

Abstract
Acute lung injury (ALI) is well defined in humans, but there is no agreement as to the main features of acute lung injury in animal models. A Committee was organized to determine the main features that characterize ALI in animal models and to identify the most relevant methods to assess these features. We used a Delphi approach in which a series of questionnaires were distributed to a panel of experts in experimental lung injury. The Committee concluded that the main features of experimental ALI include histological evidence of tissue injury, alteration of the alveolar capillary barrier, presence of an inflammatory response, and evidence of physiological dysfunction; they recommended that, to determine if ALI has occurred, at least three of these four main features of ALI should be present. The Committee also identified key “very relevant” and “somewhat relevant” measurements for each of the main features of ALI and recommended the use of least one “very relevant” measurement and preferably one or two additional separate measurements to determine if a main feature of ALI is present. Finally, the Committee emphasized that not all of the measurements listed can or should be performed in every study, and that measurements not included in the list are by no means “irrelevant.” Our list of features and measurements of ALI is intended as a guide for investigators, and ultimately investigators should choose the particular measurements that best suit the experimental questions being addressed as well as take into consideration any unique aspects of the experimental design.
Keywords: acute lung injury, animal model, disease models
CONTENTS
I. Statement of the Problem
II. What Is Being Modeled?
III. Methodology
IV. Results: Features and Measurements of ALI in Animals
V. Practical Aspects of Measuring Ali in Animals
VI. Critical Assessment of Selected Common Models of Lung Injury
VII. Limitations
VIII. Summary and Conclusions
I. STATEMENT OF THE PROBLEM
What is acute lung injury (ALI) in an animal? One would think that the answer to this question is well established, considering that a PubMed search in mid-2008 retrieved more than 14,000 entries for the query “Lung Injury AND Animal,” and that considerable resources have been invested in the study of experimental ALI during past decades. However, a closer look at the published literature reveals that there is no universal agreement as to the precise definition of ALI in experimental animal models. Is an increase in the concentration of lung cytokines sufficient to indicate lung injury, or should there also be evidence of cellular inflammation? If so, does it suffice to have inflammatory cells in the interstitium, or should they also be present in the airspaces? Or perhaps lung injury should, instead, be determined based on changes in alveolar epithelial permeability? There is no consensus in the scientific community as to what exactly constitutes ALI in an animal, in part because there is no single marker or parameter that has sufficient sensitivity and specificity to identify the occurrence of all forms and severities of ALI. As a result, it is difficult for investigators to determine if they have achieved (or prevented) lung injury in an experimental model. For example, if an experimental drug decreases neutrophil migration into the lungs but has no effect on permeability changes, does it prevent lung injury or not? In addition, a comparison of studies is difficult because of the diversity of assays used by investigators to assess lung injury. The same experimental challenge may or may not result in lung injury, depending on how lung injury is defined by the investigators. Clearly, answers to such questions are relevant in an area of research that is as important to patient care as ALI.
In humans, the definition of ALI is based on the following well-defined set of clinical parameters developed by the American European Consensus Conference: acute onset, radiological evidence of diffuse bilateral pulmonary infiltrates, a ratio of the partial pressure of arterial oxygen to the fraction of inspired oxygen (PaO2/FiO2) of less than 300, and no clinical evidence for elevated pulmonary arterial pressure (1). However, these criteria cannot be directly translated to experimental animals. Although arterial blood gases, chest radiographs, cardiac echocardiography, and even catheterization can be performed in small experimental animals, the equipment required for these measurements is available only in a few laboratories. Furthermore, such measurements may be incompatible with the design of many experimental systems. Thus, it is not practical to use the American European Consensus Conference criteria in animal studies, particularly in small animals. An alternative approach would be to define ALI in animals based on histopathological criteria similar to those seen in humans with ALI. In humans, the pathological correlate of ALI is diffuse alveolar damage (DAD), characterized by inflammatory infiltrates, thickened alveolar septae, and deposition of hyaline membranes (2). However, as discussed below, none of the available animal models of ALI reproduce all of the pathologic features of DAD in humans. Therefore, the criteria used to define ALI in humans cannot be directly translated to most animal models of ALI.
The goal of the present workshop was to determine the main features that characterize ALI in animals and then to identify the most relevant measurements to assess these features. We used a Delphi approach in which a series of questionnaires were distributed among a panel of experts in experimental lung injury. In this report, we discuss the main features of human ALI that are being modeled in animals, describe the methodology used to identify the defining features of lung injury in animals, discuss common methods of measuring lung injury, critically assess standard models of lung injury, critically assess standard models of lung injury, and finally, discuss the limitations of this approach.
II. WHAT IS BEING MODELED?
Ideally, an animal model of ALI should capture one or more features of human ALI, including rapid onset (hours) after an inciting stimulus, evidence of pulmonary physiological dysfunction (e.g., abnormalities of gas exchange, decreased lung compliance), histological evidence of injury to the lung parenchyma (endothelium, interstitium, epithelium), and evidence of increased permeability of the alveolocapillary membrane. Any one animal model is unlikely to encompass all of the salient features of ALI/ARDS observed in humans. Thus, a key question is how many of the features of human ALI need to be present in an animal model for it to be classified as demonstrating ALI? This is a complex issue without a right or wrong answer and is dependent on many factors, including the specific experimental question being addressed. To understand these issues, it is important to review certain differences between animals and humans that are pertinent to ALI and then to compare humans and animals in terms of these variables.
Differences between Animals and Humans Relevant to ALI
There are many anatomical and physiological differences between animals and humans that influence the response of the lung to an acute injurious stimulus and affect the evaluation of lung injury (3). Some of these are obvious; for example, the respiratory rate of mice (250–300 bpm) far exceeds that of adult humans (12–16 bpm), rendering absolute respiratory rate inadequate as a parameter of ALI in mice (4, 5). There are important differences in the gross and microscopic anatomy between rodent and human lungs. Mice have no bronchial arteries. Both the alveoli and thickness of the blood–gas barrier are smaller in the lungs of mice and rats compared with humans. Mice and rat lungs have a lobar anatomy distinct from that of humans and their lungs have fewer branches of the conducting airways proximal to the terminal bronchioles. At the microscopic level, murine lungs differ from human lungs in that they have a larger number of Clara cells in the distal airways; extensive bronchial-associated lymphoid tissue, and a virtual absence of submucosal glands beyond the proximal trachea (6). In the setting of ALI, murine lungs rarely demonstrate typical hyaline membranes in situations where there is clearly enhanced permeability of the alveolocapillary membrane. There are also important differences in key elements of the inflammatory response, both cellular and humoral, between rodents and humans. For example, mice have fewer circulating neutrophils (10–25%) than humans (50–70%) and do not express defensins (6). The neutrophil repertoire of CXC chemokines differs between rodents and humans (keratinocyte-derived chemokine KC [CXCL1] and macrophage inflammatory protein [MIP-2] in mice, cytokine induced neutrophil chemoattractant [CINC] in rats, and IL-8 in humans) (7). In addition, critically ill humans develop ALI/ARDS in the setting of a number of interventions such as prolonged ventilation or hemodynamic support, which are difficult to reproduce in animals. Finally, animal studies frequently use young mice with no comorbidities, whereas human patients tend to be older and have multiple medical problems such as diabetes, coronary artery disease, renal or hepatic insufficiency, and so forth. For all of these reasons, the responses of animal and human lungs to an injurious stimulus cannot be expected to be identical or perhaps even similar.
Comparison of Specific Parameters in the Assessment of ALI in Animal Models
1. Assessment of the kinetics of lung injury.
Given the relatively controlled nature of experimental models of ALI, the time of exposure of the inciting stimulus (e.g., lipopolysaccharide, acid aspiration, hemorrhagic shock, or injurious mechanical ventilation) is usually known with precision. This differs from the human situation where the time of exposure to an inciting stimulus (e.g., bacterial infection with sepsis) is often less clear. Nonetheless, in animal systems that seek to model human ALI, maximal lung injury should be evident within 24 hours of exposure to the inciting stimulus to distinguish those conditions that reflect more subacute or chronic lung injury.
2. Radiographic assessment of lung injury.
The ability to assess lung injury radiographically in animal models is constrained by several factors, including the small size of rodents and the limited availability of radiographic facilities for animals in most laboratories. Nonetheless, if available, radiographic demonstration (plain X-ray, micro-computer tomography) of bilateral and diffuse pulmonary infiltrates in animals represents one parameter to assist in assessing the extent of lung injury. This is more feasible for larger animals such as ferrets, rabbits, dogs, or sheep.
3. Physiological assessment of lung injury.
(i) Abnormalities of gas exchange. In humans, an elevated a–a gradient, reflecting compromise of the gas exchange function of the lungs, is one of the principal parameters used to diagnose, stratify, and monitor patients with ALI or acute respiratory distress syndrome (ARDS). Although this is feasible and indeed routine in larger animals, it is more difficult in rodents, and especially mice, because of the difficulty of obtaining a sufficient sample of arterialized blood without introducing other variables (e.g., trauma during sample collection or hypovolemia from removal of a substantial fraction of the circulating blood volume). Noninvasive continuous monitoring of capillary or tissue oxygen saturation using conventional microscopic or fluorescence enhanced oximetry or electron paramagnetic resonance spectroscopy is technically possible and is a useful parameter to monitor if the appropriate instrumentation is available (8, 9).
(ii) Decreased lung compliance. Decreased lung compliance due to pulmonary edema and atelectasis is a hallmark of human ALI/ARDS and is an important and easily assessed parameter in animal models. In mechanically ventilated animals, respiratory system compliance can be assessed in vivo during mechanical ventilation in a similar fashion to how it is measured in humans or by determining the pressure–volume curve after a recruitment maneuver. A particularly useful method is to use commercially available ventilator systems that allow automated measurement of respiratory mechanics and generate pressure-volume curves and measurement of compliance (10). These systems can be programmed to automatically perform measurements at predetermined time intervals. These systems are effective but remain expensive. Measurement of lung compliance in vivo requires an estimate of pleural pressure. An equally acceptable method is to measure the pressure–volume curve of lungs ex vivo.
4. Assessment of increased permeability of the alveolo-capillary membrane.
Change in alveolocapillary membrane permeability is one of the critical parameters that defines the pathophysiology of ALI in humans and animals, but its measurement is fraught with technical pitfalls that vary based on the method used. During the genesis of ALI/ARDS the selective barrier function of the pulmonary endothelium and/or epithelium is lost due to injury or dysfunction. This is manifest by leakage of protein-rich fluid from the vascular to the interstitial and/or alveolar space. Many methods have been developed to assess this alteration. The most commonly used methods include (1) measurement of the concentration of albumin or other high molecular weight proteins such as IgM in bronchoalveolar lavage (BAL) fluid compared with that in plasma, (2) assessment of the leakage of an exogenous labeled high molecular weight molecule (e.g., 125I-labeled albumin or fluorescent high molecular weight dextran) or Evans blue dye (which binds to albumin) into the alveolar space, and (3) the wet to dry weight of the excised lungs. In larger animals, especially sheep, older studies measured increased lymphatic flow and protein concentration in lymphatic fluid as an indicator of interstitial edema (presumably an early phase of pulmonary edema). Each of these is a valid method provided that appropriate corrections for dilution of BAL fluid and/or intravascular blood volume are made. Measurement of the total protein concentration of the BAL fluid may reflect an increase in the permeability of the alveolocapillary membrane, but it should be used with caution as the sole measure of increased permeability, as further discussed in Section V.2 below.
