

Here, Szigety et al. investigated the function of histone deacetylases in epidermal development, and they found that HDAC3 operates in conjunction with KLF4 to repress inappropriate expression of Tgm1, Krt16, and Aqp3, and suppresses expression of inflammatory cytokines through a Rela-dependent mechanism. Their data identify HDAC3 as a hub coordinating multiple aspects of epidermal barrier acquisition.
Keywords: epidermis, histone deacetylase, mouse, KLF4, HDAC3, NCoR, SMRT, chromatin, epigenetic, epidermal barrier
Abstract
Chromatin modifiers play critical roles in epidermal development, but the functions of histone deacetylases in this context are poorly understood. The class I HDAC, HDAC3, is of particular interest because it plays divergent roles in different tissues by partnering with tissue-specific transcription factors. We found that HDAC3 is expressed broadly in embryonic epidermis and is required for its orderly stepwise stratification. HDAC3 protein stability in vivo relies on NCoR and SMRT, which function redundantly in epidermal development. However, point mutations in the NCoR and SMRT deacetylase-activating domains, which are required for HDAC3's enzymatic function, permit normal stratification, indicating that HDAC3's roles in this context are largely independent of its histone deacetylase activity. HDAC3-bound sites are significantly enriched for predicted binding motifs for critical epidermal transcription factors including AP1, GRHL, and KLF family members. Our results suggest that among these, HDAC3 operates in conjunction with KLF4 to repress inappropriate expression of Tgm1, Krt16, and Aqp3. In parallel, HDAC3 suppresses expression of inflammatory cytokines through a Rela-dependent mechanism. These data identify HDAC3 as a hub coordinating multiple aspects of epidermal barrier acquisition.
The epidermis functions as a barrier, serving to protect the organism from external insults and to retain moisture. This is achieved through its structure as a stratified epithelium, in which a relatively undifferentiated proliferative basal cell layer successively gives rise to suprabasal, granular, and enucleated cornified cell layers, each of which is characterized by expression of a unique set of genes (Eckert 1989; Feil et al. 1996; Fuchs 2007). In addition to forming the physical structure of the epidermis, keratinocytes are sentinels of the innate immune system, producing proinflammatory alarmins including cytokines and chemokines, as well as antimicrobial peptides and structural proteins, in response to barrier breach (Eckert et al. 2004; Oppenheim and Yang 2005; Lai and Gallo 2009; Nestle et al. 2009). Intriguingly, exposure of keratinocytes to amniotic fluid in utero leads to up-regulation of short proline-rich region proteins, demonstrating that barrier dysfunction can be sensed prior to birth (Huebner et al. 2012).
Global regulation of gene expression is critical for the development of proper epidermal structure and function. An important mechanism of control is the regulation of chromatin structure. In the epidermis, DNA methyltransferases such as DNMT1; histone methyltransferases including EZH1, EZH2, and Setd8; histone demethylases such as JMJD3; and ATP-dependent chromatin remodelers including Brg1 play important roles in regulating epidermal proliferation, differentiation, and progenitor cell activity (Sen et al. 2008, 2010; Ezhkova et al. 2011; Driskell et al. 2012; Xiong et al. 2013; Mardaryev et al. 2014). Nonenzyme proteins can also regulate epidermal chromatin structure. For example, Sin3A, a scaffolding protein known to interact with histone deacetylases 1 and 2, suppresses epidermal proliferation (Nascimento et al. 2011).
The functions of histone deacetylases (HDACs) are of particular interest, given the use of class I HDAC inhibitors in treatment of cutaneous malignancies (Duvic 2015). HDACs regulate gene expression by removing histone acetyl marks, resulting in chromatin compaction and transcriptional repression (Seto and Yoshida 2014). HDACs also deacetylate transcription factors, such as p53, to regulate their activity (Higashitsuji et al. 2007; Tang et al. 2008). These enzymes do not bind DNA directly but are instead recruited to chromatin as members of multi-protein complexes. HDACs 1 and 2 participate in the NuRD, Sin3, and CoREST complexes, whereas HDAC3 is unique in its association with the NCoR and SMRT scaffolding proteins (Yang and Seto 2008). As different class I HDAC family members participate in different complexes, their target genes are likely distinct. Broad inhibition of all class I HDACs is likely unnecessary for therapeutic efficacy and results in undesirable side effects.
Characterizing the functions of individual HDACs will improve understanding of epigenetic regulation of epidermal gene expression and can serve to guide rational drug design. To this end, we previously characterized the roles of HDAC1/2 in embryonic epidermal development (LeBoeuf et al. 2010). Loss of HDAC1/2 leads to failure of epidermal stratification and proliferation through loss of p63 repressive activity as well as gain of p53 activity. The role of a third member of the class I HDAC family, HDAC3, in epidermal development has not yet been characterized.
Global loss of HDAC3 leads to lethality prior to E9.5 due to gastrulation defects (Bhaskara et al. 2008; Montgomery et al. 2008). Tissue-specific deletion has revealed diverse roles for HDAC3 in different contexts. In intestinal epithelium, HDAC3 regulates expression of genes involved in barrier function as well as the inflammatory response to commensal bacteria (Alenghat et al. 2013). HDAC3 also controls inflammatory NF-κB signaling in macrophages and T cells (Chen et al. 2001; Yan et al. 2012). Additional functions for HDAC3 have been described in hepatic metabolism, heart and lung development, neuronal cell fate, pancreatic β-cell activity, skeletal muscle metabolism, brown adipose thermogenesis, and bone remodeling (Feng et al. 2011; Carpio et al. 2016; Wang et al. 2016; Zhang et al. 2016; Emmett et al. 2017; Hong et al. 2017; Poleshko et al. 2017; Remsberg et al. 2017). Importantly, HDAC3 regulates gene expression in a highly tissue-dependent manner, suggesting that its mechanisms of action vary among different tissues (Emmett and Lazar 2019).
HDAC3 participates in complexes scaffolded by nuclear receptor corepressor 1 (NCoR) or its homolog silencing mediator of retinoic and thyroid receptors (SMRT) (Li et al. 2000; Wen et al. 2000). In vitro studies revealed that HDAC3 interaction with the deacetylase-activating domain (DAD) of NCoR/SMRT is specifically required for activation of its histone deacetylase activity (Guenther et al. 2001). Mice bearing loss-of-function point mutations in the NCoR/SMRT DAD express normal levels of HDAC3 protein but have no detectable HDAC3 histone deacetylase activity (You et al. 2013). In contrast to global loss of Hdac3, which results in early embryonic lethality, mice lacking HDAC3 histone deacetylase activity due to mutations in the NCoR/SMRT DAD survive to adulthood, suggesting histone deacetylase-independent functions for HDAC3. Consistent with this, HDAC3 performs scaffolding functions in conjunction with NCoR in the liver and heart (Sun et al. 2013; Lewandowski et al. 2015). Like HDAC3, NCoR/SMRT do not bind DNA directly, and the transcription factors that recruit these proteins to their target genes in the developing epidermis are unknown.
Here, we show that HDAC3, in complex with NCoR/SMRT, forms a hub for epigenetic regulation of differentiation and innate immune functions of the embryonic epidermis. Loss of Hdac3 or codeletion of Ncor1 (encoding NCoR) and Ncor2 (encoding SMRT) leads to expansion of basal cells and premature expression of terminal differentiation markers, demonstrating that these factors are required for orderly stratification of the epidermis. Genes implicated in the epidermal danger response, including genes encoding structural proteins as well as cytokines and chemokines, are up-regulated in both Hdac3 and Ncor1/Ncor2 epidermal mutants. Interestingly, codeletion of Rela rescues expression of inflammatory genes but not dysregulated structural gene expression or stratification defects, indicating that these phenotypes are not secondary to an NF-κB-mediated innate immune response. Deletion of epidermal Ncor1 or Ncor2 alone does not result in an obvious phenotype, suggesting functional redundancy of NCoR and SMRT in embryonic epidermis, which is distinct from nonredundant roles described in other tissues (Ghisletti et al. 2009; Sun et al. 2013). Codeletion of both Ncor1 and Ncor2 causes loss of HDAC3 protein, providing evidence that NCoR/SMRT stabilize HDAC3 in the epidermis in vivo. Importantly, HDAC3-mediated regulation of epidermal structure is independent of its histone deacetylase activity, as epidermis in mice homozygous for loss-of-function point mutations in the NCoR and SMRT DADs is indistinguishable from that of controls. HDAC3-bound sites are significantly enriched for predicted binding motifs for the transcription factors AP1, GRHL, and KLF, which are known to play critical functions in epidermal development. Our data show that HDAC3 operates both in conjunction with, and independently of, KLF4 to repress expression of different epidermal stress and differentiation genes. Interestingly, for several genes coregulated by HDAC3 and KLF4, HDAC3 binds its target sites in the absence of KLF4, suggesting importance of additional members of the transcriptional repressive complex. In parallel, HDAC3 suppresses expression of inflammatory cytokines through a Rela-dependent mechanism. These data demonstrate how HDAC3 cooperates with specific transcriptional partners to coordinate diverse transcriptional programs necessary for establishing barrier function.
Results
Deletion of Hdac3 in embryonic epidermis disrupts barrier development and causes perinatal lethality
Analysis of HDAC3 expression by immunofluorescence demonstrated that HDAC3 is expressed broadly in developing epidermis (Supplemental Fig. S1). To delineate the roles of HDAC3 in embryonic epidermal development, we used mice carrying an Hdac3 conditional loss-of-function allele together with a K5-rtTA transgene in which expression of a reverse tetracycline-controlled transactivator is driven by the keratin 5 (K5) promoter, and a tetO-Cre transgene in which expression of Cre recombinase is under the control of a tetracycline-responsive promoter element. This system allows for temporal and spatial control of Hdac3 deletion, such that Cre recombinase is expressed only in the epidermis upon doxycycline administration. Pregnant dams were placed on doxycycline chow at E8.5, 1 d prior to activation of the K5 promoter (Byrne et al. 1994), until pups were born. Quantitative RT-PCR (qPCR) for Hdac3 transcripts at E18.5 confirmed efficient recombination of epidermal Hdac3 (Fig. 1A) and immunostaining confirmed loss of HDAC3 protein (Fig. 1B,C). Hdac3 mutant animals were born at expected Mendelian ratios but displayed physical abnormalities including erythematous, tight skin, and failed eyelid fusion, and died perinatally.
Figure 1.
