Connexin 37 (Cx37; protein product of GJA4) expression profoundly suppresses proliferation of rat insulinoma (Rin) cells in a manner dependent on gap junction channel (GJCh) functionality and the presence and phosphorylation status of its C-terminus (CT). In Rin cells, growth is arrested upon induced Cx37 expression and serine 319 (S319) is frequently phosphorylated. Here, we show that preventing phosphorylation at this site (alanine substitution; S319A) relieved Cx37 of its growth-suppressive effect whereas mimicking phosphorylation at this site (aspartate substitution; S319D) enhanced the growth-suppressive properties of Cx37. Like wild-type Cx37 (Cx37-WT), Cx37-S319D GJChs and hemichannels (HChs) preferred the closed state, rarely opening fully, and gated slowly. In contrast, Cx37-S319A channels preferred open states, opened fully and gated rapidly. These data indicate that phosphorylation-dependent conformational differences in Cx37 protein and channel function underlie Cx37-induced growth arrest versus growth-permissive phenotypes. That the closed state of Cx37-WT and Cx37-S319D GJChs and HChs favors growth arrest suggests that rather than specific permeants mediating cell cycle arrest, the closed conformation instead supports interaction of Cx37 with growth regulatory proteins that result in growth arrest.
KEY WORDS: Connexin, Cell cycle, Gap junction channel, Hemichannel, Gating
Summary: Cx37-mediated switching between proliferation and growth arrest depends on phosphorylation at serine 319, which induces a protein conformation that likely supports unique interactions with cell cycle regulatory proteins.
INTRODUCTION
Cell-to-cell communication is critical for proper growth, differentiation and maintenance of homeostasis in virtually all tissues of the body. This direct intercellular communication involves the regulated diffusive exchange of ions and small metabolites through gap junction channels (GJChs) composed of connexin (Cx) proteins, which comprise a gene family with 21 members (Morel, 2014; Sohl and Willecke, 2004). Although differentially expressed in specific tissues, all Cxs share a common secondary structure consisting of four transmembrane domains connected by two extracellular loops, an intracellular loop, and cytosolic N-terminus (NT) and C-terminus (CT). Cx monomers assemble into hexamers, called connexons; docking of connexons between two neighboring cells forms a complete GJCh (Beyer et al., 1990; Zimmer et al., 1987). Disruption of GJCh communication has long been implicated in tumorigenesis and enhanced cell proliferation (Eghbali et al., 1991). In the past twenty or so years multiple investigators have shown that Cxs additionally participate in growth control of many cell types via GJCh-independent functions, specifically via transmembrane signaling through functional connexons, called hemichannels (HChs), and via protein–protein interactions with growth regulatory proteins (channel permeation-independent) (Dang et al., 2006; Goodenough and Paul, 2003; Kardami et al., 2007; Zhang et al., 2001, 2003).
Cx37 (product of the GJA4 gene) is expressed in endothelial and smooth muscle cells of the arterial vasculature (Gabriels and Paul, 1998; Reed et al., 1993) where it participates in regulating cell cycle progression and programmed cell death during development and in response to injury (Fang et al., 2011, 2017) and disease (Allagnat et al., 2017; Pfenniger et al., 2012). Cx37 is also expressed in monocytes and macrophages, influencing their invasive behavior (Chanson and Kwak, 2007; Wong et al., 2006). We have previously demonstrated that Cx37, but not Cx43 or Cx40 (products of GJA1 and GJA5 genes, respectively), affects cell cycle progression and cell death in rat insulinoma (Rin) and other Cx-deficient tumor cells (Burt et al., 2008), making the Rin cells an ideal system in which to explore the mechanisms underlying Cx37-mediated growth regulation. In Rin cells, the growth-suppressive effect of Cx37 requires the protein be able to form functional GJChs regulated by the Cx37 CT (Good et al., 2012, 2014, 2011; Nelson et al., 2013). Recent studies have demonstrated that regulation by the CT likely involves its differential phosphorylation (Jacobsen et al., 2019, 2017), as is the case for growth regulation by Cx43 (Moreno et al., 2002; Shin et al., 2001; Solan and Lampe, 2007, 2009). Moreover, this differential phosphorylation alters the intra-domain interactions of the end-tail and mid-tail regions of the CT (residues 318–333 and 274–317, respectively), and the interaction of these regions is known to be required for survival of cells expressing Cx37 (Jacobsen et al., 2019).
Cx37 has been reported to be a phosphoprotein by several investigators (Larson et al., 2000; Morel et al., 2010; Traub et al., 1998), but the precise residues phosphorylated by cells (as opposed to kinases in in vitro assays) have not been identified. We reported previously the results of mass spectrometry data that revealed phosphorylation of Cx37 at serine residues 275, 319 (most abundantly), 321, 325 and 328 in iRin37 cells growth arrested following induction of Cx37 expression (Jacobsen et al., 2019). In humans the codon for residue 319 is polymorphic, C1019T, and results in either a proline (C-allele) or serine (T-allele) at this position. The C-allele frequency is ∼70% in the human population, with ∼43% of the population homozygous for the C-allele and 13% homozygous for the T-allele (Boerma et al., 1999). The gnomAD database (https://gnomad.broadinstitute.org/), with over 140,000 genomes, now estimates that individuals homozygous for the T-allele may be as low as 5–7%, but given the range of the 1019T allele frequency in different ancestral populations, 17–53%, there clearly would be a range of homozygosity as well. Boerma and colleagues showed that the C-allele is over-represented in atherosclerotic plaques of Swedish males, leading to the supposition that Cx37-319P predisposes to this disease. Comparable studies in various human populations (including Chinese, Japanese, Taiwanese, and Sicilian, Swiss, and Czech Caucasians; reviewed in Wen et al., 2014), have shown that, in some populations, the C-allele is associated with coronary artery disease (CAD), myocardial infarction (MI), or both, and with acute myocardial infarction (AMI), while in other populations either the T-allele is predictive of these diseases or the Cx37 allele type was not correlated with these diseases. Meta-analysis using both dominant and recessive models (Wen et al., 2014) of data from 3498 MI and associated 3986 ‘control’ subjects and of 1808 CAD and associated 1197 ‘control’ subjects concluded that the 1019T isoform was a risk factor for MI but a protective factor for CAD. Mice are homozygous for the T-allele, always expressing Cx37 with serine in the 319 position. Wild-type mice (and rodents in general) are not susceptible to atherosclerosis, even when fed a high-fat diet; however, Apoe−/− mice fed a high-fat diet develop plaques that are far more numerous and extensive in mice also deficient in Cx37 (Wong et al., 2006), indicating that Cx37 (with serine at residue 319) is protective against plaque development. We showed that Cx37 exerts a growth-suppressive effect on vascular development and vascular response to injury (Fang et al., 2011; 2012; 2017). Since vascular development and response to injury, and diseases such as atherosclerosis and myocardial infarction are multifactorial, these (and other) human and mouse Cx37 data implicate differential regulation of and/or by Cx37 and its binding partners in disease susceptibility and progression, and growth control.
We hypothesized that phosphorylation of S319 alters the conformation of Cx37 such that it mediates growth suppression. We provide here mass spectrometry data showing abundant phosphorylation at S319 in iRin37 cells that display arrested growth owing to induced Cx37 expression. We compare the growth effects and channel properties (an indicator of possible differences in channel conformation) of Cx37 with alanine or aspartate substituted at S319, substitutions that mimic the dephosphorylated (dpS319) and phosphorylated (pS319) states, respectively. We show that Cx37-S319D is more growth suppressive than wild-type Cx37 (Cx37-WT) and Cx37-S319A is not detectably growth suppressive. Although both mutants form functional GJChs and HChs, the growth-arresting Cx37-S319D prefers the closed state, as previously predicted (Jacobsen et al., 2017), which suggests that phosphorylation at S319 of Cx37 induces a protein conformation that favors interaction with protein partners that arrest cell cycling rather than supporting permeation of anti-proliferative molecules.
RESULTS
Cx37 is phosphorylated at S319 in Rin cells where growth is arrested
For Cx37 to regulate cell cycle progression, it must be able to form GJChs (Good et al., 2011), whose function is regulated by the Cx37 CT (Nelson et al., 2013) in a phosphorylation-dependent manner (Jacobsen et al., 2017). Data from several multi-site dephospho- and phospho-mimetic mutants (Jacobsen et al., 2017) indicated three possible Cx37-induced phenotypes in Rin cells: proliferative, growth arrested, and death. A previous mass spectrometry of Cx37-WT isolated from iRin37 cells with arrested growth as a consequence of induced Cx37 expression (Jacobsen et al., 2019) showed a high incidence (36%; 204 of 559 fragments) of phosphorylation at S319 (see data deposited under PRIDE partner repository accession no. PXD012191). Many spectra comparable to that shown in Fig. 1A enabled unambiguous identification of phosphorylation at S319, and not at the other serine residues in this 11-mer peptide. This conclusion is evident from the mass, 1197 Da, of the 11-mer parent fragment, which includes the mass of a phosphate group (95 Da). The fragment mass table (Fig. 1B) shows the predicted mass of the expected b- and y-fragments without and with an additional 2H, or missing either an NH3 or H2O group (the blue and yellow highlighted fragments were detected in the spectra, many of which are labeled in Fig. 1A). The identification of S319 as phosphorylated (as opposed to one of the other serine residues in this fragment) is indicated by: (1) the fragments containing serine 319 (b1-b11 and y11) have a mass 95 Da greater than the mass predicted without the phosphate group, and (2) the mass of all fragments missing S319 is equal to that predicted by non-phosphorylated serine residues at positions other than 319.
