

Abstract
Microvascular endothelial cell (EC) injury and the subsequent blood‐brain barrier (BBB) breakdown are frequently seen in many neurological disorders, including stroke. We have previously documented that peroxisome proliferator‐activated receptor gamma (PPARγ)‐mediated cerebral protection during ischemic insults needs Krüppel‐like factor 11 (KLF11) as a critical coactivator. However, the role of endothelial KLF11 in cerebrovascular function and stroke outcome is unclear. This study is aimed at investigating the regulatory role of endothelial KLF11 in BBB preservation and neurovascular protection after ischemic stroke. EC‐targeted overexpression of KLF11 significantly mitigated BBB leakage in ischemic brains, evidenced by significantly reduced extravasation of BBB tracers and infiltration of peripheral immune cells, and less brain water content. Endothelial cell‐selective KLF11 transgenic (EC‐KLF11 Tg) mice also exhibited smaller brain infarct and improved neurological function in response to ischemic insults. Furthermore, EC‐targeted transgenic overexpression of KLF11 preserved cerebral tight junction (TJ) levels and attenuated the expression of pro‐inflammatory factors in mice after ischemic stroke. Mechanistically, we demonstrated that KLF11 directly binds to the promoter of major endothelial TJ proteins including occludin and ZO‐1 to promote their activities. Our data indicate that KLF11 functions at the EC level to preserve BBB structural and functional integrity, and therefore, confers brain protection in ischemic stroke. KLF11 may be a novel therapeutic target for the treatment of ischemic stroke and other neurological conditions involving BBB breakdown and neuroinflammation.
Keywords: blood‐brain barrier, endothelial cell, ischemic brain injury, Krüppel‐like factor, tight junction
Abbreviations
- BBB
blood‐brain barrier
- BMECs
brain microvascular endothelial cells
- CBF
cerebral blood flow
- EC
endothelial cell
- KLF
Krüppel‐like factor
- MAP2
microtubule‐associated protein 2
- MCAO
middle cerebral artery occlusion
- OGD
oxygen‐glucose deprivation
- PPARγ
peroxisome proliferator‐activated receptor gamma
- TEER
transepithelial/transendothelial electrical resistance
- TJ
tight junction
- TTC
2,3,5‐triphenyltetrazolium chloride
Introduction
Stroke ranks as the fifth leading cause of death and accounts for 1 out of every 20 deaths in the United States (6, 60). Ischemic stroke leads to cerebrovascular and neuronal damage, both of which contribute to ischemic injury and dictate stroke outcomes (24, 66). Although studies have implied the effectiveness of neuroprotectants in animal stroke models (24, 32), there has been very little progress made in the past three decades concerning the successful translation of neuroprotective strategies to the clinical setting. The lack of clinical translation implies that focusing on neuroprotection only is insufficient. Non‐neuronal cells and the local microenvironment of the surviving neurons could also serve as therapeutic targets to alleviate ischemic brain injury (61, 65). Cerebral vascular endothelial cells (ECs) are major components of brain microvasculature and play critical roles in maintaining the integrity of the blood‐brain barrier (BBB, a brain‐specific microvascular structure) and cerebral homeostasis under physiological conditions (1, 5, 50). Cerebral ECs are tightly connected by adherens junctions (AJs) and tight junctions (TJs), including occludin, claudins and junctional adhesion molecule (JAM), which are anchored to the actin cytoskeleton by scaffold proteins such as zonula occludens (ZO)‐1, AF6 and cingulin. Together they form a diffusion barrier that selectively blocks the passage of most compounds through the blood to brain compartments (4, 16, 39). When BBB integrity is compromised, proinflammatory factors may pass through the BBB to attract circulating immune cells to the injured brain, leading to secondary ischemic brain parenchymal injuries (15, 33, 34, 35, 46, 54, 58). Therefore, inhibition of BBB disruption by stabilizing TJs and protecting brain endothelium has become promising therapeutic strategies for ischemic stroke (18, 22, 45).
Krüppel‐like factors (KLFs) are a large family of zinc‐finger transcription factors that are known to trans‐activate or trans‐repress gene expression in various organisms (63). Recent studies have reported the involvement of KLF members in various developmental and pathological vascular processes (2, 52). A recent RNA sequencing study showed that in human endothelium, KLF11 has modest mRNA expression (20). KLF11 has been reported to play functional roles in the regulation of cell growth and differentiation, cholesterol metabolism and cell death (21, 25, 42, 52). Moreover, population genetics studies have demonstrated that mutations in the KLF11 gene are strongly associated with type 2 diabetes (21, 42). Previous studies indicated that KLF11 inhibits endothelial activation in response to inflammatory stimuli (19). In addition, vascular smooth muscle cell‐selective deletion of KLF11 aggravates arterial thrombosis in mice (36). However, the role of KLF11 in cerebrovascular biology remains largely undetermined.
A previous study by our group reported that KLF11 functions as a peroxisome proliferator‐activated receptor‐γ coregulator to attenuate middle cerebral artery occlusion (MCAO)‐induced ischemic brain injury (62). In conventional KLF11 knockout mice, we observed larger brain infarct, increased sensorimotor loss, aggravated BBB leakage, higher brain edema and lower cerebral blood flow (CBF) following cerebral ischemic insult (53). These studies suggest that KLF11 plays a protective role in the pathogenesis of ischemic stroke. However, the molecular events and regulatory roles of endothelial KLF11 itself in stroke‐induced cerebral EC injury and BBB disruption remain virtually unclear. Thus, the present study sought to test the hypothesis that KLF11 stabilizes the cerebrovascular endothelial structure while simultaneously reducing stroke‐induced inflammatory events by preserving the integrity of TJs in cerebral ECs.
Materials and Methods
All procedures using laboratory animals were approved by the University of Pittsburgh Institutional Animal Care and Use Committee, and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All mice were randomly assigned to various experimental groups using a lottery box. All surgical preparation, stroke outcome assessments, neurobehavioral tests and data analysis were performed in a blinded manner.
EC‐KLF11 Tg and littermate wild‐type control (WT) mice (male, 8–10 weeks‐old, body weight 23–25 g) were housed in a temperature‐ and humidity‐controlled animal facility with a 12 h light/dark cycle and with unlimited access to food and water. Animals that did not show a more than 75% CBF reduction or a less than 60% reperfusion over baseline levels or died after ischemic induction (~10% of stroke animals) were excluded from further experimentation. In this study, we used 138 MCAO‐operated mice (WT, n = 69; EC‐KLF11 Tg, n = 69). Sham‐operated mice were used as controls (WT, n = 28; EC‐KLF11 Tg, n = 28).
Generation of EC‐selective KLF11 transgenic (EC‐KLF11 Tg) mice
Transgenic mice with endothelial cell‐selective KLF11 overexpression were generated as described (64). A 1539‐bp DNA fragment containing a KLF11 coding sequence was PCR amplified from human genomic DNA using the primers 5′‐GATATCATGCACACGCCGGACTTCGCAGGCC‐3′ (Forward) and 5′‐GATATCTCAGGCAGAGGCTGGCATGCTCACC‐3′ (Reverse). The DNA fragment was then cloned into the EcoRV site of the pBluescript II SK (+) vector to generate a pBlue‐KLF11 plasmid. To generate the Tie‐2 promoter‐driven pBlue‐KLF11 construct, we then inserted the Tie‐2 promoter (2089 bp) (a generous gift from Dr. Sato, (47)) into the HindIII site and the polyA plus full Tie‐2 enhancer (10 367 bp) (a generous gift from Dr. Sato, (47)) into the Xba I/Not I cloning site on pBlue‐KLF11 plasmid, respectively. The construct was injected into fertilized C57 mouse oocytes and implanted into pseudo‐pregnant female mice. Transgenic founder mice were identified by PCR genotyping using primers 5′‐CTGTGCTCAGACAGAAATGAGAC‐3′ (forward) and 5′‐ATCATCTGGCAAAGGACAGG‐3′ (reverse). The PCR amplification conditions were 94°C × 5 minutes, 40 cycles of 94°C × 30 s, 55°C × 30 s, 72°C × 80 s, followed by 72°C × 5 minutes. This produced a 1.2 kb DNA fragment that contains both plasmid and Tie‐2 promoter sequences. EC‐KLF11 Tg mice are viable, fertile, normal in size and do not display any gross physical or behavioral abnormalities.
Mouse model of transient focal cerebral ischemia
Focal cerebral ischemia was induced in adult male mice (8–10 weeks old, 25–30 g) by intraluminal middle cerebral artery occlusion (MCAO) as described previously (53, 59, 61, 67). Briefly, mice were anesthetized with 1.5%–3% isoflurane (Henry Schein Animal Health). A 2‐cm length of a 7‐0 rounded tip nylon suture was gently advanced from the internal carotid artery up to the origin of the middle cerebral artery (MCA) until regional cerebral blood flow (rCBF) was reduced to less than 25% of baseline. After 60 minutes of MCAO, blood flow was restored by removing the suture and mice were allowed to recover for 1–7 days. In sham‐operated mice, the same surgical procedure was performed except for suture insertion. Changes in rCBF, arterial blood gases, mean arterial pressure and heart rate were monitored in animals 15 minutes before, during and 15 minutes after MCAO. Animals that did not show a CBF reduction of at least 75% over baseline levels were excluded from further experimentation. Approximately 90% survival rate was observed in EC‐KLF11 Tg or WT mice at 1–7 days after MCAO. Animals that died after ischemic induction were also excluded. The rectal temperature was controlled at 37.0 ± 0.5°C during surgery.
Measurement of infarct volume, neurological deficits and sensorimotor function
2,3,5‐triphenyltetrazolium (TTC) staining was performed to measure brain infarct after MCAO in some mice. These mice were sacrificed and the brains were harvested 24 h after MCAO. The forebrain was sliced into seven coronal sections, each 1 mm thick. Sections were stained with 2% TTC in 0.9% NaCl for 20 minutes, followed by fixation with 4% paraformaldehyde in PBS. Infarct volume was determined with ImageJ (National Institute of Health) as the volume of the contralateral hemisphere minus the non‐infarcted volume of the ipsilateral hemisphere.
Neurobehavioral deficits were determined by the adhesive removal test, foot fault test and rotarod test 1–3 days before MCAO and also at 3, 5 and 7 days of reperfusion after MCAO (53, 55, 59, 67). Following cerebral ischemia, mice were also tested for neurological deficits and scored on a 5‐point scale: 0, no observable neurological deficits (normal); 1, failure to extend right forepaw (mild); 2, circling to the contralateral side (moderate); 3, falling to the right (severe); 4, mice could not walk spontaneously; 5, depressed level of consciousness (very severe).
Immunohistochemistry and image analysis
At different time points following MCAO, EC‐KLF11 Tg and WT mice were anesthetized with carbon dioxide and transcardially perfused with 0.9% NaCl followed by 4% paraformaldehyde in PBS. Brains were harvested and cryoprotected in 30% sucrose in PBS, and frozen serial coronal brain sections (30 µm thick) were prepared on a cryostat (CM1900, Leica). Brain sections were blocked with 5% normal goat serum in PBS for 1 h, followed by overnight incubation (4°C) with the following primary antibodies: mouse anti‐MAP2 (1:200; EMD Millipore, Billerica, MA), rabbit anti‐NeuN (1:500; EMD Millipore), rat anti‐CD31 (1:200; BD Biosciences, San Jose, CA), mouse anti‐ZO‐1 (1:100; Invitrogen, Carlsbad, CA), rat anti‐Ly‐6B (1:100; Abcam, Cambridge, UK) and rabbit anti‐Occludin (1:100; Thermo Fisher Scientific, Waltham, MA) (Supporting Table S1). Secondary antibodies: Cy3‐conjugated goat anti‐rat IgG, Alexa Fluor 488‐conjugated goat anti‐rabbit IgG and goat anti‐mouse IgG (all at 1:400; The Jackson ImmunoResearch Lab). Images were collected by confocal microscopy (FV1000‐II; Olympus; Center for Biological Imaging, University of Pittsburgh Medical School) and processed in Adobe Photoshop for compositions (17). Five different sections from each animal were photographed and six ROIs were randomly selected from the infarct core and inner infarct border, respectively, on each section. Infarct volume was measured on seven equally spaced MAP2‐stained sections encompassing the MCA territory using ImageJ. Capillary densities were examined by counting the number of capillaries stained with the anti‐CD31 antibody as previously described (56).