5. Histological assessment of lung injury.
One of the most challenging aspects of using animal models of ALI pertains to the histological assessment of lung injury. The pathological hallmark of ALI in humans is diffuse alveolar damage (DAD) (2, 11, 12). In humans, DAD is characterized by: an early exudative phase featuring neutrophil accumulation in the vascular, interstitial, and alveolar spaces known as neutrophilic alveolitis); deposition of hyaline membranes composed of fibrin and other proteinaceous debris as evidence that serum proteins have entered and precipitated in the airspaces (i.e., disruption of the alveolocapillary membrane); interstitial thickening; and the formation of microthrombi (evidence of endothelial injury and intraluminal activation of the coagulation cascade). This is followed by a proliferative phase characterized by alveolar epithelial cell hyperplasia and interstitial fibrosis. It is reasonable to expect that animal models of ALI/ARDS should reflect some of these features. However, no animal model completely reproduces all of the histological features of DAD in humans (7, 13). The presence of only a single histologic feature of ALI in an experimental animal (e.g., neutrophilic alveolitis) does not necessarily indicate a bona fide model of ALI and conversely, the absence of one or more features does not exclude ALI. Despite these differences, a semi-quantitative or quantitative assessment of multiple histological features of ALI is critical for evaluating the extent of lung injury in animal models. Notably, reliable histological assessment of lung injury requires that the lungs be properly fixed at functional residual capacity, which can be achieved by inflating the lungs with a pressure of 15–20 cm H2O.
Other Considerations
An important consideration is that animal models, because of their very reductionist nature, are typically used to investigate one specific aspect of lung injury (e.g., mechanisms of leukocyte activation, endothelial or epithelial injury, acid-induced lung injury) in contrast to the human situation where there are often multiple inciting insults, comorbid conditions, and therapeutic interventions that all contribute to the clinical picture. Thus, although animal models of ALI should reflect one or more of the clinical features observed in humans, no single animal model can encompass all of them. On the other hand, the more restricted the animal model is (i.e., possessing fewer features of the human situation), the less representative it is of human disease.
Given these considerations, how can an investigator decide on an appropriate model system, and which features of ALI are “required” for it to be a “valid” model? The primary consideration in the choice of an animal model of ALI should be the experimental question to be addressed. For example, if the primary question pertains to sepsis-induced ALI, then models incorporating elements of bacterial infection such as intraperitoneal or intravenous administration of lipopolysaccharide or live bacteria, or cecal ligation and perforation, should be considered and the relevant endpoints assessed (14). If the primary question pertains to the mechanisms of lung injury induced by mechanical ventilation, then various modes of injurious mechanical ventilation can be imposed on the experimental animals in the presence or absence of other inciting stimuli such as sepsis, hypotension, or acid-aspiration. If the primary question relates to the mechanisms of leukocyte-induced endothelial or epithelial injury, then inciting stimuli can be given intravenously (e.g., complement activating agents) or via the airways (chemoattractants, lipopolysaccharide, or hyperoxic gas mixtures). Some challenges imposed on the animal model (e.g., extremely large tidal volumes in certain models of ventilator-induced lung injury or massive doses of lipopolysaccharide in certain models of sepsis) may be so extreme as to be nonrepresentative of the range of conditions present in humans with ALI/ARDS, but may be useful to answer very specific scientific questions. Finally, models in which more than one inciting stimulus for ALI is present are probably more reflective of the human situation in which a single inciting stimulus is rarely present (“two-hit hypothesis”).
The question remains as to what constitutes the minimal criteria for the diagnosis of ALI in animal models. Although there is no consensus about this issue (indeed, this is one of the primary purposes of this work), several general comments can be made. First, models in which only one aspect of lung injury is prominent (e.g., excess neutrophils in the alveolar space without evidence of physiological pulmonary dysfunction or increased permeability of the alveolocapillary membrane) are not reflective of the human situation and should probably not be labeled as ALI but may still be appropriate to answer specific scientific questions. Second, depending on the specific features of the model, it is possible that there may be a divergence between different aspects of lung injury. For example, under certain experimental conditions it is possible to induce large numbers of neutrophils to migrate into the alveolar space with minimal or only transient evidence of physiological pulmonary dysfunction or increases in permeability of the alveolocapillary membrane. Thus, it is prudent to use more than one independent measurement that reliably assesses lung injury in any model of ALI.
III. METHODOLOGY
A series of targeted questionnaires was transmitted by e-mail to a panel of experts. We requested participation from a total of 29 individuals selected with the goal of generating an international panel of investigators with broad expertise in lung injury. Of these 29 individuals, two declined, three did not reply, and two failed to return conflict-of-interest statements, leaving the final number of participants at 22. The first questionnaire was designed to generate a list of main features (categories) of ALI in animals, as well as specific measurements (criteria) that can be used to determine if a main feature is present. Questionnaire One yielded 70 individual measurements grouped under eight main features. The results of Questionnaire One were compiled and returned to the participants who were asked to rank each measurement as 1.0 (very important), 2.0 (somewhat important), or 3.0 (not important). The results were analyzed and the means and SD of each measurement were calculated. All measurements with a mean greater than 2 were removed and redundant measurements were combined, leaving 32 individual measurements grouped under “main features.” Of the original nine main features, four were removed because none of the measurements reached a mean of two; these four main features were: “evidence of cellular injury,” “conversion to a fibrotic response,” “radiological changes,” and “other.” An additional main feature was “clinical evidence of disease”; within this feature only one measurement, “rapid onset (within 24 hours),” received a score of two or less; this measurement was incorporated into the definition of ALI. Next, the panelists were asked to determine the relevance of each of these measurements to lung injury, using a scale of 1.0 to 3.0, with 1 being “very relevant,” 2 being “somewhat relevant,” and 3 being “not relevant.” Of the 32 measurements, 17 were considered “very relevant” and 15 were considered “somewhat relevant.” Finally, in addition to the questionnaires, there were three in-detail face-to-face meetings that allowed members of the panel to discuss the results of each step, including combining redundant criteria. The final results are summarized below, and the questionnaires (including responses) are shown in the online supplement.
IV. RESULTS: FEATURES AND MEASUREMENTS OF ALI IN ANIMALS
1. Main Features of Experimental ALI
The main features of experimental ALI in animals are rapid onset (within 24 h) and:
1.1 Histological evidence of tissue injury
1.2 Alteration of the alveolar capillary barrier
1.3 An inflammatory response
1.4 Evidence of physiological dysfunction
Of these, the most relevant features were considered to be evidence of tissue injury and evidence of alteration of the alveolar capillary barrier. The presence of the main features of experimental lung injury can be established with the following measurements:
2. Measurements of Histological Evidence of Tissue Injury
Very relevant
2.1 Accumulation of neutrophils in the alveolar or the interstitial space
2.2 Formation of hyaline membranes
2.3 Presence of proteinaceous debris in the alveolar space (such as fibrin strands)
2.4 Thickening of the alveolar wall
2.5 Enhanced injury as measured by a standardized histology score
Somewhat relevant
2.6 Evidence of hemorrhage
2.7 Areas of atelectasis
2.8 Gross macroscopic changes such as discoloration of the lungs
3. Measurements of Alteration of the Alveolar Capillary Barrier
Very relevant
3.1 An increase in extravascular lung water content
3.2 Accumulation of an exogenous protein or tracer in the airspaces or the extra vascular compartment
3.3 Increase in total bronchoalveolar (BAL) protein concentration
3.4 Increase in concentration of high molecular weight proteins in BAL fluid (e.g., albumin, IgM)
3.5 Increase in the microvascular filtration coefficient
Somewhat relevant
3.6 Increase in lung wet/dry weight ratio
3.7 Translocation of a protein from the airspaces into plasma
3.8 Increased lung lymph flow
3.9 High lymph protein concentration
4. Measurements of the Inflammatory Response
Very relevant
4.1 Increase in the absolute number of neutrophils in BAL fluid
4.2 Increase in lung myeloperoxidase (MPO) activity or protein concentration
4.3 Increase in the concentrations of proinflammatory cytokines in lung tissue or BAL fluid
Somewhat relevant
4.4 Increases in procoagulatory activity
4.5 Increased expression of adhesion molecules
4.6 Conversion of the neutrophilic alveolitis into a mononuclear alveolitis with time
4.7 Increase in levels of complement factors and matrix metalloproteinases
5. Measurements of Physiological Dysfunction
Very relevant
5.1 Hypoxemia
5.2 Increased alveolar–arterial oxygen difference
Somewhat relevant
5.3 PaO2/FiO2 < 200
5.4 Increase in spontaneous minute ventilation
5.5 Increase in spontaneous respiratory rate
To determine if ALI has occurred, we recommend that at least three of the four “main features” of ALI be identified. To determine if any of the main features of ALI are present, we recommend using at least one of the “very relevant” measurements listed above, and preferably, one or two additional separate measurements to confirm the results.
We would like to emphasize that not all of the measurements listed can or should be performed in every study, and that measurements not included in the list are by no means irrelevant. For example, there are many measurements that are useful for assessing lung injury, but can only be performed in specialized laboratories (e.g., animal computer tomography). We have tried to include measurements that are within the reach of most laboratories located at major universities and health care facilities. We would also like to point out that, depending on the experimental question, the fact that fully developed ALI has occurred may not be necessary or relevant to establish. For example, a model with alveolar neutrophilia without changes in alveolar permeability is suitable for studying the mechanisms of neutrophil migration, even if such a model does not reproduce all of the “main features” of ALI. Thus, our list of features and measurements of ALI is intended as a guide for investigators, but ultimately investigators should choose any particular measurement based on their unique experimental questions and the characteristics of their experimental design.
One additional aspect of the proposed features is that they emphasize functional parameters of injury over individual molecular pathways. This highlights an important distinction between evidence of activation of a particular signaling pathway (e.g., the MAP kinase pathway) and lung injury. In this regard, there may be activation of (one or multiple) signaling pathways without concomitant lung injury. Conversely, there may be evidence of lung injury without activation of particular signaling pathways. This is not to say that mechanistic studies aimed at dissecting the role of signaling pathways in lung injury are not important but rather that the presence or absence of evidence of activation of these pathways is distinct from whether or not there is evidence of ALI.