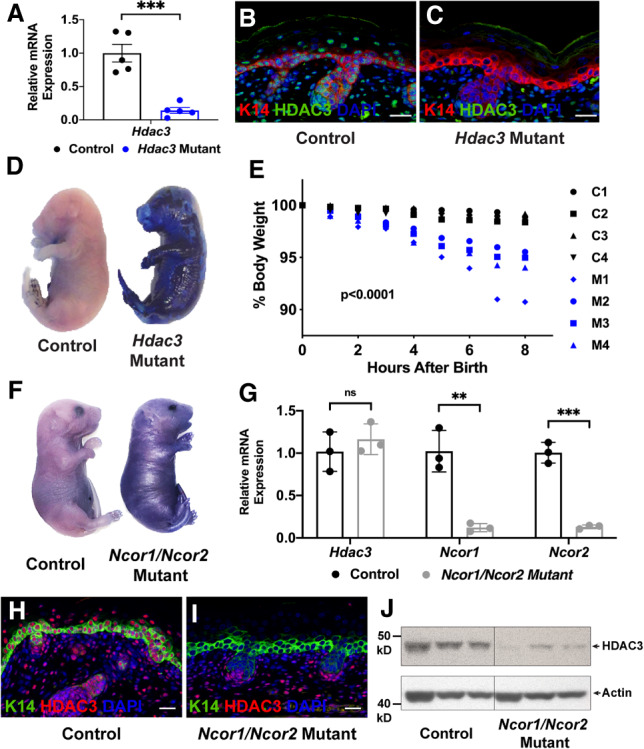
HDAC3 and NCoR/SMRT are necessary for embryonic epidermal barrier function and HDAC3 protein stability requires NCoR/SMRT. (A) qPCR analysis demonstrates that Hdac3 is efficiently recombined using the K5-rtTA tetO-Cre Hdac3fl/fl-inducible system; n = 5. (***) P < 0.001, unpaired two-tailed Student's t-test. Error bars indicate SEM. (B,C) Immunofluorescence for K14 and HDAC3 demonstrates that HDAC3 protein is lost in Hdac3 mutant epidermis at E18.5. (D) Dye exclusion assay shows that Hdac3 mutant epidermis fails to exclude dye at E18.5. (E) Hdac3 mutant animals lose weight more rapidly after birth than control littermates; n = 4. P = 0.0015, two-way ANOVA with Geisser-Greenhouse correction. (F) Dye exclusion assay shows that Ncor1/Ncor2 mutant epidermis fails to exclude dye at E18.5. (G) qPCR analysis demonstrates that Ncor1 and Ncor2 are efficiently recombined using the K5-rtTA tetO-Cre Ncor1fl/fl Ncor2fl/fl-inducible system, while Hdac3 mRNA expression is unchanged; n = 3. (**) P < 0.01; (***) P < 0.001, unpaired two-tailed Student's t-test. Error bars indicate SEM. (H,I) Immunofluorescence for K14 and HDAC3 reveals that HDAC3 protein is lost in Ncor1/Ncor2 mutant epidermis. (J) Western blot demonstrates that HDAC3 protein is severely diminished in Ncor1/Ncor2 mutant epidermis. Each lane contains lysate generated from a single embryo; n = 3 mutant and control littermates. Scale bars, 25 µm.
This phenotype suggested that Hdac3 mutant animals had an epidermal barrier defect. The epidermis functions both as an “outside-in” barrier, blocking external substances from entering the body, and an “inside-out” barrier, preventing body contents such as water from escaping. To assess “outside-in” barrier function, we performed dye exclusion assays and found that E18.5 Hdac3 mutant embryos failed to exclude dye appropriately (Fig. 1D). To assess the “inside-out” barrier, we evaluated the ability of the epidermis to retain moisture by measuring body weight after birth. Hdac3 mutant neonates lost weight more rapidly than their control littermates, indicating that they lost more water across the skin (Fig. 1E). Together, these assays demonstrated that deletion of epidermal HDAC3 results in both “outside-in” and “inside-out” barrier dysfunction. In line with these observations, cornified envelopes isolated from Hdac3 mutant epidermis were rough and fragile in appearance (Supplemental Fig. S2A,B). Transmission electron microscopy of E18.5 Hdac3 mutant stratum corneum revealed irregular spacing of layers and retention of cellular contents (Supplemental Fig. S2C–F).
Codeletion of epidermal Ncor1 and Ncor2 phenocopies Hdac3 deletion
HDAC3 interacts with NCoR/SMRT to regulate target gene expression in multiple cellular contexts (Emmett and Lazar 2019). However, the functions of NCoR/SMRT in developing epidermis are unknown. To begin to address this question, we used the K5-rtTA tetO-Cre system in conjunction with conditional Ncor1 and Ncor2 loss-of-function alleles. We found that deletion of either Ncor1 or Ncor2 alone did not result in a gross phenotype in embryonic epidermis: Single Ncor1 and Ncor2 mutants were indistinguishable from their control littermates at E18.5, demonstrating appropriate stratification and normal expression of keratinocyte differentiation markers (Supplemental Fig. S3). These data indicate that NCoR and SMRT are functionally redundant in embryonic keratinocytes, unlike in other cell types such as macrophages (Ghisletti et al. 2009). In contrast to deletion of Ncor1 or Ncor2 alone, simultaneous deletion of both Ncor1 and Ncor2 resulted in perinatal lethality. As with Hdac3 mutants, Ncor1/Ncor2 double mutants were born at expected numbers, but displayed erythematous, tight skin, and failure of eyelid fusion. Dye exclusion assay performed at E18.5 showed that Ncor1/Ncor2 double mutants failed to appropriately exclude dye, revealing failure of epidermal barrier function in these animals (Fig. 1F). Quantitative RT-PCR for Ncor1 and Ncor2 confirmed efficient recombination of both alleles (Fig. 1G).
Quantitative RT-PCR for Hdac3 showed that mRNA expression was unaltered in double-Ncor1/Ncor2 mutants compared with controls (Fig. 1G). Strikingly, however, immunofluorescence and immunoblotting revealed that the level of HDAC3 protein was severely diminished in Ncor1/Ncor2-null epidermis, demonstrating that NCoR/SMRT stabilize HDAC3 protein in vivo without altering Hdac3 transcription (Fig. 1H–J). In line with this finding, previous studies have shown that HDAC3 is unstable in the absence of NCoR/SMRT in vitro (Guo et al. 2012).
Perinatal lethality in Hdac3 and Ncor1/Ncor2 mutants is associated with disordered epidermal stratification
To determine whether the barrier defect observed in Hdac3 and Ncor1/Ncor2 epidermal mutants was due to aberrant epidermal structure, we carried out histological analysis, which revealed similar architectural abnormalities in Hdac3 and Ncor1/Ncor2 mutant epidermis, including loss of the basket weave structure of the cornified layer, the presence of enlarged immediately suprabasal cells that failed to flatten appropriately, and underdeveloped hair follicles (Fig. 2A,B, I,J). Despite their unusual appearance, enlarged cells did not display evidence of chromatin damage, although this was observed in the more superficial layers of both Hdac3 and Ncor1/Ncor2 mutant epidermis (Supplemental Fig. S4).
Figure 2.

Epidermal stratification is disrupted by loss of HDAC3 or NCoR/SMRT but does not require HDAC3's deacetylase activity. (A,B) H&E staining at E18.5 reveals abnormalities in Hdac3 mutant epidermal structure, including a dense, compact cornified layer (cf. red arrowheads) and enlarged immediately suprabasal cells with perinuclear clearing (yellow arrowheads). (C–H) Immunofluorescence for the basal layer marker K14, spinous layer marker K10 (C,D), and granular layer markers LOR (E,F) and FLG (G,H) demonstrates expansion of K14-positive cells, increased K14/K10-double-positive cells (D, white arrowheads), expanded LOR expression (F), less tightly compacted FLG expression (H), decreased K14/FLG-double-negative cells in E18.5 Hdac3 mutant epidermis (H) compared with control (G) and occasional K14/LOR- and K14/FLG-double-positive cells (arrowheads in F,H). (I,J) H&E staining at E18.5 demonstrates structural abnormalities in Ncor1/Ncor2 mutant epidermis, including a dense, compact cornified layer (cf. red arrowheads) and enlarged immediately suprabasal cells with perinuclear clearing (yellow arrowheads). (K–P) Immunofluorescence for K14, K10 (K,L), LOR (M,N), and FLG (O,P) demonstrates expanded K14 expression and premature expression of terminal differentiation markers in E18.5 Ncor1/Ncor2 mutant epidermis, evidenced by K14/K10-, K14/LOR-, and K14/FLG-double-positive cells (white arrowheads). Filaggrin granules are less tightly compacted in Ncor1/Ncor2 mutant (P) compared with control (O) epidermis, and extend into hair follicles. (Q,R) H&E staining shows NCoR/SMRT-DAD double-mutant (NS-DADm) E18.5 epidermis is unremarkable. (S–X) Immunofluorescence for K14, K10 (S,T), LOR (U,V), and FLG (W,X) demonstrates appropriate stratification and expression of terminal differentiation markers in E18.5 NCoR/SMRT DAD double-mutant epidermis. Scale bars, 25 µm.
Immunostaining for the basal marker keratin 14 (K14), the spinous cell marker keratin 10 (K10), and the granular cell proteins loricrin (LOR) and filaggrin (FLG), revealed that Hdac3 and Ncor1/Ncor2 mutant epidermis stratified but failed to do so in a tightly controlled fashion (Fig. 2C–H,K–P). K14 expression was expanded, evidenced by an increased number of K14/K10-double-positive cells (Fig. 2C,D,K,L), as well as absence of K14-negative cells immediately below the LOR/FLG-positive cell layer (Fig. 2E–H,M–P). LOR staining was expanded compared with controls (Fig. 2E,F, M,N), FLG granules were less tightly compacted (Fig. 2G,H, O,P), even extending into hair follicles (Fig. 2P), and occasional K14/LOR- and K14/FLG-double-positive cells were observed (arrowheads in Fig. 2F,H,N,P). To ensure that our findings were not an artifact of the K5-rtTA tetO-Cre deletion system, we also deleted Hdac3 in the epidermis using a constitutively active K14-Cre transgene. Epidermal Hdac3 was efficiently deleted using the K14-Cre system (Supplemental Fig. S5C,D). K14-Cre Hdac3fl/fl mice died perinatally with similar histological and differentiation defects to those observed in induced K5-rtTA tetO-Cre Hdac3fl/fl mice (Supplemental Fig. S5A,B,E–J). Thus, stepwise differentiation is poorly controlled in both Hdac3 and Ncor1/Ncor2 mutant epidermis, resulting in abnormal stratification. Together, these data demonstrate that HDAC3 depends on NCoR/SMRT for its stability and functions in embryonic epidermis. Despite the extensive overlap in phenotypes, Ncor1/Ncor2-null epidermis differed from Hdac3-null epidermis in that it was thicker than that of controls. This is likely due to loss of NCoR/SMRT interactions with proteins other than HDAC3.
HDAC3-mediated regulation of epidermal stratification is independent of its histone deacetylase activity
HDAC3 has enzyme-independent scaffolding functions in hepatic and cardiac tissue (Sun et al. 2013; Lewandowski et al. 2015). To test whether HDAC3-mediated gene regulation in the epidermis depends on its histone deacetylase activity, we used mice homozygous for point mutations in the deacetylase-activating domain (DAD) of both NCoR and SMRT, referred to as NS-DADm mice. NCoR and SMRT DAD mutant proteins maintain stability of HDAC3 protein but are unable to activate its histone deacetylase activity (You et al. 2013). As previously reported, NS-DADm mice survived to adulthood, in striking contrast to the perinatal lethality observed in epidermal Ncor1/Ncor2 double mutants. Histological analysis and immunofluorescent staining for K14, K10, LOR, and FLG revealed that NS-DADm epidermis was indistinguishable from control epidermis at E18.5 (Fig. 2Q–X). Thus, epidermal stratification does not require HDAC3's histone deacetylation functions, and instead may rely on its ability to act as a scaffolding protein.