Quantification of BBB permeability/leakage after MCAO
For the analysis of cerebrovascular permeability by Evans Blue extravasation, mice were injected with 100 µl of 4% Evans Blue (EB) (Sigma‐Aldrich, St. Louis, MO) through tail vein 23 h after MCAO. After 1 h, mice were perfused with 0.9% NaCl, mouse brains were then removed and separated into ipsilateral and contralateral hemispheres. Each hemisphere was homogenized in N,N‐dimethylformamide (Sigma‐Aldrich) and centrifuged for 45 minutes at 25 000 rcf. The supernatants were collected and EB levels in each hemisphere were calculated using the formula: (A620 nm − (A500 nm + A740 nm)/2)/mg wet weight. Background EB levels in the nonischemic hemisphere were subtracted from the ischemic hemisphere ipsilateral to the MCAO.
For the analysis of cerebrovascular permeability by the intravenous injection and detection of fluorescent‐labeled Dextran and fluorescent tracer, mice were subjected to tail vein injection of either Tetramethylrhodamine‐Dextran (TMR‐Dextran, 70 kDa; 0.1 mg/g body weight, Invitrogen) or Alexa Fluor 555‐conjugated cadaverine (0.95 kDa; Invitrogen) 30 minutes before sacrifice. Mice were anesthetized and transcardially perfused with 0.9% NaCl followed by 4% paraformaldehyde in PBS. Mouse brains were collected and cryoprotected in 30% sucrose in PBS and frozen serial coronal brain sections (30 µm thick) were prepared on a cryostat. Brain sections were visualized directly under a fluorescent microscope. In parallel, brain hemispheres were homogenized in 1% Triton X‐100 (Sigma‐Aldrich) in PBS and fluorescent intensity was quantified by a SpectraMax i3x plate reader (Molecular Devices, San Jose, CA) using 555‐nm excitation and 580‐nm emission.
To measure the extravasation of endogenous IgG, sections were blocked in avidin and biotin solution (two drops of avidin/biotin solution into 10 mL of PBS; Vector Laboratories, Burlingame, CA) for 15 minutes each, followed by blocking in 5% NDS for 1 h. Sections were then incubated with biotinylated anti‐mouse IgG (1:500; Vector Laboratories) at 4°C overnight. Sections were then incubated with Alexa 488 Streptavidin (1:1000; The Jackson ImmunoResearch Labs). Whole‐section images were acquired using an inverted Nikon fluorescence microscope. Six equally spaced sections encompassing the MCA territory were quantified for the cross‐sectional area of fluorescence. These areas were summed and multiplied by the distance between sections (1 mm) to yield a volume of leakage (in mm3).
Adenovirus‐mediated gain‐ or loss‐ of‐ KLF11 expression in BMECs
To generate adenoviral vectors overexpressing KLF11 (19), the coding sequences of human KLF11 were PCR‐amplified with primers 5′‐ATGCACACGCCGGACTTCGCAGG‐3′ (Forward) and 5′‐TCAGGCA GAGGCTGGCATGCTCA‐3′ (Reverse), and subcloned into the pCR8⁄GW⁄TOPO entry vector (Invitrogen). To generate adenoviral vectors for overexpressing LacZ, the coding sequences of Escherichia coli LacZ were PCR amplified with primers 5′‐ATGTCGTTTACTTTGACCAACA‐3′ (Forward) and 5′‐TTATTTTTGACACCAGACCAACT‐3′ (Reverse), and subcloned into the pCR8⁄GW⁄TOPO entry vector (Invitrogen). After sequencing, the LR recombination reactions were carried out between the entry clone pCR8⁄GW⁄TOPO/KLF11 and the destination vector pAd/CMV/V5‐DEST according to the manufacturer's protocol (Invitrogen). For knockdown experiments, an siRNA oligo, which targets a region 100% conserved between human and mouse, was purchased from Invitrogen. To prepare adenovirus‐containing shRNA for KLF11 or LacZ, synthesized oligos were annealed and inserted into the BLOCK‐iT U6 entry vector. The U6 promoter and shRNA were cloned into the adenoviral plasmid pAd/BLOCK‐iT‐DEST according to the manufacturer's instructions. The sequences for shRNA were as follows:
shKLF11, 5′‐CACCGGGTAGACTTTTCCCGAAGGCGAACCTTCGGGAAAAGTCTACC‐3′,
5′‐AAAAGGTAGACTTTTCCCGAAGGTTCG CCTTCGGGAAAAGTCTACCC‐3′
shLacZ, 5′‐CACCGCTACACAAATCAGCGATTTCGAAAAATCGCTGATTTGTGTAG‐3′,
5′‐AAAACTACACAAATCAGCGATTTTTCGAAATCGCTGATTTGTGTAGC‐3′
To package the adenoviruses, adenoviral vectors were linearized with PacI and transfected into HEK293 AD cells using Lipofectamine 2000. The recombinant adenoviruses were purified by CsCl2 density gradient ultracentrifugation. Adenovirus genomic DNA was purified with NucleoSpin Virus Kit (Macherey Nagel), the adenovirus titration was determined using the Adeno‐X™ qPCR Titration Kit (Clontech). The generated adenovirus was used to infect BMECs for 48–72 h.
Two cell‐based in vitro BBB model
Mouse astrocytes were purchased from the ScienCell research laboratories (M1800‐57). Astrocytes (2–8 passages) were grown in astrocyte medium (AM‐a 1831; ScienCell) and were seeded in a regular 12‐well plate at 37 ºC in a humidified incubator until reaching confluence. Transwell chamber inserts with PET membranes (0.4 µm pore; Corning) were coated with collagen type IV (0.3 mg/mL) and fibronectin (0.5 mg/mL). Mouse BMECs were seeded onto the membrane at a density of 1 × 105 cells per insert. The inserts were then placed into the 12‐well plates containing confluent astrocytes (Figure 7A) and co‐cultures were maintained in endothelial growth medium (M1168; Cell Biologics) at 37 ºC in a humidified incubator overnight. The inserts seeded with mBMECs were then separated from the co‐culture system and mBMECs were infected with either Ad. LacZ/Ad. KLF11 or Ad. shLacZ/Ad. shKLF11 in endothelial cell infection medium without phenol red and antibiotics (M1168PF; Cell Biologics) for 6 h. The inserts were placed back to the 12‐well plate containing confluent astrocytes and co‐cultures were maintained in the endothelial growth medium to reach confluence.
Figure 7.
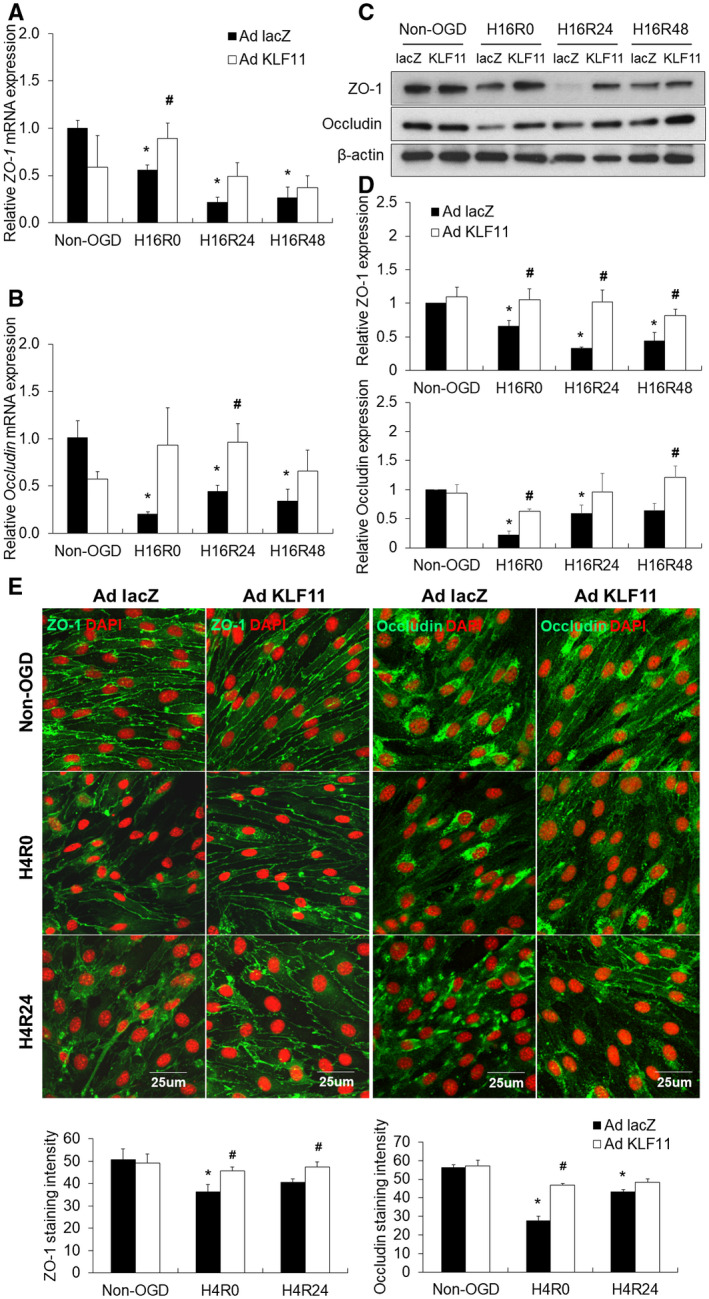
Adenovirus‐mediated KLF11 overexpression in mouse BMEC cultures preserves the integrity of tight junction proteins after OGD. Cultured mBMECs were infected with Ad. LacZ or Ad. KLF11 for 48 h prior to 16 h or 4 h OGD followed by 24 h or 48 h reperfusion. A and B. The mRNA expression levels of ZO‐1 and occludin were determined by qPCR and normalized to cyclophilin (n = 3 per group). C and D. Expression levels of tight junction proteins, ZO‐1 and occludin were evaluated by western blotting. β‐Actin was used as an internal loading control. Data represent three independent experiments and representative blots are displayed. n = 3 per group. *P < 0.05 vs. Ad. LacZ + non‐OGD group, # P < 0.05 vs. Ad. LacZ + OGD group. E. Representative images and quantification of ZO‐1 (green) and occludin (green) in cultured mBMECs after 4 h OGD, followed by 24 h reoxygenation. (Scale bar: 25 µm.) Adenoviral KLF11 overexpression suppressed OGD‐induced disruption of junctional proteins, ZO‐1 and occludin. n = 3 per group. *P < 0.05 vs. Ad. LacZ + non‐OGD group, # P < 0.05 vs. Ad. LacZ + OGD group. H16R0 = 16 h OGD + 0 h reperfusion, H16R24 = 16 h OGD + 24 h reperfusion, H16R48 = 16 h OGD + 48 h reperfusion, H4R0 = 4 h OGD + 0 h reperfusion, H4R24 = 4 h OGD + 24 h reperfusion.
Changes in transendothelial electrical resistance (TEER) at designated time points or conditions were used to detect permeability changes in the BBB. The reading of total resistance (R Total) was measured with an Epithelial Volt/Ohm Meter (WPI, FL) at room temperature. The value of each sample (R TEER) was corrected by the reading of an empty coated‐insert cell (R Blank) and calculated with the polyester membrane area (S Membrane) using the following formula and TEER values were reported in units of Ω‐cm2:
To assess paracellular permeability, Dextran Alexa Fluor 488 (3000 MW) was added to the luminal chamber at a concentration of 1 µg/1 mL of medium. At 0.5, 1, 2, 4, 8 and 24 h after OGD treatment, 50 µL medium was collected from the abluminal chamber. Fluorescence intensity was measured with a SpectraMax i3x Multi‐Mode Detection Platform. The accumulated fluorescence intensity of abluminal Dextran Alexa Fluor 488 was measured at each time point and corrected by the respective blank (same treatment without adding Dextran in the luminal). The relative fluorescence unit (RFU) was recorded.
Cerebral microvessel isolation
Cerebral microvessels from the mouse brain were isolated for determining the expression of KLF11, as previously described with modification (61). Briefly, EC‐KLF11 Tg and littermate WT control mice were anesthetized with carbon dioxide and transcardially perfused with ice‐cold 0.9% NaCl. The brain was immediately removed from the skull and immersed in ice‐cold PBS. Brainstem, meninges and pia vessels were quickly removed, and the brain was cut into 1mm block and transfer to a 15 mL of Dounce Tissue Grinder Tube (Kimble Chase, TN) with 5 mL of buffer TE (a mixture of 0.25% Trypsin‐EDTA and DMEM (Invitrogen, CA) at 1:1). The brain was homogenized by uf5 strokes with a small clearance pestle. The homogenate was mixed with another 5 mL of buffer TE and incubate at 37°C for 1 h with occasional agitation. After triturating 10–20 times with a 5 mL pipette, the creamy texture was centrifuged at 500 × g for 10 minutes at room temperature. The pellet was collected and dissolved in 8 mL of HBSS (Invitrogen, CA) by mixing twice with a 5 mL pipette and the homogenate was suspended in 11 mL HBSS dissolved 25% BSA by gently triturate five times with a 10 mL pipette. The homogenate was centrifuged at 3000 × g for 15 minutes at 4°C to separate the lipid and the capillary fraction. The pellet was collected and rinsed with 20 mL of PBS once followed by another centrifuge at 1800 × g for 10 minutes. The pellet was rinsed with 1 mL of PBS, transferred to a 1.5 ml centrifuge tube and spun down at 16 000 × g for 1 minute. The final microvessel pellet was stored at −80°C freezer until use.