V. PRACTICAL ASPECTS OF MEASURING ALI IN ANIMALS
1. Measuring Histological Evidence of Tissue Injury
Histological evidence of tissue injury was identified as the most relevant defining feature of ALI. The normal lung is characterized by thin alveolar walls with occasional intra-alveolar macrophages and vanishingly few neutrophils (Figure 1A−1B). Commonly, during the dehydration process associated with the preparation of tissues, the peribronchial space may appear expanded (Figure 1A). This finding must not be confused with peribronchial edema. The intratracheal administration of injurious substances is usually associated with patchy injury, and large areas of the lung may be spared (Figure 1C). To facilitate the identification of areas that have been exposed to the instillate, colloidal carbon (1:100 dilution) can be added to the instillate; the colloidal carbon will be phagocytosed by resident macrophages and appear as intracellular black dots (Figure 1). This can be particularly useful when a “negative” experiment raises doubts as to whether the instillate could have been inadvertently administered into the esophagus. Some types of injury are primarily associated with neutrophil infiltration and deposition of proteinaceous debris or fibrin strands in the airspaces but with normal alveolar walls (Figure 1D). In contrast, other types of injury may be associated with marked thickening of the alveolar walls, which may contain abundant neutrophils but no evidence of intra-alveolar neutrophil infiltrates or protein deposition (Figure 1E). Hyaline membranes, when seen, appear as pink deposits on the alveolar walls stained with hematoxylin and eosin (Figure 1F). It is important to note that the presence of increased neutrophil numbers in the BAL fluid is unlikely to be associated with obvious intra-alveolar neutrophil infiltrates unless the total BAL neutrophil counts are greater than 106, because the BAL procedure collects neutrophils from the entire lung. Also, in certain animals the neutrophils may display light staining; for example, in rabbits neutrophils are slightly eosinophilic and therefore not strictly a “neutrophil” in the sense of a “cell that does not stain” (Figure 2C−2D).
Figure 1.
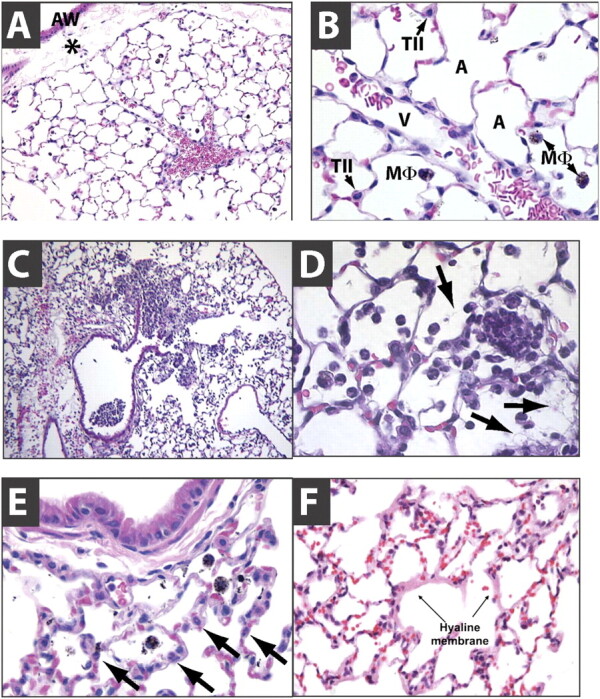
Normal and injured lungs. (A, B) Normal mouse lungs stained with HandE (A, 200×; B, 630×). The alveolar walls are very thin and the alveoli contain occasional alveolar macrophages. Note that the artifactual expansion of the peribronchial tissue (asterisk), which commonly results from tissue processing, does not represent edema. A, airspace; V, blood vessel; TII, type II cell; M, alveolar macrophage; AW, airway. (C and D) show patchy neutrophilic infiltrates with deposition of fibrin strands (arrows in D). Note that the alveolar walls are thin. (E) shows thickened alveolar walls with intramural neutrophils (arrows). Note the absence of neutrophils inside the alveoli. The black substance inside the macrophages is colloidal carbon. (F) shows hyaline membranes (arrows).
Figure 2.

Comparison of neutrophils from three mammalian species. (A) Human neutrophils show a typical multi-lobed nucleus interconnected by thin chromatin strands, and a colorless cytoplasm on Wright-Giemsa stains. Notice the presence of two eosinophils (open arrows) that can be distinguished by their pink cytoplasm and bilobulated nuclei (peripheral blood cytospin, Diff-Quick, 400×). (B) Murine neutrophils exhibit characteristic ring- or pretzeloid-shaped nucleus with drumstick shaped nodules containing pericentric heterochromatin; as in humans, the cytoplasm is colorless, whereas eosinophils are pink and have a bilobulated nucleus (peripheral blood cytospin, Diff Quick, 400×). In contrast, (C and D) rabbit “neutrophils” are notorious for their pink coloration (black arrows in D), and can be easily confused with the “eosinophil” of other mammalian species; they can be recognized because of their multilobulated nucleus, which is very similar to that of the human neutrophil (C, lung from a rabbit with ventilator-induced lung injury, HandE, 630×; and D, further magnification from the section in C). Note the different color of a macrophage (D, thick black arrow).(A) and (B) are courtesy of Dr. Jatinder Juss, University of Cambridge, UK; (C) and (D) are courtesy of Dr. G. Matute-Bello, University of Washington, Seattle, WA.
The Committee identified the need for a scoring system to quantify the extent of histologic lung injury. Any scoring system must be approached with caution, however, as it may not function similarly across different model systems, the sensitivity is limited, and there may be considerable interobserver variation. It is therefore not surprising that the scoring of injury in the research setting has met with limited success despite a number of equally valid approaches. Furthermore, appropriate scoring of tissue sections requires a considerable amount of time, including the period required to validate the system. The simplest tissue injury scoring system is a binary approach in which sections are categorized as either injured or normal. A number of more complex systems have been used in the literature, each incorporating features of ALI tissue to variable degrees. The use of strict morphometric criteria maximizes the reproducibility of the data, but adherence to strict morphometric standards is difficult to achieve in many laboratories. In this section, we propose a histologic ALI scoring system incorporating criteria that are accessible to both laboratory researchers and pathologists (Table 1). To mitigate against interobserver variability, we constructed a relatively simple scheme that uses coarse gradations of severity for each separate finding, which differs from previous scoring systems (15, 16).
TABLE 1.
LUNG INJURY SCORING SYSTEM
|
Score per field |
|||||
|---|---|---|---|---|---|
| Parameter |
0 |
1 |
2 |
||
| A. Neutrophils in the alveolar space | none | 1–5 | >5 | ||
| B. Neutrophils in the interstitial space | none | 1–5 | >5 | ||
| C. Hyaline membranes | none | 1 | >1 | ||
| D. Proteinaceous debris filling the airspaces | none | 1 | >1 | ||
| E. Alveolar septal thickening |
<2x |
2x–4x |
>4x |
||
Score = [(20 × A) + (14 × B) + (7 × C) + (7 × D) + (2 × E)]/(number of fields × 100)
Slides for evaluation should be prepared from suitably inflated (15–25 cm H2O), formalin-fixed paraffin-embedded tissue that has been stained with hematoxylin and eosin. Due to the patchy nature of ALI, at least 20 random high-power fields (400× total magnification) should be independently scored in a blinded fashion for each condition. The selection of random fields typically involves successive random displacements (each at least one high-power field in length) from the current position. More rigorous sampling may use a random function to generate (x, y) coordinates on the slide. Sophisticated strategies, such as stratified sampling and Poisson-disk distributions, may allow for more even sampling of the tissue and reject overly close samples. Furthermore, at least 50% of each field should be occupied by lung alveoli; fields that consist predominately of the lumen of large airways or vessels should be rejected. Each of five histological findings is graded using a three-tiered schema summarized in Table 1 and outlined in detail below.
Neutrophils are counted in the alveolar airspaces, alveolar walls, and interstitium. Alveolar wall neutrophils include cells that have entered the interstitium as well as those that are circulating or adhere to the alveolar capillaries. It should be noted that, as mentioned earlier, the neutrophils of some animals differ significantly in morphology from those in humans. For instance, murine models typically show ring-form neutrophils, and rabbit neutrophils appear “eosinophilic” on a Wright-Giemsa stain (i.e., they look pink rather than neutral in color). If neutrophils are not visible within the field, a score of zero is recorded for that field. One to five neutrophils give a score of one, and more than five neutrophils give a score of two.
Hyaline membranes, the hallmark of ALI in humans, are not routinely observed in many animal systems, most notably in murine models. The presence of even a single well-formed eosinophilic band of fibrin within the airspace earns a score of one, whereas multiple membranes visible in the field are scored as two. Pink proteinaceous debris filling the airspace is considered in an analogous manner to hyaline membranes.
The evaluation of septal thickening is highly dependent on the uniformity of inflation throughout the lung. If regions are underinflated, either due to errors in technique or variations in airway geometry, septae may appear artifactually thickened. Therefore, only septal thickening that is equal or greater than twice normal is considered in these criteria. Furthermore, septal thickness should not be evaluated in alveolar septa that are directly adjacent to a blood vessel or airway because these are normally thickened by the collagen present in the peribronchovascular bundle.
To generate a lung injury score, the sum of each of the five independent variables shown in Table 1 are weighted according to the relevance ascribed to each feature by the Committee, and then are normalized to the number of fields evaluated. The resulting injury score is a continuous value between zero and one (inclusive).
2. Measuring Alterations in Permeability
There are several potential approaches for measuring changes in lung vascular and epithelial permeability (17, 23). In large animals, such as sheep and goats, it is possible to collect lung lymph and measure the concentration of total protein in the lymph and in the plasma. The normal ratio is approximately 0.50 (24). If the left atrial pressure increases, then the lymph to plasma total protein ratio declines, because more salt and water are being filtered out of the lung capillaries than protein. If an animal is given an injury that increases permeability, such as oleic acid, endotoxin, live bacteria, or acid, then the lymph-to-plasma total protein ratio will normally not change. The lung lymph flow will increase, but the maintenance of a normal lymph-to-plasma total protein ratio indicates that the mechanism for the increase in fluid filtration is primarily increased permeability. The one caveat is that an increase in cardiac output with an increase in the surface area of lung perfusion will result in an increase in lung lymph flow with no change in lymph-to-plasma total protein ratio, so this could be misinterpreted as an increased permeability when it is really higher pulmonary blood flow with greater surface area of perfusion.
Another experimental approach that is useful for the isolated perfused lung is to measure the microvascular filtration coefficient, which can be interpreted as reflecting a change in lung vascular permeability. However, it is important to realize that the commonly utilized gravimetric measurement procedure depends on several assumptions such as an isogravimetric state (weight equilibrium) before and after the applied increment in hydrostatic pressure, the absence of alveolar edema and a constant vascular surface area (25). If these conditions cannot be met, it may be more appropriate to use the phrase “apparent filtration rate.” Thus, to calculate a filtration coefficient appropriately, the vascular filling component must be excluded from the analyses, applied pressure increments should be small, alveolar edema should be avoided, and lymph flow should be excluded.