Microarray analysis of Hdac3 and Ncor1/Ncor2 mutants reveals disruption of diverse cellular processes and identifies potential HDAC3 binding partners
To delineate the molecular mechanisms by which HDAC3 controls embryonic epidermal development, we performed microarray analysis of epidermis from five Hdac3 mutants and five control littermates at E18.5 (Supplemental Tables S1,S2). Functional annotation analysis of dysregulated genes using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) (Huang et al. 2009a,b) revealed disruption of keratinocyte differentiation, lipid, cholesterol and steroid metabolism, inflammatory signaling, oxidoreductase activity and cell–cell adhesion upon loss of Hdac3.
To determine whether the overlapping gross and histological phenotypes observed in Hdac3 and Ncor1/Ncor2 mutant epidermis correlated with similar transcriptional changes, we performed microarray analysis of epidermis from four Ncor1/Ncor2 mutants and four control littermates at E18.5 and compared the results with those from Hdac3 mutants and controls (Supplemental Tables S3,S4). Sixty-three percent of the transcript cluster IDs up-regulated upon deletion of Hdac3 were also up-regulated upon deletion of Ncor1/Ncor2 and 61% of those down-regulated upon deletion of Hdac3 were also down-regulated upon deletion of Ncor1/Ncor2 (Fig. 3A,B). Conversely, 39% of the transcript cluster IDs up-regulated upon deletion of Ncor1/Ncor2 were up-regulated upon deletion of Hdac3, and 45% of those down-regulated upon deletion of Ncor1/Ncor2 were down-regulated upon deletion of Hdac3. The overlapping transcriptional changes upon loss of HDAC3 and loss of NCoR/SMRT were highly statistically significant, consistent with HDAC3 functioning in complex with NCoR/SMRT to regulate target gene expression.
Figure 3.

Microarray analysis demonstrates significant overlap in Hdac3 and Ncor1/Ncor2 mutant epidermis and reveals potential HDAC3-binding partners. (A) Venn diagram demonstrating significant overlap in transcript cluster IDs up-regulated upon Hdac3 deletion and up-regulated upon Ncor1/Ncor2 deletion; P < 0.0001, two-tailed χ2 test with Yates correction. (B) Venn diagram demonstrating significant overlap in transcript cluster IDs down-regulated upon Hdac3 deletion and down-regulated upon Ncor1/Ncor2 deletion; P < 0.0001, two-tailed χ2 test with Yates correction. (C) DAVID analysis for genes up-regulated in Hdac3 mutant epidermis reveals that keratinocyte differentiation, lipid biosynthesis, cell–cell adhesion, and inflammatory signaling are disrupted upon loss of HDAC3. (D) Homer cis-regulatory motif analysis demonstrates that multiple transcription factor-binding motifs are enriched in the promoters of genes up-regulated upon Hdac3 deletion. (E) Homer cis-regulatory motif analysis demonstrates that multiple transcription factor-binding motifs are enriched in the promoters of genes up-regulated upon Ncor1/Ncor2 deletion and that these largely overlap with those enriched in genes up-regulated upon Hdac3 deletion (D).
The lack of an epidermal phenotype in NS-DAD mutants implies that HDAC3's functions in epidermal development are largely independent of its deacetylase activity, suggesting that HDAC3 could scaffold activating as well as repressive transcriptional complexes. To begin to investigate this, we performed DAVID analyses of genes that were either down-regulated or up-regulated upon deletion of Hdac3. Down-regulated genes were most significantly associated with broad functional terms such as glycoprotein, transmembrane region and signal peptide (Supplemental Fig. S6A); however, up-regulated genes revealed enrichment of terms related to keratinocyte differentiation and inflammatory signaling, which were clearly implicated in the Hdac3 mutant phenotype (Fig. 3C). While not excluding possible functions for HDAC3 in transcriptional activation, these data are consistent with a major role in transcriptional repression.
HDAC3 and NCoR/SMRT do not bind chromatin directly but are instead recruited to genomic loci in large transcriptional complexes that include DNA-binding proteins. To find potential HDAC3-binding partners, we performed cis-regulatory motif analysis to identify transcription factor-binding motifs that were enriched in the promoters of genes that were up-regulated upon Hdac3 deletion. This analysis showed that NF-κB, KLF, Sp, and AP1 motifs were the most significantly enriched motifs (Fig. 3D; Supplemental Table S5). Consistent with HDAC3 acting in complex with NCoR/SMRT, we found that all of these motifs were also enriched in the promoters of genes up-regulated in Ncor1/Ncor2 mutant epidermis (Fig. 3E; Supplemental Table S6). Transcription factor-binding motifs enriched in the promoters of genes down-regulated upon loss of Hdac3 or Ncor1/Ncor2 were largely distinct from those in up-regulated genes, and included predicted binding sites for DLX and LHX family members (Supplemental Fig. S6B,C; Supplemental Tables S7, S8). The retinoic acid receptor (RAR) and retinoid X receptor (RXR) families are thought to play roles in epidermal lipid processing and barrier function (Attar et al. 1997); therefore, dysregulated RAR/RXR target gene expression could account for elevated expression of lipid processing genes in Hdac3 and Ncor1/Ncor2 mutants. However, the RXR:RAR motif was not significantly enriched in the promoter regions of genes that were up-regulated in Hdac3 mutant embryonic epidermis.
Codeletion of Rela rescues expression of inflammatory genes in Hdac3 mutants but does not ameliorate dysregulated structural gene expression or stratification defects
The NF-κB motif was one of the most significantly enriched motifs in the promoters of genes up-regulated upon Hdac3 deletion. As NF-κB plays major roles in controlling expression of inflammatory genes in other tissues (Chen et al. 2001; Yan et al. 2012), we asked whether the phenotype of Hdac3 mutant epidermis might be secondary to NF-κB target gene dysregulation. We found that homozygous codeletion of Rela, encoding the RelA subunit of NF-κB, led to a dramatic rescue of Ccl20 and Tnf expression in Hdac3 mutant epidermis to almost normal levels (Fig. 4A). Consistent with these findings, studies in postnatal epidermis have shown that HDAC3–NCoR/SMRT complexes are required for glucocorticoid receptor-mediated NF-κB target gene transrepression, including repression of Tnf (Hua et al. 2016a,b). In contrast, the structural genes Sprr1a and Tgm1 remained significantly up-regulated in embryos with homozygous codeletion of Hdac3 and Rela, indicating that expression of these genes is largely independent of canonical NF-κB signaling (Fig. 4A). Consistent with a Rela-independent mechanism of structural gene dysregulation, Hdac3/Rela double mutants died perinatally and H&E staining demonstrated that deletion of Rela did not rescue architectural abnormalities in Hdac3 mutant epidermis including compaction of the stratum corneum and presence of enlarged cells (Fig. 4B–D). Additionally, immunostaining for K14, K10, LOR, and FLG demonstrated disorganized expression of differentiation markers in Hdac3/Rela mutant epidermis, evidenced by K14/K10-, K14/LOR-, and K14/FLG-double-positive cells (Fig. 4E–P). Together, these data demonstrate that up-regulated structural gene expression, disordered stratification, and barrier defects in Hdac3 mutant epidermis are not secondary to an NF-κB-mediated innate immune response.
Figure 4.
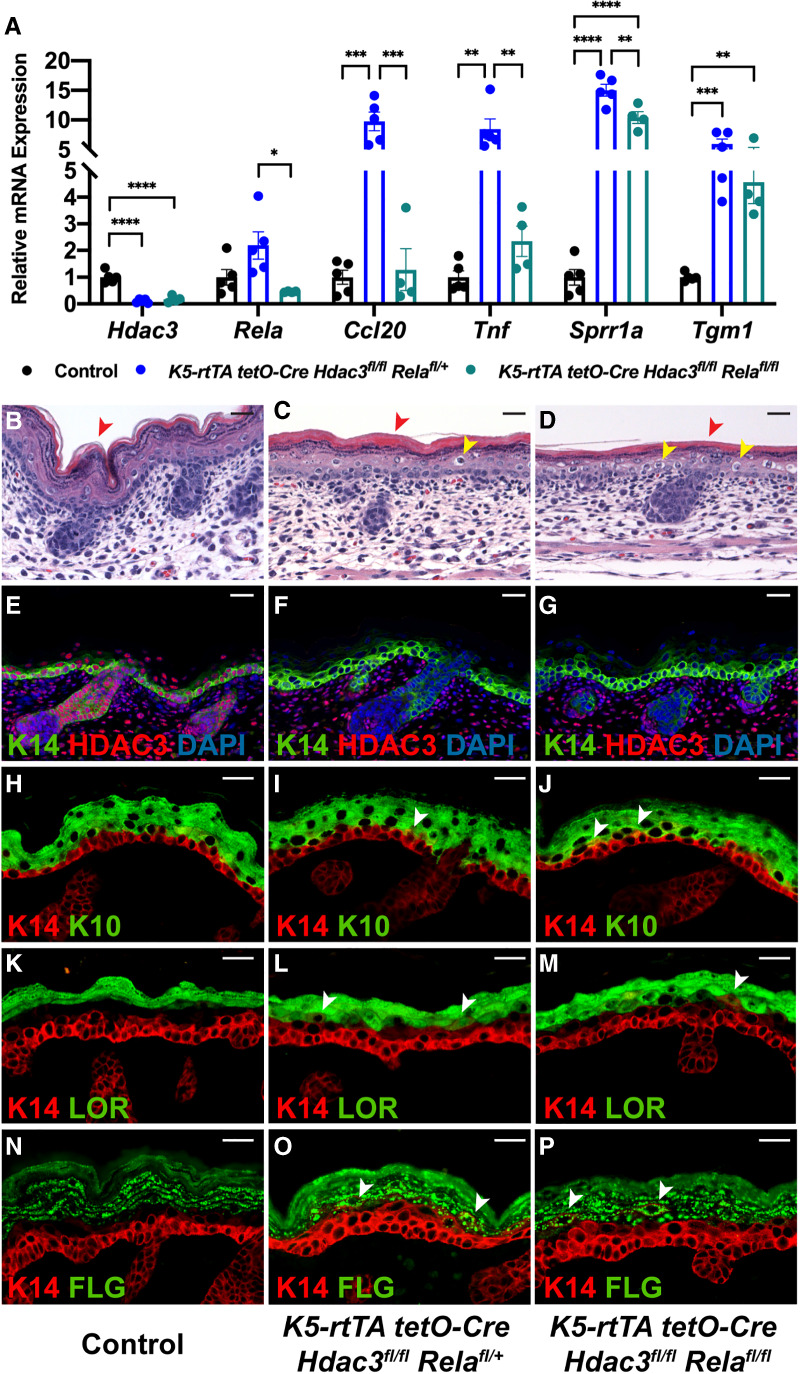
Deletion of Rela prevents up-regulation of inflammatory genes but does not rescue structural abnormalities in Hdac3 mutant epidermis. (A) qPCR analysis demonstrates that deletion of Rela prevents up-regulation of inflammatory genes Ccl20 and Tnf but not up-regulation of structural genes Sprr1a and Tgm1 in Hdac3-deleted epidermis; n = 5 control; n = 5 induced K5-rtTA tetO-Cre Hdac3fl/fl Relafl/+; n = 4 induced K5-rtTA tetO-Cre Hdac3fl/fl Relafl/fl animals. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001, ordinary one-way ANOVA with multiple comparisons. Error bars indicate SEM. (B–D) H&E staining shows that deletion of Rela does not rescue structural abnormalities in Hdac3 mutant epidermis, including compaction of the stratum corneum (cf. red arrowheads) and enlarged suprabasal cells (yellow arrowheads). (E–G) Immunofluorescence for K14 and HDAC3 demonstrates that HDAC3 protein is deleted in induced K5-rtTA tetO-Cre Hdac3fl/fl Relafl/+ and K5-rtTA tetO-Cre Hdac3fl/fl Relafl/fl epidermis. (H–P) Immunofluorescence for the basal layer marker K14, spinous layer marker K10 (H–J) and granular layer markers LOR (K–M) and FLG (N–P) show that deletion of Rela does not rescue expanded K14 expression and appearance of K14/K10-, K14/LOR-, and K14/FLG-double-positive cells in Hdac3 mutant epidermis (white arrowheads). Scale bars, 25 µm.