Brain water content
Brain water content was measured by the dry‐wet method as described previously (53). Following MCAO, mice were sacrificed by exposure to CO2. The weights of the ipsilateral and contralateral hemispheres were recorded separately as wet weights. The dry weights of the ipsilateral and contralateral hemispheres were obtained after being heated at 100°C in an oven for 24 h. Brain content was calculated by the following formula: Brain content = (Wet weight − Dry weight)/Wet weight × 100%.
Cell cultures and oxygen‐glucose deprivation (OGD)
Mouse primary brain microvascular endothelial cells (mBMECs) were purchased from Cell Biologics (C57‐6023). Mouse brain microvascular endothelial cells, bEnd.3 were purchased from American Type Culture Collection (CRL‐2299, ATCC, Manassas, VA). Mouse BMECs and bEnd.3 cells (2–8 passages) were grown to 85%–95% confluency before using for studies. To mimic ischemia‐like conditions in vitro, mouse BMEC cultures were exposed to OGD for up to 16 h. Briefly, culture medium was replaced with deoxygenated glucose‐free Dulbecco's Modified Eagle Media (DMEM; Gibco, Grand Island, NY) and cultures were flushed with 95% N2 and 5% CO2 for 5 minutes in a Billups‐Rothenberg modular incubator chamber (Del Mar, San Diego, CA). The modular incubator chamber was then sealed and placed in a water‐jacketed incubator (Forma, Thermo Fisher Scientific, Waltham, MA) at 37 ˚C for the indicated period of time in each experiment before returning to humidified 95% air and 5% CO2 and glucose‐containing medium (62, 67).
Immunocytochemistry
Mouse BMEC cells were seeded in 4‐chamber polystyrene vessel tissue culture treated glass slides (Corning, NY) and infected with Ad. LacZ or Ad. KLF11. Seventy‐two hours after infection, BMEC cultures were subjected to OGD treatment and reperfusion for indicated time points. The cells were then fixed in 4% paraformaldehyde followed by blocking with 5% normal goat serum in PBST for 1 h at room temperature. The cells were then incubated with the following primary antibodies overnight at 4°C: mouse anti‐ZO‐1 (1:100; Invitrogen, Carlsbad, CA), rabbit anti‐Occludin (1:100; Thermo Fisher Scientific, Waltham, MA). After rinse in PBS, cells were incubated with secondary antibodies: Alexa Fluor 488‐conjugated goat anti‐rabbit IgG and goat anti‐mouse IgG (all at 1:400; The Jackson ImmunoResearch Laboratories). After PBS rinses, cells were counterstained with DAPI for nuclear labeling and mounted with antifade VECTASHIELD solution (Vector Laboratories). Images were collected on an Olympus FV1000‐II confocal microscope and processed in Adobe Photoshop for compositions (17). Immunofluorescence intensities of ZO‐1 and occludin were quantified by ImageJ. Three ROIs were randomly selected from each chamber and 2–3 chambers were quantified for each experimental condition in each independent culture.
Molecular cloning
The pGL4.10 [luc2] vector was purchased from Promega. To make pGL4_mZO‐1 and pGL4_mOccludin promoter‐reporter constructs, a 1013 bp fragment of the promoter region of mouse ZO‐1 or a 1014 bp fragment of mouse occludin promoter region containing the putative KLF11 binding sequence was PCR‐amplified from mouse genomic DNA using primers 5‐CGAGACGCTAGCCTTGACTTTGAAACCTTAATTGATG and 5‐CTGGACCTCGAGGCAAAACCTGCCGGACCGGGCCACT, or 5‐CGAGACGCTAGCAGATAGTTAACTAACAAGAACTAAAATCTC and 5‐CTGGACCTCGAGCCTACCCCCGGGCATGCGCACCAATT. Both promoter fragments were then subcloned into the pGL4.10[luc2] vector, respectively. Mutant promoter constructs where all three putative KLF11 binding sequences were deleted were generated by QuikChange XL Site‐directed Mutagenesis kit (Stratagene, Santa Clara, CA) (64). All constructs were validated by DNA sequencing (Genewiz, South Plainfield, NJ). NCBI RefSeq ID: ZO‐1 (mouse): NP_001157046.1, occludin (mouse): NP_001347465.1. Supporting Table S2.
Dual‐luciferase reporter assays
Mouse bEnd.3 cells (ATCC, Manassas, VA) were seeded at 0.5 × 105 cells/well in 24‐well plates. After overnight incubation, cells were infected with different adenoviruses aimed at achieving KLF11 overexpression or knockdown. The cells were also co‐transfected with pGL4_mZO‐1/ΔmZO‐1 or pGL4_mOccludin/ΔmOccludin luciferase reporter vector and a Renilla luciferase control reporter vector (pRL‐TK; Promega), along with Lipofectamine 2000 (Invitrogen) for 5 h. Luciferase activity was measured 48 h after transfection by Dual‐Luciferase assay kits (Promega) using a SpectraMax i3x Multi‐Mode Detection Platform. Individual luciferase activity was normalized to the corresponding Renilla‐luciferase activity (62, 64).
Quantitative real‐time PCR
Total RNA was isolated from BMEC cultures or cerebral cortex by RNeasy Mini Kit (Qiagen, Valencia, CA) or TRIzol (Invitrogen, Carlsbad, CA). Quantitative real‐time reverse‐transcriptase polymerase chain reaction (RT‐PCR) was carried out with a Bio‐Rad CFX Connect thermocycler, iScript cDNA synthesis kit and iTaq Universal SYBR green supermix (Bio‐Rad, Hercules, CA). Specific primers used for the reactions are ZO‐1 Forward, 5′‐gccgctaagagcacagcaa‐3′; ZO‐1 Reverse, 5′‐tccccactctgaaaatgagga‐3′. Occludin Forward, 5′‐tgaaagtccacctccttacaga‐3′; Occludin Reverse, 5′‐ccgcataaaaagagtacgctgg‐3′. TNF‐α Forward, 5′‐ctcctcacccacaccgtcagc‐3′; TNF‐α Reverse, 5′‐aacacccattcccttcacagagca‐3′. IL‐1β Forward, 5′‐aggagaaccaagcaacgacaaaatac‐3′; IL‐1β Reverse, 5′‐tggggaactctgcagactcaaact‐3′. MCP‐1 Forward, 5′‐gcaccagcaccagccaactctcact‐3′; MCP‐1 Reverse, 5′‐cattccttcttggggtcagcacag‐3′. IL‐6 Forward, 5′‐agttgccttcttgggactga‐3′; IL‐6 Reverse, 5′‐tccacgatttcccagagaac‐3′. Icam‐1 Forward, 5′‐ttcacactgaatgccagctc‐3′; Icam‐2 Reverse, 5′‐gtctgctgagacccctcttg‐3′. P‐selectin Forward, 5′‐gtccacggagagtttggtgt‐3′; P‐selectin Reverse, 5′‐aagtggtgttcggaccaaag‐3′. Cyclophilin Forward, 5′‐actcctcatttagatgggcatca‐3′; Cyclophilin Reverse, 5′‐gagtatccgtacctccgcaaa‐3′ (Supporting Table S3). The relative mRNA expression was normalized to cyclophilin RNA levels. PCR experiments were repeated three times, each using separate sets of cultures (53, 59, 67).
Western blot analysis
Samples from the 0.9% NaCl perfused mouse cerebral cortex or cultured mBMECs were homogenized in T‐PER Tissue Protein Extraction Reagent (Pierce, IL) containing Mini Protease Inhibitor Cocktail (cOmpleteTM, Sigma) by Dounce Tissue Grinder or sonication for 30 s with 30% pulse on ice, respectively. The tissue or cell lysates were centrifuged at 10 000 × g for 5 minutes to pellet tissue or cell debris. Then supernatants were collected to measure protein concentrations by the Bio‐Rad Protein Assay (Bradford, Bio‐Rad). As described previously (53, 59, 67), equal amounts of protein were loaded into 4%–15% precast gels (Bio‐Rad) and transferred to polyvinylidene difluoride (PVDF, Bio‐Rad) membranes. Membranes were blocked for 1 h with TBS/0.1%‐Tween buffer plus 5% (w/v) non‐fat dried milk and incubated overnight at 4°C with primary antibodies diluted in the same blocking buffer. Membranes were then incubated with secondary antibodies diluted in blocking buffer for 1 h and developed using a Pierce® ECL Western blotting detection kit (Thermo Scientific) and Amersham High‐Performance Chemiluminescence Films (GE Healthcare). Various primary antibodies were used and are listed in Supporting Table S1, including mouse anti‐KLF11 (1:500; NovusBio), rabbit anti‐occludin (1:1000; Invitrogen), rabbit anti‐ZO‐1 (1:250; Abcam) and mouse anti‐β‐actin (1:2000; Sigma‐Aldrich).
Enzyme‐linked immunosorbent assay (ELISA)
Cerebral cortices of mice were collected and sonicated by an ultrasound homogenizer. After centrifugation, supernatants were collected and concentrations of a series of pro‐inflammatory chemokines, cytokines and adhesive molecules in the supernatants were determined by Quantikine ELISA kits (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions (67). All assays were performed in triplicates.
Statistical analysis
Quantitative data are expressed as mean ± SEM or ± SD. Differences among three or more groups were statistically analyzed by one or two‐way ANOVA followed by the Bonferroni's post hoc test. Comparisons between two experimental groups were conducted by the two‐tailed Student's t‐test. A P‐value less than 0.05 was considered significant. Statistical analyses and graphic representations were obtained with GraphPad Prism 7.0 software.
Results
Generation and characterization of EC‐selective KLF11 transgenic mice
ECs are major components of the BBB and are very sensitive to oxidative stress and ischemic insults (23). Previously, KLF11 expression was found to be significantly decreased in cultured mouse primary brain microvascular endothelial cells (BMECs) after 4 or 16 h oxygen‐glucose deprivation (OGD) treatment (Figure S1A) as well as in isolated cerebral microvessels from C57BL/6J mice that were subjected to 1 h MCAO followed by 24 h reperfusion (Figure S1B). We also reported that the genetic deletion of KLF11 in mice significantly augmented MCAO‐induced BBB disruption by exaggerating cerebrovascular permeability and edema (53). However, whether KLF11 maintains the integrity of the BBB at the very early stages (1–3 h) after ischemic insults and whether endothelial KLF11 plays distinct roles in the pathogenesis of ischemic brain injury remain unknown. To explore the specific role of KLF11 in endothelial cells after ischemic injury, we generated transgenic mice with vascular endothelial cell‐selective overexpression of KLF11 on a C57BL/6J background. The EC‐specific KLF11 transgenic construct contains the Tie 2 promoter and Tie 2 enhancer (47, 64) to drive expression of the full‐length KLF11 cDNA sequence (Figure 1A). The Tie 2 promoter allows selective targeting of KLF11 to vascular ECs. The transgenic founder mice with KLF11 overexpression (EC‐KLF11 Tg) were identified by genomic PCR genotyping (Figure 1B). To confirm the overexpression of KLF11 in the brain microvasculature, we isolated cerebral microvessels from EC‐KLF11 Tg and WT mouse brains. We found a significant upregulation of the cerebrovascular KLF11 protein level in EC‐KLF11 Tg mice than that of WT controls (Figure 1C left). Moreover, KLF11 was not overexpressed in the cortical protein extracts from the EC‐KLF11 Tg mouse brain in comparison with WT controls (Figure 1C right). Of note, in sham‐operated mice, no significant differences were observed in CD31‐labeled microvasculature of EC‐KLF11 Tg mice compared to WT controls, as indicated by branch points, capillary numbers and branch length (Figure 1D,E). We also noticed that regional CBF did not differ by the transgenic overexpression of KLF11 in endothelium 15 minutes before ischemia, 15 minutes after ischemia, or 15 minutes after reperfusion (Figure 3D,E), suggesting that the transgenic manipulation of KLF11 in endothelium does not cause possible cerebrovascular structural (collateral) changes in the brains of EC‐KLF11 Tg mice. Thus, EC‐KLF11 Tg mice can be used as a powerful tool to investigate the functional role of KLF11 in vascular endothelial biology and BBB pathologies in vivo.
