

Abstract
Pulmonary fibrosis is characterized by fibroblasts persisting in an activated form, producing excessive fibrous material that destroys alveolar structure. The second messenger molecule cyclic 3′,5′-adenosine monophosphate (cAMP) has antifibrotic properties, and prostaglandin E2 (PGE2) can stimulate cAMP production through prostaglandin E (EP)2 and EP4 receptors. Although EP receptors are attractive therapeutic targets, the effects of long-term exposure to PGE2 have not been characterized. To determine the effects of long-term exposure of lung fibroblasts to PGE2, human fetal lung (HFL)-1 cells were treated for 24 h with 100 nM PGE2 or other cAMP-elevating agents. cAMP levels stimulated by acute exposure to PGE2 were measured using a fluorescent biosensor. Pretreatment for 24 h with PGE2 shifted the concentration-response curve to PGE2 rightward by approximately 22-fold but did not affect responses to the beta-adrenoceptor agonist isoproterenol. Neither isoproterenol nor forskolin pretreatment altered PGE2 responses, implying that other cAMP-elevating agents do not induce desensitization. Use of EP2- and EP4-selective agonists and antagonists suggested that PGE2-stimulated cAMP responses in HFL-1 cells are mediated by EP2 receptors. EP2 receptors are resistant to classical mechanisms of agonist-specific receptor desensitization, so we hypothesized that increased PDE activity mediates the loss of signaling after PGE2 pretreatment. PGE2 treatment upregulated messenger RNA for PDE3A, PDE3B, PDE4B, and PDE4D and increased overall PDE activity. The PDE4 inhibitor rolipram partially reversed PGE2- mediated desensitization and PDE4 activity was increased, but rolipram did not alter responses to isoproterenol. The PDE3 inhibitor cilostazol had minimal effect. These results show that long-term exposure to PGE2 causes agonist-specific desensitization of EP2 receptor-stimulated cAMP signaling through the increased expression of PDE isozymes, most likely of the PDE4 family.
Keywords: PGE2, Pulmonary fibrosis, Phosphodiesterase, cAMP, EP2 receptors
Introduction
Pulmonary fibrosis (PF) is characterized by continuous scarring of the lungs (Dackor et al. 2011). Previously, PF was thought to result from an inflammatory response (Liu et al. 2004). However, anti-inflammatory treatments show little to no effect in slowing the progression of PF and clinical out-comes were not improved (Ostrom 2014). Current understanding of PF has shifted from it being an inflammatory process to a focus on tissue damage with defects in subsequent healing and repair processes and the cellular signaling pathways that regulate them (Liu et al. 2004).
The second messenger molecule cyclic 3′,5′-adenosine monophosphate (cAMP) will (1) slow fibroblast migration to the site of injury, (2) prevent differentiation of fibroblasts into myofibroblasts, (3) reduce the amount of collagen 1α(II) and 5α(I), and (4) increase the activity of certain matrix metalloproteinases, which are responsible for degrading ECM proteins (Yamaguchi et al. 1988; Kohyama et al. 2002a; Kolodsick et al. 2003; Liu et al. 2004). Thus, cAMP is an antifibrotic second messenger. cAMP is generated in the cell when certain agonists bind to their cognate G protein–coupled receptors (GPCRs) to activate the G protein Gαs, which, in turn, stimulates adenylyl cyclase (AC) (Liu et al. 2004). Once activated, AC converts adenosine triphosphate (ATP) into cAMP, which subsequently binds to protein kinase A (PKA) and the exchange protein activated by cAMP (Epac) (Liu et al. 2010; Insel et al. 2012). This, in turn, leads to regulation of a myriad of different proteins and pathways to produce different downstream responses (Liu et al. 2010). cAMP signaling is terminated by phosphodiesterase (PDE) enzymes that degrade cAMP and halt the signaling cascade (Liu et al. 2004). Although Gα has several different isoforms, only Gαs is responsible for stimulating cAMP production in the cell, and hence, the GαS-AC-cAMP pathway is pertinent to the study of cAMP and its antifibrotic effects (Liu et al. 2004).
Elevating cAMP levels for 24 h via treatment with forskolin, isoproterenol, prostaglandin E2 (PGE2), butaprost, or beraprost, or by overexpression of adenylyl cyclase 6 (AC6), inhibits fibroblast proliferation and collagen synthesis (Liu et al. 2004, 2008, 2010). Moreover, increasing cAMP levels over 24 h lowers messenger RNA (mRNA) levels of collagen types 1α(II) and 5α(I) (Liu et al. 2004). Exposure to these cAMP-elevating agents for 24 h also increases expression of matrix metalloproteinase-2 (Liu et al. 2004). PGE2, acting via prostaglandin E (EP)2 receptors stimulating cAMP in lung fibroblasts, disrupts calcium signaling and induces an antifibrotic phenotype (Mukherjee et al. 2019). Activation of cAMP response element–binding (CREB) protein via phosphorylation inhibits the profibrotic TGF-β signaling pathway by inhibiting SMAD-mediated transcriptional activation of fibrotic genes (Liu et al. 2005). cAMP elevation by prostacyclin receptors inhibits the transcription cofactors YAP/TAZ to oppose TGF-β–stimulated fibrotic responses (Zmajkovicova et al. 2019). Thus, cAMP induces diverse antifibrotic effects and approaches for increasing cAMP levels should be considered for PF therapy.