HDAC3 peaks are enriched for AP1-, GRHL-, KLF-, and C/EBP-binding motifs
To identify sites of HDAC3 localization in E18.5 wild-type epidermal chromatin, we performed chromatin immunoprecipitation followed by DNA sequencing (ChIP-seq) (Supplemental Table S9). We found that the majority of HDAC3-binding sites were in introns and intergenic regions, while a minority of sites were found in exons, promoters, and transcriptional termination sites (Fig. 5A), consistent with previously published HDAC3 cistromes (Feng et al. 2011; Remsberg et al. 2017). Out of 2184 dysregulated genes, 1506 were associated with HDAC3 peaks, suggesting them as candidate direct targets. Predicted binding motifs for AP1, GRHL, KLF, and C/EBP were enriched in HDAC3 peaks as well as in the promoters of genes up-regulated in Hdac3 and Ncor1/Ncor2 epidermal mutants (Figs. 3D,E, 5B; Supplemental Tables S5, S6, S10). DLX and LHX transcription factor binding motifs, which were enriched in the promoters of down-regulated genes (Supplemental Fig. S6B,C; Supplemental Tables S7, S8), were not among the most highly significantly enriched motifs in HDAC3 binding peaks, suggesting that HDAC3's effects on genes down-regulated in Hdac3 mutants may mainly be indirect. These observations, together with our analyses of the predicted functions of up-regulated versus down-regulated genes in Hdac3 mutants, are consistent with the notion that HDAC3 acts predominantly as a transcriptional repressor in embryonic epidermis. Together, these data imply important functional interactions between HDAC3 and AP1, GRHL, KLF, and C/EBP family members and suggest that one or more of these transcription factors may recruit HDAC3 to its target sites in epidermal chromatin.
Figure 5.
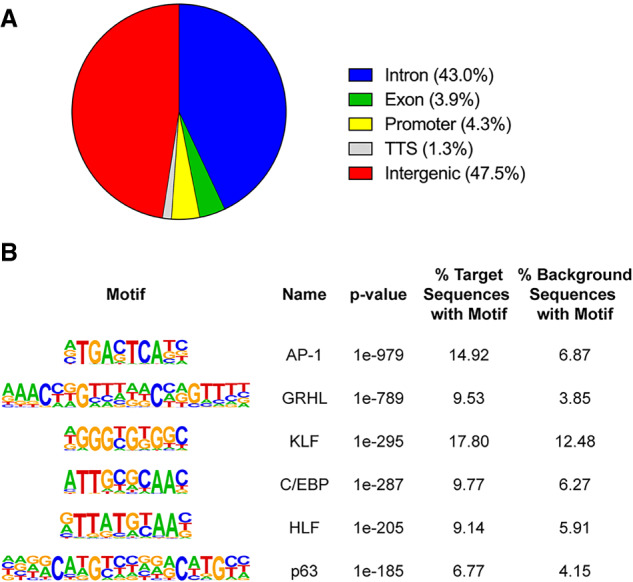
ChIP-seq for HDAC3 in E18.5 wild-type epidermis reveals sites of HDAC3 localization and identifies putative HDAC3-binding partners. (A) Distribution of HDAC3-binding sites relative to known genes. (B) AP1, GRHL, KLF, and C/EBP transcription factor-binding motifs are the most significantly enriched motifs in HDAC3 peaks.
Epidermal deletion of Klf4 partially phenocopies Hdac3 deletion
The data presented above suggest possible interactions between HDAC3 and AP1, GRHL, KLF, and C/EBP transcription factors in epidermal development. Among these, the KLF family member KLF4 is well established as a critical regulator of embryonic epidermal differentiation. Global loss of KLF4 results in perinatal lethality secondary to epidermal barrier dysfunction, while ectopic expression of KLF4 in embryonic epidermal basal cells causes premature barrier formation (Segre et al. 1999; Jaubert et al. 2003). Importantly, KLF family members can act as both transcriptional activators and repressors, depending on the target gene and cellular context. For example, in intestinal epithelial cells, KLF4 promotes expression of intestinal alkaline phosphatase through recruitment of the histone acetyltransferase p300 and represses expression of cyclin B1 through recruitment of HDAC3 (Evans et al. 2007). We therefore chose to interrogate KLF4 as a potential HDAC3 binding partner.
To ask whether epidermal-specific deletion of Klf4 resembled the Hdac3 mutant phenotype, we used the K5-rtTA tetO-Cre system in conjunction with a Klf4 conditional loss-of-function allele. Quantitative RT-PCR (Fig. 6A) and immunofluorescence confirmed efficient KLF4 deletion in mutant epidermis (Fig. 6E,F). Importantly, expression of HDAC3 was unaffected by Klf4 deletion (Fig. 6A,G,H), and KLF4 expression was not perturbed by loss of HDAC3 (Fig. 6O,P) or NCoR/SMRT (Fig. 6Q,R). Epidermal Klf4 knockouts were born at expected numbers but died perinatally. Dye exclusion assays demonstrated that loss of epidermal KLF4 resulted in barrier dysfunction (Fig. 6B). Thus, epidermal-specific deletion of Klf4 phenocopied global deletion, demonstrating that the barrier defect observed in global knockouts is a primary defect, rather than secondary to loss of KLF4 in another tissue.
Figure 6.

Epidermal deletion of Klf4 partially phenocopies Hdac3 deletion and KLF4 complexes with HDAC3 in suprabasal nuclei. (A) qRT-PCR analysis demonstrates that Klf4 is efficiently recombined using the K5-rtTA tetO-Cre Klf4fl/fl-inducible system. n = 5. (**) P < 0.01, unpaired two-tailed Student's t-test. Error bars indicate SEM. (B) Dye exclusion assay shows that Klf4 mutant epidermis fails to exclude dye at E18.5. (C,D) H&E staining demonstrates compaction of the stratum corneum (cf. red arrowheads) in E18.5 Klf4 mutant epidermis. (E,F) Immunofluorescence for K14 and KLF4 demonstrates that KLF4 protein is lost in E18.5 Klf4 mutant epidermis. (G,H) Immunofluorescence for K14 and HDAC3 demonstrates that HDAC3 protein expression is unchanged in E18.5 Klf4 mutant epidermis. (I–N) Immunofluorescence for the basal layer marker K14, spinous layer marker K10 (I,J) and granular layer markers LOR (K,L), and FLG (M,N) demonstrates that E18.5 Klf4 mutant epidermis stratifies and expresses terminal differentiation markers, but LOR and FLG expression are decreased and patchy. (O,P) Immunofluorescence for K14 and KLF4 in E18.5 Hdac3 mutant and control epidermis demonstrates that KLF4 expression is not affected by loss of HDAC3. (Q,R) Immunofluorescence for K14 and KLF4 in E18.5 Ncor1/Ncor2 mutant and control epidermis demonstrates that KLF4 expression is not affected by loss of NCoR/SMRT. (S) Venn diagram demonstrating significant overlap in transcript cluster IDs up-regulated upon Hdac3 deletion and up-regulated upon Klf4 deletion. P < 0.0001, two-tailed χ2 test with Yates correction. (T) DAVID analysis for genes up-regulated in both Hdac3 mutant epidermis and Klf4 mutant epidermis reveals that these commonly up-regulated genes are involved in keratinocyte differentiation, inflammation, cell–cell adhesion and lipid biosynthesis. (U–U′′) HDAC3 and KLF4 colocalize to suprabasal nuclei in wild-type E18.5 epidermis. Scale bars, 25 µm. (V) HDAC3 and KLF4 coimmunoprecipitate in wild-type E18.5 epidermis. (W–Z) Proximity ligation assay demonstrates that HDAC3 and KLF4 lie within 40 nm of each other in suprabasal nuclei of wild-type E18.5 epidermis (Z). Positive control using two different antibodies to HDAC3 (W) and negative controls using a single antibody (X) and Hdac3-deleted epidermis (Y) provide assay validation.
Klf4 mutant epidermis showed compaction of the cornified layer, which was also observed in Hdac3 and Ncor1/Ncor2 mutants (Fig. 6C,D). Klf4 mutant epidermis stratified and expressed markers of the basal, spinous, and granular layers (Fig. 6I–N). Notably, LOR expression was patchy and decreased (Fig. 6K,L). FLG expression was disorganized and also diminished (Fig. 6M,N). The decreased expression of both LOR and FLG is consistent with previously published work establishing a role for KLF4 in promoting expression of a subset of epidermal differentiation genes (Boxer et al. 2014). These data indicate that KLF4's functions in the epidermis partially overlap with those of HDAC3 and are also consistent with additional roles for KLF4 as a transcriptional activator.
To determine whether KLF4 and HDAC3 share a subset of target genes, we performed transcriptional profiling of epidermis from five Klf4 mutants and six control littermates at E18.5 and compared the results with the gene expression changes found in Hdac3 mutant epidermis (Supplemental Tables S11, S12). Strikingly, over 40% of transcript cluster IDs up-regulated in Hdac3 mutants were also up-regulated in Klf4 mutants, which was highly statistically significant (Fig. 6S). We performed DAVID functional annotation analyses on commonly up-regulated genes and found that these genes are involved in multiple biologic processes including keratinocyte differentiation and inflammatory signaling (Fig. 6T).