Figure 1.
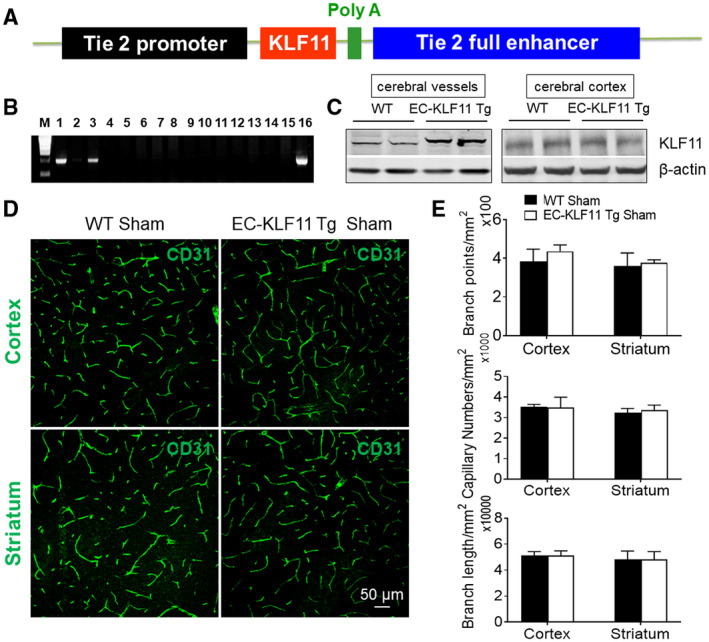
Generation of endothelial cell (EC)‐selective KLF11 transgenic mice. A. Schematic diagram of EC‐selective KLF11 transgenic structure. The transgene cassette is composed of a ~ 2.1‐kb Tie‐2 promoter, a ~ 10.4‐kb Tie‐2 enhancer and a 1539‐bp DNA fragment containing the human KLF11 coding sequence. B. Genomic PCR for genotyping EC‐selective KLF11 transgenic mice (EC‐KLF11 Tg). A 960 bp band is expected from EC‐KLF11 transgenics (Lanes 1, 3 and 16). C. Representative western blotting images indicated enhanced protein levels of KLF11 in the cerebral microvessels (cerebral vessels, left) but not in brain parenchyma (cerebral cortex, right) of EC‐KLF11 Tg mice than WT controls. n = 5 mice per group. D. Representative immunofluorescent images show CD31‐positive capillaries in the cortex and striatum of both WT and EC‐KLF11 Tg mice. n = 5 mice per group. Scale bar: 50 µm. E. Quantification of CD31‐labeled branch points, capillary numbers and branch length is shown. In comparison with WT controls, EC‐targeted transgenic overexpression of KLF11 does not affect cerebral vascular density and structure in mice. Data are expressed as mean ± SEM.
Figure 3.
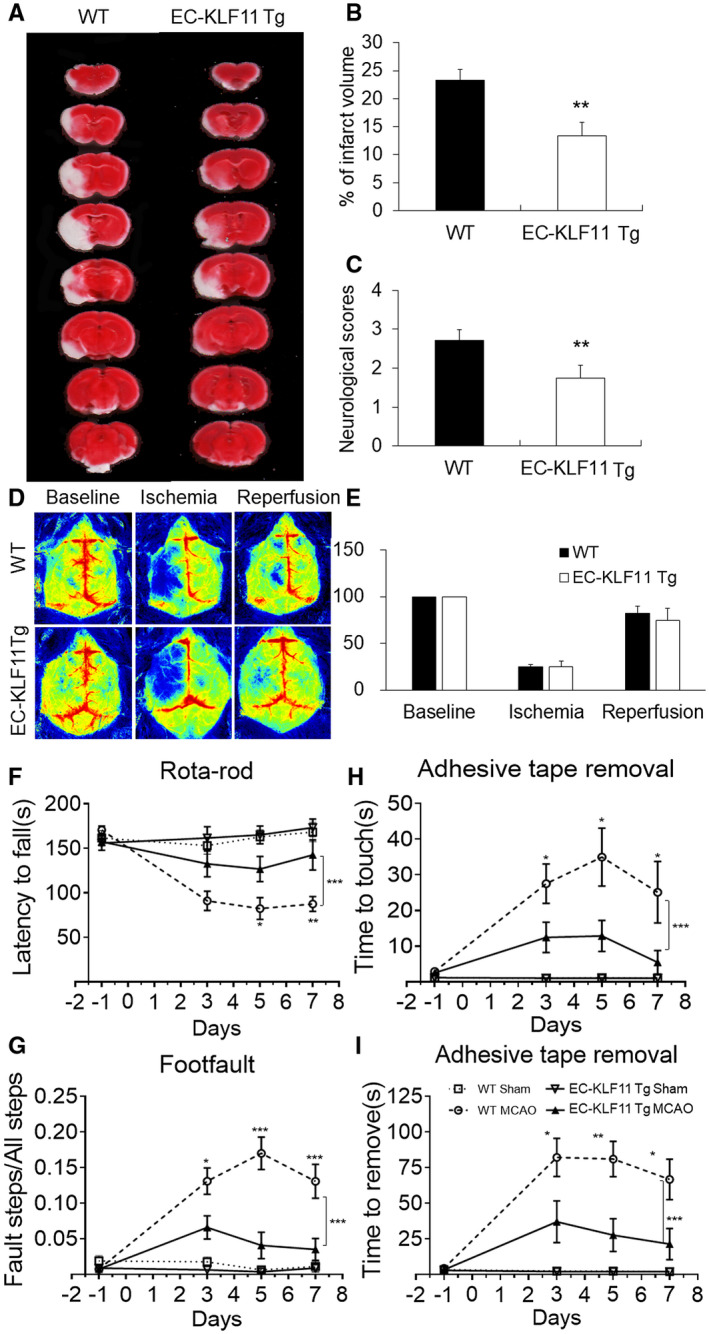
Transgenic overexpression of KLF11 in endothelium improves short‐term histological and functional outcomes after focal cerebral ischemia. A. EC‐KLF11 Tg mice and WT controls were subjected to 1 h MCAO and 72 h reperfusion. Two percent TTC‐stained coronal sections were shown at different brain levels from the frontal to the posterior pole. B. Quantitative analysis was performed on infarct volume and (C) neurological deficits in these mice after ischemic stroke. n = 8 mice per group. D. Regional CBF was measured by a laser speckle imager. EC‐KLF11 Tg and WT mice were subjected to 1 h MCAO followed by 72 h reperfusion. Representative CBF images were shown at 15 minutes before MCAO (baseline), 15 minutes after the onset of MCAO (ischemia) and 15 minutes after the onset of reperfusion (reperfusion). E. Two identical elliptical ROIs were selected as indicated on the same brain region of the ipsilateral and contralateral hemispheres. The relative CBF was first determined as the ratio of ischemic to nonischemic cerebral blood flow and then as the percentage value normalized to the presurgical baseline for each animal. n = 6 each group. F–I. Sensorimotor deficits were assessed before and up to 7 days after MCAO by a battery of behavioral tests, including Rotarod test (F), Foot fault (G) and Adhesive tape removal test (H–I). Compared with WT controls, mice with endothelial‐specific overexpression of KLF11 showed dramatically improved sensorimotor function (increased staying latency in the rotarod test, lower rate of fault steps in the foot fault test and shorter touching or removing time in the adhesive tape removal test. Data are represented as mean ± SD. n = 12 per group. *P < 0.05, **P < 0.01, ***P < 0.001 EC‐KLF11 Tg MCAO vs. WT MCAO by one‐way ANOVA (individual time point) or two‐way ANOVA (bracket).
EC‐selective KLF11 transgenic overexpression ameliorates post‐ischemic cerebrovascular permeability
Next, we performed Evans Blue and TMR‐Dextran extravasation assays to observe and quantify changes in BBB permeability. EC‐KLF11 Tg and WT mice were subjected to 1 h MCAO followed by 1, 3 or 24 h reperfusion. The results showed that EC‐targeted transgenic overexpression of KFL11 markedly reduced, but not completely blocked the leakage of both Evans blue dye and TMR‐dextran into the ischemic brain after 24 h reperfusion (Figure 2A‐D). To assess BBB permeability at earlier stages (1–3 h) after ischemic insult, we also analyzed the extravasation of a small fluorescent tracer, Alexa 555 cadaverine (0.95 kDa), which was injected through the tail vein and the relatively larger endogenous plasma IgG (150 kDa) into the ischemic brain parenchyma (48, 49). The extravasation of the small molecule cadaverine in both the ipsilateral cortex and striatum was detected as early as 1 h reperfusion, whereas endogenous IgG was detected in the same area at 3 h reperfusion (Figure 2E). Compared with WT controls, EC‐KLF11 Tg mice consistently showed significantly reduced the extravasation of cadaverine (at 1, 3 and 24 h reperfusion; Figure 2F) and IgG (at 3 and 24 h reperfusion; Figure 2G). These results suggest that early‐onset BBB impairment occurred following ischemic insult and endothelial‐specific KLF11 transgenic overexpression protects against MCAO‐induced early‐onset and progressive (late‐onset) BBB impairments. Moreover, anti‐MAP2 immunostaining indicated that the brain regions which present signs of BBB breakdown at earlier stages after MCAO progressed into infarct zones at 24 h after MCAO (Figure 2E). Quantification analysis of anti‐microtubule‐associated protein 2 (MAP2) immunostaining showed that EC‐KLF11 Tg mice had significantly reduced brain infarction compared with WT controls (Figure 2H). We further measured and quantified the water content in ipsilateral and contralateral hemispheres of EC‐KLF11 Tg and WT mice 72 h after MCAO. The results demonstrated that the brain water content in the ipsilateral hemisphere of EC‐KLF11 Tg mice was significantly less than that in the WT group (Figure 2I), whereas the water content in the contralateral hemisphere from both groups showed no significant differences (Figure 2I). It is known that blood neutrophils can migrate through an impaired BBB and carry proinflammatory mediators into the injured brain, thereby causing more serious BBB damage and secondary expansion of ischemic brain parenchymal injury (49). We used immunohistochemistry methods to detect neutrophils with a labeled Ly‐6B antibody (Figure 2J). Quantification analysis of Ly‐6B immunostaining confirmed that in comparison with WT controls, MCAO‐induced increases in brain infiltration of Ly‐6B+ neutrophils were significantly reduced in EC‐KLF11 Tg mouse brains (Figure 2K). Taken together, these results suggest that endothelial‐selective KLF11 transgenic overexpression not only protects against ischemia‐induced BBB damage but also provides further neurovascular protection against ischemic stroke.
Figure 2.
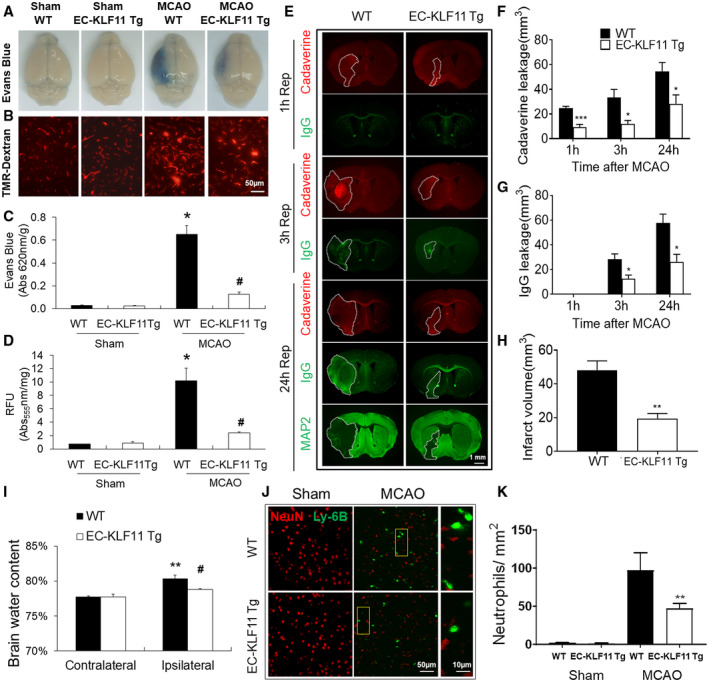
Targeted overexpression of KLF11 in endothelium ameliorates both early‐ and late‐onset BBB impairments, edema and neutrophil infiltration. EC‐KLF11 Tg mice and WT controls were subjected to 1 h MCA occlusion followed by 1, 3, or 24 h reperfusion. A. Extravasation of Evans blue dye and (B) 70 kDa TMR‐Dextran 24 h after MCAO in whole brains or coronal sections was shown. Scale bar, 50 µm. C and D. Quantification of Evans blue in panel A and TMR‐Dextran in panel B. n = 6 mice per group. E. Representative images demonstrate the extravasation of Alexa Fluor 555 (0.95 kDa, red) or endogenous plasma IgG (~150 kDa, green) into brain parenchyma 1, 3, or 24 h after MCAO. At 24 h after MCAO, the area with loss of microtubule‐associated protein 2 (MAP2) immunofluorescence illustrates the infarct zone on adjacent sections from the same brains. (Scale bar: 1 mm). F and G. Quantitative analysis of the volume of the brain with leakage of cadaverine and IgG at indicated times of reperfusion after MCAO. n = 5 mice per group. H. Brain infarct volume at 24 h after MCAO was quantitatively measured on MAP2‐stained coronal sections. n = 5 mice per group. I. Brain water content was measured by the wet/dry weight protocol as described in the “Material and Methods.” n = 10 per group, **P < 0.01 vs. WT contralateral hemisphere, # P < 0.05 vs. WT ipsilateral hemisphere. J. Representative images taken from the ipsilateral periinfarct cortex after MCAO or the corresponding region after sham operation; markers used: Ly‐6B (neutrophil) and NeuN (neuron) (Scale bar: 50 μm). Rectangle: the region enlarged in high‐power images (third column). (Scale bar: 10 µm.) K. Ly‐6B + cells were counted in the areas described in (J) and data are expressed as the number of cells/mm2. After MCAO, the number of infiltrated neutrophils is significantly less in EC‐KLF11 Tg mouse brains compared with WT controls. n = 5 mice per group. Data are expressed as mean ± SEM. *P < 0.05 vs. WT + sham group, # P < 0.05 vs. WT + MCAO group.