Fig. 1.

Cx37 is phosphorylated at S319 in Rin cells with arrested growth consequent to induced Cx37 expression. (A) Spectra of a peptide extending from S319 to K329 with b and y ion fragments identified. (B) Table of predicted fragment masses for the parent ion with sequence shown in amino acid (AA) column. The observed b-fragment (N-terminal) and y-fragment (C-terminal) ions, which are highlighted in blue or yellow, allowed unambiguous localization of the phosphorylation event at S319. The masses of all fragments containing S319, B1-11 and Y11, include the mass of a phosphate group (95 Da); in contrast, the mass of all fragments that do not include S319 (Y1-10) do not include the mass of a phosphate group, which indicates that only S319 is phosphorylated. pS319 was identified in 204 of 559 fragments (Jacobsen et al., 2019).
Cx37-S319 mutants localize to the membrane and form functional channels
To determine whether phosphorylation at S319 is necessary or sufficient for growth regulation by Cx37, we generated single-site mutations in the CT of Cx37 that mimic either the phosphorylated or dephosphorylated state of the protein (Huang and Erikson, 1994; McCulloch et al., 2001). Alanine substitution was used to prevent phosphorylation at the site (Cx37-S319A) and aspartate, which mimics the negative charge of a phosphate group, was used to mimic the phosphorylated protein (Cx37-S319D). After stable transfection of Cx37-S319A and -S319D into iRin cells, the cells were dilution cloned and Cx37 expression as a function of doxycycline (dox) concentration and induction time assessed (clones are denoted iRin37-S319A and iRin37-S319D). Dox induced maximal and sustained expression of Cx37-WT at 2 µg/ml after 24 h, Cx37-S319A at 2 µg/ml after 48 h, and Cx37-S319D at 1 µg/ml after 48 h. From these qualitative dox dose response studies, two clones for each mutant were selected for further study. We quantified protein expression (see Fig. S1 for examples of quantitative western and slot blot results) by each clone and by Cx37-WT cells (at the indicated dox concentration and induction time); the data are summarized in Fig. 2A and show that expression levels in the iRin37-S319A and -S319D clones and -WT did not differ (ANOVA; P=0.44). It is worth noting that growth arrest of iRin cells still occurs when Cx37-WT expression levels are 20-fold lower than in this WT clone (Burt et al., 2008; Good et al., 2012, 2014, 2011). Immunofluorescence labeling of Cx37-S319A and Cx37-S319D revealed significant intracellularly localized Cx37 as well as punctate labeling at what appears to be appositional membranes of dox-induced iRin37-S319A and -S319D clones (Fig. 2B; images not shown for clone 3E5 and 2B7) but not in non-induced cells, as previously shown for the iRin37 cells (Burt et al., 2008; Good et al., 2012, 2014, 2011). Finally, since growth suppression by Cx37 requires the protein be able to form functional GJChs, although the functional state of the channel that is growth suppressive remains uncertain (Good et al., 2014, 2011), we verified that each clone formed functional gap junctions; junctional conductance did not differ (ANOVA; P=0.6834) between the iRin37-S319A, -S319D and -WT cells (Fig. 2C).
Fig. 2.

Cx37-S319 mutants localize to appositional membrane and form functional gap junction channels. (A) Maximal expression levels for Cx37 in iRin37-WT, iRin37-S319A and iRin37-S319D cells were not different (mean±s.e.m., n=4 samples in triplicate; P=0.44). Data are from slot blot analysis. (B) Pseudo-DIC and immunofluorescence images of iRin37-S319A (3D8) and iRin37-S319D (2C1) induced (bottom) or not (top) to express Cx37. Cells were stained (Hoechst) to reveal nuclei (blue) of all cells in the field of view, and with antibody against Cx37 protein (green). Punctate labeling at appositional membranes (arrowheads), suggestive of gap junction plaques, was evident in the dox+ but not dox− setting. Scale bars: 100 μm. (C) The conductance of junctions formed by each clone (and isoform) were comparable (P=0.6834). Results are mean±s.e.m. [WT, 3.4±0.69 nS (n=30); iRin37-S319A 3D8 and 3E5, 3.9±0.92 nS (n=17) and 4.4±0.73 nS (n=14); iRin37-S319D 2B7 and 2C1 cells, 5.2±0.94 nS (n=9) and 4.2±0.92 nS (n=11)].
Cx37-S319A relieves and Cx37-S319D augments the growth-suppressive effect of Cx37
To determine whether preventing phosphorylation at S319 eliminated the growth-suppressive effect of Cx37-WT in Rin cells, we compared the proliferation of the Cx37-S319A clones and Cx37-WT cells that were dox-induced or not (expressing versus not expressing) over 21 days (Fig. 3A). The data show that unlike Cx37-WT, Cx37-S319A was not growth suppressive as the proliferation of non-expressing and expressing cells was not significantly different for each clone [slopes of semi-log regression lines for dox+ (Cx37-S319A clones) versus dox− (all clones) are not different, P=0.1582; Fig. 3A, inset]; in contrast, slopes of Cx37-WT dox+ and dox– proliferation differ significantly (P<0.0001), with the slope of dox+ not different from zero (P=0.1218). These data indicate that alanine substitution for S319 relieved Cx37 of its growth-suppressive properties. Since there were no differences in proliferation between the iRin37 Cx37-S319A clones (3D8 and 3E5), data from the two clones were combined in the remaining datasets.
Fig. 3.

Cx37-S319A alleviates whereas -S319D enhances Cx37 mediated growth arrest. (A) Proliferation of expressing (dox+) and non-expressing (dox−) iRin37-S319A clones and iRin37-WT (n=3 for each) did not differ over 21 days (P=0.1582); only for Cx37-WT do the dox+ and dox− curves differ (P<0.0001), indicating lack of growth suppression by the -S319A mutant. The semi-log plot of these data (inset) shows that non-expressing iRin37-WT and both expressing and non-expressing iRin37-S319A mutant cells proliferate at comparable rates, whereas WT-expressing cells do not proliferate. *P<0.0001 significantly different from non-expressing (dox−) cells; ns, not significantly different. (B) The proliferation of iRin cells expressing Cx37-S319D or Cx37-WT is comparable and significantly (*P<0.0001) reduced compared to non-expressing cells of the same clone (n=3 for each). Inset, semi-log plot of proliferation over 6 days (n=3) showing that cell number increases, but does not double, in iRin37-S319D cells. †P=0.0024 from WT; ns, not significantly different. (C) 21-day proliferation curves in which Cx37-WT or -S319D expression was induced with dox on day 12 (n=3 for each clone) show suppressed proliferation at higher cell density (than in B). Inset, semi-log plot and regression analysis showing slowed proliferation after versus before induced expression of both Cx37-WT and Cx37-S319D isoforms. *P<0.05 from dox− growth period (2C1 P=0.0026; 2B7 P=0.0229; WT P=0.0024); ns, not significantly different. Double headed arrows in C indicate the period of dox exposure. All error bars denote s.e.m.; statistical differences were assessed using linear regression analysis.
To determine the effect of simulated phosphorylation at S319 on Cx37-induced growth phenotype, we compared proliferation of the expressing (dox+) and non-expressing (dox−) iRin37-S319D clones (2B7 and 2C1) and -WT over a 21-day period (Fig. 3B). Compared to their non-expressing counterparts (dox−), cells expressing Cx37-S319D or Cx37-WT were growth suppressed (P<0.0001; Fig. 3B). To better resolve possible differences between the growth-suppressive effect of Cx37-S319D versus -WT, we assessed proliferation over a 6-day period, evaluating cell number daily. The semi-log plots and regression analysis of proliferation in these experiments (Fig. 3B, inset) showed that the iRin37-S319D cells failed to double in number over the 6-day period, which represents a significantly (P<0.0024) reduced rate of cell cycling compared to Cx37-WT-expressing cells. In addition, like Cx37-WT, Cx37-S319D slowed proliferation irrespective of cell density, as indicated by slowed proliferation of cells induced to express after 12 days of proliferation in the absence of Cx37 expression (wherein cell density increased from ∼3100 to ∼52,000 cells/cm2; Fig. 3C and inset). Proliferation after addition of dox (from 15–21 days) was significantly slowed for both iRin37-S319D clones and the -WT cells (2C1 P=0.0026; 2B7 P=0.0229; WT P=0.0024). This slowing is specific to the Cx37 isoform, not to doxycycline exposure, as shown previously (Burt et al., 2008; Jacobsen et al., 2019). Taken together, these data indicate that aspartate substitution for serine 319 resulted in growth arrest irrespective of whether GJCh formation was possible (i.e. cell–cell contact). As there were no significant differences between the iRin37-S319D clones (2B7 and 2C1), data from these clones were combined in the remaining experiments.