HDAC3 and KLF4 interact in embryonic epidermis and show overlapping binding at genomic loci
Immunofluorescence for HDAC3 (Fig. 6U) and KLF4 (Fig. 6U′) in wild-type E18.5 epidermis showed that these proteins are coexpressed in suprabasal cell nuclei (Fig. 6U′′), and coimmunoprecipitation demonstrated that HDAC3 complexes with KLF4 (Fig. 6V). In line with these results, proximity ligation assay, which detects in situ protein–protein interaction (Fredriksson et al. 2002; Bagchi et al. 2015), showed that HDAC3 and KLF4 proteins lie within 40 nm of each other in wild-type E18.5 suprabasal nuclei (Fig. 6W–Z). This finding is consistent with studies demonstrating that KLF4 complexes with HDAC3 to repress target gene expression in intestinal epithelial cells and odontoblasts (Evans et al. 2007; Tao et al. 2019).
To determine whether HDAC3 and KLF4 localize to overlapping sites in the genome, we performed ChIP-seq for KLF4 in wild-type E18.5 epidermis (Supplemental Table S13) and compared the results with our HDAC3 ChIP-seq data (Supplemental Table S9). We found that >25% of HDAC3 binding sites were also occupied by KLF4, which was highly statistically significant (Fig. 7A; Supplemental Table S14). HOMER motif enrichment analysis showed that the KLF motif was enriched at shared HDAC3/KLF4 peaks, validating our ChIP-seq data. In addition, GRHL, AP1, and C/EBP motifs were enriched at these binding sites, indicating the possible existence of multi-protein repressive complexes at a subset of target genes (Fig. 7B; Supplemental Table S15). In line with this, the AP1 predicted binding site was among the most significantly enriched motifs in the promoters of genes up-regulated in Hdac3 mutants (Fig. 3D; Supplemental Table S5) and the GRHL and C/EBP motifs were also significantly enriched in this data set (Supplemental Table S5).
Figure 7.

HDAC3 and KLF4 coregulate a subset of target genes, but HDAC3 binding to these targets is independent of KLF4. (A) Venn diagram demonstrating significant overlap in HDAC3 and KLF4 ChIP-seq peaks in wild-type E18.5 epidermis. P < 0.0001, two-tailed χ2 test with Yates correction. (B) HOMER motif analysis demonstrates that the GRHL, AP1, KLF, and C/EBP motifs are enriched in shared HDAC3/KLF4 ChIP-seq peaks. (C–E) ChIP-seq tracts for HDAC3 (blue) and KLF4 (red) showing overlap in HDAC3- and KLF4-binding sites at Tgm1 (C), Krt16 (D), and Aqp3 (E). HOMER-called peaks are indicated by filled blue (HDAC3) or red (KLF4) rectangles under the ChIP-seq tracts. AP1-binding motifs are indicated by a circle, GRHL-binding motifs are indicated by a triangle, and KLF-binding motifs are indicated by a star. C/EBP binding motifs were not present at these called peaks. (F,G) RNAscope for Krt14 and Tgm1 demonstrates that Tgm1 expression is up-regulated and expanded in E18.5 Hdac3 mutant epidermis; white arrowheads indicate inappropriate expression in basal and immediately suprabasal cells. (H,I) RNAscope for Krt14 and Tgm1 demonstrates that Tgm1 expression is up-regulated and expanded in E18.5 Klf4 mutant epidermis; white arrowheads indicate inappropriate expression in basal and immediately suprabasal cells. (J,K) Immunostaining for K14 and K16 reveals that K16 expression is increased and expanded in suprabasal cells in Hdac3 mutant epidermis. (L,M) Immunostaining for K14 and K16 reveals that K16 expression is increased and expanded in suprabasal cells in Klf4 mutant epidermis. (N,O) Immunostaining for K14 and aquaporin 3 (AQP3) reveals that AQP3 expression is increased in suprabasal cells in Hdac3 mutant epidermis. (P,Q) Immunostaining for K14 and AQP3 reveals that AQP3 expression is increased in suprabasal cells in Klf4 mutant epidermis. (R) ChIP-qPCR for IgG (open symbols) and KLF4 (closed symbols) in control (black symbols) and Klf4-null (red symbols) epidermis validates KLF4 binding at two different loci at Tgm1, as well as binding at Krt16 and Aqp3; n > 3 independent biological samples for each experiment. (*) P < 0.05; (**) P < 0.01; (****) P < 0.0001, ordinary one-way ANOVA with multiple comparisons. Error bars indicate SEM. (S) ChIP-qPCR for IgG (open symbols) and HDAC3 (closed symbols) in control (black symbols) and Klf4-null (blue symbols) epidermal chromatin demonstrates that HDAC3 binding at two different loci at Tgm1, as well as binding at Krt16 and Aqp3, is enriched in control epidermis relative to control IgG and is not dependent on KLF4. A sequence within exon 2 of the insulin gene (Ins1) was used as a negative control locus. n = 3 independent biological samples in each experiment. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001, ordinary one-way ANOVA with multiple comparisons. Error bars indicate SEM. ChIP-seq and all ChIP-qPCR experiments were performed on epidermis that had been separated from whole skin. Scale bars, 10 µm.
HDAC3 and KLF4 function both together and independently to regulate a subset of differentiation genes
To identify candidate genes that might be coregulated by HDAC3 and KLF4, we focused on those that were up-regulated upon both Hdac3 deletion and Klf4 deletion and had overlapping peaks for HDAC3 and KLF4 binding in the vicinity of the gene body (Supplemental Table S16). Genes that met these criteria included many epidermal differentiation genes, including envoplakin, transglutaminase 1, β-defensin 1, and members of the late cornified envelope (LCE), short protein-rich repeat protein (SPRR) and S100 families; genes that are up-regulated in response to damage, including keratins 6a, 16, and 17 (McGowan and Coulombe 1998); and aquaporin-3 (Apq3), encoding a glycerol and water channel that shows abnormal expression in multiple skin disorders (Voss et al. 2011).
To determine whether HDAC3 and KLF4 directly coregulate gene expression, we first determined whether putative shared targets, including Tgm1, Krt16, Aqp3, Sprr1a, and Krt17 (Fig. 7C–E; Supplemental Figs. S7A,B), were up-regulated in the same cell types in both mutants. RNAscope (Wang et al. 2012) demonstrated that Tgm1 was prematurely expressed in suprabasal cells in both mutants (Fig. 7F–I), and immunofluorescence demonstrated that expression of both keratin 16 (Fig. 7J–M) and AQP3 (Fig. 7N–Q) was increased in suprabasal cells in both mutants. These data are consistent with a model in which KLF4 and HDAC3 interact in suprabasal epidermis to corepress Tgm1, Krt16, and Aqp3.
Like Tgm1, Krt16, and Aqp3, Sprr1a and Krt17 were highly up-regulated in both Hdac3 and Klf4 mutant epidermis. However, we found that Sprr1a was prematurely expressed in suprabasal cells in Hdac3 mutants but was up-regulated in its normal location in the granular layer in Klf4 mutants (Supplemental Fig. S7C–F). Krt17 expression, which is normally restricted to hair follicles, was broadly expressed in the basal layer of Hdac3 mutant epidermis, but was up-regulated only in hair follicle-associated cells in Klf4 mutants (Supplemental Fig. S7G–J). These data suggest that, unlike their coordinate actions at the Tgm1, Krt16, and Aqp3 genes, HDAC3 and KLF4 act independently and in different cell types to repress Sprr1a and Krt17.
Analysis of NS-DAD mutants indicated that HDAC3's requirements in epidermal stratification are largely independent of its histone deacetylase activity. Previous findings showed that, while histone hyperacetylation is increased and target genes are up-regulated in Hdac3-deleted liver cells, introduction of deacetylase-dead HDAC3 rescued HDAC3-mediated transcriptional repression but not histone hyperacetylation (Sun et al. 2013). Thus, although not playing a major role in transcriptional repression, HDAC3's enzymic activity can still cause deacetylation of histones at its target genes. In line with this, we found evidence of increased acetylation of H3K9 at Tgm1, Krt16, and Aqp3 loci in Hdac3 mutant compared with control epidermal chromatin (Supplemental Fig. S8).
ChIP-qPCR for KLF4 (Fig. 7R) and HDAC3 (Fig. 7S) confirmed that these factors localize specifically to regions of Tgm1, Krt16, and Aqp3 that contain overlapping ChIP-seq peaks for HDAC3 and KLF4. We therefore asked whether localization of HDAC3 to these loci is dependent on KLF4. ChIP-qPCR for HDAC3 showed that HDAC3 binding was enhanced relative to control IgG at these sites in both control and Klf4-null epidermis (Fig. 7S), demonstrating that KLF4 is not required to recruit HDAC3 to these targets. Interestingly, several of the interrogated loci also contain binding motifs for AP1 and/or GRHL transcription factors (Fig. 7C,E; Supplemental Fig. S7A,B), consistent with the motif enrichment analysis of HDAC3 peaks and HDAC3/KLF4 shared peaks (Figs. 5B, 7B; Supplemental Tables S10, S15). While these data do not preclude the existence of additional sites that require KLF4 for HDAC3 localization, our results suggest that HDAC3 and KLF4 can participate in large multiprotein complexes containing AP1 and/or GRHL family members, and indicate that HDAC3 recruitment to a subset of its targets could be dependent on AP1 or GRHL transcription factors, rather than KLF4.
Discussion
Unlike other class I HDACs that have broad general functions, HDAC3 operates in a highly tissue-specific manner by associating with cell type-specific transcription factors (Emmett and Lazar 2019). Here, we show that HDAC3 plays an essential role in establishing the epidermal barrier during embryonic development. Epidermal deletion of either HDAC3 or its corepressors NCoR and SMRT results in failure of the normal, orderly process of epidermal stratification, expansion of basal layer markers, and premature expression of differentiation genes. We found that stability of epidermal HDAC3 in vivo is dependent on NCoR and SMRT; however, HDAC3's functions in regulating epidermal stratification appear to be independent of its histone deacetylase activity as mice homozygous for point mutations in the NCoR and SMRT deacetylase-activating domains are viable and lack epidermal differentiation defects. Our data show that HDAC3-bound sites are enriched for predicted binding motifs for critical epidermal transcription factors including AP1, GRHL, and KLF family members. Among these, HDAC3 functions with KLF4 to corepress Tgm1, Krt16, and Aqp3, and independently from KLF4 in repressing Sprr1a and Krt17. Interestingly, HDAC3 localization to the shared HDAC3/KLF4 target sites analyzed in this study was independent of KLF4, indicating importance of additional factors and suggesting that these function in multi-protein complexes to mediate repression.
The epidermal phenotypes caused by Hdac3 or Ncor1/Ncor2 deletion differed from the effects of codeletion of two other members of the class I HDAC family, Hdac1 and Hdac2. In contrast to disordered differentiation in Hdac3 or Ncor1/Ncor2 mutants, Hdac1/Hdac2 double-mutant epidermis completely failed to stratify and exhibited progressive failure of proliferation and survival of epidermal progenitor cells that could be explained in part by derepression of the senescence gene p16/Ink4 and hyperacetylation of p53 (LeBoeuf et al. 2010). These observations are in line with the known association of HDAC1/2 and HDAC3 with distinct transcriptional repressor complexes (Yang and Seto 2008).
