

Dystonia‐parkinsonism, associated with inherited errors in manganese metabolism, is caused by biallelic pathogenic variants in the SLC30A10 1 or SLC39A14 2 genes. This rare and potentially treatable disease was almost exclusively reported in children2, 3, 4. Chelation therapy is administered for this condition based on few reports indicating clinical improvement in young patients with a progressive disease.2 To demonstrate the possible benign course of some patients with this condition and the difficulty in recommending chelation therapy in elderly diagnosed patients with clinically stable disease, we present here a patient with manganese brain accumulation attributed to a SLC39A14 homozygous mutation diagnosed in late adulthood.
A 65‐year‐old woman was evaluated in our clinic for long‐term dysarthria and general dystonia. The patient is from a consanguineous family of Ashkenazi origin, with no other family members affected. She reported a change in her handwriting at the age of 18 years as her first noticeable symptom and impaired gait and balance, frequent falls, and unintelligible speech gradually developing in the subsequent year. This deterioration was followed by 4 decades of clinical stability and even certain improvement of her speech following the administration of a low dose of lamotrigine.
Neurological examination revealed impaired ocular convergence, deep nasolabial folds, comprehensible dysarthria, and moderate bradykinesia that was more pronounced on the right side of the body, dystonia of her right limbs, and spastic gait with dystonic features. Brain magnetic resonance imaging revealed a prominent T1 hyperintense, diffuse, and nonenhancing signal involving the basal ganglia and subcortical white matter (Fig. 1) compatible with manganese accumulation.
Figure 1.
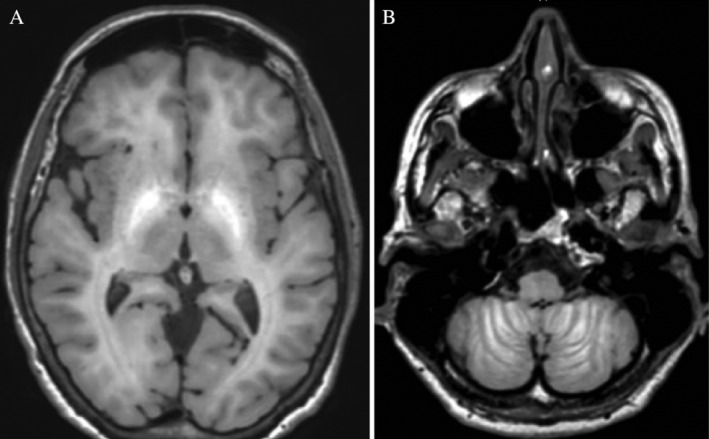
Brain magnetic resonance imaging demonstrates hyperintense T1 signal of the pallidum (A) as well as in the cerebellar folds (B). To a lesser degree, hyperintense T1 signals were also observed in the subcortical white matter, anterior midbrain, and vermis. Fluid attenuated inversion recovery and susceptibility weighted images were normal.
Whole‐exome sequencing revealed a homozygous missense variant, g.8:22273712G>A (GRCh37) c.1066G>A (p.G356S), almost exclusively found in Ashkenazi Jews (gnomAD v3 frequency 1:3316, 1/42004 in Africans), situated in a highly evolutionarily conserved position (GERP = 5.9899) in the SLC39A14 gene. This variant is predicted to damage the translated protein by SIFT, Mutation Taster, and PolyPhen. An increased manganese blood level was also demonstrated (60.4 μg/L, normal range 4.2–16.5 μg/L).
Chelation therapy by either intravenous EDTA or oral para‐aminosalicylic acid5 was recommended to the patient. The patient chose to avoid these therapies because of her long‐standing clinical stability. The lack of clear evidence in the medical literature regarding the efficacy of these treatments in elderly patients with stable disease also played a role in her decision. A daily oral iron supplement was commenced instead. A clinical follow‐up lasting 2 years and repeated brain magnetic resonance imaging scan performed 2 years after diagnosis did not demonstrate any clinical progression or a change in T1 signal or brain volume.
The natural history of dystonia‐parkinsonism secondary to pathogenic variants of SLC39A14 is unknown. Although the clinical benefit of chelation therapy was anecdotally reported,2 the long‐term need of this therapy was not established. Intravenous EDTA therapy is considered safe but requires repetitive, monthly infusions. Data regarding its safety in chronic conditions are also limited. We show that in some clinically stable cases, withholding chelating therapy is a reasonable course of action. This case report raises the need for further research to establish the natural history of patients with a genetic error in manganese metabolism and the appropriate conditions for administering chelation therapy.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
M.N.: 1B, 1C, 2A
M.B.: 1B, 1C, 2A
H.M.S.: 1C, 2A, 2B
S.B.B.: 1C, 2B
D.R.: 1C, 2B
L.J.O.: 1C, 2B
D.A.: 1A, 1C, 2A, 2B
Disclosures
Ethical Compliance Statement: The authors confirm that an approval of an institutional review board and patient consent were not required for this work. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: This study was not funded, and no conflicts of interest exist.
Financial Disclosures for the previous 12 months: M.N., M.B., and H.M.S. have no financial disclosures. S.B.B. is supported by the Michael J. Fox Foundation and the National Institutes of Health. D.R. is supported by grants from the Michael J. Fox Foundation and from the National Institute of Neurological Disorders and Stroke. L.J.O. receive grants from National Institutes of Health and patent royalties related to DYT1 and DYT6 dystonia from Athena Diagnostics Inc. D.A. is supported by a research grant from the Israeli Ministry of Science.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Quadri M, Federico A, Zhao T, et al. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet 2012;90(3):467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tuschl K, Meyer E, Valdivia LE, et al. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood‐onset parkinsonism‐dystonia. Nat Commun 2016;7:11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Juneja M, Shamim U, Joshi A, et al. A novel mutation in SLC39A14 causing hypermanganesemia associated with infantile onset dystonia. J Gene Med 2018;20(4):e3012. [DOI] [PubMed] [Google Scholar]
- 4. Zeglam A, Abugrara A, Kabuka M. Autosomal‐recessive iron deficiency anemia, dystonia and hypermanganesemia caused by new variant mutation of the manganese transporter gene SLC39A14 . Acta Neurol Belg 2019;119(3):379–384. [DOI] [PubMed] [Google Scholar]
- 5. Zheng W, Jiang Y‐M, Zhang Y, Jiang W, Wang X, Cowan DM. Chelation therapy of manganese intoxication with para‐aminosalicylic acid (PAS) in Sprague‐Dawley rats. Neurotoxicology 2009;30(2):240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]