EC‐targeted KLF11 overexpression reduces ischemic brain injury and improves short‐term neurological function
The protective role of EC‐KLF11 overexpression against ischemic insult was further confirmed by 2% 2,3,5‐triphenyltetrazolium chloride (TTC) staining in another cohort of mice, in which neurological deficits were also scored. Compared with WT controls, EC‐KLF11 Tg mice presented much smaller cerebral infarcts (Figure 3A,B) and significantly reduced neurological scores (Figure 3C), indicating better neurological outcome after stroke. Of note, regional CBF showed similar changes between EC‐KLF11 Tg and WT mice 15 minutes before ischemia, 15 minutes after ischemia, or 15 minutes after reperfusion (Figure 3D,E). In humans, cerebral ischemia always leads to sensorimotor dysfunction. We then investigated whether EC‐targeted KLF11 overexpression affects post‐stroke functional recovery by a battery of three different behavioral tests: adhesive tape removal, foot fault and rotarod tests before and up to 7 days following MCAO (Figure 3F–I). Compared with WT control mice, EC‐KLF11 Tg mice showed better recovery of sensorimotor function: longer staying latency in the rotarod test (Figure 3F), lower rate of fault steps in the foot fault test (Figure 3G) and shorter touching or removing time in the adhesive tape removal test (Figure 3H‐I). These cumulative findings suggest that endothelial‐specific overexpression of KLF11 facilitates spontaneous sensorimotor recovery after ischemic brain insult.
EC‐selective KLF11 overexpression preserves the integrity of junctional proteins after ischemic brain injury
TJ proteins are essential for maintaining the integrity of the BBB (28). Therefore, we further examined the underlying mechanism of endothelial KLF11‐mediated BBB protection in vivo. EC‐KLF11 Tg and WT mice were subjected to 1 h MCAO followed by 1 or 3 days of reperfusion. Mouse brain tissue was harvested at the indicated time points after MCAO and subjected to total RNA isolation and qPCR analysis (Figure 4A,B) or western blotting analysis (Figure 4C–E). Endothelial‐targeted KLF11 transgenic overexpression significantly preserved the expression of TJ proteins, ZO‐1 and occludin in ischemic brain tissue at both the mRNA (Figure 4A,B) and protein (Figure 4C–E) levels, consistent with the protective role of EC‐KLF11 overexpression in MCAO‐induced BBB damage. Reduction in ZO‐1 and occludin expression in vascular endothelial cells (labeled by CD31) following ischemic brain insults were further confirmed by immunofluorescent staining (Figure 4F). As expected, EC‐KLF11 overexpression markedly retained their in situ expressions (Figure 4F). These results indicate that endothelial‐targeted KLF11 overexpression robustly protected the structural integrity of the BBB.
Figure 4.
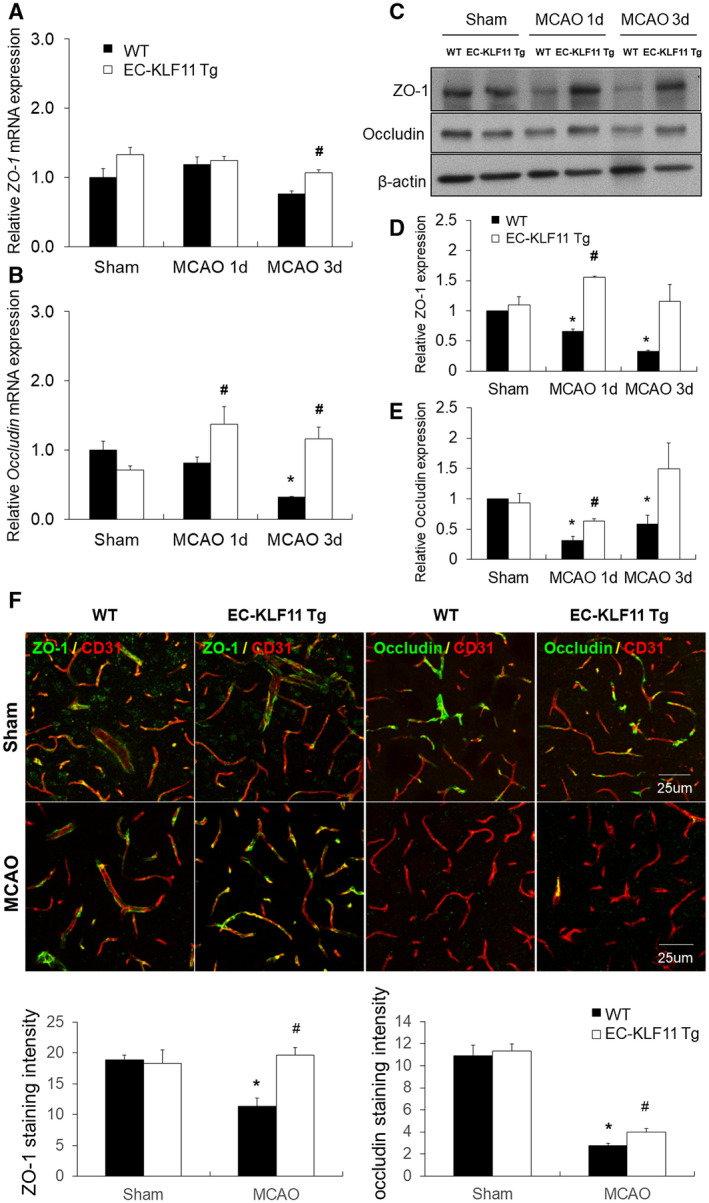
EC‐targeted KLF11 overexpression preserves the integrity of tight junctions after focal cerebral ischemia. EC‐KLF11 Tg mice and WT controls were subjected to 1 h MCA occlusion and 1d or 3d reperfusion. A and B. Total RNA was extracted from the ipsilateral cortex of EC‐KLF11 Tg and WT mice 1d and 3d after MCAO. ZO‐1 and occludin mRNA expression levels were determined by qPCR and normalized to cyclophilin (n = 6 per group). C. Total protein was extracted and subjected to gel electrophoresis. The protein levels of ZO‐1 and occludin were determined with β‐actin as the loading control. D and E. Quantification of ZO‐1 and occludin protein. Experiments were repeated three times and representative blots are displayed. n = 6 per group. F. Representative images and quantification of ZO‐1 (green) and occludin (green) on CD31 + microvessels (red) in the cortex of the ischemic hemisphere at 1d after MCAO. (Scale bar: 25 µm.) Endothelial KLF11 overexpression suppressed MCAO‐induced disruption of junctional proteins ZO‐1 and occludin. n = 6 mice per group. *P < 0.05 vs. WT + sham group, # P < 0.05 vs. WT + MCAO group.
EC‐targeted KLF11 transgenic overexpression mitigates inflammatory activity in ischemic brain regions
Neuroinflammation following ischemic stroke contributes to BBB damage (57). To test whether EC‐targeted KLF11 overexpression alleviates MCAO‐induced inflammatory activities in the ischemic brain, we measured a panel of inflammatory mediators in the cortex of the ipsilateral brain hemisphere at one or three days after MCAO. The mRNA expression levels of six pro‐inflammatory mediators: TNF‐α, IL‐1β, MCP‐1, IL‐6, ICAM‐1 and P‐selectin markedly elevated following MCAO and EC‐targeted KLF11 overexpression attenuated this elevation (Figure 5A–F). We then measured the expression of the abovementioned inflammatory factors using Quantikine ELISA kits. Consistent with our previous observations, the six inflammatory mediators all reduced in EC‐KLF11 Tg mice when compared with WT controls following MCAO (Figure 5G–L). Taken together, these results demonstrate that KLF11 transgenic overexpression in endothelium mitigates post‐stroke inflammatory responses.
Figure 5.
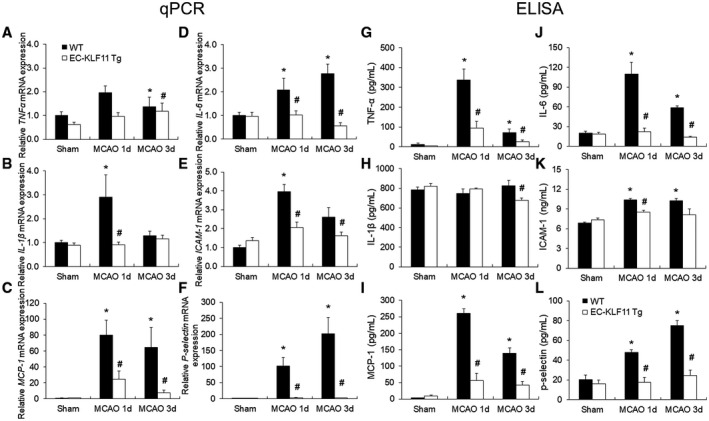
Effects of EC‐targeted overexpression of KLF11 on inflammatory mediators in mouse cortex after focal cerebral ischemia. EC‐KLF11 Tg and WT mice were subjected to sham operation or 1 h of MCAO followed by 1d or 3d of reperfusion. A–F. mRNA expression levels of different inflammatory markers were measured in the ipsilateral cortex by qPCR and normalized to cyclophilin. G and L. Protein levels of different inflammatory markers were analyzed by ELISA. EC‐targeted overexpression of KLF11 inhibited MCAO‐induced upregulation of TNFα, IL‐1β, MCP‐1, IL‐6, ICAM‐1 and P‐selectin expression. n = 6 mice per group. *P < 0.05 vs. WT + sham group, # P < 0.05 vs. WT + MCAO group.
KLF11 alleviates OGD‐induced BBB leakage in an in vitro BBB model
To further investigate endothelial KLF11‐mediated BBB protection against ischemic insults, we employed a two‐cell‐based in vitro BBB model. The co‐culture system consists of mouse astrocytes that are seeded on the bottom of a 12‐well plate and adenovirus‐transduced mouse BMECs that are seeded on the polyester membrane of transwell inserts (Figure 6A). The co‐culture system was then subjected to ischemic‐like insult of OGD for 16 h, followed by 2 h reoxygenation. Transepithelial/transendothelial electrical resistance (TEER) was measured to monitor the functional integrity of the cellular barrier (51). When KLF11 was overexpressed in mBMECs by adenovirus‐mediated infection before being subjected to OGD, the barrier functional disruption induced by OGD was attenuated in the Ad. KLF11group when compared with Ad. LacZ control group (Figure 6B). In contrast, OGD‐induced barrier functional disruption was worsened in the adenoviral KLF11 knockdown group (Ad. shKLF11) compared with Ad. shLacZ control group (Figure 6C). Next, luminal‐to‐abluminal barrier permeability (paracellular permeability) of the small fluorescent tracer (3 kDa Dextran) was measured at indicated time points after OGD. When KLF11 was upregulated by adenovirus in mBMECs prior to OGD, leakage of the 3 kDa tracer was significantly reduced, but not completely blocked, at 2–8 h post‐OGD reoxygenation (Figure 6D). In contrast, when KLF11 was downregulated in mBMECs by adenovirus‐mediated infection (Ad. shKLF11) prior to OGD, leakage of the 3 kDa tracer was significantly increased at 2–24 h post‐OGD reoxygenation compared to the Ad. shLacZ group (Figure 6E). Based on the above observations, we conclude that KLF11 overexpression attenuates OGD‐induced endothelial barrier functional disruption in vitro, while KLF11 knockdown reverses this effect.