Unlike HDAC1/2, HDAC3's functions in embryonic epidermal stratification appear to be independent of its histone deacetylase activity, suggesting that it may function primarily as a scaffolding factor in this context. While our studies focused on embryonic epidermal development, it will be important in the future to elucidate whether or not HDAC3's roles in postnatal epidermis in homeostasis and in hyperproliferative skin conditions require its enzymatic functions. If not, therapeutics designed to block the deacetylase activity of class I HDACs may partially mimic the effects of genetic loss of Hdac1/2 in the epidermis but would not be expected to reproduce phenotypes caused by Hdac3 deletion.
Our results showed highly significant overlap in genes dysregulated in Hdac3 and Ncor1/Ncor2 mutant epidermis. However, premature expression of differentiation genes was more pronounced in Ncor1/Ncor2 epidermal mutants than in Hdac3 mutants, and Ncor1/Ncor2 mutants displayed an additional phenotype of epidermal thickening that was not apparent in Hdac3 mutants. These observations indicate that NCoR/SMRT perform additional, HDAC3-independent roles in the epidermis. This will be an interesting area for future studies.
Embryonic epidermis lacking either HDAC3 or NCoR/SMRT was characterized by increased expression of inflammatory genes such as Ccl20 and Tnf that are known to be regulated in part through NF-κB signaling. In line with this, NF-κB predicted binding sites were enriched in the promoters of up-regulated genes in Hdac3 mutant epidermis, and enhanced expression of inflammatory genes in Hdac3 mutants was rescued by codeletion of Rela. Importantly, Rela deletion did not rescue perinatal lethality, disordered stratification, or premature expression of differentiation markers in Hdac3 mutant epidermis, indicating that these phenotypes are not caused by dysregulated expression of inflammatory molecules.
HDAC3 peaks and the promoters of genes up-regulated upon loss of Hdac3 were enriched for the AP1-, GRHL-, KLF-, and C/EBP-binding motifs, suggesting functional interactions between HDAC3 and members of these transcription factor families. We chose to focus on potential interactions of HDAC3 with KLF4, which has a well-established role in epidermal development. Our data showed that HDAC3 and KLF4 coregulate a subset of target genes, including Tgm1, Krt16, and Aqp3. Our finding that HDAC3 represses expression of Aqp3 is in line with previous studies performed in cultured primary murine keratinocytes (Choudhary et al. 2017); our results further suggest a novel function for KLF4 as a corepressive partner for HDAC3 in regulation of Aqp3 expression.
Interestingly, we found that KLF4 is not required for HDAC3 stability or for its binding at these loci, suggesting that HDAC3 localization may depend on additional members of a multiprotein repressive complex. In agreement with motif enrichment analyses, most of the HDAC3/KLF4 binding sites that we studied in detail contain predicted AP1 and/or GRHL consensus motifs. For example, one of the Tgm1 loci we examined contains a predicted GRHL binding site, suggesting that both HDAC3 and KLF4 may be targeted to this site by a GRHL family member. In line with this, GRHL3 is known to bind the Tgm1 gene locus and control its expression (Boglev et al. 2011). Predicted AP1 and C/EBP binding sites were also highly significantly enriched in HDAC3 binding peaks, including at some sites that are also bound by KLF4; thus, in addition to GRHL family members, AP1 and C/EBP are candidates as factors that target HDAC3 to chromatin. Consistent with this, recent evidence shows that AP1, GRHL, and C/EBP family members function to establish chromatin accessibility by interacting with chromatin remodeling factors, and can recruit additional transcription factors to their target sites in various tissues (Biddie et al. 2011; Grøntved et al. 2013; Vierbuchen et al. 2017; Chen et al. 2018; Jacobs et al. 2018). In future studies, it will be important to determine whether one or more of these transcription factors is required for HDAC3 recruitment to its targets. In addition, further studies may identify additional sites in epidermal chromatin that require KLF4, and/or other KLF family members, for HDAC3 localization.
Our studies highlight the role of KLF4 in repressing aberrant expression of a subset of epidermal genes, and are in line with previous data demonstrating up-regulation of Sprr family members in Klf4-deleted epidermis and cornea (Patel et al. 2003; Swamynathan et al. 2008). However, KLF4 also functions to activate specific sets of epidermal differentiation genes (Sen et al. 2012; Boxer et al. 2014). Our microarray data are consistent with these dual roles for KLF4, as we identified a substantial number of down-regulated genes as well as upregulated genes in Klf4 mutant epidermis.
In summary, our data suggest a model in which HDAC3 represses its targets by scaffolding a multiprotein complex that includes NCoR and SMRT, which are required for HDAC3 stability, and interacts with KLF4 to repress a subset of its transcriptional targets. However, KLF4 is not required for HDAC3's recruitment to several target sites examined in this study; thus, additional transcription factor(s) are likely to recruit HDAC3 to these sites. HDAC3 binding peaks, and genes up-regulated in Hdac3 and Ncor1/Ncor2 mutants, are highly significantly enriched for AP1, GRHL, and C/EBP, as well as KLF, binding motifs, identifying members of these transcription factor families as candidate factors that target HDAC3 to its binding sites. Our detailed analyses of several HDAC3 binding peaks reveal that these often contain motifs for several different transcription factor families, suggesting that repression is achieved by complexes containing specific combinations of multiple transcription factors. This unexpected complexity sheds new light on the mechanisms by which HDAC3 achieves target specificity. While the current study focused on the developing epidermis, our findings provide a paradigm for understanding HDAC3's functions and mechanisms of action in other tissues.
Materials and methods
Generation of mice
Mice were maintained on a mixed C57BL/6/FVB/N/SJL background in a specific pathogen-free barrier facility on a standard light–dark cycle. Mice carrying the Hdac3 floxed allele (McQuown et al. 2011) were crossed with mice carrying the K5-rtTA (Diamond et al. 2000) and tetO-Cre transgenes (Perl et al. 2002). K5-rtTA tetO-Cre Hdac3fl/fl animals were considered Hdac3 mutants. Littermates homozygous for the Hdac3 floxed allele lacking K5-rtTA, tetO-Cre, or both were used as controls. For experiments using the K14-Cre transgene (Dassule et al. 2000), K14-Cre Hdac3fl/fl animals were considered mutants; K14-Cre Hdac3fl/+ and Hdac3fl/fl littermates were used as controls. The same experimental setup as described for the K5-rtTA tetO-Cre Hdac3 floxed experiments was used for the Klf4 floxed (Katz et al. 2002), Ncor1 floxed (Yamamoto et al. 2011), Ncor2 floxed (Shimizu et al. 2015), and Ncor1 floxed/Ncor2 floxed mice. For the Hdac3 floxed/Rela floxed (Heise et al. 2014) experiments, K5-rtTA tetO-Cre Hdac3fl/fl Relafl/+ males were crossed with Hdac3fl/fl Relafl/fl females to generate K5-rtTA tetO-Cre Hdac3fl/fl Relafl/fl genetic rescue animals, K5-rtTA tetO-Cre Hdac3fl/fl Relafl/+ Hdac3 mutant animals and control animals lacking K5-rtTA, tetO-Cre or both. For experiments using NCoR deacetylase-activating domain (DAD) mutant mice (Alenghat et al. 2008) and SMRT DAD mutant mice (You et al. 2013), experimental animals were homozygous for both mutant alleles. Crossing double-heterozygous animals to generate double-homozygous mutants and wild-type littermate controls is highly inefficient, so double-homozygous and wild-type lines were bred separately to generate experimental and control animals.
For timed mating experiments, embryonic ages were determined from the time of appearance of a vaginal plug (E0.5). To induce gene deletion, females were fed chow containing 1 g/kg doxycycline from E8.5 to the time of embryo collection. All experiments were performed following approved animal protocols according to institutional guidelines established by the University of Pennsylvania IACUC committee. Embryos of both sexes were analyzed, and no sex-specific differences were observed.
Dye exclusion assay
Dye exclusion assays were performed as described previously (Hébert et al. 1994).
Histology and immunostaining
Immunostaining was performed as described previously (Zhang et al. 2009). See Supplemental Table S17 for antibody information.
Epidermal isolation
Following euthanasia, skin was removed and floated dermis side down on 2.4 U/mL Dispase II (Gibco) in PBS for 75 min at 37°C. Epidermis was peeled from dermis using fine forceps.
Preparation of cell envelopes
Epidermis was isolated and heated for 10 min to 100°C in the presence of 2% SDS and 25 mM DTT. Corneocytes were examined and photographed by phase-contrast microscopy.
Transmission electron microscopy
Fresh skin was dissected into small pieces and fixed by immersion in 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M PBS (pH 7.4) at room temperature. Tissue was postfixed with 0.2% ruthenium tetroxide, dehydrated, and embedded in agar 100 resin. Ultrathin sections were contrasted with uranyl acetate and lead citrate and examined with a transmission electron microscope.
Proximity ligation assay
Proximity ligation assay was performed as described (Bagchi et al. 2015), with modifications (see the Supplemental Material).
Microarray analysis
Epidermis was collected as described and homogenized in TRIzol reagent (Invitrogen). Total RNA was extracted using the RNeasy kit (Qiagen). Microarray services were provided by the UPENN Molecular Profiling Facility, including quality control tests of the total RNA samples by Agilent Bioanalyzer and Nanodrop spectrophotometry. All protocols were conducted as described in the Affymetrix WT Plus reagent kit manual and the Affymetrix GeneChip expression analysis technical manual. See the Supplemental Material for detailed protocols and a description of bioinformatic analyses.
Microarray data sets are available in the GEO repository (GSE137233 and GSE137234).
Quantification of transcript levels by qPCR
Epidermis was collected and total RNA was extracted as described. RNA (0.5–2 µg) was reverse-transcribed using the high-capacity cDNA reverse transcription kit (Applied Biosystems). Complementary DNA was analyzed by qPCR with technical replicates in triplicate using the ViiA 7 system and software (Applied Biosystems) and Power SYBR Green PCR master mix (Applied Biosystems). Data were normalized to 18s expression. Primer sequences are listed in Supplemental Table S18.
RNAscope
RNAscope was performed on frozen tissue sections following the user manual provided by Advanced Cell Diagnostics. The following probes were used: Mm 3-plex-positive control probe (ACD 320881), 3-plex-negative control (ACD 320871), Mm-Krt14 (ACD 422521), Mm-Sprr1a-C2 (ACD 426871-C2), and Mm-Tgm1-C3 (custom). Fluorescein was used at 1:1500 for C1 probe and Cy3 was used at 1:1500 for C2 and C3 probes.
Immunoprecipitation and Western blotting from isolated epidermis
Epidermis was isolated as described. Samples for Western blot analysis were homogenized in RIPA buffer (Santa Cruz Biotechnology 24948). Coimmunoprecipitations were performed using the nuclear complex co-IP kit (Active Motif 54001). See Supplemental Table S17 for antibody information.