Since the 21- and 6-day proliferation data showed that Cx37-S319D arrested Rin cell growth, we evaluated cell cycle position and cell number as a function of Cx37 expression time. Fig. 4A shows the number of iRin37-S319D, -WT and -S319A cells over a 5-day dox-induction period in the presence of serum (comparable to proliferation curves); only the -S319A cells proliferated (4.60-fold increase in cell number, P=0.014). Fig. 4B–D shows the corresponding distribution of cells in the cell cycle for each Cx37 isoform. The distribution of the proliferating -S319A cells did not change significantly, suggesting no effect of -S319A expression on Rin cell cycling. The distribution of the anti-proliferative -WT cells also did not change, consistent with a prolongation of all phases, such that doubling time increased from 2 to 9 days, as previously reported (Burt et al., 2008). The -S319D cells accumulated in G1 over the 5-day period, increasing significantly from 73.7±0.7% in G1 to 79.4±1.78% in G1 after 5 days (mean±s.e.m., P=0.0151). Since the percentage of -S319D cells in S phase did not change (P=0.779) and the percentage in G2 declined from 9.51±2.1% to 4.61±1.1% (P=0.053), it appears that cells in G2 when -S319D expression was induced were able to complete the cell cycle and were then arrested in G1, joining those already unable to pass through the G1-S check point. It should be noted that FACSs data obtained from cells grown in serum, revealed no debris or dead cells (peak to the left of G1 peak) at any time point for any of these Cx37 isoforms (data not shown).
Fig. 4.
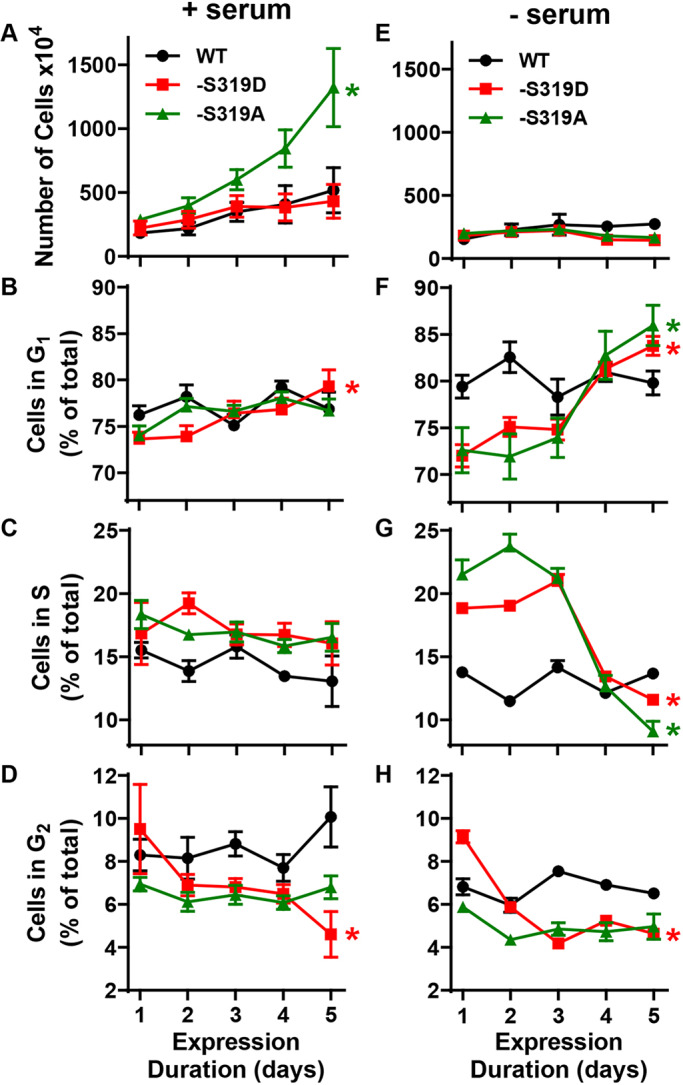
iRin37-S319D cells are growth arrested and accumulate in G0/G1. (A) In the presence of serum, Cx37-WT and Cx37-S319D suppressed the proliferation of Rin cells; Cx37-S319A had no growth-suppressive effect. Error bars denote s.e.m.; *P<0.05 (one-way ANOVA with Tukey's post-hoc test). (B–D) In the presence of serum, the percentage of Cx37-S319D cells in G0 or G1 (labeled G1) increased as the percentage in G2 decreased, suggesting that cells in G2, when CX37-S319D expression was initiated, were able to complete the cell cycle and were subsequently arrested in G0 or G1. (E) Serum deprivation arrested proliferation in an isoform independent manner. (F–H) All phases of the cell cycle were extended in Cx37-WT expressing cells, with no significant increase in any phase over the 5-day period. In contrast, the percentage of S319D cells in G0 or G1 increased while simultaneously the percentage in both S and G2 decreased significantly, suggesting that S and G2 cells are able to complete the cell cycle and arrest in G0 or G1. The Cx37-S319A cells also accumulate in G0, but for this isoform the percentage in S decreases substantially while G2 percentage is constant. All error bars denote s.e.m.; *P<0.05 between day 1 vs 5, P values stated in text.
We previously reported that growth of Cx37-WT-expressing cells was arrested in serum-free medium and the cells accumulated in G1 following only 24 h of Cx37-WT expression (Burt et al., 2008). Shown in Fig. 4E and F–H are proliferation and cell cycle data for cells maintained continuously in serum-free medium and expressing for 1 to 5 days Cx37-WT, -S319A or -S319D. None of the Cx37 isoform-expressing cell types proliferated in the absence of serum; however, their cell cycle distribution patterns differed. The percentage of Cx37-WT-expressing cells in all phases of the cell cycle was relatively constant over the 5-day period (no significant differences between day 5 and day 1 for any phase), with the percentage in G1 somewhat elevated compared to cells in serum-containing medium (79±1.2% versus 75.8±0.88%). In contrast, the percentage of -S319A and -S319D cells in G1 increased significantly over the 5-day period (-S319A: 72.6±2.2% to 86.0±2.0%, P=0.0012; -S319D: 72±1.1% to 83.8±0.93%, P<0.0001), with a significant decrease in both S (38%, P<0.0001) and G2 (49%, P<0.0001) percentages for the -S319D cells and a significant decrease in the S phase (58%, P=0.0028) percentage for the -S319A cells. These changes in cell cycle distribution in the serum-free condition indicate that both -S319A and -S319D cells differ from the -WT cells, suggesting that phosphorylation at S319 may change as a function of cell cycle progression.
Gap junction channel behavior
A change in protein conformation for channel-forming proteins can often be detected as a change in channel properties, such as conductance, preferred open state, open probability and gating (Beyer and Berthoud, 2017; Ismailov and Benos, 1995; Liu et al., 2006; Su et al., 2012). We previously (Jacobsen et al., 2017) hypothesized that GJChs formed by growth-permissive isoforms would display a strong preference for a 75–100 pS subconductance state (substate) and transition frequently from this state to multiple more conductive open states. In contrast, we suggested that GJChs formed by growth-suppressive isoforms would prefer the closed state, but would transition between closed and the same 75–100 pS substate observed in the GJChs of proliferative isoforms. To assess whether Cx37-S319A versus -S319D GJChs adhered to this prediction we used dual whole-cell voltage clamp to examine channel behavior in iRin37-S319A versus -S319D cells. Representative single channel recordings, collected in the absence of halothane (sample traces displaying transitions corresponding to ∼350pS for Cx37-S319A and -S319D are shown in Fig. S2), for Cx37-WT, -S319A, and -S319D are shown in Fig. 5. The Cx37-WT trace (Fig. 5A) shows that this channel has a strong preference for the closed state (0 pS peak in all points histogram on the right), as previously described (Jacobsen et al., 2017), and rarely displays transitions between fully open and closed (∼350 pS), as is evident in the relative frequency transition histogram (Fig. 5D; Jacobsen et al., 2017). In contrast, the growth-permissive Cx37-S319A (Fig. 5B) responds rapidly to the same magnitude change in Vj (transjunctional voltage; 25 mV) and prefers the 60–120 pS subconductance state (see inset), with rare closures and rare openings to the fully open state. The Cx37-S319D (Fig. 5C) channel opens to an intermediate conductance state of ∼220 pS and slowly responds (like WT) to the change in Vj by closing. Of note, this Cx37-S319D record shows three open channels at the start of the pulse (peak at 652 pS) that close sequentially (producing the peaks at 440 and 192 pS) such that only one channel remains open at the end of the ∼50 s record. These traces suggest that our previous predictions (Jacobsen et al., 2017) were correct; the growth-suppressive Cx37-WT and -S319D channels prefer the closed state, whereas the growth-permissive -S319A channel prefers the ∼100 pS substate and transitions from that substate to multiple open states, including fully open. The relative frequency histograms (Fig. 5D) suggest both Cx37-S319A and -S319D differ from -WT (wherein some channels are likely phosphorylated and others not) by having fewer transitions in the 70–110 pS range. GJCh open probabilities (Po) could not be quantitatively determined for these mutants as too few records [none for iRin37-S319D clones; one (shown) for iRin37-S319A clones] contained only a single active channel.
Fig. 5.