In terms of smaller animal studies (rats, rabbits, and mice), a precise way to quantify an increase in lung vascular and epithelial permeability is to use a protein labeled with a radioactive isotope such as 125I-albumin or 131I-albumin, fluorescent-labeled albumin, or a fluorescent-labeled high molecular weight dextran, and then to demonstrate that there is accumulation of this tracer in the extravascular compartments of the lung by either (1) homogenizing the intact lung and subtracting the blood volume or by (2) measuring the accumulation of this labeled protein in the distal airspaces of the lung using bronchoalveolar lavage (BAL). Experimental interventions that reduce permeability should result in a reduced quantity of labeled protein accumulation in the airspaces and/or the extravascular compartments of the lung. It is important to verify that the label has remained bound to the protein or dextran molecule. The possibility of dissociation of the label is particularly important when using albumin bound to radioactive iodine. This can be determined by actively dissociating the isotope from the albumin using 20% trichloroacetic acid and then comparing the total free radioactivity with the radioactivity of the untreated sample (26). Also, the permeability of the alveolar epithelium can be measured by instilling a labeled protein or dextran into the air spaces and measuring its accumulation in the plasma and its loss from the lung. One emerging problem with albumin-based markers is transport by transcytosis, which may lead to increased tissue albumin in the absence of increased permeability (27).
A popular method of measuring changes in capillary permeability involves the use of Evans Blue dye, a fluorescent azo dye that was originally described in the 1930s as a tool to determine blood volume using fluorometry (28). Evans Blue dye was subsequently used to determine changes in capillary permeability in tissues (29). The method requires the intravenous injection of Evans Blue dye, followed by determination of the fluorescence of formamide-treated lung extracts at excitation = 620 nm, emission = 680 nm (30, 31). The determination of the Evans blue dye concentration (EBD) in a sample should include a correction for contaminating heme pigments using the formula E620(EBD) = E620 − (1.426 × E740 + 0.030) (30). Evans blue dye is commonly used because it allows for the determination of permeability changes without the use of radioactive isotopes. This is similar to fluorescently labeled dextrans, although the later have the advantage that multiple sizes of dextran can be selected and the amount of processing required is minimal.
A crude method to determine increased lung water is to calculate wet-to-dry ratios. The wet lung is weighed, and then placed in an oven and weighed daily until its weight stabilizes (dry weight). The wet-to-dry ratio then becomes an estimate of the total water content of the lung. One problem with this method is that with very small lungs, small errors in weight measurement may result in a large variability in the ratios, so particular care should be exercised when measuring the lung weight.
Another approach is to measure the total protein concentration in the BAL, which should reflect the accumulation of extravascular protein primarily from an increase in permeability. In this case, the total quantity of protein is determined by multiplying the concentration of the protein in the BAL fluid by the volume of the lavage. Hydrostatic stress will displace some intravascular protein into the interstitial and alveolar compartments, so if needed, controls can be done with elevated left atrial pressure or hydrostatic stress to show that the quantity of protein under these control hydrostatic conditions is less than under conditions of increased permeability. Furthermore, the BAL total protein concentration can be influenced by the protein derived from inflammatory and lung epithelial cells that are present in the alveolar space, or by changes in the ability of the alveolar epithelium to clear water from the airspaces (28, 29, 31). For instance, the alveolar protein concentration can increase dramatically in nonperfused lungs (30). It is possible to test for the presence of a very high molecular weight molecule such as IgM (900kD) as a marker of increased lung permeability. As BAL measurements are notoriously difficult to standardize, we recommend that the recovery of the BAL fluid be reported.
The collection of pulmonary edema fluid from animals can also serve as a useful measurement (32). If edema fluid is collected in the early phase (first 30 min) after the development of experimental or clinical pulmonary edema, then the concentration of total protein in the edema fluid can be compared with time-matched plasma samples. If the ratio is greater than 0.65, then it is likely to reflect an increase in permeability. If the edema fluid sample is collected more than 15–30 minutes after the onset of alveolar edema, then reabsorption of alveolar fluid may occur, resulting in concentration of the alveolar protein and providing a misleading result regarding the mechanism of the pulmonary edema. For example, if hydrostatic pressure is raised sufficiently to cause hydrostatic edema with alveolar flooding, then the initial concentration of alveolar protein will be less than 65% of plasma protein. However, if the elevated hydrostatic pressure is returned to normal, then reabsorption of salt in the water will occur rapidly, driven by active ion transport across the lung epithelium and resulting in a elevated concentration of alveolar protein, which, when compared with the concentration of that protein in plasma, will suggest that there is an underlying increase in permeability when in fact this is not the case.
The measurement of extravascular lung water is valuable in studies of mechanisms of increased permeability pulmonary edema. However, it should be recognized that an increase in extravascular lung water alone does not categorize the lung edema as hydrostatic or increased permeability edema. Also, investigators should remember than an increase in lung vascular pressure can markedly increase the extent of pulmonary edema in the face of an increase in lung vascular permeability so that two mechanisms for edema formation may occur at the same time.
3. Measuring Inflammation
ALI is most frequently accompanied by an inflammatory response within the lungs. Among possible measurements of inflammation, three were identified by the panel as “very relevant.” These included total neutrophil counts in the BAL fluid, lung myeloperoxidase (MPO) (a surrogate for neutrophil influx), and measurements of proinflammatory cytokines. Each of these parameters is easily measured in most research laboratories by the following methodologies.
Neutrophils in the BAL fluid.
The quantification of neutrophils in BAL fluid reflects migration of neutrophils into the airspaces of the lungs. This measurement is most readily accomplished by performing a differential analysis of cells collected from the BAL. The addition of ethylenediamine tetraacetic acid (EDTA) to the fluid used to perform the BAL (e.g., 0.9% NaCl or PBS) will prevent clumping of the cells and facilitate counting. After collection, the samples should be kept on ice and analyzed as soon as possible to prevent cell death. Animals can be killed at relevant time points (e.g., 1, 3, 5, and 7 d postinsult), the trachea can be cannulated and saline (generally a volume of 1 ml for mice) is instilled into lungs and then recovered. This procedure can be repeated several times. The total cell pellet is then collected by centrifugation and cells are sedimented by centrifugation (cytospun) onto glass slides and stained with Wright-Giemsa or hematoxylin and eosin. Neutrophils are identified using light microscopy by their characteristic nuclei (Figure 3). A minimum of 300 total cells should be counted to obtain a reliable neutrophil percentage in the BAL. This percentage can be multiplied by the total cell numbers recovered to arrive at an absolute count in the BAL fluid. It is always preferable to report the total number of neutrophils in the BAL fluid, along with, or instead, of the percentages. This is because a decrease in the percentage of BAL neutrophils in response to a given treatment may reflect an increase in the number of macrophages, without any real change in the number of total neutrophils. Alternatively, neutrophils can be readily identified in the BAL fluid by flow cytometry using specific antibodies against Ly6G (33, 34). Neutrophils, being granular, have a characteristic side versus forward scatter profile, which is evident using this technique.
Figure 3.

Neutrophils are readily identified by their nuclear morphology. A composite of cells types found in the alveolar space and identified by differential staining with Wright-Geimsa. Neutrophils (black arrowheads) are identified by their segmented nuclear morphology. Alveolar macrophages (white arrowheads) are identified by their large size and high cytoplasm to nuclear ratio. The cytoplasm of alveolar macrophages can also be quite granular. Lymphocytes (yellow arrowheads) are identified by their prominent nuclear staining with little cytoplasm. Magnification ×1,000 under oil.
Myeloperoxidase (MPO) activity.
MPO is a glycoprotein expressed in all cells of the myeloid lineage, but found most abundantly in azurophilic granules of neutrophils (35). MPO is released by activated neutrophils and, as such, measurement of MPO in cell-free BAL fluid serves as a surrogate for the accumulation of activated neutrophils in the airspaces of the lung. Alternatively, measurement of MPO in whole lung homogenates reflects accumulation of neutrophils in the lungs, and as such may be a useful complement to the measurement of neutrophils in the BAL, which reflects migration of neutrophils into the airspaces. MPO can be measured effectively either by commercial ELISA kits according to manufacturer's directions or by a colorimetric assay which correlates with MPO activity as described previously (33, 36, 37).
When MPO activity is measured in whole lung supernatants, it is important to add a buffer that is designed to preserve the activity of the enzyme (e.g., see Reference [38]). The determination of MPO activity may be associated with greater variability than the ELISA methods.
Concentrations of proinflammatory mediators.
ALI is almost invariably associated with elevations in proinflammatory mediators including tumor necrosis factor (TNF), IL-1, IL-6, macrophage inhibitory factor (MIF), transforming growth factor (TGF), platelet activating factor (PAF) and the leukotriene LTB4 (reviewed in [39–43]), although anti-inflammatory cytokines such as IL-10 are usually also elevated (44). In humans, IL-8 is a prominent neutrophil-recruiting chemokine. The murine orthologs of this molecule are KC (CXCL1) and MIP-2 (CXCL2) (39, 45). These mediators are readily measured by commercial ELISAs or multiplex assays using either BAL fluid or lung homogenates. Measurement of LTB4 in biological fluids is best accomplished by first extracting the lipids using C18 cartridges from the biological sample, drying them under nitrogen, and then performing the ELISA on the reconstituted sample (46). Mediators can also be evaluated using real-time PCR assays to look for mRNA expression, or in the case of LTB4, for the up-regulation of the 5-lipoxygenase (5-LO), 5-LO activating protein, or LTA4 hydrolase enzymes involved in the lipid synthesis.
Particular care should be exercised to prevent degradation of cytokines. This can be accomplished by adding commercially available protease inhibitors to the samples. In addition, some cytokines such as TNF are extremely sensitive to freeze and thaw cycles; accordingly, we recommend storing the samples in individual aliquots and performing measurements after a single thaw. When using bead-based multiplex assays on tissue homogenates, the addition of DNase to the samples will decrease viscosity and prevent clogging of the machine.
Cytokine mRNA expression using quantitative PCR or expression arrays can be used to identify patterns of cytokine gene expression. This is a powerful technique, but requires particular attention to three key issues. The first issue is that RNA is easily degraded, and it is essential to ensure that sample collection strictly adheres to protocols designed for RNA preservation; this is particularly important when the samples are intended to be used for microarray analysis (47, 48). The second issue is the selection of the reference gene. There are increasing concerns that commonly used reference genes such as GAPDH and actin have variable expression under certain conditions, and while other genes such as HPRT and 18S RNA appear to display more stable expression, there is no consensus as to the preferred gene to use (49–51) Finally, mRNA expression does not necessarily correlate with protein expression, and key findings should be verified by measuring the actual protein.