Figure 6.
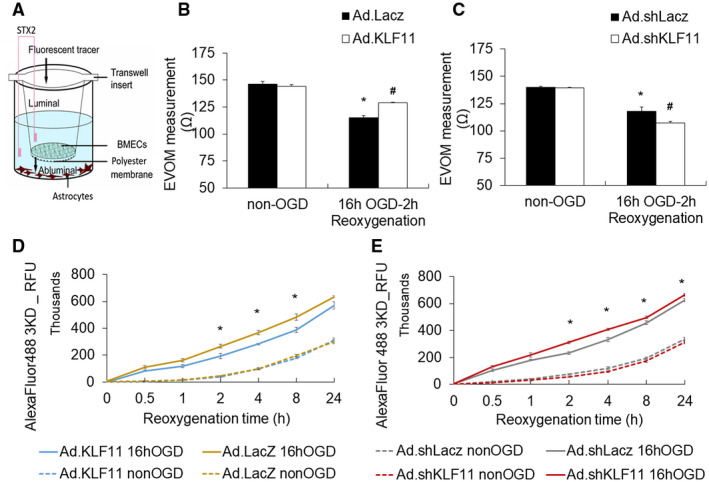
Adenoviral gain‐ or loss‐of‐KLF11 expression in mouse BMECs affects endothelial barrier function after OGD in vitro. A. Illustration of the transwell co‐culture system. Mouse astrocytes were seeded at the bottom of a 12‐well plate with transwell insert. Cultured mBMECs were seeded on top of a collagen‐ and fibronectin‐coated membrane of the insert and grown to form a confluent monolayer. The mBMECs were infected for 48 h with either an empty adenovirus (Ad. LacZ), adenoviral vector carrying mouse KLF11 (Ad. KLF11), adenoviral vector shLacZ (Ad. shLacZ), or adenoviral vector carrying short hairpin sequences targeting mouse KLF11 (Ad. shKLF11) and then subjected to OGD for 16 h before being placed back to the plate with astrocytes. B and C. Transepithelial electrical resistance (TEER) in the co‐culture model was measured at 2 h after OGD and in non‐OGD conditions. n = 3 per group, *P < 0.05 vs. Ad. LacZ/Ad. shLacZ + non‐OGD group, # P < 0.05 vs. Ad LacZ/Ad. shLacZ + 16 h OGD‐2 h Reoxygenation. D and E. Paracellular permeability was quantified at 0–24 h after OGD by measuring the fluorescence intensity of abluminal Dextran Alexa Fluor 488 (3000 MW). Adenovirus‐mediated KLF11 overexpression improved endothelial barrier function after OGD treatment, whereas KLF11 knockdown worsened endothelial barrier function. Data are expressed as mean ± SEM of three independent experiments with triplicate wells (n = 3 per group). *P < 0.05 vs. Ad. LacZ/Ad. shLacZ + 16 h OGD group.
KLF11 inhibits OGD‐induced disruption of junctional proteins in cultured brain microvascular endothelial cells
To dissect functional roles and molecular mechanisms of KLF11 in stabilizing BBB integrity in response to ischemic stimuli, mouse primary BMECs were subjected to ischemic‐like insult of OGD for 16 h, followed by 24 or 48 h reoxygenation. We found adenovirus‐mediated overexpression of KLF11 blocked OGD‐induced upregulation of pro‐inflammatory cytokines IL‐6 and MCP‐1 at both the mRNA (Figure S2A,B) and protein levels (Figure S2C,D). Moreover, OGD stimuli significantly reduced the expression of TJ proteins ZO‐1 and occludin in mouse BMECs (Figure 7A–D). As expected, adenovirus‐mediated KLF11 overexpression successfully preserved the expression of both TJ proteins at the mRNA (Figure 7A,B) and protein (Figure 7C,D) levels, consistent with previous observations in EC‐KLF11 Tg mice (Figure 4). Immunocytochemical studies also confirmed decreased ZO‐1 and occludin expression after 4 h OGD treatment (Figure 7E). This effect was markedly attenuated in mouse BMECs overexpressing KLF11 (Figure 7E), indicating KLF11 overexpression preserves the integrity of tight junction proteins after OGD.
KLF11 regulates the expression of tight junction proteins ZO‐1 and Occludin at the transcription level
As a member of the KLF family of transcription factors, KLF11 is known to bind to GC‐rich target sequences on promoters (38). We analyzed mouse ZO‐1 and occludin promoters and identified three potential KLF11 binding sequences on each respective promoter (Figure 8A), suggesting KLF11 may transcriptionally regulate ZO‐1 and occludin. To confirm that KLF11 regulates ZO‐1 and occludin through direct interaction with predicted binding sequences located in the promoter regions of these two genes, we cloned the promoter region of mouse ZO‐1 and occludin, respectively, into a dual‐luciferase reporter vector (pGL4.10 [luc2]). We also clone the deleted promoters of mouse ZO‐1 and occludin (without the three potential KLF11 binding sites) into the pGL4.10 [luc2] vector (Figure 8B,E). We then co‐transfected mouse bEnd.3 cells with these luciferase reporter constructs and a Renilla luciferase control reporter vector (pRL‐TK). We found adenovirus‐mediated KLF11 overexpression significantly increased luciferase activity of the reporter constructs containing wild type ZO‐1 (pGL4_mZO‐1) and occludin (pGL4_mOccludin) promoter sequences (Figure 8C,F), whereas adenovirus‐mediated KLF11 knockdown significantly decreased the luciferase activity of these two promoter constructs (Figure 8D,G). In addition, neither adenovirus‐mediated KLF11 overexpression nor KLF11 knockdown affects the luciferase activities of the two deleted promoter constructs (pGL4_Δ mZO‐1, pGL4_Δ mOccludin. Taken together, these results confirm that KLF11 transactivates ZO‐1 and occludin by directly binding to their promoter region.
Figure 8.
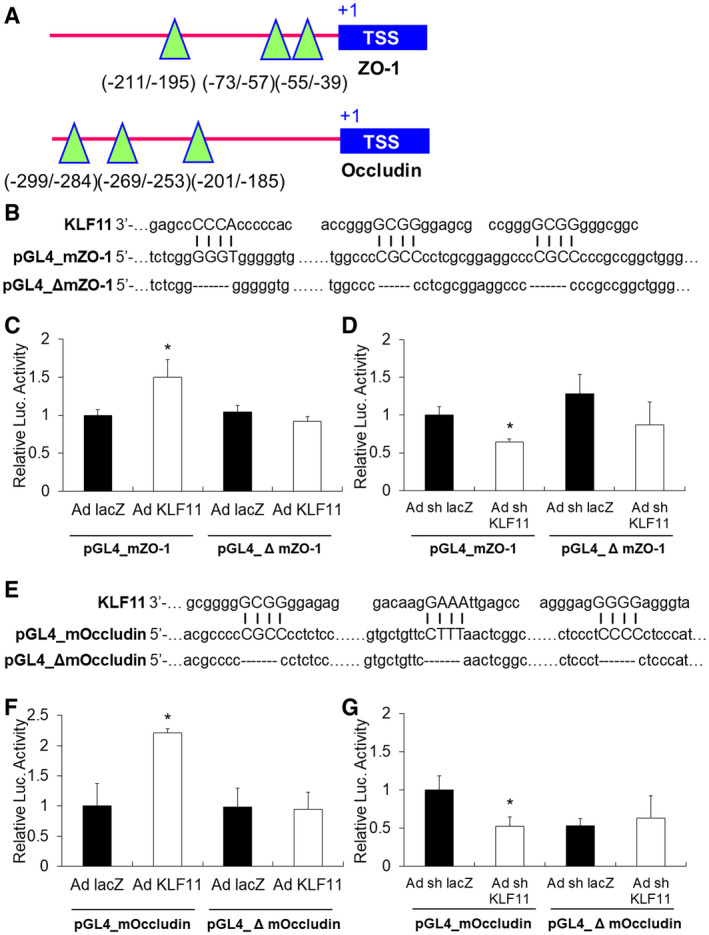
KLF11 transcriptionally activates ZO‐1 and occludin. A. Potential KLF11 binding site in the promoter region of mouse ZO‐1 and occludin. TSS: transcription start site. The green triangles mark the binding sites of KLF11. The numbers indicate the relative distance to TSS, which is labeled as + 1. B. About 1kb of the mouse ZO‐1 promoter or a mutated ZO‐1 promoter (deleted all three KLF11 binding sequences, Δ mZO‐1) was cloned into the pGL4.10[luc2] vector. The pGL4 promoter constructs were then transfected into bEnd.3 cultures. bEnd.3 cells were also co‐transduced with an empty adenovirus (Ad. LacZ) or adenoviral vectors carrying mouse KLF11 (Ad. KLF11) for 48–72 h prior to performing luciferase reporter activity assays. C. KLF11 overexpression in bEnd.3 cells significantly increased luciferase activity of pGL4_mZO‐1, which contains all three KLF11 binding sequences (n = 4 per group). D. In contrast, the luciferase activity was significantly reduced in the pGL4_mZO‐1 group co‐transfected with Ad. shKLF11 (n = 4 per group). E. The occludin promoter or a mutated occludin promoter (deleted all three KLF11 binding sequences, Δ mOccludin) was cloned into the pGL4.10[luc2] vector. F. KLF11 overexpression significantly increased luciferase activity of pGL4_mOccludin, but not pGL4_Δ mOccludin (n = 4 per group). G. In contrast, luciferase activity was significantly reduced in the pGL4_mOccludin group co‐transfected with Ad. shKLF11 (n = 4 per group). Results shown are representative of three separate experiments with similar results. Data are expressed as mean ± SEM. *P < 0.05 vs. Ad. LacZ group or Ad. shLacZ group.
Discussion
This study investigated the protective function of endothelial KLF11 against BBB damage in a mouse ischemic stroke model. The results indicate that KLF11 transgenic overexpression in endothelium attenuates post‐ischemic cerebrovascular permeability, brain water accumulation and brain infarction, presumably by blocking the disruption of the junctional proteins, ZO‐1 and occludin. By preserving the integrity of the BBB after ischemic insult, EC‐targeted KLF11 overexpression effectively reduces the infiltration of neutrophils into brain parenchyma and the production of proinflammatory factors in the injured brain, thereby eliciting neurovascular protection against neurological deficits. Our results also demonstrate that KLF11 transactivates ZO‐1 and occludin through direct binding to their promoters. Taken together, our findings suggest EC‐targeted KLF11 transgenic overexpression is effective in mitigating BBB disruption and improving overall stroke outcome.
The functional involvement of KLF family members has been reported in various developmental and pathological vascular processes (2, 52). KLF2 has been demonstrated to regulate endothelial proliferation, migration and angiogenesis (8, 9, 13). KLF2 has also been reported to regulate vasoreactivity and vascular tone (13, 14, 43). Another KLF family member, KLF4, has been reported as an endothelial regulator in response to pro‐inflammatory stimuli (27) and shear stress (40). A study using EC‐targeted KLF4 transgenic and conditional knockout mouse models demonstrated vascular anti‐inflammatory and anti‐atherothrombotic functions of KLF4 (68). KLF4 was also reported to activate VE‐cadherin at the transcriptional level and thereby maintain normal endothelial barrier function (11). In addition, KLF6 has been demonstrated to play functional roles in regulating vascular development, remodeling and responses to injury (10, 12). KLF14 was reported to mitigate atherosclerosis through modulating hepatic ApoA‐I production in mice (26). Moreover, KLF14 was demonstrated to reduce endothelial inflammation by inhibiting the NF‐κB signaling cascade (30). However, KLFs are less studied in the context of cerebrovascular biology and diseases such as cerebral ischemia.
We have previously performed genome‐wide screening for PPARγ coregulators and identified KLF11 as a novel PPARγ coregulator (62). We further confirmed a physical interaction between PPARγ and KLF11, and the regulatory effects of KLF11 on PPARγ‐mediated cerebrovascular protection in primary BMEC cultures and mouse brain after in vitro (OGD) and in vivo (MCAO) ischemic insults. Later (53), we provided direct evidence of KLF11 itself in the regulation of ischemic brain injury. We found that conventional KLF11 KO mice had larger brain infarcts, along with worsened neurobehavioral performance, increased edema and greater BBB disruption compared with WT mice (53). Our studies suggest KLF11 as an endogenous protective mediator of ischemic stroke. However, the regulatory role of vascular KLF11 and the underlying mechanisms of KLF11 itself in mediating protection against BBB leakage, especially at the early onset (1–3 h) following ischemic insult, remain unknown.