Gap junction channels formed by growth-arresting isoforms of Cx37 prefer the closed state. (A–C) Representative single-channel traces from the indicated genotypes suggesting that Cx37-S319D and -WT prefer the closed state whereas Cx37-S319A prefers a 60–120 pS substate. For the Cx37-WT and -S319A traces, only one channel is active and its closed state corresponds to the 0 pS line indicated by long dashes. For the Cx37-S319D trace, three channels are open at the start of the pulse (652 pS); their successive stable closures result in the peaks at 440 pS (2 channels open) and 192 pS (1 channel open). The size of observed transitions is shown on the left of each trace; all-points histograms, on the right, indicate the frequency of each conductance state for the illustrated trace. ↓ and ↑ indicate polarity and onset/end of pulse in the partner cell. (D) Relative mean±s.e.m. transition frequencies for all three Cx37 isoforms showing transitions between multiple open states and a closed state, including fully open to closed. Together the traces and histogram suggest Cx37-WT channels frequently transition between the ∼90 pS substate and the closed state [traces and previously published studies (Jacobsen et al., 2017) suggest the closed state is preferred]. The Cx37-S319D channels close frequently from multiple open states, but most commonly from the ∼200 pS conductance state (traces suggest closed state is preferred over the many open states). Cx37-WT: n=17, event no.=1917; Cx37-S319A: n=12, event no.=1280; Cx37-S319D: n=20, event no.=4493.
The different time courses for response to imposed Vj evident in Fig. 5 suggested that the voltage-dependent gating of Cx37-S319D might differ from that of -S319A. To examine this possibility, we explored Vj-gating for the Cx37-S319D and -S319A isoforms. In Fig. 6A we show representative junctional current traces for both isoforms, and in Fig. 6B we show the Boltzmann fits (Spray et al., 1984) of data from multiple cell pairs [gj (representing the junctional conductance): -S319D 6±0.6 nS (n=4); -S319A 4.2±0.91 nS (n=9)]. Comparisons of the fits for the negative and positive voltage ranges of each mutant revealed that the fits were not different (Cx37-S319D, P=0.9720; Cx37-S319A, P=0.479). Comparison of fits for -S319A versus -S319D for negative voltages as well as positive voltages, revealed significant differences between the mutants (P<0.001 for both voltage ranges). Gmin (representing the minimum conductance percentage) did not differ between the mutants (P=0.3475 negative voltages; P=0.0738 positive ranges), but V0 (representing the voltage at which Po is 0.5) differed significantly in both the positive and negative range comparison (+V0: -S319D 45±5 mV versus -S319A 30±1.4 mV, P=0.0013; −V0: -S319D −49±5 mV versus -S319A −33±2.0, P=0.0036). Voltage-dependent gating data for the Cx37-WT protein are included in Fig. S3, and comparison of the Boltzmann fits for these mutants and Cx37-WT data is summarized in Fig. 6C. These differences in gating behavior and open characteristics strongly suggest that the conformations of Cx37-S319A and -S319D GJChs differ.
Fig. 6.

Voltage-dependent gating of Cx37-S319D is diminished compared to Cx37-S319A. (A) Voltage protocol and current traces derived from Cx37-S319D and -S319A mutants, red trace shows response to ±110 mV pulses. (B) Boltzmann fits for Cx37-S319D (n=4) and -S319A (n=9). Fits for positive and negative voltage ranges did not differ for either mutant (or WT). Results are mean±s.e.m. (C) Boltzmann fit parameters for mutants and WT pulsed negative versus positive and compared using ANOVA analysis (negative range, P=0.0008; positive range, P=0.0002). Differences between mutants and between mutants and WT were tested (least squares); where significant differences were found they are indicated in the row labeled Fit.
Cx37-S319A and Cx37-S319D hemichannel activity
Both Cx37-S319A and -S319D formed functional HChs in standard external solution (containing 1 mM Ca2+). Segments of HCh records for Cx37-S319A and -S319D, as well as -WT, are shown in Fig. 7A. The percentage of cells found to have active channels for -WT, -S319A and -S319D was 45% (n=49), 50% (n=60) and 56% (n=25), respectively. Relative frequency amplitude histograms for transitions of Cx37-S319A and -S319D HChs are shown in Fig. 7B. The transition amplitudes of Cx37-S319D HChs were concentrated in the 300–600 pS range whereas transition amplitudes of Cx37-S319A HChs were dispersed throughout the conductance range (100–800 pS). Transition amplitudes of Cx37-WT HChs also occur throughout the conductance range (Jacobsen et al., 2017), consistent with the presence of phosphorylated and non-phosphorylated channels.
Fig. 7.

Behavior of hemichannels formed by Cx37-S319D and -S319A differ. (A) Representative traces show that Cx37-S319A HChs spend more time in open states than either Cx37-S319D or Cx37-WT HChs. The size of transitions is shown on the left of each recording; all-points histograms, on the right, show the frequency of each conductance state for the illustrated trace. (B) Transition frequency (mean±s.e.m. for each conductance bin) and open probability (Po) for HChs formed by Cx37-S319D and Cx37-S319A. Transitions of multiple amplitudes between fully open (∼750–800 pS) and closed were observed for both isoforms, but their open probabilities differed. Cx37-S319D preferred the closed state and a 60–90 pS subconductance state, whereas Cx37-S319A HChs preferred open states. Cx37-S319A (n=32, event no.=240) and Cx37-S319D (n=35, event no.=1521).
To determine the conductance state in which the HChs predominantly dwelled, open probabilities for cells with only one active channel were determined. The results are plotted on the transition amplitude histograms in Fig. 7B and open probabilities compared in Fig. 8. For Cx37-S319D, Fig. 7B shows that the channels transition from open states of ∼330–420 pS to either the closed or ∼100 pS subconductance states, where they tended to dwell for extended periods. Fig. 7B also shows that there were fewer Cx37-S319A channel events, which may reflect long dwell times in the 60–120 pS open state, as suggested by the significantly fewer events per unit time for this isoform (Cx37-S319A: 2.2±0.4 events/min in 174 min of record, n=32; Cx37-S319D: 6.4±0.9 events/min in 244 min of record, n=44; Student's t-test; P<0.0001). This conclusion is further substantiated by the open probability analysis shown in Fig. 8. The closed probability and open probability of the 105 pS conductance state of Cx37-S319A channels were significantly less than those of Cx37-S319D channels, and the open probabilities for conductance states larger than the ∼100 pS state were significantly greater for Cx37-S319A than observed for Cx37-S319D. These data, along with our previously published data on other growth-arresting and growth-permissive mutants (Jacobsen et al., 2017), suggest that growth suppression is mediated by the closed conformation of Cx37 GJChs and HChs rather than permeants unique to the briefly open channels.
Fig. 8.
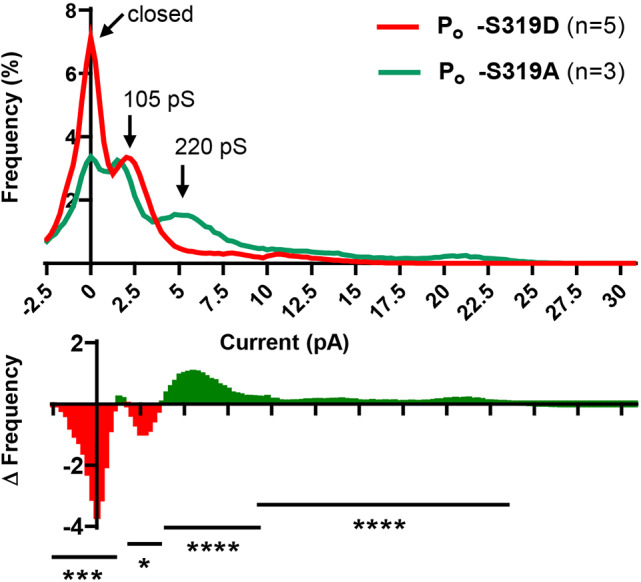
Hemichannels formed by Cx37-S319D prefer the closed state. Summary of open state/closed state probability (Po) for HChs formed by Cx37-S319D (n=5) and -S319A (n=3) and a difference plot showing where the two isoforms differed. Data derived from cells in which a single active channel was detected. *P<0.01; ***P<0.001; ****P<0.0001 (t test).
DISCUSSION
Cx37 exerts a growth-suppressive effect in vitro in cultured cell systems (Burt et al., 2008; Morel et al., 2010), and in vivo in monocytes and macrophages (Chanson and Kwak, 2007), in vasculogenesis (Allagnat et al., 2017; Fang et al., 2017) and in the vascular response to ischemic injury (Fang et al., 2011). In cultured cell systems, growth suppression involves prolongation of all phases of the cell cycle and in both in vitro and in vivo settings involves cell cycle arrest in G0 or G1 (Burt et al., 2008; Fang et al., 2017). Mutations of Cx37 that interfere with its ability to form functional GJChs (Good et al., 2011), regulated by the Cx37 CT (Nelson et al., 2013), eliminate its growth-suppressive effect, although which functional state of the GJCh (an open state or the closed state) is required for growth regulation remains uncertain. Regulation by the CT of phenotypic switching between growth arrest, proliferation and death involves differential phosphorylation (Jacobsen et al., 2017) and phosphorylation-dependent intra-CT-domain interactions (Jacobsen et al., 2019). These intra-domain interactions, between residues 273–317 and 318–333 (Jacobsen et al., 2019), appear to hinge near S319, a residue that is polymorphic in human Cx37, with the polymorphic forms displaying different channel properties and growth-suppressive effects (Derouette et al., 2009; Morel et al., 2010). We recently reported that in Rin cells growth arrested following induced expression of Cx37, S319 was frequently (36%) identified as being phosphorylated (Jacobsen et al., 2019). Here, we confirmed the unambiguous identification of this residue as phosphorylated in Cx37-induced growth-arrested cells, and, determined that aspartate substitution (to mimic the charge effects of phosphorylation) for S319 was sufficient to arrest growth of Rin cells. We also explored the mechanistic basis (channel permeation-dependent versus -independent) for Cx37-mediated growth suppression.