Additional criteria that were considered somewhat relevant for the measurement of ALI included measurements of procoagulant activity (52), expression of adhesion molecules (53), conversion of the neutrophilic alveolitis to a monocytic alveolitis with time, and elevations in the levels of specific complement factors (54) and matrix metalloproteinases (55).
4. Physiological Dysfunction
Measurements of gas exchange.
The main purpose of the lung is to absorb oxygen and remove carbon dioxide. As such, it is noteworthy that hypoxemia and an increased alveolar–arterial oxygen difference [(Aa)DO2] were listed as the most relevant measures of physiological dysfunction in models of lung injury. Despite this, it is important to point out that hypoxemia is not a direct measure of injury per se, but is often a manifestation of injury. Hypoxemia can occur due to many physiological mechanisms not associated with ALI (see below). Thus, measurement of oxygenation should not be the only variable that is assessed in determining if an animal has lung injury, but measuring oxygenation can be very useful in helping to assess the degree and time course of injury.
Hypoxemia and alveolar–arterial oxygen difference [(a-a)DO2]: Hypoxemia can be assessed by measurement of the arterial partial pressure of oxygen (PaO2) from arterial blood gases, or from oximetry; typically, hypoxemia is defined as PaO2 < ∼60 mm Hg, or SpO2 < 90%. Measurement of arterial blood gases are usually more accurate and have the added advantage of providing a value for PaO2, which is important in assessing the extent to which any decrease in PaO2 is due to hypoventilation (increased PaCO2). This can be calculated using the (simplified) alveolar gas equation [PAO2 = (PB-PH2O)·FiO2 – PaCO2/R], where PaO2 is the alveolar partial pressure of oxygen, PB is the atmospheric pressure, PH2O is the saturated vapor pressure of water, FiO2 is the inspired fraction of oxygen, PaCO2 is the arterial partial pressure of carbon dioxide, and R is the respiratory quotient. The alveolar–arterial oxygen difference [AaDO2 = (PAO2 – PaO2)] can be used to correct for differences in PaCO2, but can vary substantially with FiO2 depending on the degree of ventilation/perfusion (V/Q) mismatch versus shunt. As such, if one wants to compare oxygenation at different time points and/or among animals, the same FiO2 (often chosen at an FiO2 of 0.5 or 1.0) should be used.
As can easily be deduced by the alveolar air equation, oxygenation also varies with barometric pressure, and respiratory quotient. Taking into account the barometric pressure is important if one is doing experiments at a high altitude or comparing results in laboratories at different altitudes.
Arterial oxygenation can vary substantially with a number of physiological variables, many of which are controlled by the investigator. In animals with ALI, oxygenation may vary with changes in mean airway pressure, which is dependent upon the inspired fraction of oxygen (FiO2), the level of positive end-expiratory pressure (PEEP), tidal volume, and the inspiratory to expiratory ratio. Oxygenation can also vary with changes in the stiffness of the chest wall or in body position (e.g., prone vs. supine). As such, depending on the purpose of the measurement, estimates of oxygenation should be made by keeping as many of these variables as constant as possible.
In animals that are being mechanically ventilated, oxygenation can also depend on lung volume history, (i.e., the size of the breaths preceding the oxygen measurement), especially in models of injury characterized by substantial atelectasis. One approach that is used to mitigate this effect is to provide a relatively uniform volume history by performing a recruitment maneuver immediately before measurement of the oxygenation variable(s).
If there is significant pulmonary shunt, arterial oxygenation can decrease with decreases in cardiac output because of a decrease in mixed venous oxygenation. This is important if one is using arterial oxygenation as a measure of the degree of lung injury, because a decreasing PaO2 may not necessarily signify a worsening of lung injury but may signify a worsening in hemodynamics.
The PaO2/FiO2 ratio (P/F) is used to define (along with other characteristics) human acute respiratory distress syndrome (P/F < 200 mm Hg) and ALI (P/F < 300 mm Hg), based on the American European Consensus Committee. There are no agreed-upon diagnostic criteria of different lung injury entities (ALI vs. ARDS) in animal models of lung injury, and indeed this workshop proposed that the PaO2/FiO2 ratio should only be considered “somewhat relevant” for assessing physiological function in animal models of ALI. This may be because the Committee members felt that it is easier to make measurements under more identical controlled FiO2 in animal models, in which case the alveolar–arterial oxygen difference would suffice.
Measurement of arterial blood gases are reasonably easy to measure in relatively large animals but may be quite difficult to measure in very small animals (e.g., mice), especially repeatedly during the course of an experiment. As such, the workshop participants felt that minute ventilation and respiratory frequency might be variables that are somewhat relevant in assessing lung injury. These variables diverge substantially among species of different sizes, which may not be an issue in comparisons to studies on the same species. Most importantly, they are affected by a number of factors that may bear no relationship to ALI, including the degree of sedation, pain, body temperature, acid–base balance, and so forth.
Some general considerations.
Animal studies are fraught with high variability in the results, and this is true even under strictly controlled conditions and when all the animals are genetically identical. One common source of variability is animal stress, and this is particularly important in mice, which may be affected by even minor changes in light/dark cycles or by minor handling. Care should be taken to ensure that sources of stress, including minor handling, are reduced to a minimum, and that control and experimental animals are all handled in exactly the same way. In addition, the performance of animal studies is almost always associated with a learning curve, and even very experienced technicians will be better at specific techniques toward the end of a study. It is extremely important to avoid working with all the experimental animals first and all the controls at the end of a study; instead, controls and experimental animals should be alternated throughout the study and if at all possible, the investigators should be blinded to the experimental conditions. Study protocols should ensure humane treatment of all animals, including the use of protocolized monitoring to identify evidence of animal discomfort, the use of clear criteria to minimize discomfort by providing sedatives or anesthesia, and the use of prospective criteria to identify animals that should be subject to early euthanasia. All animal studies should only be performed after approval by the investigator's Institutional Animal Care and Use Committee (IACUC) or equivalent regulatory body.
VI. CRITICAL ASSESSMENT OF SELECTED COMMON MODELS OF LUNG INJURY
We evaluated the presence of criteria considered as “very relevant” in selected common models of lung injury based on representative studies from the literature. A literature search over the last five years ranked mechanical ventilation with high tidal volumes or high peak inspiratory pressures, pulmonary or systemic administration of endotoxin, and inhalation or instillation of live bacteria as the three most commonly applied models of lung injury in animals. Evidence of the main features of ALI by the defined measurements was analyzed for these three models, as summarized in Table 2. A more extensive review of animal models of lung injury is beyond the scope of this manuscript, but can be found elsewhere (7).
TABLE 2.
PRESENCE OF “VERY RELEVANT” CRITERIA IN STANDARD MODELS OF ACUTE LUNG INJURY
|
|
VILI |
LPS |
Bacteria |
O2 |
OA |
Acid |
I/R |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Histological evidence of tissue injury | ||||||||||||||
| Accumulation of neutrophils in the alveolar/interstitial space | + | + | + | +(1) | +(1) | +(1) | +(2) | |||||||
| Formation of hyaline membranes | + | + | + | +(3) | +(4) | +(5) | +(2) | |||||||
| Proteinaceous debris in alveolar space | + | + | + | +(6) | +(1) | +(1) | +(7) | |||||||
| Thickening of the alveolar wall | + | + | + | +(3) | +(8) | + | +(9) | |||||||
| Injury by a standardised histology score | + | + | + | +(3) | +(10) | +(11) | +(2) | |||||||
| Alteration of the alveolar capillary barrier | ||||||||||||||
| Increased extravascular lung water content | + | + | + | +(1) | +(1) | +(12) | +(13) | |||||||
| Accumulation of protein/tracer in airspaces/extravascular space | + | + | + | +(1) | +(10) | +(14) | +(13) | |||||||
| Total BAL protein concentration | + | + | + | +(15) | +(16) | +(12) | +(2) | |||||||
| BAL concentration of high molecular weight proteins | + | + | + | +(17) | +(8) | +(5) | +(18) | |||||||
| (Micro-) Vascular filtration coefficient (Kf) | + | (+) | (+) | (+)(19,20)* | +(21) | +(12) | +(2) | |||||||
| Inflammation | ||||||||||||||
| BAL total neutrophil counts | + | + | + | +(1) | +(16) | +(12) | +(22) | |||||||
| Lung MPO activity | + | + | + | +(1) | +(16) | +(16) | +(13) | |||||||
| Concentrations of cytokines | (+) | + | + | +(1) | +(16) | +(5) | +(13) | |||||||
| Physiological dysfunction | ||||||||||||||
| Hypoxemia | + | + | + | −(23)† | +(1) | +(16) | +(9) | |||||||
| Increased a-a oxygen difference |
+ |
+ |
+ |
+(1) |
+(16) |
+(9) |
||||||||
Definition of abbreviations: a–a, alveolar-arterial; BAL, bronchoalveolar lavage; I/R, ischemia-reperfusion; MPO, myeloperoxidase; OA, oleic acid; VILI, ventilator-induced lung injury.
+ or (+), the criterion is present in virtually all or the majority of studies using this model.
−, criterion is absent.
Ventilator-Induced Lung Injury
Ventilator-induced lung injury (VILI) models typically present characteristic features of tissue injury such as interstitial thickening and alveolar infiltration of neutrophils (56). Hyaline membranes are seen in larger animals, but are rare in rats or mice unless the tidal volumes are very elevated (Figure 1F: rat lungs ventilated for 60 minutes with tidal volumes = 35 cc/kg and PEEP = 0). Tissue injury is also documented by blinded assessments of histopathological scores based on edema severity, hemorrhage, septal thickening, and interstitial or alveolar infiltration of inflammatory cells in different species ventilated with high tidal volumes of 20–30 ml/kg body weight for several hours (57, 58). VILI also causes prominent changes in water and protein permeability as evidenced in isolated rat and mouse lungs by increased wet-to-dry weight ratios and microvascular filtration coefficients (59, 60). In vivo, ventilation with tidal volumes of 20 to 30 ml/kg body weight results in an accumulation of Evans blue–labeled albumin in the lung or the extravasation of intravascular radiolabeled plasma proteins such as albumin or transferrin into the extravascular space (57, 61, 62). Increased concentrations of albumin and total protein are detectable in the BAL (63, 64). Recruitment of inflammatory cells as assessed by lung MPO assay or BAL neutrophil counts is generally evident (58, 64). Increased formation and release of cytokines from overventilated lungs has been reported in mouse and rat studies using isolated lung preparations, but may not be present in all models of VILI (30, 65, 66). Last, but not least, over-ventilation causes a steady decline in arterial oxygen saturation which typically results in severe hypoxemia within several hours (56, 67).