ChIP-seq library preparation and sequencing
ChIP was performed independently on samples of separated epidermis from at least 12 different embryos, and the input DNA and precipitated DNA samples were pooled separately. Ten nanograms of pooled DNA was then amplified according to the ChIP sequencing sample preparation guide by Illumina, using adaptor oligomers and primers from Illumina and enzymes from New England Biolabs. The resulting libraries were purified with the PCR purification kit (Qiagen) and MinElute kit (Qiagen). Libraries were assayed for overall quality and sequenced on an Illumina HiSeq 2000 sequencer. See the Supplemental Material for detailed protocols and a description of bioinformatic analyses.
ChIP-seq data sets are available in the GEO repository (GSE137232).
ChIP-qPCR
Epidermis was separated from dermis as described above and ChIP was performed as previously described (Emmett et al. 2017) with modifications (see the Supplemental Material). See Supplemental Table S17 for antibody information.
DNA was analyzed by quantitative PCR with technical replicates in triplicate using the ViiA 7 system and software and Power SYBR Green PCR Master Mix. A sequence from the insulin gene (Ins1) was used as a negative control (Feng et al. 2011). See Supplemental Table S18 for primer information.
Quantification and statistical analysis
We used n = 5 control and n = 5 experimental mice wherever possible, providing 80% power at a two-sided significance level of 0.05 to detect a difference (effect size) of 2.0s where s is the standard deviation; at a minimum, we used n = 3 control and n = 3 experimental mice, providing 80% power at a two-sided significance level of 0.05 to detect a difference of 2.8s. Statistical tests were chosen based on experimental parameters and are described in the relevant figure legends.
Supplementary Material
Acknowledgments
We thank Stephen Prouty and Tzvete Dentchev for technical assistance with histology and RNAscope, Donna Brennan-Crispi for technical support with RNAscope, David Steger for guidance related to ChIP-seq experiments, and Yong Kim for assistance with ChIP-qPCR. Additionally, we thank Kristen Lynch, George Cotsarelis, Edward Morrisey, and Elizabeth Grice for their helpful advice. This work was supported by National Institutes of Health (NIH) grants R01AR063146 and R01AR076428 (SEM), NIH grant R01DK43806 (MAL), NIH grants F31AR072461 and T32HL007971 (KMS), and the Penn Skin Biology and Diseases Resource-based Center NIH grant P30AR069589.
Author contributions: K.M.S., F.L., M.X., and S.E.M. designed the study with help from J.W.T., M.A.L., J.P.K., K.H.K., and R.N.C. K.M.S., F.L., C.Y.Y., J.H., H.R.G., D.J.M., and M.X. performed experiments. K.M.S., F.L., J.T.S., J.W.T., M.X., and S.E.M. interpreted data. M.A.L., J.P.K., K.H.K., and R.N.C. provided mouse models. K.M.S. and S.E.M. wrote the manuscript with input from co-authors.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.333674.119.
References
- Alenghat T, Meyers K, Mullican SE, Leitner K, Adeniji-Adele A, Avila J, Bućan M, Ahima RS, Kaestner KH, Lazar MA. 2008. Nuclear receptor corepressor and histone deacetylase 3 govern circadian metabolic physiology. Nature 456: 997–1000. 10.1038/nature07541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alenghat T, Osborne LC, Saenz SA, Kobuley D, Ziegler CG, Mullican SE, Choi I, Grunberg S, Sinha R, Wynosky-Dolfi M, et al. 2013. Histone deacetylase 3 coordinates commensal-bacteria-dependent intestinal homeostasis. Nature 504: 153–157. 10.1038/nature12687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attar AS, Wertz PW, MacArthur M, Imakado S, Bickenbach JR, Roop DR. 1997. Inhibition of retinoid signaling in transgenic mice alters lipid processing and disrupts epidermal barrier function. Mol Endocrinol 11: 792–800. 10.1210/mend.11.6.0010 [DOI] [PubMed] [Google Scholar]
- Bagchi S, Fredriksson R, Wallén-Mackenzie Å. 2015. In situ proximity ligation assay (PLA). Methods Mol Biol 1318: 149–159. 10.1007/978-1-4939-2742-5_15 [DOI] [PubMed] [Google Scholar]
- Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, Hiebert SW. 2008. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell 30: 61–72. 10.1016/j.molcel.2008.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung MH, Trump S, Lightman SL, et al. 2011. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell 43: 145–155. 10.1016/j.molcel.2011.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boglev Y, Wilanowski T, Caddy J, Parekh V, Auden A, Darido C, Hislop NR, Cangkrama M, Ting SB, Jane SM. 2011. The unique and cooperative roles of the Grainy head-like transcription factors in epidermal development reflect unexpected target gene specificity. Dev Biol 349: 512–522. 10.1016/j.ydbio.2010.11.011 [DOI] [PubMed] [Google Scholar]
- Boxer LD, Barajas B, Tao S, Zhang J, Khavari PA. 2014. ZNF750 interacts with KLF4 and RCOR1, KDM1A, and CTBP1/2 chromatin regulators to repress epidermal progenitor genes and induce differentiation genes. Genes Dev 28: 2013–2026. 10.1101/gad.246579.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne C, Tainsky M, Fuchs E. 1994. Programming gene expression in developing epidermis. Development 120: 2369–2383. [DOI] [PubMed] [Google Scholar]
- Carpio LR, Bradley EW, McGee-Lawrence ME, Weivoda MM, Poston DD, Dudakovic A, Xu M, Tchkonia T, Kirkland JL, van Wijnen AJ, et al. 2016. Histone deacetylase 3 supports endochondral bone formation by controlling cytokine signaling and matrix remodeling. Sci Signal 9: ra79 10.1126/scisignal.aaf3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Fischle W, Verdin E, Greene WC. 2001. Duration of nuclear NF-κB action regulated by reversible acetylation. Science 293: 1653–1657. 10.1126/science.1062374 [DOI] [PubMed] [Google Scholar]
- Chen AF, Liu AJ, Krishnakumar R, Freimer JW, DeVeale B, Blelloch R. 2018. GRHL2-dependent enhancer switching maintains a pluripotent stem cell transcriptional subnetwork after exit from naive pluripotency. Cell Stem Cell 23: 226–238.e4. 10.1016/j.stem.2018.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary V, Olala LO, Kagha K, Pan ZQ, Chen X, Yang R, Cline A, Helwa I, Marshall L, Kaddour-Djebbar I, et al. 2017. Regulation of the glycerol transporter, Aquaporin-3, by histone deacetylase-3 and p53 in keratinocytes. J Invest Dermatol 137: 1935–1944. 10.1016/j.jid.2017.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassule HR, Lewis P, Bei M, Mass R, McMahon AP. 2000. Sonic hedgehog regulates growth and morphogenesis of the tooth. Development 127: 4775–4785. [DOI] [PubMed] [Google Scholar]
- Diamond I, Owolabi T, Marco M, Lam C, Glick A. 2000. Conditional gene expression in the epidermis of transgenic mice using the tetracycline-regulated transactivators tTA and rTA linked to the keratin 5 promoter. J Invest Dermatol 115: 788–794. 10.1046/j.1523-1747.2000.00144.x [DOI] [PubMed] [Google Scholar]
- Driskell I, Oda H, Blanco S, Nascimento E, Humphreys P, Frye M. 2012. The histone methyltransferase Setd8 acts in concert with c-Myc and is required to maintain skin. EMBO J 31: 616–629. 10.1038/emboj.2011.421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvic M. 2015. Histone deacetylase inhibitors for cutaneous T-cell lymphoma. Dermatol Clin 33: 757–764. 10.1016/j.det.2015.05.010 [DOI] [PubMed] [Google Scholar]
- Eckert RL. 1989. Structure, function, and differentiation of the keratinocyte. Physiol Rev 69: 1316–1346. 10.1152/physrev.1989.69.4.1316 [DOI] [PubMed] [Google Scholar]
- Eckert RL, Broome AM, Ruse M, Robinson N, Ryan D, Lee K. 2004. S100 proteins in the epidermis. J Invest Dermatol 123: 23–33. 10.1111/j.0022-202X.2004.22719.x [DOI] [PubMed] [Google Scholar]
- Emmett MJ, Lazar MA. 2019. Integrative regulation of physiology by histone deacetylase 3. Nat Rev Mol Cell Biol 20: 102–115. 10.1038/s41580-018-0076-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmett MJ, Lim HW, Jager J, Richter HJ, Adlanmerini M, Peed LC, Briggs ER, Steger DJ, Ma T, Sims CA, et al. 2017. Histone deacetylase 3 prepares brown adipose tissue for acute thermogenic challenge. Nature 546: 544–548. 10.1038/nature22819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PM, Zhang W, Chen X, Yang J, Bhakat KK, Liu C. 2007. Kruppel-like factor 4 is acetylated by p300 and regulates gene transcription via modulation of histone acetylation. J Biol Chem 282: 33994–34002. 10.1074/jbc.M701847200 [DOI] [PubMed] [Google Scholar]
- Ezhkova E, Lien WH, Stokes N, Pasolli HA, Silva JM, Fuchs E. 2011. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev 25: 485–498. 10.1101/gad.2019811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil R, Brocard J, Mascrez B, LeMeur M, Metzger D, Chambon P. 1996. Ligand-activated site-specific recombination in mice. Proc Natl Acad Sci 93: 10887–10890. 10.1073/pnas.93.20.10887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat E, Liu XS, Lazar MA. 2011. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science 331: 1315–1319. 10.1126/science.1198125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Östman A, Gústafsdóttir SM, Landegren U. 2002. Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol 20: 473–477. 10.1038/nbt0502-473 [DOI] [PubMed] [Google Scholar]
- Fuchs E. 2007. Scratching the surface of skin development. Nature 445: 834–842. 10.1038/nature05659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisletti S, Huang W, Jepsen K, Benner C, Hardiman G, Rosenfeld MG, Glass CK. 2009. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev 23: 681–693. 10.1101/gad.1773109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grøntved L, John S, Baek S, Liu Y, Buckley JR, Vinson C, Aguilera G, Hager GL. 2013. C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J 32: 1568–1583. 10.1038/emboj.2013.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Barak O, Lazar MA. 2001. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol 21: 6091–6101. 10.1128/MCB.21.18.6091-6101.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Gow CH, Li Y, Gardner A, Khan S, Zhang J. 2012. Regulated clearance of histone deacetylase 3 protects independent formation of nuclear receptor corepressor complexes. J Biol Chem 287: 12111–12120. 10.1074/jbc.M111.327023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert JM, Rosenquist T, Götz J, Martin GR. 1994. FGF5 as a regulator of the hair growth cycle: evidence from targeted and spontaneous mutations. Cell 78: 1017–1025. 10.1016/0092-8674(94)90276-3 [DOI] [PubMed] [Google Scholar]
- Heise N, De Silva NS, Silva K, Carette A, Simonetti G, Pasparakis M, Klein U. 2014. Germinal center B cell maintenance and differentiation are controlled by distinct NF-κB transcription factor subunits. J Exp Med 211: 2103–2118. 10.1084/jem.20132613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashitsuji H, Higashitsuji H, Masuda T, Liu Y, Itoh K, Fujita J. 2007. Enhanced deacetylation of p53 by the anti-apoptotic protein HSCO in association with histone deacetylase 1. J Biol Chem 282: 13716–13725. 10.1074/jbc.M609751200 [DOI] [PubMed] [Google Scholar]