In this manuscript, we found that KLF11 expression was significantly downregulated in cultured BMECs and mouse cerebral microvessels after in vitro (OGD) and in vivo (MCAO) ischemic insults (Figure S1). Brain microvascular ECs are the major components of the BBB and along with pericytes, astrocytes, perivascular microglia and the basal lamina, they form the neurovascular unit (29, 31). Given the importance of brain microvascular ECs in the formation of the BBB as well as in the pathogenesis of stroke, we, therefore, generated transgenic mice with vascular EC‐targeted transgenic overexpression of KLF11 driven by the Tie‐2 promoter. Our results have shown that KLF11 overexpression in ECs not only reduces MCAO‐induced BBB permeability and disruption (Figure 2) but also leads to better neurological outcomes (Figure 3). Moreover, in our two‐cell‐based in vitro BBB model, we determined that KLF11 overexpression effectively preserved endothelial barrier integrity following ischemic‐like conditions (Figure 6). These findings suggest that endothelial KLF11 contributes to its overall protective effects against ischemic insults, therefore, opening the possibility of potential therapeutic applications for KLF11.
There are numerous well‐studied mechanisms that contribute to ischemic‐induced BBB disruption, such as increased oxidative stress, vascular inflammation, activation of matrix metalloproteinases (MMPs), loss of BBB cellular components and abnormal pathologies of endothelial tight junctions (33, 37, 44, 46, 49, 54, 58). A recent study demonstrated that MCAO‐induced early structural disruption in brain microvascular ECs, including actin stress fiber formation and redistribution of junctional proteins, resulted in impaired BBB integrity (49). This early BBB disruption further facilitates MMP‐mediated (especially gelatinase B/MMP‐9) degradation of EC tight junctions/basal lamina and the consequent infiltration of circulating immune cells, leading to further BBB breakdown and secondary expansion of ischemic injury (41, 49). In our current study, we found that EC‐targeted KLF11 transgenic overexpression ameliorates MCAO‐induced early (1–3 h) and late (24 h) onset of BBB impairment (Figure 2E), by preserving the integrity of tight junction proteins, ZO‐1 and occludin (Figure 5). It remains to be determined whether and how endothelial KLF11 affects MMP‐mediated late‐stage BBB disruption.
Brain ECs express TJ proteins at relatively high levels, thereby maintaining low permeability between blood components and the cerebrospinal fluid (7). The three primary TJ proteins expressed in brain ECs, occludin, claudins and JAM, are tethered to the actin cytoskeleton by scaffold proteins such as ZO‐1, AF6 and cingulin (28). As a member of the zinc finger family of transcription factors, KLF11 binds to GC‐rich regions on gene promoters (38). We analyzed the promoter regions of all TJ proteins and discovered three KFL11 binding motifs on mouse ZO‐1 and occludin promoters. Further, we confirmed the direct binding of KLF11 to these promoters and transactivation activities on ZO‐1 and occludin by dual‐luciferase assays (Figure 8). Although in the current study we did not take into consideration that TJs are known substrates of MMPs (37), whether EC‐KFL11 mediated transactivation and upregulation of ZO‐1 and occludin are further regulated by MMPs need further investigation. Moreover, increased transcytosis in CNS endothelial cells may also lead to BBB disruption (3). Ben‐Zvi et al reported that genetic deletion of major facilitator superfamily domain‐containing 2a that is specifically expressed in CNS ECs results in a leaky BBB due to increased transcytotic vesicles within ECs, without affecting tight junctions (7). In our studies, EC‐KLF11 Tg mice preserved relatively intact TJs but still developed BBB breakdown to some extent after MCAO, suggesting the possible involvement of the transcellular pathway. Whether KLF11 plays a role in transcytosis needs further investigation.
Loosening of EC junctions due to ischemic insult facilitates the infiltration of circulating immune cells and inflammatory factors, thereby producing serious clinical consequences other than ischemic injuries, such as vasogenic edema and hemorrhagic transformation (49). We previously observed that KLF11 KO mice show aggravated brain edema after ischemic insult when compared with WT controls (53), whereas in the current study, EC‐targeted KLF11 transgenic overexpression alleviates edema in the ischemic brain (Figure 2I). We also observed less infiltration of neutrophils into the ischemic brain of EC‐KLF11 Tg mice (Figure 2K). Moreover, the expression of six inflammatory factors (TNF‐α, IL‐1β, MCP‐1, IL‐6, ICAM‐1, p‐selectin) that are frequently upregulated after ischemic insult were found to be significantly reduced in the ischemic brain of EC‐KLF11 Tg mice compared with WT controls (Figure 5). This is consistent with our previous report that the genetic deletion of KLF11 in mice further aggravated MCAO‐induced upregulation of IL‐6 (53), a major stroke‐related pro‐inflammatory cytokine. Our observations are also consistent with one previous publication that demonstrated lipopolysaccharide (LPS)‐induced endothelial inflammation can be inhibited by KLF11 (19). They reported that compared with WT mice, LPS‐induced leukocyte rolling and adhesion on postcapillary venular endothelium were further augmented in KLF11 KO mice. In addition, the expression levels of VCAM‐1 and E‐selectin were significantly increased in the aortas of KLF11 KO mice (19). Therefore, KLF11‐triggered anti‐inflammation mechanisms may contribute to its overall protective functions against BBB damage and ischemic neurovascular injuries. Taken together, our results demonstrate that endothelial KLF11‐mediated improvements in BBB tightness or stabilization limited the infiltration of blood neutrophils and reduced the release of inflammatory mediators into the ischemic brain, ultimately reducing secondary brain injuries caused by neuroinflammation. Also, the regulatory effects of vascular KLF11 on other blood immune cells need further attention.
In conclusion, our study demonstrates that EC‐targeted KLF11 transgenic overexpression mitigates both early and late BBB disruption by preserving the integrity of TJ proteins and hindering the progression of neuroinflammation, leading to an overall functional improvement after ischemic stroke. Our results may have potential clinical applications because cerebral endothelial barrier disruption is an important contributor to several pathological processes in the brain, especially those associated with inflammatory brain disorders. Therefore, the findings of this study may be applied to other disease processes including but not limiting to ischemic stroke.
Author Contribution Statement
X.Z., X.T., Y.E.C. and K.‐J.Y. designed the research; X.Z., X.T., F.M., Y.F., T.Z. and J.Z. performed the research; X.Z., X.T., F.M. and K.‐J.Y. analyzed the data; and X.Z., M.H.H and K.‐J.Y. wrote the paper.
Competing Interests
None.
Supporting information
Supplementary Material
Figure S1. KLF11 expression in mouse BMEC cultures and cerebral microvessels after in vitro and in vivo ischemic stimuli. A. Mouse BMEC cultures were subjected to 4 or 16 h OGD treatment. Total RNA was then isolated and KLF11 mRNA levels were examined by real‐time PCR. *P< 0.05 vs. non‐OGD group. B. Cerebral microvessels were isolated from C57BL/6J mice after 1 h MCAO followed by 24 h reperfusion. Total RNA was isolated and KLF11 levels were examined by real‐time PCR. Cerebral ischemia resulted in a significant decrease in KLF11 expression. Data are expressed as mean ±SEM. *P < 0.05 vs. sham group.
Figure S2. Effects of KLF11 overexpression on pro‐inflammatory cytokines in mouse BMEC cultures after OGD. Cultured mBMECs were transduced with Ad. LacZ or Ad. KLF11 for 48 h before subjected to 16 h OGD, followed by 24 or 48 h reperfusion. Total RNA and culture medium were collected, and a series of pro‐inflammatory chemokines, cytokines and adhesive molecules were analyzed by qPCR (A,B) and ELISA (C,D). *P < 0.05 vs. Ad. LacZ + non‐OGD group, # P < 0.05 vs. Ad. LacZ + OGD group. n=3 per group.
Table S1. List of primary antibodies. Rat anti‐CD31/MAP‐2/Ly‐6B, rabbit anti‐NeuN/occludin and mouse anti‐ZO‐1 antibodies were used for immunofluorescence staining. Rabbit anti‐occludin and mouse anti‐ZO‐1/β‐actin/KLF11 antibodies were used in western blotting.
Table S2. The constructs used for dual‐luciferase assays.
Table S3. The specific primers designed for real‐time PCR.
Acknowledgments
This work was supported by National Institutes of Health Grants NS094930, NS091175 and NS086820 to K.‐J.Y, HL134569 to Y.E.C and HL138139 to J.Z.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and from the corresponding author upon reasonable request.
Reference
- 1. Abbott NJ, Ronnback L, Hansson E (2006) Astrocyte‐endothelial interactions at the blood‐brain barrier. Nat Rev Neurosci 7:41–53. [DOI] [PubMed] [Google Scholar]
- 2. Atkins GB, Jain MK (2007) Role of Kruppel‐like transcription factors in endothelial biology. Circ Res 100:1686–1695. [DOI] [PubMed] [Google Scholar]
- 3. Ayloo S, Gu C (2019) Transcytosis at the blood‐brain barrier. Curr Opin Neurobiol 57:32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ballabh P, Braun A, Nedergaard M (2004) The blood‐brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis 16:1–13. [DOI] [PubMed] [Google Scholar]
- 5. Begley DJ, Brightman MW (2003) Structural and functional aspects of the blood‐brain barrier. Prog Drug Res 61:39–78. [DOI] [PubMed] [Google Scholar]
- 6. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R , et al. (2017) Heart disease and stroke statistics‐2017 update: a report from the American Heart Association. Circulation 135:e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ben‐Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, Gu C (2014) Mfsd2a is critical for the formation and function of the blood‐brain barrier. Nature 509:507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bhattacharya R, Senbanerjee S, Lin Z, Mir S, Hamik A, Wang P , et al. (2005) Inhibition of vascular permeability factor/vascular endothelial growth factor‐mediated angiogenesis by the Kruppel‐like factor KLF2. J Biol Chem 280:28848–28851. [DOI] [PubMed] [Google Scholar]
- 9. Bielenberg DR, Hida Y, Shimizu A, Kaipainen A, Kreuter M, Kim CC, Klagsbrun M (2004) Semaphorin 3F, a chemorepulsant for endothelial cells, induces a poorly vascularized, encapsulated, nonmetastatic tumor phenotype. J Clin Invest 114:1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Botella LM, Sanchez‐Elsner T, Sanz‐Rodriguez F, Kojima S, Shimada J, Guerrero‐Esteo M , et al. (2002) Transcriptional activation of endoglin and transforming growth factor‐beta signaling components by cooperative interaction between Sp1 and KLF6: their potential role in the response to vascular injury. Blood 100:4001–4010. [DOI] [PubMed] [Google Scholar]
- 11. Cowan CE, Kohler EE, Dugan TA, Mirza MK, Malik AB, Wary KK (2010) Kruppel‐like factor‐4 transcriptionally regulates VE‐cadherin expression and endothelial barrier function. Circ Res 107:959–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Das A, Fernandez‐Zapico ME, Cao S, Yao J, Fiorucci S, Hebbel RP , et al. (2006) Disruption of an SP2/KLF6 repression complex by SHP is required for farnesoid X receptor‐induced endothelial cell migration. J Biol Chem 281:39105–39113. [DOI] [PubMed] [Google Scholar]
- 13. Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW , et al. (2006) KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood 107:4354–4363. [DOI] [PubMed] [Google Scholar]
- 14. Dekker RJ, van Thienen JV, Rohlena J, de Jager SC, Elderkamp YW, Seppen J , et al. (2005) Endothelial KLF2 links local arterial shear stress levels to the expression of vascular tone‐regulating genes. Am J Pathol 167:609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. del Zoppo GJ, Hallenbeck JM (2000) Advances in the vascular pathophysiology of ischemic stroke. Thromb Res 98:73–81. [DOI] [PubMed] [Google Scholar]
- 16. Dilling C, Roewer N, Forster CY, Burek M (2017) Multiple protocadherins are expressed in brain microvascular endothelial cells and might play a role in tight junction protein regulation. J Cereb Blood Flow Metab 37:3391–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dong W, Zhang X, Liu W, Chen YJ, Huang J, Austin E , et al. (2015) A conserved polybasic domain mediates plasma membrane targeting of Lgl and its regulation by hypoxia. J Cell Biol 211:273–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fagan SC, Hess DC, Hohnadel EJ, Pollock DM, Ergul A (2004) Targets for vascular protection after acute ischemic stroke. Stroke 35:2220–2225. [DOI] [PubMed] [Google Scholar]