Endotoxin-Induced Lung Injury
Endotoxin challenge by inhalation, intratracheal instillation, or intravenous infusion of various lipopolysaccharides typically results in considerable tissue injury, which can be evident within less than 1 hour and is characterized by neutrophil accumulation in the alveolar and interstitial space, alveolar wall thickening, and accumulation of proteinaceous edema and detritus in the alveolar space (34, 68–72). Endotoxin-dependent tissue injury has also been confirmed by semi-quantitative histological scores based on signs of inflammation and hemorrhage in the alveolar and interstitial space, edema, atelectasis, hyaline membranes, and cellular necrosis (70, 72, 73). Increased wet-to-dry weight ratios, Evans blue accumulation in the lung, and increased albumin and total protein concentrations in the BAL fluid after endotoxin challenge demonstrate acute alterations in water and protein permeability (70, 73–77). Of note, changes in lung vascular filtration coefficient (Kf) are present in some but not all models of endotoxin-induced lung injury. Increased Kf values were detected after endotoxin challenge in vivo (78) and also after infusion of LPS in buffer-perfused isolated rat and piglet lungs (79, 80), but other studies found increased Kf only in serum-perfused but not in plasma-perfused isolated rat lungs (81), whereas no changes were detected in goat lungs perfused with autologous blood (82). In blood-perfused rat lungs, endotoxin only increased microvascular permeability in the presence of a nitric oxide synthase inhibitor (83). Increased neutrophil counts in the BAL, high MPO activity, and lung cytokine levels are hallmarks of the inflammatory response in virtually all models of endotoxin-induced lung injury (72, 73, 76, 77, 79, 84), whereas hypoxemia is a less consistent finding (69, 70, 84, 85).
Lung Injury by Live Bacteria
Microbial infection of the lungs by inhalation of aerosolized bacteria or direct intranasal, endotracheal, or endobronchial instillation is used extensively as an experimental model of pneumonia in animals (86). Tissue injury in this model is striking with accumulation of neutrophils in the alveolar and interstitial space but also includes alveolar wall thickening and hyaline membrane formation (86–94); this can be assessed semi-quantitatively by histological scores. Permeability is commonly increased as demonstrated by elevated wet-to-dry weight ratios, leakage of radioactive protein tracers from the vascular space or accumulation of Evans blue dye, increased protein levels in the BAL, and elevated vascular filtration coefficients (88, 90, 93, 95–100). Yet, in individual cases, for example, in a mouse model of intratracheal challenge with E. coli, changes in the filtration coefficient and lung weight gain have been reported to be minimal over a period of up to 12 hours (92). Inflammation markers, that is, increased neutrophil counts in the lavage, MPO activity and cytokine levels, as well as systemic hypoxemia, are in general uniformly present in lung injury that is induced by live bacteria (88, 92, 97, 99–101).
Taken together, the most frequently applied standard models demonstrate the main features of ALI described in this article. Yet, not all criteria are consistently met by all models. It is therefore important to note that failure to comply with a single criterion does not necessarily compromise the validity of an established model.
VII. LIMITATIONS
This document represents a consensus reached among a diverse group of investigators actively involved in the study of ALI. However, it does not reflect unanimity, and there were clearly areas of diverging opinions indicating the complexities involved in many of these measurements. As such, this document is not meant to be prescriptive or to suggest that there is only one way of assessing ALI, insofar as different model systems and local availability of instrumentation may be the overriding factors that dictate the variables to be measured. As discussed above, it is prudent to measure several independent variables that reflect various aspects of ALI rather than relying on a single measurement. Finally, a number of the recommendations in this report were made despite a paucity of data. As such, we used an iterative process to reach consensus and leave open the possibility that future studies may refute these recommendations.
VIII. SUMMARY AND CONCLUSIONS
In summary, we present an initial attempt at defining lung injury in animals. We have identified four major features of experimental ALI as well as measurements for each of those features. We hope that this work will be of help to investigators and will also become a first step toward a definition of ALI in animals.
This official summary of an ATS Workshop Proceedings was prepared by an ad hoc subcommittee of the Assembly on Microbiology, Tuberculosis, and Pulmonary Infections.
Writing Committee:
Gustavo Matute-Bello, M.D. (Chair)
Gregory P. Downey, M.D., F.R.C.P(C). (Chair)
Bethany B. Moore, Ph.D.
Steve D. Groshong, M.D., Ph.D
Michael A. Matthay, M.D.
Arthur S. Slutsky, M.D.
Wolfgang M. Kuebler, M.D.
The members of the subcommittee were:
Gustavo Matute-Bello, M.D. (Chair)
Gregory P. Downey, M.D., F.R.C.P(C). (Chair)
Edwin R. Chilvers, Ph.D., FMedSci.
Claire Doerschuck, M.D.
Didier Dreyfuss, M.D.
Peter Q. Eichacker M.D.
Jack A. Elias, M.D.
Andres Esteban, M.D.
Niall D. Ferguson, M.D., F.R.C.P.C.
Robb W. Glenny, M.D.
Steve D. Groshong, M.D., Ph.D.
Peter Henson, Ph.D., M.D.
Wolfgang M. Kuebler, M.D.
José A. Lorente, M.D.
Thomas R. Martin, M.D.
Michael A. Matthay, M.D.
Bethany B. Moore, Ph.D.
Jerry A. Nick, M.D.
Peter Rimensberger, M.D.
Arthur S. Slutsky, M.D.
Stefan Uhlig, M.D.
Tom van der Poll, M.D.
Jesus Villar, M.D., Ph.D.
Acknowledgments
The authors thank Pam Henderson for her administrative assistance and Mark Looney and Guy Zimmerman for reviewing a draft of the manuscript.
Author Disclosure: G.M-B. reported research support from the American Heart Association ($50,001–$100,000). G.P.D. reported no commercial interests or non-governmental, non-commercial interests relevant to subject matter. B.B.M. consulted with Centocor (up to $1,000) and received research support from the American Lung Association ($100,000 or more), the Pulmonary Fibrosis Foundation ($50,001–$100,000), and Centocor ($100,001 or more). S.D.G. and M.A.M. each reported no commercial interests or non-governmental, non-commercial interests relevant to subject matter. A.S.S. served on advisory committees of Apeiron ($5,001–$10,000) and Maquet Medical ($10,001–$50,000) and received royalties from Maquet Medical ($10,001–$50,000). W.M.K. served on advisory committees of Revotar ($1,001–$5,000) and Hoffman-LaRoche ($1,001–$5,000), received lecture fees from Nycomed ($1,001–$5,000), and received research support from GlaxoSmithKline ($10,001–$50,000), Hoffman-Laroche ($50,001–$100,000) and Pfizer ($50,001–$100,000). Note: the above reflects disclosure by workshop report authors during period of document development. Governmental support is not reported consistent with changes in American Thoracic Society disclosure requirements effective September 2010. Other workshop contributors also disclosed to ATS, in compliance with ATS policies; summaries are available upon request.
Contributor Information
Collaborators: on behalf of the Acute Lung Injury in Animals Study Group
References
- 1.Bernard GA, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, LeGall JR, Morris A, Spragg R. Report of the American-European consensus conference on acute respiratory distress syndrome: Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Consensus Committee. J Crit Care 1994;9:72–81. [DOI] [PubMed] [Google Scholar]
- 2.Katzenstein AL, Bloor CM, Leibow AA. Diffuse alveolar damage–the role of oxygen, shock, and related factors. A review. Am J Pathol 1976;85:209–228. [PMC free article] [PubMed] [Google Scholar]
- 3.Irvin CG, Bates JH. Measuring the lung function in the mouse: the challenge of size. Respir Res 2003;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res 1993;10:1093–1095. [DOI] [PubMed] [Google Scholar]
- 5.Hamelmann E, Schwarze J, Takeda K, Oshiba A, Larsen GL, Irvin CG, Gelfand EW. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med 1997;156:766–775. [DOI] [PubMed] [Google Scholar]
- 6.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol 2004;172:2731–2738. [DOI] [PubMed] [Google Scholar]
- 7.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 2008;295:L379–L399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swartz HM, Clarkson RB. The measurement of oxygen in vivo using EPR techniques. Phys Med Biol 1998;43:1957–1975. [DOI] [PubMed] [Google Scholar]
- 9.Bambot SB, Rao G, Romauld M, Carter GM, Sipior J, Terpetchnig E, Lakowicz JR. Sensing oxygen through skin using a red diode laser and fluorescence lifetimes. Biosens Bioelectron 1995;10:643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin EL, Sheikh TA, Leco KJ, Lewis JF, Veldhuizen RA. Contribution of alveolar macrophages to the response of the timp-3 null lung during a septic insult. Am J Physiol Lung Cell Mol Physiol 2007;293:L779–L789. [DOI] [PubMed] [Google Scholar]
- 11.Bachofen M, Weibel ER. Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med 1982;3:35–56. [PubMed] [Google Scholar]
- 12.Bachofen A, Weibel ER. Alterations of the gas exchange apparatus in adult respiratory insufficiency associated with septicemia. Am Rev Respir Dis 1977;116:589–615. [DOI] [PubMed] [Google Scholar]
- 13.Villar J, Edelson JD, Post M, Mullen JB, Slutsky AS. Induction of heat stress proteins is associated with decreased mortality in an animal model of acute lung injury. Am Rev Respir Dis 1993;147:177–181. [DOI] [PubMed] [Google Scholar]
- 14.Herrera MT, Toledo C, Valladares F, Muros M, Diaz-Flores L, Flores C, Villar J. Positive end-expiratory pressure modulates local and systemic inflammatory responses in a sepsis-induced lung injury model. Intensive Care Med 2003;29:1345–1353. [DOI] [PubMed] [Google Scholar]