We previously suggested that phenotypic switching mediated by Cx37 involved differential phosphorylation of the CT of this protein (Jacobsen et al., 2017). This suggestion was based on the different phenotypes induced by either alanine or aspartate substitutions at as many as three or seven serine sites in the CT. Since the likelihood of all seven or even three sites being simultaneously phosphorylated is low, we used mass spectrometry to identify phosphorylated residues in Cx37 immunoprecipitated from cells growth arrested by induced Cx37 expression and found that S319 was phosphorylated at a high frequency. This residue is amongst the seven substituted with either alanine or aspartate (Cx37-S7A7 and Cx37-S7D7 mutants, respectively) in our 2017 study (Jacobsen et al., 2017). Since the Cx37-S7A7 mutant induces growth arrest in subconfluent cells, where gap junction formation is not possible due to lack of cell–cell contact, we hypothesized that absence of phosphorylation at one or more of these sites might be growth suppressive and phosphorylation growth permissive. The high frequency of phosphorylated (p)S319 observed in Cx37 immunoprecipitated from cells growth arrested by induced Cx37 expression suggested this interpretation might be incorrect. The current data, comparing the growth phenotypes of Rin cells expressing either Cx37-S319D or Cx37-S319A, indicate that phosphorylation at S319 is both necessary and sufficient for induction of growth arrest. Both mutations likely eliminate dpS319- or pS319-specific protein binding, but the unique tertiary structures and functions conferred by dpS319 and pS319 may be mimicked (Huang and Erikson, 1994; McCulloch et al., 2001) by alanine and aspartate substitutions, respectively. Thus, it is possible that phosphorylation at S319 (or aspartate substitution) exposes additional phosphorylation targets (or limits their exposure) and/or reveals binding sites necessary for Cx37-mediated growth arrest. That the Cx37-S319A and Cx37-S319D isoforms differ in conformation is supported by the electrophysiological data, as discussed next.
We determined that both the Cx37-S319A and -S319D isoforms formed functional channels, with WT and mutant clones displaying comparable levels of junctional conductance (gj), which indicates that the product of channel conductance (γj), channel open probability (Po) and channel number (N) for the three isoforms was similar. It should be noted that channels in their closed state are not detected by electrophysiological approaches, so the number of channels contributing to gj is challenging to evaluate. The transition amplitude histograms for all three isoforms had comparable conductance ranges, suggesting that the conductance of the fully open GJCh formed by all three isoforms was comparable despite rare transitions between closed and fully open. Voltage-dependent gating behavior differed between these isoforms as did the preferred open state at Vj=25 mV, both suggesting conformational differences between the isoforms. The GJCh formed by Cx37-WT preferred the closed state and a ∼90 pS substate with many transitions between these states, but also showed many short duration transitions between these states and other open states (Jacobsen et al., 2017), such as the 268 pS state, as illustrated in our current traces. The Cx37-S319D GJChs also appeared to prefer open states other than the fully open state (∼200 pS, rather than 350–400 pS), but these channels closed completely rather than to the 90 pS substate evident in Cx37-WT GJChs. Fully open 350–400 pS channels were evident in Cx37-S319A records, but this channel preferred the ∼60–120 pS substate, rarely ever closing completely. These differences in channel behavior demonstrate conformational differences between the isoforms, and strongly suggest that growth-arresting isoforms prefer the closed state, despite being able to open.
Because Cx37-WT, -S319D and -S7A7 (Jacobsen et al., 2017) exert growth-suppressive properties at cell densities where GJChs cannot form, we also examined the properties of the HCh formed by Cx37-WT, -S319D and -S319A isoforms. The behavior of the HChs formed by these isoforms paralleled the behavior of their GJChs. The fully open HChs formed by each had comparable conductances (750–800 pS), but the preferred open states differed. Cx37-WT and -S319D HChs preferred the closed state, whereas -S319A channels preferred substates of 70–280 pS. It is noteworthy that each of the isoforms displayed HCh activity in the presence of 1 mM extracellular [Ca2+]. Thus, like the GJCh, the HCh data show that the three isoforms differ in conformation, with growth-arresting isoforms forming HChs that prefer the closed state, despite opening occasionally to comparable conductance open states.
That the closed state of these channels appears to mediate growth suppression suggests that the closed conformation of the channel is critical to the growth-suppressive function of Cx37, rather than possible differences in channel permeants. This latter possibility cannot be ruled out, however, given the different preferred conductance states, and therefore permeation properties displayed by the GJChs and HChs. Since aspartate substitution at S319 is unlikely to mimic pS319-specific binding [e.g. phosphospecific antibodies do not recognize the non-phosphorylated or aspartate substituted residues; see for example (Solan et al., 2007)], our data suggest that phosphorylation at S319 induces a conformation of Cx37 that favors the closed state of both GJChs and HChs and supports interaction of the closed channels with other proteins necessary for growth-suppressive effect. Which proteins and the specific domains of Cx37 actually interact with those proteins remain to be discovered.
We concluded recently that a phosphorylation-dependent intradomain interaction within the CT of Cx37 was necessary for cell survival (Jacobsen et al., 2019). The present data suggest that S319 may serve as a hinge in the CT, altering protein conformation in a phosphorylation-dependent manner to effect the switch between the growth arrest and proliferative phenotypes. That phospho-mimetic mutation at S319 appears to prevent the cells from cycling suggests that the effect of phosphorylation at this site must be relieved or ‘countered’ by phosphorylation at additional sites for cell cycle progression. This possibility is supported by the altered phenotypes of Rin cells expressing double and triple mutants that include S319D. Cx37-S319D renders cells at low and high density (HChs versus GJChs and HChs, respectively) growth suppressed indefinitely (as much as 21 days). As noted previously, cells expressing Cx37-S7A7 (alanine substituted for serine residues 275, 285, 302, 319, 321, 325 and 328) are growth suppressed at low, but not high cell density where cell–cell contact supports formation of GJChs. Cells expressing Cx37-S275D,S319D show a similar growth pattern (Weyker, R., N.L.J., T.K.P., J.M.B., unpublished) to Cx37-S7A7 expressing cells, and cells expressing Cx37-S275D,S319D,S328D proliferate without restraint at all plating densities (Zehri, A., N.L.J., T.K.P., J.M.B., unpublished). Importantly, Cx37-S275D induces cell death at low density, but is growth permissive at high cell density and Cx37-S275A limits growth at low cell density, but is growth permissive at high cell density (Jacobsen et al., 2019). Also, Cx37-S328A and -S328D are death inducing at all cell densities (Jacobsen et al., 2019). These results show that these three sites, S275, S319 and S328 influence, collaboratively, the growth phenotype of Rin cells and suggest phosphorylation-dependent differences in protein conformation and binding partners.
In summary, the different phenotypes exhibited by the single site S319A and S319D mutants in comparison to multi-site mutants (Good et al., 2012, 2014, 2011; Jacobsen et al., 2019, 2017) indicate that the coordination of phosphorylation events in the CT of Cx37 is likely involved in growth phenotype regulation by Cx37. Furthermore, the data strongly suggest that unique, phosphorylation-dependent conformations of the CT, involving both intradomain interactions as well as interactions with phenotype-specific binding partners, are necessary for Cx37-mediated phenotypic switching. Finally, it appears that Cx37-mediated phenotypic switching involves channel permeation-dependent and -independent mechanisms. That Cx37 serves as a molecular switch between different growth phenotypes would suggest a promising use of Cx37 in therapeutic applications in vascular diseases, such as to prevent the development of angiosarcomas or to enhance vascular repair and remodeling after injury.
MATERIALS AND METHODS
Antibodies and reagents
All general chemicals were purchased from Sigma-Aldrich (St Louis, MO), unless otherwise noted. Cx37 polyclonal primary antibody, anti-Cx37 antibody [αCx37-18264, a gift from Dr Alexander Simon, University of Arizona (Simon et al., 2006)] was used for immunoblotting (1:5000 dilution) and immunofluorescence (1:400 dilution) for all mutants. Cx37–GST fusion protein (rat sequence, residues 229–333; Simon et al., 2006) was used as a positive control. Caspase-3 primary antibody (Cell Signaling Technology, #9662; Danvers, MA) was used for immunoblotting (1:1000 dilution) for caspase-3. Horseradish peroxidase-conjugated anti-rabbit-IgG secondary antibodies and enhanced chemiluminescence strategies were used in all immunoblots (SuperSignal West Pico or Femto Systems (Thermo Fisher Scientific, Waltham, MA), as appropriate. Cy3-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch #711-165-152; West Grove, PA) was used for immunofluorescence (1:400 dilution).