- 19. Fan Y, Guo Y, Zhang J, Subramaniam M, Song CZ, Urrutia R, Chen YE (2012) Kruppel‐like factor‐11, a transcription factor involved in diabetes mellitus, suppresses endothelial cell activation via the nuclear factor‐kappaB signaling pathway. Arterioscler Thromb Vasc Biol 32:2981–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fan Y, Lu H, Liang W, Hu W, Zhang J, Chen YE (2017) Kruppel‐like factors and vascular wall homeostasis. J Mol Cell Biol 9:352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fernandez‐Zapico ME, van Velkinburgh JC, Gutierrez‐Aguilar R, Neve B, Froguel P, Urrutia R, Stein R (2009) MODY7 gene, KLF11, is a novel p300‐dependent regulator of Pdx‐1 (MODY4) transcription in pancreatic islet beta cells. J Biol Chem 284:36482–36490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fisher M (2008) Injuries to the vascular endothelium: vascular wall and endothelial dysfunction. Rev Neurol Dis 5(Suppl. 1):S4–S11. [PubMed] [Google Scholar]
- 23. Freeman LR, Keller JN (2012) Oxidative stress and cerebral endothelial cells: regulation of the blood‐brain‐barrier and antioxidant based interventions. Biochim Biophys Acta.1822:822–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ginsberg MD (2009) Current status of neuroprotection for cerebral ischemia: synoptic overview. Stroke 40(3, Suppl. 1):S111–S114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gohla G, Krieglstein K, Spittau B (2008) Tieg3/Klf11 induces apoptosis in OLI‐neu cells and enhances the TGF‐beta signaling pathway by transcriptional repression of Smad7. J Cell Biochem 104:850–861. [DOI] [PubMed] [Google Scholar]
- 26. Guo Y, Fan Y, Zhang J, Lomberk GA, Zhou Z, Sun L , et al. (2015) Perhexiline activates KLF14 and reduces atherosclerosis by modulating ApoA‐I production. J Clin Investig 125:3819–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hamik A, Lin Z, Kumar A, Balcells M, Sinha S, Katz J , et al. (2007) Kruppel‐like factor 4 regulates endothelial inflammation. J Biol Chem 282:13769–13779. [DOI] [PubMed] [Google Scholar]
- 28. Hartsock A, Nelson WJ (2008) Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta 1778:660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hawkins BT, Davis TP (2005) The blood‐brain barrier/neurovascular unit in health and disease. Pharmacol Rev 57:173–185. [DOI] [PubMed] [Google Scholar]
- 30. Hu W, Lu H, Zhang J, Fan Y, Chang Z, Liang W , et al. (2018) Kruppel‐like factor 14, a coronary artery disease associated transcription factor, inhibits endothelial inflammation via NF‐kappaB signaling pathway. Atherosclerosis 278:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Iadecola C (2017) The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron 96:17–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Iadecola C, Anrather J (2011) Stroke research at a crossroad: asking the brain for directions. Nat Neurosci 14:1363–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ishikawa M, Zhang JH, Nanda A, Granger DN (2004) Inflammatory responses to ischemia and reperfusion in the cerebral microcirculation. Fron Biosci 9:1339–1347. [DOI] [PubMed] [Google Scholar]
- 34. Jackman K, Kahles T, Lane D, Garcia‐Bonilla L, Abe T, Capone C , et al. (2013) Progranulin deficiency promotes post‐ischemic blood‐brain barrier disruption. J Neurosci 33:19579–19589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leak RK, Zhang L, Stetler RA, Weng Z, Li P, Atkins GB , et al. (2013) HSP27 protects the blood‐brain barrier against ischemia‐induced loss of integrity. CNS Neurol Disord Drug Targets 12:325–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liang W, Fan Y, Lu H, Chang Z, Hu W, Sun J , et al. (2019) KLF11 (Kruppel‐like factor 11) inhibits arterial thrombosis via suppression of tissue factor in the vascular wall. Arterioscler Thromb Vasc Biol 39:402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu J, Jin X, Liu KJ, Liu W (2012) Matrix metalloproteinase‐2‐mediated occludin degradation and caveolin‐1‐mediated claudin‐5 redistribution contribute to blood‐brain barrier damage in early ischemic stroke stage. J Neurosci 32:3044–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lomberk G, Urrutia R (2005) The family feud: turning off Sp1 by Sp1‐like KLF proteins. Biochem J 392(Pt 1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luissint AC, Artus C, Glacial F, Ganeshamoorthy K, Couraud PO (2012) Tight junctions at the blood brain barrier: physiological architecture and disease‐associated dysregulation. Fluids Barriers CNS 9:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McCormick SM, Eskin SG, McIntire LV, Teng CL, Lu CM, Russell CG, Chittur KK (2001) DNA microarray reveals changes in gene expression of shear stressed human umbilical vein endothelial cells. Proc Natl Acad Sci U S A 98:8955–8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Neumann‐Haefelin T, Kastrup A, de Crespigny A, Yenari MA, Ringer T, Sun GH, Moseley ME (2000) Serial MRI after transient focal cerebral ischemia in rats: dynamics of tissue injury, blood‐brain barrier damage, and edema formation. Stroke 31:1965–1972; discussion 1972–1973. [DOI] [PubMed] [Google Scholar]
- 42. Neve B, Fernandez‐Zapico ME, Ashkenazi‐Katalan V, Dina C, Hamid YH, Joly E , et al. (2005) Role of transcription factor KLF11 and its diabetes‐associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci U S A 102:4807–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN , et al. (2006) Integration of flow‐dependent endothelial phenotypes by Kruppel‐like factor 2. J Clin Invest 116:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rempe RG, Hartz AMS, Bauer B (2016) Matrix metalloproteinases in the brain and blood‐brain barrier: versatile breakers and makers. J Cereb Blood Flow Metab 36:1481–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rodriguez‐Yanez M, Castellanos M, Blanco M, Mosquera E, Castillo J (2006) Vascular protection in brain ischemia. Cerebrovasc Dis 21(Suppl. 2):21–29. [DOI] [PubMed] [Google Scholar]
- 46. Sandoval KE, Witt KA (2008) Blood‐brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis 32:200–219. [DOI] [PubMed] [Google Scholar]
- 47. Schlaeger TM, Bartunkova S, Lawitts JA, Teichmann G, Risau W, Deutsch U, Sato TN (1997) Uniform vascular‐endothelial‐cell‐specific gene expression in both embryonic and adult transgenic mice. Proc Natl Acad Sci U S A 94:3058–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shi Y, Jiang X, Zhang L, Pu H, Hu X, Zhang W , et al. (2017) Endothelium‐targeted overexpression of heat shock protein 27 ameliorates blood‐brain barrier disruption after ischemic brain injury. Proc Natl Acad Sci U S A 114:E1243–E1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shi Y, Zhang L, Pu H, Mao L, Hu X, Jiang X , et al. (2016) Rapid endothelial cytoskeletal reorganization enables early blood‐brain barrier disruption and long‐term ischaemic reperfusion brain injury. Nat Commun 7:10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sivandzade F, Cucullo L (2018) In‐vitro blood‐brain barrier modeling: A review of modern and fast‐advancing technologies. J Cereb Blood Flow Metab 38:1667–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Srinivasan B, Kolli AR, Esch MB, Abaci HE, Shuler ML, Hickman JJ (2015) TEER measurement techniques for in vitro barrier model systems. J Lab Autom 20:107–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Suzuki T, Aizawa K, Matsumura T, Nagai R (2005) Vascular implications of the Kruppel‐like family of transcription factors. Arterioscler Thromb Vasc Biol 25:1135–1141. [DOI] [PubMed] [Google Scholar]
- 53. Tang X, Liu K, Hamblin MH, Xu Y, Yin KJ (2018) Genetic deletion of Kruppel‐Like factor 11 aggravates ischemic brain injury. Mol Neurobiol 55:2911–2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang X, Lo EH (2003) Triggers and mediators of hemorrhagic transformation in cerebral ischemia. Mol Neurobiol 28:229–244. [DOI] [PubMed] [Google Scholar]
- 55. Wang J, Shi Y, Zhang L, Zhang F, Hu X, Zhang W , et al. (2014) Omega‐3 polyunsaturated fatty acids enhance cerebral angiogenesis and provide long‐term protection after stroke. Neurobiol Dis 68:91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yamahara K, Itoh H, Chun TH, Ogawa Y, Yamashita J, Sawada N , et al. (2003) Significance and therapeutic potential of the natriuretic peptides/cGMP/cGMP‐dependent protein kinase pathway in vascular regeneration. Proc Natl Acad Sci U S A 100:3404–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang C, Hawkins KE, Dore S, Candelario‐Jalil E (2019) Neuroinflammatory mechanisms of blood‐brain barrier damage in ischemic stroke. Am J Physiol Cell Physiol 316:C135–C153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yang Y, Rosenberg GA (2011) Blood‐brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 42:3323–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yang X, Tang X, Sun P, Shi Y, Liu K, Hassan SH , et al. (2017) MicroRNA‐15a/16‐1 antagomir ameliorates ischemic brain injury in experimental stroke. Stroke 48:1941–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang Q, Tong X, Schieb L, Vaughan A, Gillespie C, Wiltz JL , et al. (2017) Vital signs: recent trends in stroke death rates — United States, 2000–2015. Morb MortalWkly Rep 66:933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yin KJ, Deng Z, Hamblin M, Xiang Y, Huang H, Zhang J , et al. (2010) Peroxisome proliferator‐activated receptor delta regulation of miR‐15a in ischemia‐induced cerebral vascular endothelial injury. J Neurosci 30:6398–6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yin KJ, Fan Y, Hamblin M, Zhang J, Zhu T, Li S , et al. (2013) KLF11 mediates PPARgamma cerebrovascular protection in ischaemic stroke. Brain 136(Pt 4):1274–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yin KJ, Hamblin M, Fan Y, Zhang J, Chen YE (2015) Krupple‐like factors in the central nervous system: novel mediators in stroke. Metab Brain Dis 30:401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yin KJ, Olsen K, Hamblin M, Zhang J, Schwendeman SP, Chen YE (2012) Vascular endothelial cell‐specific microRNA‐15a inhibits angiogenesis in hindlimb ischemia. J Biol Chem 287:27055–27064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang JH, Badaut J, Tang J, Obenaus A, Hartman R, Pearce WJ (2012) The vascular neural network–a new paradigm in stroke pathophysiology. Nat Rev Neurol 8:711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang X, Hamblin MH, Yin KJ (2019) Noncoding RNAs and stroke. Neuroscientist 25:22–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang X, Tang X, Liu K, Hamblin MH, Yin KJ (2017) Long noncoding RNA Malat1 regulates cerebrovascular pathologies in ischemic stroke. J Neurosci 37:1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhou G, Hamik A, Nayak L, Tian H, Shi H, Lu Y , et al. (2012) Endothelial Kruppel‐like factor 4 protects against atherothrombosis in mice. J Clin Invest 122:4727–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Figure S1. KLF11 expression in mouse BMEC cultures and cerebral microvessels after in vitro and in vivo ischemic stimuli. A. Mouse BMEC cultures were subjected to 4 or 16 h OGD treatment. Total RNA was then isolated and KLF11 mRNA levels were examined by real‐time PCR. *P< 0.05 vs. non‐OGD group. B. Cerebral microvessels were isolated from C57BL/6J mice after 1 h MCAO followed by 24 h reperfusion. Total RNA was isolated and KLF11 levels were examined by real‐time PCR. Cerebral ischemia resulted in a significant decrease in KLF11 expression. Data are expressed as mean ±SEM. *P < 0.05 vs. sham group.
Figure S2. Effects of KLF11 overexpression on pro‐inflammatory cytokines in mouse BMEC cultures after OGD. Cultured mBMECs were transduced with Ad. LacZ or Ad. KLF11 for 48 h before subjected to 16 h OGD, followed by 24 or 48 h reperfusion. Total RNA and culture medium were collected, and a series of pro‐inflammatory chemokines, cytokines and adhesive molecules were analyzed by qPCR (A,B) and ELISA (C,D). *P < 0.05 vs. Ad. LacZ + non‐OGD group, # P < 0.05 vs. Ad. LacZ + OGD group. n=3 per group.
Table S1. List of primary antibodies. Rat anti‐CD31/MAP‐2/Ly‐6B, rabbit anti‐NeuN/occludin and mouse anti‐ZO‐1 antibodies were used for immunofluorescence staining. Rabbit anti‐occludin and mouse anti‐ZO‐1/β‐actin/KLF11 antibodies were used in western blotting.
Table S2. The constructs used for dual‐luciferase assays.
Table S3. The specific primers designed for real‐time PCR.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and from the corresponding author upon reasonable request.