- 15.Sterner-Kock A, Vesely KR, Stovall MY, Schelegle ES, Green JF, Hyde DM. Neonatal capsaicin treatment increases the severity of ozone-induced lung injury. Am J Respir Crit Care Med 1996;153:436–443. [DOI] [PubMed] [Google Scholar]
- 16.Matute-Bello G, Winn RK, Jonas M, Chi EY, Martin TR, Liles WC. Fas (CD95) induces alveolar epithelial cell apoptosis in vivo: implications for acute pulmonary inflammation. Am J Pathol 2001;158:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Looney MR, Su X, Van Ziffle JA, Lowell CA, Matthay MA. Neutrophils and their fc gamma receptors are essential in a mouse model of transfusion-related acute lung injury. J Clin Invest 2006;116:1615–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin TR, Pistorese BP, Chi EY, Goodman RB, Matthay MA. Effects of leukotriene b4 in the human lung. Recruitment of neutrophils into the alveolar spaces without a change in protein permeability. J Clin Invest 1989;84:1609–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiener-Kronish JP, Albertine KH, Matthay MA. Differential responses of the endothelial and epithelial barriers of the lung in sheep to escherichia coli endotoxin. J Clin Invest 1991;88:864–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berthiaume Y, Staub NC, Matthay MA. Beta-adrenergic agonists increase lung liquid clearance in anesthetized sheep. J Clin Invest 1987;79:335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erdmann AJ III, Vaughan TR Jr, Brigham KL, Woolverton WC, Staub NC. Effect of increased vascular pressure on lung fluid balance in unanesthetized sheep. Circ Res 1975;37:271–284. [DOI] [PubMed] [Google Scholar]
- 22.Vreim CR, Snashall PD, Demling RH, Staub NC. Lung lymph and free interstitial fluid protein composition in sheep with edema. Am J Physiol 1976;230:1650–1653. [DOI] [PubMed] [Google Scholar]
- 23.Vreim CE, Snashall PD, Staub NC. Protein composition of lung fluids in anesthetized dogs with acute cardiogenic edema. Am J Physiol 1976;231:1466–1469. [DOI] [PubMed] [Google Scholar]
- 24.Brigham KL, Bowers R, Haynes J. Increased sheep lung vascular permeability caused by escherichia coli endotoxin. Circ Res 1979;45:292–297. [DOI] [PubMed] [Google Scholar]
- 25.Uhlig S, von Bethmann AN. Determination of vascular compliance, interstitial compliance, and capillary filtration coefficient in rat isolated perfused lungs. J Pharmacol Toxicol Methods 1997;37:119–127. [DOI] [PubMed] [Google Scholar]
- 26.Modelska K, Pittet JF, Folkesson HG, Courtney Broaddus V, Matthay MA. Acid-induced lung injury. Protective effect of anti-interleukin-8 pretreatment on alveolar epithelial barrier function in rabbits. Am J Respir Crit Care Med 1999;160:1450–1456. [DOI] [PubMed] [Google Scholar]
- 27.Predescu D, Vogel SM, Malik AB. Functional and morphological studies of protein transcytosis in continuous endothelia. Am J Physiol Lung Cell Mol Physiol 2004;287:L895–L901. [DOI] [PubMed] [Google Scholar]
- 28.Sartori C, Matthay MA. Alveolar epithelial fluid transport in acute lung injury: new insights. Eur Respir J 2002;20:1299–1313. [DOI] [PubMed] [Google Scholar]
- 29.Matthay MA, Flori HR, Conner ER, Ware LB. Alveolar epithelial fluid transport: basic mechanisms and clinical relevance. Proc Assoc Am Physicians 1998;110:496–505. [PubMed] [Google Scholar]
- 30.Tremblay L, Valenza F, Ribeiro SP, Li J, Slutsky AS. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest 1997;99:944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakuma T, Pittet JF, Jayr C, Matthay MA. Alveolar liquid and protein clearance in the absence of blood flow or ventilation in sheep. J Appl Physiol 1993;74:176–185. [DOI] [PubMed] [Google Scholar]
- 32.Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev 2002;82:569–600. [DOI] [PubMed] [Google Scholar]
- 33.Hirsh M, Carmel J, Kaplan V, Livne E, Krausz MM. Activity of lung neutrophils and matrix metalloproteinases in cyclophosphamide-treated mice with experimental sepsis. Int J Exp Pathol 2004;85:147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reutershan J, Morris MA, Burcin TL, Smith DF, Chang D, Saprito MS, Ley K. Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J Clin Invest 2006;116:695–702. Epub 2006 Feb 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nauseef WM, Olsson I, Arnljots K. Biosynthesis and processing of myeloperoxidase—a marker for myeloid cell differentiation. Eur J Haematol 1988;40:97–110. [DOI] [PubMed] [Google Scholar]
- 36.Neff TA, Guo R-F, Neff SB, Sarma JV, Speyer CL, Gao H, Bernacki KD, Huber-Lang M, McGuire S, Hoesel LM, et al. Relationship of acute lung inflammatory injury to Fas/FasL system. Am J Pathol 2005;166:685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lomas JL, Chung CS, Grutkoski PS, LeBlanc BW, Lavigne L, Reichner J, Gregory SH, Doughty LA, Cioffi WG, Ayala A. Differential effects of macrophage inflammatory chemokine-2 and keratinocyte-derived chemokine on hemorrhage-induced neutrophil priming for lung inflammation: assessment by adoptive cells transfer in mice. Shock 2003;19:358–365. [DOI] [PubMed] [Google Scholar]
- 38.Greenberger MJ, Strieter RM, Kunkel SL, Danforth JM, Goodman RE, Standiford TJ. Neutralization of IL-10 increases survival in a murine model of Klebsiella pneumonia. J Immunol 1995;155:722–729. [PubMed] [Google Scholar]
- 39.Togbe D, Schnyder-Candrian S, Schnyder B, Doz E, Noulin N, Janot L, Secher T, Gasse P, Lima C, Coelho FR, et al. Toll-like receptor and tumour necrosis factor dependent endotoxin-induced acute lung injury. Int J Exp Pathol 2007;88:387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mukhopadhyay S, Hoidal JR, Mukherjee TK. Role of TNFalpha in pulmonary pathophysiology. Respir Res 2006;7:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ward PA. Role of complement, chemokines, and regulatory cytokines in acute lung injury. Ann N Y Acad Sci 1996;796:104–112. [DOI] [PubMed] [Google Scholar]
- 42.Baugh JA, Bucala R. Macrophage migration inhibitory factor. Crit Care Med 2002;30:S27–S35. [PubMed] [Google Scholar]
- 43.Bulger EM, Maier RV. Lipid mediators in the pathophysiology of critical illness. Crit Care Med 2000;28:N27-36. [DOI] [PubMed] [Google Scholar]
- 44.Park WY, Goodman RB, Steinberg KP, Ruzinski JT, Radella F II, Park DR, Pugin J, Skerrett SJ, Hudson LD, Martin TR. Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 2001;164:1896–1903. [DOI] [PubMed] [Google Scholar]
- 45.Kobayashi Y. The role of chemokines in neutrophil biology. Front Biosci 2008;13:2400–2407. [DOI] [PubMed] [Google Scholar]
- 46.Wilborn J, Bailie M, Coffey M, Burdick M, Strieter R, Peters-Golden M. Constitutive activation of 5-lipoxygenase in the lungs of patients with idiopathic pulmonary fibrosis. J Clin Invest 1996;97:1827–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feezor RJ, Baker HV, Mindrinos M, Hayden D, Tannahill CL, Brownstein BH, Fay A, MacMillan S, Laramie J, Xiao W, et al. Whole blood and leukocyte RNA isolation for gene expression analyses. Physiol Genomics 2004;19:247–254. [DOI] [PubMed] [Google Scholar]
- 48.Fleige S, Pfaffl MW. Rna integrity and the effect on the real-time qRT-PCR performance. Mol Aspects Med 2006;27:126–139. [DOI] [PubMed] [Google Scholar]
- 49.Glare EM, Divjak M, Bailey MJ, Walters EH. Beta-actin and GAPDH housekeeping gene expression in asthmatic airways is variable and not suitable for normalising mRNA levels. Thorax 2002;57:765–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bas A, Forsberg G, Hammarstrom S, Hammarstrom ML. Utility of the housekeeping genes 18s rRNA, beta-actin and glyceraldehyde-3-phosphate-dehydrogenase for normalization in real-time quantitative reverse transcriptase-polymerase chain reaction analysis of gene expression in human T lymphocytes. Scand J Immunol 2004;59:566–573. [DOI] [PubMed] [Google Scholar]
- 51.Schmittgen TD, Zakrajsek BA. Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J Biochem Biophys Methods 2000;46:69–81. [DOI] [PubMed] [Google Scholar]
- 52.Bastarache JA, Ware LB, Bernard GR. The role of the coagulation cascade in the continuum of sepsis and acute lung injury and acute respiratory distress syndrome. Semin Respir Crit Care Med 2006;27:365–376. [DOI] [PubMed] [Google Scholar]
- 53.Reutershan J, Ley K. Bench-to-bedside review: acute respiratory distress syndrome—how neutrophils migrate into the lung. Crit Care 2003;2004:453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guo RF, Ward PA. Role of c5a in inflammatory responses. Annu Rev Immunol 2005;23:821–852. [DOI] [PubMed] [Google Scholar]
- 55.Ohbayashi H. Matrix metalloproteinases in lung diseases. Curr Protein Pept Sci 2002;3:409–421. [DOI] [PubMed] [Google Scholar]
- 56.Tsuno K, Miura K, Takeya M, Kolobow T, Morioka T. Histopathologic pulmonary changes from mechanical ventilation at high peak airway pressures. Am Rev Respir Dis 1991;143:1115–1120. [DOI] [PubMed] [Google Scholar]
- 57.Frank JA, Pittet JF, Wray C, Matthay MA. Protection from experimental ventilator-induced acute lung injury by IL-1 receptor blockade. Thorax 2008;63:147–153. [DOI] [PubMed] [Google Scholar]
- 58.Sakashita A, Nishimura Y, Nishiuma T, Takenaka K, Kobayashi K, Kotani Y, Yokoyama M. Neutrophil elastase inhibitor (sivelestat) attenuates subsequent ventilator-induced lung injury in mice. Eur J Pharmacol 2007;571:62–71. [DOI] [PubMed] [Google Scholar]
- 59.Hamanaka K, Jian MY, Weber DS, Alvarez DF, Townsley MI, Al-Mehdi AB, King JA, Liedtke W, Parker JC. Trpv4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. Am J Physiol Lung Cell Mol Physiol 2007;293:L923–L932. [DOI] [PubMed] [Google Scholar]
- 60.Parker JC, Ivey CL, Tucker JA. Gadolinium prevents high airway pressure-induced permeability increases in isolated rat lungs. J Appl Physiol 1998;84:1113–1118. [DOI] [PubMed] [Google Scholar]