Mutant connexin generation and cell culture
The QuikChange site-directed mutagenesis kit (Stratagene; San Diego, CA) was used to introduce the S319D mutation into the pTRE2hyg-mCx37 plasmid (Burt et al., 2008) using the oligonucleotide primers: 5′-CAGGGTGGCCGAAAGGATCCTAGCCGCCCCAAC-3′ and 5′-GTTGGGGCGGCTAGGATCCTTTCGGCCACCCTG-3′ (Operon Biotechnologies; Huntsville, AL). The correct sequence was confirmed by the University of Arizona Genomic Analysis and Technology Core. The S319A mutation was created in a similar manner using the following primers: 5′-CAGGGTGGCCGAAAGGCACCTAGCCGCCCCAAC-3′ and 3′-GTTGGGGCGGCTAGGTGCCTTTCGGCCACCCTG-5′. iRin cells (rat insulinoma cells with the TET-ON expression system; Burt et al., 2008) were transfected with the mutant-containing plasmids, pTRE2hyg-mCx37-S319D and pTRE2hyg-mCx37-S319A, using Lipofectamine (Thermo Fisher Scientific) according to the manufacturer's instructions. Stably expressing cells, denoted iRin37-S319A and iRin37-S319D, were selected using 100 μg/ml hygromycin and dilution subcloned. All iRin cell lines were negative for mycoplasma contamination and maintained in standard RPMI 1640 medium with 10% FetalPlex (FP) (Gemini Bioproducts; Sacramento, CA) and antibiotics (300 μg/ml G418 and 100 μg/ml hygromycin). Cx37-WT- Cx37-S319A- and Cx37-S319D-expressing cells were induced with dox (dox+) for maximum protein expression: 2 μg/ml for 24 h, 2 μg/ml for 48 h, and 1 μg/ml for 48 h, respectively.
Mass spectrometry
Mass spectrometry analyzed was from Jacobsen et al. (2019), performed as described therein; data are available from the PRIDE partner repository (Vizcaíno et al., 2013) with the identifier (PXD012191). Briefly, nearly confluent, dox-induced iRin37 or iRin37-V5 (expressing Cx37-V5; Jacobsen et al., 2019) cells were lysed, samples sonicated and precleared with Protein A/G sepharose beads (Promega), and Cx37 immunoprecipitated from the supernatants with either anti-V5 (Thermo Fisher Scientific) or anti-Cx37 antibodies. Precipitate was resuspended in sample buffer prior to heating and loading onto a 12% TGX gel (Bio-Rad, Hercules, CA) for electrophoretic separation of sample proteins. Following in-gel trypsin digestion of the gel band corresponding to Cx37, the resulting peptides were purified by C18-based desalting (Kruse et al., 2017). High-pressure liquid chromatography electrospray ionization tandem mass spectrometry (HPLC-ESI-MS/MS) was performed in positive ion mode on a Thermo Scientific Orbitrap Fusion Lumos tribrid mass spectrometer fitted with an EASY-Spray Source (Thermo Fisher Scientific). Spectra were acquired using XCalibur, version 2.3 (Thermo Fisher Scientific) (Chambers et al., 2012). The fragment mass spectra were then searched against the mouse SwissProt_2016_10 database (23,550 entries) using Mascot (Matrix Science, London, UK; version 2.4). Cross-correlation of Mascot search results with X! Tandem was accomplished with Scaffold (version Scaffold_4.8.7; Proteome Software, Portland, OR).
Immunoblotting
Whole-cell protein was isolated as previously described (Burt et al., 2008) and protein concentration was determined using the BCA assay (Pierce Chemical, Dallas, TX). Protein expression levels were determined by quantitative western blotting as previously described (see for example, Nelson et al., 2013) or using slot blot analysis. Reported herein are the results for slot blot analysis, as we were unable to consistently have our samples lie within the range defined by our standards using quantitative western blotting. Slot blot wells were loaded in triplicate with 50 µg of total cell protein for each clone and WT. Standard curve slots were also loaded in triplicate with 0.25, 0.5, 1.0, 1.34, or 2 pmol of Cx37–GST fusion protein. Blots were blocked with 5% non-fat dried milk (NFDM) and Cx37 detected with anti-Cx37 antibody, enhanced chemiluminescence strategies and a Kodak Image Station 2000. For each experiment (n=4), the slot density for triplicate samples was averaged; the Cx37 content of cell lysates was calculated from the standard curve created from the Cx37–GST fusion protein slots. All clones and WT had densities within the range defined by the standard curve. It is worth noting that Cx37-WT is growth suppressive at expression levels 20-fold lower than the level shown for the iRin37 clone quantified here (Burt et al., 2008).
Immunofluorescence
Cells were plated on 22 mm glass coverslips in six-well dishes and induced, or not, with dox for 48 h, according to dosage for maximum protein expression, before being fixed in 100% methanol for 5 min on a shaker at room temperature. Cells were treated with 0.2% Triton X-100 for 5 min with washes in PBS preceding and following permeabilization. Cells were incubated in blocking reagent (4% fish skin gelatin, 1% normal donkey serum, 0.2% Triton X-100 in PBS) for 60 min at room temperature before incubation in anti-Cx37 antibody in a humidified chamber at 4°C, overnight. Following PBS washes, cells were incubated in secondary antibody (Cy3-conjugated, diluted 1:400 in blocking reagent) at 4°C, overnight on a shaker. Cell nuclei were stained with Hoechst dye 33342 (1:10,000). Cells were visualized using a 40× fluoroplan objective on an Olympus IX71 fluorescence microscope outfitted with a Photometrics CoolSnap ES2 camera and V++ software. Because Rin cells are thicker than the focal plane of the 40× objective, and large quantities of Cx37 are found in the cytoplasm, out of focus fluorescence tends to make images appear blurry. All of each image included in Fig. 2 was sharpened to the same extent (146%) using Adobe Photoshop.
Proliferation
Cells were plated in triplicate at an initial density of 3×104 cells/well (∼3125 cells/cm2) in six-well plates for each time point. At 24 h after plating (day 0 of proliferation assays), cells were induced or not with the appropriate dox dose (Cx37-WT, 2 μg/ml; Cx37-S319A, 2 μg/ml; Cx37-S319D, 1 μg/ml). In some experiments, cells were not induced until day 12 (dox−/+). Every 48 h, medium was refreshed with or without dox, as appropriate. Every 3 days, triplicate wells were harvested and counted using a cellometer (Nexcelom, Lawrence, MA, USA) for a total of 21 days. Mutant- and WT-expressing cells were run in parallel in each proliferation experiment, and each experiment was conducted three times, which produced a sample size sufficient to detect statistically significant differences (Burt et al., 2008; Good et al., 2012, 2014, 2011; Jacobsen et al., 2019, 2017; Nelson et al., 2013).
To reveal possible differences in cell proliferation at the earlier time points, 5.5×105 cells (∼3125 cells/cm2) were plated in 150 mm dishes. The same procedure for dox induction and feeding of cells was used as described above; WT- and mutant-expressing cells were included in each of three experiments, a sample size previously demonstrated as sufficient to reveal differences when present. Every 24 h, plates were harvested and counted using a cellometer (Nexcelom) for a total of 6 days.
Serum deprivation and cell cycle analysis
For cell cycle analysis, 106 cells were plated in 100 mm dishes (∼12,730 cells/cm2) in RPMI culture medium containing 10% FP. After 24 h, plates were divided into two groups: those maintained in medium containing 10% FP (+serum) and those switched to medium without FP (−serum). For both groups, at 72 h after plating (and every 48 h thereafter for ensuing 5 days), medium was refreshed with dox-containing medium (with or without serum as appropriate to the group). Cells in each group were harvested daily for determination of cell number and analysis of cell cycle position, as previously described (Burt et al., 2008). Briefly, cells were fixed in ethanol, resuspended in PBS (2×106 cells/ml), stained with propidium iodide (50 μg in solution with 50 μg RNase A total), and 1×104 events were analyzed for cell cycle position using fluorescence-activated cell sorting (FACS) on a FACS CantoII at the AZCC/ARL (Division of Biotechnology Cytometry Core Facility at the University of Arizona). Cell cycle distribution was determined using ModFit-LT software with doublet discrimination. Because Cx37-WT has been reported to reduce cell adhesion (Wong et al., 2006) and induce apoptosis (Jacobsen et al., 2017; Seul et al., 2004), in some cell types, the position in the cell cycle of both non-adherent and adherent cells was evaluated. In every experiment (n=8), mutant- and WT-expressing cells were run in parallel.
Electrophysiology
Cells were plated in standard culture medium at low density on 25 mm glass coverslips and induced with the appropriate dose of dox for maximum protein expression. After 24–48 h, coverslips were mounted in a custom-made chamber and bathed in external solution containing (in mM): 142.5 NaCl, 4 KCl, 1 MgCl2, 5 glucose, 2 sodium pyruvate, 10 HEPES, 15 CsCl, 10 TEA-Cl, 1 BaCl2, 1 CaCl2 (pH 7.2 and osmolarity 330 mOsM). Isolated cells and cell pairs were not in external solution for more than 60 min. An Olympus inverted (IMT2) microscope with phase contrast optics was used to identify cells on the coverslip. Patch pipets were filled with internal solution containing (in mM): 124 KCl, 14 CsCl, 9 HEPES, 9 EGTA, 0.5 CaCl2, 5 glucose, 9 TEA-Cl, 3 MgCl2, 5 Na2ATP (pH 7.2 and osmolarity 326 mOsM).