- Hong S, Zhou W, Fang B, Lu W, Loro E, Damle M, Ding G, Jager J, Zhang S, Zhang Y, et al. 2017. Dissociation of muscle insulin sensitivity from exercise endurance in mice by HDAC3 depletion. Nat Med 23: 223–234. 10.1038/nm.4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua G, Ganti KP, Chambon P. 2016a. Glucocorticoid-induced tethered transrepression requires SUMOylation of GR and formation of a SUMO-SMRT/NCoR1-HDAC3 repressing complex. Proc Natl Acad Sci 113: E635–E643. 10.1073/pnas.1522826113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua G, Paulen L, Chambon P. 2016b. GR SUMOylation and formation of an SUMO–SMRT/NCoR1–HDAC3 repressing complex is mandatory for GC-induced IR nGRE-mediated transrepression. Proc Natl Acad Sci 113: E626–E634. 10.1073/pnas.1522821113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009a. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. 2009b. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Huebner AJ, Dai D, Morasso M, Schmidt EE, Schäfer M, Werner S, Roop DR. 2012. Amniotic fluid activates the nrf2/keap1 pathway to repair an epidermal barrier defect in utero. Dev Cell 23: 1238–1246. 10.1016/j.devcel.2012.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J, Atkins M, Davie K, Imrichova H, Romanelli L, Christiaens V, Hulselmans G, Potier D, Wouters J, Taskiran II, et al. 2018. The transcription factor Grainy head primes epithelial enhancers for spatiotemporal activation by displacing nucleosomes. Nat Genet 50: 1011–1020. 10.1038/s41588-018-0140-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaubert J, Cheng J, Segre JA. 2003. Ectopic expression of kruppel like factor 4 (Klf4) accelerates formation of the epidermal permeability barrier. Development 130: 2767–2777. 10.1242/dev.00477 [DOI] [PubMed] [Google Scholar]
- Katz JP, Perreault N, Goldstein BG, Lee CS, Labosky PA, Yang VW, Kaestner KH. 2002. The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development 129: 2619–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Gallo RL. 2009. AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol 30: 131–141. 10.1016/j.it.2008.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBoeuf M, Terrell A, Trivedi S, Sinha S, Epstein JA, Olson EN, Morrisey EE, Millar SE. 2010. Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev Cell 19: 807–818. 10.1016/j.devcel.2010.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski SL, Janardhan HP, Trivedi CM. 2015. Histone deacetylase 3 coordinates deacetylase-independent epigenetic silencing of Transforming growth factor-β1 (TGF-β1) to orchestrate second heart field development. J Biol Chem 290: 27067–27089. 10.1074/jbc.M115.684753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang J, Wang J, Nawaz Z, Liu JM, Qin J, Wong J. 2000. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J 19: 4342–4350. 10.1093/emboj/19.16.4342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardaryev AN, Gdula MR, Yarker JL, Emelianov VU, Poterlowicz K, Sharov AA, Sharova TY, Scarpa JA, Joffe B, Solovei I, et al. 2014. p63 and Brg1 control developmentally regulated higher-order chromatin remodelling at the epidermal differentiation complex locus in epidermal progenitor cells. Development 141: 101–111. 10.1242/dev.103200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan K, Coulombe PA. 1998. The wound repair-associated keratins 6, 16, and 17. Insights into the role of intermediate filaments in specifying keratinocyte cytoarchitecture. Subcell Biochem 31: 173–204. [PubMed] [Google Scholar]
- McQuown SC, Barrett RM, Matheos DP, Post RJ, Rogge GA, Alenghat T, Mullican SE, Jones S, Rusche JR, Lazar MA, et al. 2011. HDAC3 is a critical negative regulator of long-term memory formation. J Neurosci 31: 764–774. 10.1523/JNEUROSCI.5052-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S, Humphries KM, Richardson JA, Bassel-Duby R, Olson EN. 2008. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J Clin Invest 118: 3588–3597. 10.1172/JCI35847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento EM, Cox CL, MacArthur S, Hussain S, Trotter M, Blanco S, Suraj M, Nichols J, Kübler B, Benitah SA, et al. 2011. The opposing transcriptional functions of Sin3a and c-Myc are required to maintain tissue homeostasis. Nat Cell Biol 13: 1395–1405. 10.1038/ncb2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ. 2009. Skin immune sentinels in health and disease. Nat Rev Immunol 9: 679–691. 10.1038/nri2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim JJ, Yang D. 2005. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol 17: 359–365. 10.1016/j.coi.2005.06.002 [DOI] [PubMed] [Google Scholar]
- Patel S, Kartasova T, Segre JA. 2003. Mouse Sprr locus: a tandem array of coordinately regulated genes. Mamm Genome 14: 140–148. 10.1007/s00335-002-2205-4 [DOI] [PubMed] [Google Scholar]
- Perl AK, Wert SE, Nagy A, Lobe CG, Whitsett JA. 2002. Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc Natl Acad Sci 99: 10482–10487. 10.1073/pnas.152238499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poleshko A, Shah PP, Gupta M, Babu A, Morley MP, Manderfield LJ, Ifkovits JL, Calderon D, Aghajanian H, Sierra-Pagán JE, et al. 2017. Genome-nuclear lamina interactions regulate cardiac stem cell lineage restriction. Cell 171: 573–587.e14. 10.1016/j.cell.2017.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remsberg JR, Ediger BN, Ho WY, Damle M, Li Z, Teng C, Lanzillotta C, Stoffers DA, Lazar MA. 2017. Deletion of histone deacetylase 3 in adult β cells improves glucose tolerance via increased insulin secretion. Mol Metab 6: 30–37. 10.1016/j.molmet.2016.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segre JA, Bauer C, Fuchs E. 1999. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet 22: 356–360. 10.1038/11926 [DOI] [PubMed] [Google Scholar]
- Sen GL, Webster DE, Barragan DI, Chang HY, Khavari PA. 2008. Control of differentiation in a self-renewing mammalian tissue by the histone demethylase JMJD3. Genes Dev 22: 1865–1870. 10.1101/gad.1673508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen GL, Reuter JA, Webster DE, Zhu L, Khavari PA. 2010. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature 463: 563–567. 10.1038/nature08683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen GL, Boxer LD, Webster DE, Bussat RT, Qu K, Zarnegar BJ, Johnston D, Siprashvili Z, Khavari PA. 2012. ZNF750 is a p63 target gene that induces KLF4 to drive terminal epidermal differentiation. Dev Cell 22: 669–677. 10.1016/j.devcel.2011.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto E, Yoshida M. 2014. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol 6: a018713 10.1101/cshperspect.a018713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H, Astapova I, Ye F, Bilban M, Cohen RN, Hollenberg AN. 2015. NCoR1 and SMRT play unique roles in thyroid hormone action in vivo. Mol Cell Biol 35: 555–565. 10.1128/MCB.01208-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Feng D, Fang B, Mullican SE, You SH, Lim HW, Everett LJ, Nabel CS, Li Y, Selvakumaran V, et al. 2013. Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol Cell 52: 769–782. 10.1016/j.molcel.2013.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swamynathan SK, Davis J, Piatigorsky J. 2008. Identification of candidate Klf4 target genes reveals the molecular basis of the diverse regulatory roles of Klf4 in the mouse cornea. Invest Ophthalmol Vis Sci 49: 3360–3370. 10.1167/iovs.08-1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. 2008. Acetylation is indispensable for p53 activation. Cell 133: 612–626. 10.1016/j.cell.2008.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H, Lin H, Sun Z, Pei F, Zhang J, Chen S, Liu H, Chen Z. 2019. Klf4 promotes dentinogenesis and odontoblastic differentiation via modulation of TGF-β signaling pathway and interaction with histone acetylation. J Bone Miner Res 34: 1502–1516. 10.1002/jbmr.3716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, Greenberg ME. 2017. AP-1 Transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol Cell 68: 1067–1082.e12. 10.1016/j.molcel.2017.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss KE, Bollag RJ, Fussell N, By C, Sheehan DJ, Bollag WB. 2011. Abnormal aquaporin-3 protein expression in hyperproliferative skin disorders. Arch Dermatol Res 303: 591–600. 10.1007/s00403-011-1136-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, Wu X, Vo HT, Ma XJ, Luo Y. 2012. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn 14: 22–29. 10.1016/j.jmoldx.2011.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Frank DB, Morley MP, Zhou S, Wang X, Lu MM, Lazar MA, Morrisey EE. 2016. HDAC3-dependent epigenetic pathway controls lung alveolar epithelial cell remodeling and spreading via miR-17-92 and TGF-β signaling regulation. Dev Cell 36: 303–315. 10.1016/j.devcel.2015.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Perissi V, Staszewski LM, Yang W, Krones A, Glass CK, Rosenfeld MG, Seto E. 2000. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc Natl Acad Sci 97: 7202–7207. 10.1073/pnas.97.13.7202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Li W, Shang C, Chen RM, Han P, Yang J, Stankunas K, Wu B, Pan M, Zhou B, et al. 2013. Brg1 governs a positive feedback circuit in the hair follicle for tissue regeneration and repair. Dev Cell 25: 169–181. 10.1016/j.devcel.2013.03.015 [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Williams EG, Mouchiroud L, Cantó C, Fan W, Downes M, Héligon C, Barish GD, Desvergne B, Evans RM, et al. 2011. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell 147: 827–839. 10.1016/j.cell.2011.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Carmody RJ, Qu Z, Ruan Q, Jager J, Mullican SE, Lazar MA, Chen YH. 2012. Nuclear factor-κB binding motifs specify Toll-like receptor-induced gene repression through an inducible repressosome. Proc Natl Acad Sci 109: 14140–14145. 10.1073/pnas.1119842109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XJ, Seto E. 2008. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 9: 206–218. 10.1038/nrm2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You SH, Lim HW, Sun Z, Broache M, Won KJ, Lazar MA. 2013. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat Struct Mol Biol 20: 182–187. 10.1038/nsmb.2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tomann P, Andl T, Gallant NM, Huelsken J, Jerchow B, Birchmeier W, Paus R, Piccolo S, Mikkola ML, et al. 2009. Reciprocal requirements for EDA/EDAR/NF-κB and Wnt/β-catenin signaling pathways in hair follicle induction. Dev Cell 17: 49–61. 10.1016/j.devcel.2009.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, He X, Liu L, Jiang M, Zhao C, Wang H, He D, Zheng T, Zhou X, Hassan A, et al. 2016. Hdac3 interaction with p300 histone acetyltransferase regulates the oligodendrocyte and astrocyte lineage fate switch. Dev Cell 36: 316–330. 10.1016/j.devcel.2016.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.