- 61.de Prost N, Roux D, Dreyfuss D, Ricard JD, Le Guludec D, Saumon G. Alveolar edema dispersion and alveolar protein permeability during high volume ventilation: effect of positive end-expiratory pressure. Intensive Care Med 2007;33:711–717. [DOI] [PubMed] [Google Scholar]
- 62.Peng X, Abdulnour RE, Sammani S, Ma SF, Han EJ, Hasan EJ, Tuder R, Garcia JG, Hassoun PM. Inducible nitric oxide synthase contributes to ventilator-induced lung injury. Am J Respir Crit Care Med 2005;172:470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eyal FG, Hamm CR, Parker JC. Reduction in alveolar macrophages attenuates acute ventilator induced lung injury in rats. Intensive Care Med 2007;33:1212–1218. [DOI] [PubMed] [Google Scholar]
- 64.Yoshikawa S, Miyahara T, Reynolds SD, Stripp BR, Anghelescu M, Eyal FG, Parker JC. Clara cell secretory protein and phospholipase A2 activity modulate acute ventilator-induced lung injury in mice. J Appl Physiol 2005;98:1264–1271. [DOI] [PubMed] [Google Scholar]
- 65.von Bethmann AN, Brasch F, Nusing R, Vogt K, Volk HD, Muller KM, Wendel A, Uhlig S. Hyperventilation induces release of cytokines from perfused mouse lung. Am J Respir Crit Care Med 1998;157:263–272. [DOI] [PubMed] [Google Scholar]
- 66.Ricard J-D, Dreyfuss D, Saumon G. Production of inflammatory cytokines in ventilator-induced lung injury: a reappraisal. Am J Respir Crit Care Med 2001;163:1176–1180. [DOI] [PubMed] [Google Scholar]
- 67.Dries DJ, Adams AB, Marini JJ. Time course of physiologic variables in response to ventilator-induced lung injury. Respir Care 2007;52:31–37. [PubMed] [Google Scholar]
- 68.Lutz C, Carney D, Finck C, Picone A, Gatto LA, Paskanik A, Langenback E, Nieman G. Aerosolized surfactant improves pulmonary function in endotoxin-induced lung injury. Am J Respir Crit Care Med 1998;158:840–845. [DOI] [PubMed] [Google Scholar]
- 69.Da J, Chen L, Hedenstierna G. Nitric oxide up-regulates the glucocorticoid receptor and blunts the inflammatory reaction in porcine endotoxin sepsis. Crit Care Med 2007;35:26–32. [DOI] [PubMed] [Google Scholar]
- 70.Kemming GI, Flondor M, Hanser A, Pallivathukal S, Holtmannspotter M, Kneisel FF, Reuter DA, Kisch-Wedel H, Zwissler B. Effects of perfluorohexan vapor on gas exchange, respiratory mechanics, and lung histology in pigs with lung injury after endotoxin infusion. Anesthesiology 2005;103:585–594. [DOI] [PubMed] [Google Scholar]
- 71.Meyrick B, Brigham KL. Acute effects of escherichia coli endotoxin on the pulmonary microcirculation of anesthetized sheep structure:Function relationships. Lab Invest 1983;48:458–470. [PubMed] [Google Scholar]
- 72.Rotta AT, Steinhorn DM. Partial liquid ventilation reduces pulmonary neutrophil accumulation in an experimental model of systemic endotoxemia and acute lung injury. Crit Care Med 1998;26:1707–1715. [DOI] [PubMed] [Google Scholar]
- 73.Itoh T, Obata H, Murakami S, Hamada K, Kangawa K, Kimura H, Nagaya N. Adrenomedullin ameliorates lipopolysaccharide-induced acute lung injury in rats. Am J Physiol Lung Cell Mol Physiol 2007;293:L446–L452. [DOI] [PubMed] [Google Scholar]
- 74.Goggel R, Winoto-Morbach S, Vielhaber G, Imai Y, Lindner K, Brade L, Brade H, Ehlers S, Slutsky AS, Schutze S, et al. Paf-mediated pulmonary edema: a new role for acid sphingomyelinase and ceramide. Nat Med 2004;10:155–160. [DOI] [PubMed] [Google Scholar]
- 75.Huang YQ, Sauthoff H, Herscovici P, Pipiya T, Cheng J, Heitner S, Szentirmai O, Carter B, Hay JG. Angiopoietin-1 increases survival and reduces the development of lung edema induced by endotoxin administration in a murine model of acute lung injury. Crit Care Med 2008;36:262–267. [DOI] [PubMed] [Google Scholar]
- 76.Kolosova IA, Mirzapoiazova T, Moreno-Vinasco L, Sammani S, Garcia JG, Verin AD. Protective effect of purinergic agonist ATPgammas against acute lung injury. Am J Physiol Lung Cell Mol Physiol 2008;294:L319–L324. [DOI] [PubMed] [Google Scholar]
- 77.Tzeng HP, Ho FM, Chao KF, Kuo ML, Lin-Shiau SY, Liu SH. Beta-lapachone reduces endotoxin-induced macrophage activation and lung edema and mortality. Am J Respir Crit Care Med 2003;168:85–91. [DOI] [PubMed] [Google Scholar]
- 78.Takeoka M, Sakai A, Ueda G, Ge RL, Ishizaki T, Panos RJ, Taniguchi S. Dibutyryl camp inhibits endotoxin-induced increases in pulmonary vascular resistance and fluid filtration coefficient in the perfused rat lung. Tohoku J Exp Med 1997;183:273–284. [DOI] [PubMed] [Google Scholar]
- 79.Chu CH, David Liu D, Hsu YH, Lee KC, Chen HI. Propofol exerts protective effects on the acute lung injury induced by endotoxin in rats. Pulm Pharmacol Ther 2007;20:503–512. [DOI] [PubMed] [Google Scholar]
- 80.Urbain B, Gustin P, Ansay M. Endotoxin-induced microvascular injury in isolated and perfused pig lungs. Vet Res Commun 1992;16:453–464. [DOI] [PubMed] [Google Scholar]
- 81.Mouchawar AM, Kavanagh BP, Pearl RG. Serum but not plasma produces injury in the perfused rabbit lung. Anesth Analg 1994;79:40–45. [DOI] [PubMed] [Google Scholar]
- 82.Winn R, Nickelson S, Rice CL. Fluid filtration coefficient of isolated goat lungs was unchanged by endotoxin. J Appl Physiol 1988;64:2463–2467. [DOI] [PubMed] [Google Scholar]
- 83.Chlopicki S, Walski M, Bartus JB. Ultrastructure of immediate microvascular lung injury induced by bacterial endotoxin in the isolated, no-deficient lung perfused with full blood. J Physiol Pharmacol 2005;56:47–64. [PubMed] [Google Scholar]
- 84.Kuebler WM, Borges J, Sckell A, Kuhnle GE, Bergh K, Messmer K, Goetz AE. Role of l-selectin in leukocyte sequestration in lung capillaries in a rabbit model of endotoxemia. Am J Respir Crit Care Med 2000;161:36–43. [DOI] [PubMed] [Google Scholar]
- 85.Lutz CJ, Picone A, Gatto LA, Paskanik A, Landas S, Nieman GF. Exogenous surfactant and positive end-expiratory pressure in the treatment of endotoxin-induced lung injury. Crit Care Med 1998;26:1379–1389. [DOI] [PubMed] [Google Scholar]
- 86.Mizgerd JP, Skerrett SJ. Animal models of human pneumonia. Am J Physiol Lung Cell Mol Physiol 2008;294:L387–L398. [DOI] [PubMed] [Google Scholar]
- 87.Bryson DG, Ball HJ, McAliskey M, McConnell W, McCullough SJ. Pathological, immunocytochemical and microbiological findings in calf pneumonias associated with haemophilus somnus infection. J Comp Pathol 1990;103:433–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Choi G, Hofstra JJ, Roelofs JJ, Rijneveld AW, Bresser P, van der Zee JS, Florquin S, van der Poll T, Levi M, Schultz MJ. Antithrombin inhibits bronchoalveolar activation of coagulation and limits lung injury during Streptococcus pneumoniae pneumonia in rats. Crit Care Med 2008;36:204–210. [DOI] [PubMed] [Google Scholar]
- 89.Kondo H, Taguchi M, Abe N, Nogami Y, Yoshioka H, Ito M. Pathological changes in epidemic porcine Pneumocystis carinii pneumonia. J Comp Pathol 1993;108:261–268. [DOI] [PubMed] [Google Scholar]
- 90.Mizgerd JP, Scott ML, Spieker MR, Doerschuk CM. Functions of Iκb proteins in inflammatory responses to Escherichia coli LPS in mouse lungs. Am J Respir Cell Mol Biol 2002;27:575–582. [DOI] [PubMed] [Google Scholar]
- 91.Mizgerd JP, Spieker MR, Doerschuk CM. Early response cytokines and innate immunity: essential roles for TNF receptor 1 and type I IL-1 receptor during Escherichia coli pneumonia in mice. J Immunol 2001;166:4042–4048. [DOI] [PubMed] [Google Scholar]
- 92.Ong ES, Gao XP, Xu N, Predescu D, Rahman A, Broman MT, Jho DH, Malik ABE. Coli pneumonia induces CD18-independent airway neutrophil migration in the absence of increased lung vascular permeability. Am J Physiol Lung Cell Mol Physiol 2003;285:L879–L888. [DOI] [PubMed] [Google Scholar]
- 93.Viget NB, Guery BP, Ader F, Neviere R, Alfandari S, Creuzy C, Roussel-Delvallez M, Foucher C, Mason CM, Beaucaire G, et al. Keratinocyte growth factor protects against Pseudomonas aeruginosa-induced lung injury. Am J Physiol Lung Cell Mol Physiol 2000;279:L1199–L1209. [DOI] [PubMed] [Google Scholar]
- 94.Wang Q, Teder P, Judd NP, Noble PW, Doerschuk CM. CD44 deficiency leads to enhanced neutrophil migration and lung injury in Escherichia coli pneumonia in mice. Am J Pathol 2002;161:2219–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mizgerd JP, Lupa MM, Hjoberg J, Vallone JC, Warren HB, Butler JP, Silverman ES. Roles for early response cytokines during Escherichia coli pneumonia revealed by mice with combined deficiencies of all signaling receptors for TNF and IL-1. Am J Physiol Lung Cell Mol Physiol 2004;286:L1302–L1310. [DOI] [PubMed] [Google Scholar]
- 96.Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, Henson PM, Noble PW. Resolution of lung inflammation by CD44. Science 2002;296:155–158. [DOI] [PubMed] [Google Scholar]
- 97.Sawa T, Corry DB, Gropper MA, Ohara M, Kurahashi K, Wiener-Kronish JP. IL-10 improves lung injury and survival in Pseudomonas aeruginosa pneumonia. J Immunol 1997;159:2858–2866. [PubMed] [Google Scholar]
- 98.Seeger W, Obernitz R, Thomas M, Walmrath D, Suttorn N, Holland IB, Grimminger F, Eberspacher B, Hugo F, Bhakdi S. Lung vascular injury after administration of viable hemolysin-forming Escherichia coli in isolated rabbit lungs. Am Rev Respir Dis 1991;143:797–805. [DOI] [PubMed] [Google Scholar]
- 99.Song GW, Robertson B, Curstedt T, Gan XZ, Huang WX. Surfactant treatment in experimental Escherichia coli pneumonia. Acta Anaesthesiol Scand 1996;40:1154–1160. [DOI] [PubMed] [Google Scholar]
- 100.Su X, Robriquet L, Folkesson HG, Matthay MA. Protective effect of endogenous beta-adrenergic tone on lung fluid balance in acute bacterial pneumonia in mice. Am J Physiol Lung Cell Mol Physiol 2006;290:L769–L776. [DOI] [PubMed] [Google Scholar]
- 101.Luna CM, Baquero S, Gando S, Patron JR, Morato JG, Sibila O, Absi R, Famiglietti A, Vay CA, Von Stecher F, et al. Experimental severe Pseudomonas aeruginosa pneumonia and antibiotic therapy in piglets receiving mechanical ventilation. Chest 2007;132:523–531. [DOI] [PubMed] [Google Scholar]