Junctional conductance (gj) was determined as previously described (Burt et al., 2008; Kurjiaka et al., 1998) using dual whole-cell voltage-clamp techniques with Axopatch1D amplifiers and pClamp software (Molecular Devices, Sunnyvale, CA). gj was determined with 10 mV transjunctional pulses. The sensitivity of gj to transjunctional voltage (Vj) was determined by stepping the voltage of one cell of a pair successively from ±10 to ±110 mV in 20 mV increments. Each pulse was 5 s in duration, with 5 s between pulses. Initial and steady state currents for each pulse were measured, currents converted into conductances and the ratio of steady state to initial conductance as a function of Vj plotted. Because Vj-gating was very rapid for the Cx37-S319A mutant and there was no evident difference between initial and steady state conductance for the ±10 mV pulses, steady state conductance for all Vj values was normalized to the initial Gj measured for the ±10 mV pulses. The normalized junctional conductances (Gj) were plotted and the resulting data fit with a Boltzmann curve using Prism software.
Single-channel activity was recorded with a 25 mV transjunctional voltage difference. Halothane was used to reveal single-channel activity in pairs where gj exceeded 1 nS. Current records were digitized at 1–2 kHz, filtered at 50 Hz to reduce noise, and the amplitudes of transitions in current levels were measured. Transition amplitudes were converted into conductances (pS), grouped into 10 pS bins, and the relative frequency of events in each bin calculated for each cell pair. Mean data were then plotted as a relative frequency histogram. Histograms of all-digitized-points were included for displayed current traces.
HCh activity and open probability (Po) were evaluated as previously described (Jacobsen et al., 2017). For HCh activity, representative current traces are illustrated with accompanying all-digitized-points histograms (indicative of dwell time at each current level for the illustrated trace). Transition amplitudes were measured (binned into 10 pS bins) for each cell and the bin means from multiple cells plotted as relative frequency histograms. For HCh Po, isolated cells were whole-cell voltage-clamped using the following pulse protocol: 30 s at +25 mV, 2 s at 0 mV, and 270 s at +25 mV. Cells with only one active channel in both +25 mV periods and no current drift over the 5-min pulse protocol were evaluated for Po. The unfiltered record was decimated (to 400 Hz sampling frequency), filtered (50 Hz), and the baseline subtracted before determining the open probability (Po) by compiling all digitized points from 240 s of the long pulse into 0.25 pA bins and calculating the relative frequency of each bin. Mean binned data from multiple cells were then plotted as a relative frequency Po histogram.
Statistical analysis
Significance of difference in protein expression levels and junctional conductance was tested by ANOVA. Significance of difference in proliferation was tested by linear regression analysis of slopes. Significance of differences in HCh Po in specific open states was calculated as described previously (Jacobsen et al., 2017). Briefly, the relative frequency Po histogram data for S319A and S319D isoforms of Cx37 were plotted together to determine ‘cross-over’ points; the summed relative frequencies of all bins between current cross-over points (at 1.25, 3.75, 9.5 pA) were compared (−2.5 to 1.25 pA for the closed state, and 1.5–3.75, 4–9.5, 9.75–23.75 pA for open states) using paired Student's t-test. All statistical comparisons were performed using Prism software.
Supplementary Material
Acknowledgements
The authors would like to thank Drs John D. Kanady, José Ek-Vitorín, and Maura Cotter for their additional technical support and helpful discussions, and ARL/UACC Cytometry Core Facility for assistance with FACS analysis.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: N.L.J., T.K.P., J.M.B.; Methodology: S.-S.Z.T., N.L.J., T.K.P., P.R.L., J.M.B.; Software: P.R.L.; Validation: S.-S.Z.T., N.L.J., P.R.L., J.M.B.; Formal analysis: S.-S.Z.T., N.L.J., T.K.P., P.R.L., J.M.B.; Investigation: S.-S.Z.T., N.L.J., T.K.P., P.R.L., J.M.B.; Resources: J.M.B.; Data curation: S.-S.Z.T., N.L.J., T.K.P., P.R.L., J.M.B.; Writing - original draft: S.-S.Z.T.; Writing - review & editing: N.L.J., T.K.P., J.M.B.; Visualization: J.M.B.; Supervision: N.L.J., T.K.P., J.M.B.; Project administration: J.M.B.; Funding acquisition: J.M.B.
Funding
This work was supported by grants from the National Institutes of Health (HL3017960 to J.M.B.; HL007249 to J.M.B. and N.L.J.; DK098493 to P.R.L.) and the American Heart Association (16PRE27500011 to N.L.J.). The content of this report is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.240721.supplemental
References
- Allagnat F., Dubuis C., Lambelet M., Le Gal L., Alonso F., Corpataux J.-M., Déglise S. and Haefliger J.-A. (2017). Connexin37 reduces smooth muscle cell proliferation and intimal hyperplasia in a mouse model of carotid artery ligation. Cardiovasc. Res. 113, 805-816. 10.1093/cvr/cvx079 [DOI] [PubMed] [Google Scholar]
- Beyer E. C. and Berthoud V. M. (2017). Gap junction structure: unraveled, but not fully revealed. F1000Res 6, 568 10.12688/f1000research.10490.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer E. C., Paul D. L. and Goodenough D. A. (1990). Connexin family of gap junction proteins. J. Membr. Biol. 116, 187-194. 10.1007/BF01868459 [DOI] [PubMed] [Google Scholar]
- Boerma M., Forsberg L., Van Zeijl L., Morgenstern R., De Faire U., Lemne C., Erlinge D., Thulin T., Hong Y. and Cotgreave I. A. (1999). A genetic polymorphism in connexin 37 as a prognostic marker for atherosclerotic plaque development. J. Intern. Med. 246, 211-218. 10.1046/j.1365-2796.1999.00564.x [DOI] [PubMed] [Google Scholar]
- Burt J. M., Nelson T. K., Simon A. M. and Fang J. S. (2008). Connexin 37 profoundly slows cell cycle progression in rat insulinoma cells. Am. J. Physiol Cell Physiol. 295, C1103-C1112. 10.1152/ajpcell.299.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers M. C., Maclean B., Burke R., Amodei D., Ruderman D. L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J. et al. (2012). A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918-920. 10.1038/nbt.2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanson M. and Kwak B. R. (2007). Connexin37: a potential modifier gene of inflammatory disease. J. Mol. Med. (Berlin, Germany) 85, 787-795. 10.1007/s00109-007-0169-2 [DOI] [PubMed] [Google Scholar]
- Dang X., Jeyaraman M. and Kardami E. (2006). Regulation of connexin-43-mediated growth inhibition by a phosphorylatable amino-acid is independent of gap junction-forming ability. Mol. Cell Biochem. 289, 201-207. 10.1007/s11010-006-9162-2 [DOI] [PubMed] [Google Scholar]
- Derouette J. P., Desplantez T., Wong C. W., Roth I., Kwak B. R. and Weingart R. (2009). Functional differences between human Cx37 polymorphic hemichannels. J. Mol. Cell Cardiol. 46, 499-507. 10.1016/j.yjmcc.2008.12.018 [DOI] [PubMed] [Google Scholar]
- Eghbali B., Kessler J. A., Reid L. M., Roy C. and Spray D. C. (1991). Involvement of gap junctions in tumorigenesis: Transfection of tumor cells with connexin 32 cDNA retards growth in vivo. Proc. Natl. Acad. Sci. USA 88, 10701-10705. 10.1073/pnas.88.23.10701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J. S., Angelov S. N., Simon A. M. and Burt J. M. (2011). Cx37 deletion enhances vascular growth and facilitates ischemic limb recovery. Am. J. Physiol Heart Circ. Physiol. 301, H1872-H1881. 10.1152/ajpheart.00683.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J. S., Angelov S. N., Simon A. M. and Burt J. M. (2012). Cx40 is required for, and cx37 limits, postischemic hindlimb perfusion, survival and recovery. J. Vasc. Res. 49, 2-12. 10.1159/000329616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J. S., Coon B. G., Gillis N., Chen Z., Qiu J., Chittenden T. W., Burt J. M., Schwartz M. A. and Hirschi K. K. (2017). Shear-induced Notch-Cx37-p27 axis arrests endothelial cell cycle to enable arterial specification. Nat. Commun. 8, 2149 10.1038/s41467-017-01742-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriels J. E. and Paul D. L. (1998). Connexin43 is highly localized to sites of disturbed flow in rat aortic endothelium but connexin37 and connexin40 are more uniformly distributed [see comments]. Circ. Res. 83, 636-643. 10.1161/01.RES.83.6.636 [DOI] [PubMed] [Google Scholar]
- Good M. E., Nelson T. K., Simon A. M. and Burt J. M. (2011). A functional channel is necessary for growth suppression by Cx37. J. Cell Sci. 124, 2448-2456. 10.1242/jcs.081695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good M. E., Ek-Vitorin J. F. and Burt J. M. (2012). Extracellular loop cysteine mutant of cx37 fails to suppress proliferation of rat insulinoma cells. J. Membr. Biol. 245, 369-380. 10.1007/s00232-012-9459-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good M. E., Ek Vitorín J. F. and Burt J. M. (2014). Structural determinants and proliferative consequences of connexin 37 hemichannel function in insulinoma cells. J. Biol. Chem. 289, 30379-30386. 10.1074/jbc.M114.583054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenough D. A. and Paul D. L. (2003). Beyond the gap: functions of unpaired connexon channels. Nat. Rev. Mol. Cell Biol. 4, 285-294. 10.1038/nrm1072 [DOI] [PubMed] [Google Scholar]
- Huang W. and Erikson R. L. (1994). Constitutive activation of Mek1 by mutation of serine phosphorylation sites. Proc. Natl. Acad. Sci. USA 91, 8960-8963. 10.1073/pnas.91.19.8960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismailov I. I. and Benos D. J. (1995). Effects of phosphorylation on ion channel function. Kidney Int. 48, 1167-1179. 10.1038/ki.1995.400 [DOI] [PubMed] [Google Scholar]
- Jacobsen N. L., Pontifex T. K., Li H., Solan J. L., Lampe P. D., Sorgen P. L. and Burt J. M. (2017). Regulation of Cx37 channel and growth-suppressive properties by phosphorylation. J. Cell Sci. 130, 3308-3321. 10.1242/jcs.202572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen N. L., Pontifex T. K., Langlais P. R. and Burt J. M. (2019). Phosphorylation-dependent intra-domain interaction of the Cx37 carboxyl-terminus controls cell survival. Cancers 11, E188 10.3390/cancers11020188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardami E., Dang X., Iacobas D. A., Nickel B. E., Jeyaraman M., Srisakuldee W., Makazan J., Tanguy S. and Spray D. C. (2007). The role of connexins in controlling cell growth and gene expression. Prog. Biophys. Mol. Biol. 94, 245-264. 10.1016/j.pbiomolbio.2007.03.009 [DOI] [PubMed] [Google Scholar]
- Kruse R., Krantz J., Barker N., Coletta R. L., Rafikov R., Luo M., Hojlund K., Mandarino L. J. and Langlais P. R. (2017). Characterization of the CLASP2 protein interaction network identifies SOGA1 as a microtubule-associated protein. Mol. Cell. Proteomics 16, 1718-1735. 10.1074/mcp.RA117.000011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurjiaka D. T., Steele T. D., Olsen M. V. and Burt J. M. (1998). Gap junction permeability is diminished in proliferating vascular smooth muscle cells. Am. J. Physiol. 275, C1674-C1682. 10.1152/ajpcell.1998.275.6.C1674 [DOI] [PubMed] [Google Scholar]
- Larson D. M., Seul K. H., Berthoud V. M., Lau A. F., Sagar G. D. and Beyer E. C. (2000). Functional expression and biochemical characterization of an epitope-tagged connexin37. Mol. Cell Biol. Res. Commun. 3, 115-121. 10.1006/mcbr.2000.0200 [DOI] [PubMed] [Google Scholar]
- Liu J., Asuncion-Chin M., Liu P. and Dopico A. M. (2006). CaM kinase II phosphorylation of slo Thr107 regulates activity and ethanol responses of BK channels. Nat. Neurosci. 9, 41-49. 10.1038/nn1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch R. I., Daubner S. C. and Fitzpatrick P. F. (2001). Effects of substitution at serine 40 of tyrosine hydroxylase on catecholamine binding. Biochemistry 40, 7273-7278. 10.1021/bi010546d [DOI] [PubMed] [Google Scholar]
- Morel S. (2014). Multiple roles of connexins in atherosclerosis- and restenosis-induced vascular remodelling. J. Vasc. Res. 51, 149-161. 10.1159/000362122 [DOI] [PubMed] [Google Scholar]
- Morel S., Burnier L., Roatti A., Chassot A., Roth I., Sutter E., Galan K., Pfenniger A., Chanson M. and Kwak B. R. (2010). Unexpected role for the human Cx37 C1019T polymorphism in tumour cell proliferation. Carcinogenesis 31, 1922-1931. 10.1093/carcin/bgq170 [DOI] [PubMed] [Google Scholar]
- Moreno A. P., Chanson M., Elenes S., Anumonwo J., Scerri I., Gu H., Taffet S. M. and Delmar M. (2002). Role of the carboxyl terminal of connexin43 in transjunctional fast voltage gating. Circ. Res. 90, 450-457. 10.1161/hh0402.105667 [DOI] [PubMed] [Google Scholar]
- Nelson T. K., Sorgen P. L. and Burt J. M. (2013). Carboxy terminus and pore-forming domain properties specific to Cx37 are necessary for Cx37-mediated suppression of insulinoma cell proliferation. Am. J. Physiol Cell Physiol. 305, C1246-C1256. 10.1152/ajpcell.00159.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfenniger A., Wong C., Sutter E., Cuhlmann S., Dunoyer-Geindre S., Mach F., Horrevoets A. J., Evans P. C., Krams R. and Kwak B. R. (2012). Shear stress modulates the expression of the atheroprotective protein Cx37 in endothelial cells. J. Mol. Cell Cardiol. 53, 299-309. 10.1016/j.yjmcc.2012.05.011 [DOI] [PubMed] [Google Scholar]
- Reed K. E., Westphale E. M., Larson D. M., Wang H.-Z., Veenstra R. D. and Beyer E. C. (1993). Molecular cloning and functional expression of human connexin37, an endothelial cell gap junction protein. J. Clin. Invest. 91, 997-1004. 10.1172/JCI116321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seul K. H., Kang K. Y., Lee K. S., Kim S. H. and Beyer E. C. (2004). Adenoviral delivery of human connexin37 induces endothelial cell death through apoptosis. Biochem. Biophys. Res. Commun. 319, 1144-1151. 10.1016/j.bbrc.2004.05.097 [DOI] [PubMed] [Google Scholar]
- Shin J.-L., Solan J. L. and Lampe P. D. (2001). The regulatory role of the C-terminal domain of connexin43. Cell Commun. Adhes 8, 271-275. 10.3109/15419060109080736 [DOI] [PubMed] [Google Scholar]
- Simon A. M., Chen H. and Jackson C. L. (2006). Cx37 and Cx43 localize to zona pellucida in mouse ovarian follicles. Cell Commun. Adhes 13, 61-77. 10.1080/15419060600631748 [DOI] [PubMed] [Google Scholar]
- Sohl G. and Willecke K. (2004). Gap junctions and the connexin protein family. Cardiovasc. Res. 62, 228-232. 10.1016/j.cardiores.2003.11.013 [DOI] [PubMed] [Google Scholar]
- Solan J. L. and Lampe P. D. (2007). Key connexin 43 phosphorylation events regulate the gap junction life cycle. J. Membr. Biol. 217, 35-41. 10.1007/s00232-007-9035-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan J. L. and Lampe P. D. (2009). Connexin43 phosphorylation: structural changes and biological effects. Biochem. J. 419, 261-272. 10.1042/BJ20082319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan J. L., Marquez-Rosado L., Sorgen P. L., Thornton P. J., Gafken P. R. and Lampe P. D. (2007). Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J. Cell Biol. 179, 1301-1309. 10.1083/jcb.200707060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spray D. C., White R. L., de Carvalho A. C., Harris A. L. and Bennett M. V. (1984). Gating of gap junction channels. Biophys. J. 45, 219-230. 10.1016/S0006-3495(84)84150-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S. C., Seo J., Pan J. Q., Samuels B. A., Rudenko A., Ericsson M., Neve R. L., Yue D. T. and Tsai L.-H. (2012). Regulation of N-type voltage-gated calcium channels and presynaptic function by cyclin-dependent kinase 5. Neuron 75, 675-687. 10.1016/j.neuron.2012.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub O., Hertlein B., Kasper M., Eckert R., Krisciukaitis A., Hülser D. and Willecke K. (1998). Characterization of the gap junction protein connexin37 in murine endothelium, respiratory epithelium, and after transfection in human HeLa cells. Eur. J. Cell Biol. 77, 313-322. 10.1016/S0171-9335(98)80090-3 [DOI] [PubMed] [Google Scholar]
- Vizcaíno J. A., Côté R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J. et al. (2013). The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063-D1069. 10.1093/nar/gks1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen D., Du X., Nie S.-P., Dong J.-Z. and Ma C.-S. (2014). Association of Connexin37 C1019T with myocardial infarction and coronary artery disease: a meta-analysis. Exp. Gerontol. 58, 203-207. 10.1016/j.exger.2014.06.011 [DOI] [PubMed] [Google Scholar]
- Wong C. W., Christen T., Roth I., Chadjichristos C. E., Derouette J.-P., Foglia B. F., Chanson M., Goodenough D. A. and Kwak B. R. (2006). Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat. Med. 12, 950-954. 10.1038/nm1441 [DOI] [PubMed] [Google Scholar]
- Zhang Y.-W., Morita I., Ikeda M., Ma K.-W. and Murota S. (2001). Connexin43 suppresses proliferation of osteosarcoma U2OS cells through post-transcriptional regulation of p27. Oncogene 20, 4138-4149. 10.1038/sj.onc.1204563 [DOI] [PubMed] [Google Scholar]
- Zhang Y. W., Nakayama K., Nakayama K. and Morita I. (2003). A novel route for connexin 43 to inhibit cell proliferation: negative regulation of S-phase kinase-associated protein (Skp 2). Cancer Res. 63, 1623-1630. [PubMed] [Google Scholar]
- Zimmer D. B., Green C. R., Evans W. H. and Gilula N. B. (1987). Topological analysis of the major protein in isolated intact rat liver gap junctions and gap junction-derived single membrane structures. J. Biol. Chem. 262, 7751-7763. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.