PGE2 is associated with inflammation and with repair of tissue injury (Liu et al. 2004). PGE2 has antifibrotic properties due to activation of the Gαs-AC-cAMP pathway (Liu et al. 2004). PGE2 activates all EP receptors, of which EP2 and EP4 isoforms couple to the Gαs-AC-cAMP pathway (Bozyk and Moore 2011). EP receptor subtypes EP2 and EP4 are involved in antifibrotic activity in fibroblast cells from various organs. Given the therapeutic potential of targeting these receptors for PF, understanding the effects of long-term exposure of receptors to PGE2 represents a novel and important basic research question. Furthermore, PF fibroblast cells are refractory to PGE2 receptor responses (Bozyk and Moore 2011), presumably diminishing the antifibrotic action of PGE2. Fibroblasts isolated from patients with chronic obstructive pulmonary disease also have altered responses to PGE2 as compared to those from normal subjects (Michalski et al. 2012). Thus, we hypothesize that PGE2 effects are self-limiting because of desensitization caused by prolonged agonist activation of EP receptors. Thus, identifying the EP receptors involved in pulmonary fibroblast cAMP signaling, establishing whether they desensitize upon prolonged agonist exposure, and identifying the mechanism involved in any desensitization are all critical for developing novel therapeutic approaches for PF.
GPCRs that signal via cAMP are desensitized by two primary mechanisms: GRK/β-arrestin–mediated uncoupling, internalization, and eventual downregulation of receptors, and/or increased expression of PDE activity. EP2 receptors have a short C-terminal tail, lack the residues for GRK phosphorylation, and thus do not internalize (Desai et al. 2000). Moreover, later studies established that EP2 receptors are not subject to other classical mechanisms of GRK/arrestin-mediated desensitization of GPCRs (Penn et al. 2001; Deshpande et al. 2008). Conversely, EP4 receptors possess classical regulatory sites on their C-termini and so undergo β-arrestin–mediated internalization (Desai and Ashby 2001). One or both of these EP receptors may be expressed in human fetal lung (HFL)-1 cells, so diminished PGE2-stimulated cAMP levels could result from either decreased receptor activation via receptor desensitization or increased PDE activity or both. In the current study, we tested whether long-term PGE2 exposure causes desensitization, identified the specific EP receptor subtypes inducing this desensitization, and established whether upregulation of specific PDE isoforms is responsible for the desensitization.
Materials and methods
Cell culture
HFL-1 fibroblasts (ATCC, Manassas, VA, USA) were grown in Ham’s F12K medium (Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA, USA) and 1% antibiotic-antimycotic solution. Cells with a passage number of 13 to 25 were used and were grown to 70–90% confluency for all studies. Cells were grown in an incubator at 37 °C and 5% CO2 levels. Medium was aspirated and cells were washed once with PBS. After aspiration of PBS, 5 mL of 0.25% trypsin-EDTA (Thermo Fisher Scientific, Waltham, MA, USA) was added for 5 min to induce cell detachment. Media was added to the flask and gently mixed to suspend detached cells, transferred to tubes, centrifuged at 500g for 5 min, then washed to remove trypsin. The cell pellet was resuspended in growth medium, and approximately 9000 sells were plated per well on 96-well plates. Cells were pretreated with PGE2, other drugs, or vehicle prior to experiments by adding 10× concentration of drug to growth media. Medium was aspirated after 24 h, and each well was washed once with warm Dulbecco’s phosphate-buffered saline before performing cADDis assay for cAMP.
cADDis assay for cAMP
The cADDis assay was purchased from Montana Molecular (Bozeman, MT, USA). HFL-1 cells were incubated with a recombinant mammalianized baculovirus (BacMam) engineered to express a genetically modified protein that is a fusion of EPAC, a cAMP effector, with the green fluorescent protein (GFP). This protein serves as a convenient and quantitative cAMP sensor. In its unbound form, the protein fluoresces, but its fluorescence is quenched upon binding of cAMP. When seeding the cells in a 96-well plate, the following components were added: 138 μL of cells plus media, 40 μL BacMam solution, and 2 μL of trichostatin A (an inhibitor of histone deacetylase) (Sigma®, St. Louis, MO, USA). In the 96-well plate format, approximately 8900 cells were seeded per well and 2.95 × 108 viral particles per well were added. Trichostatin A stock concentration was 100 μM, giving a final concentration of 1 μM in the final volume of 200 μL per well. Transduced HFL-1 cells were incubated for 24 h at 37 °C and 5% CO2 levels. After incubation, medium was aspirated and replaced with 180 μL per well of 1× Dulbecco’s phosphate-buffered saline without calcium and magnesium (Thermo Fisher Scientific, Waltham, MA, USA). The 96-well plate was then covered with aluminum foil and incubated at 37 °C for 30 min. The plate was placed in a SpectraMax M5 plate reader (Molecular Devices, San Jose, CA, USA), and fluorescence was read from the bottom of the plate with an excitation wavelength of 494 nm and an emission wavelength of 522 nm for 5 min. Once variability of fluorescence was ≤ 5% in all wells, different concentrations ofPGE2 or other agents were added with a multi-pipette and the changes in fluorescence were measured every 30 s for 30 min.
Data from kinetic studies of fluorescence were normalized to the original level of fluorescence (ΔF/F0) and then fit to a single-site decay model using GraphPad Prism 7.0 (GraphPad Software Inc., San Diego, CA, USA). The plateau of each decay curve was then used as the maximal response to that given concentration of drug (Emax), and concentration-response curves were generated using nonlinear regression analysis (log [agonist] vs. response, variable slope, four parameters) using GraphPad Prism 7.0. Concentration-response curves in multiple conditions were compared using two-way analysis of variance (2-way ANOVA) with Tukey’s multiple comparison test, and significance (p value) is reported for the effect of different conditions. Statistical comparison tests (t tests, 1-way and 2-way ANOVA) were performed using GraphPad Prism.
Assay of basal cAMP
HFL-1 cells were washed three times with serum- and NaHCO3-free DMEM supplemented with 20 mM HEPES, pH 7.4 (DMEH), then incubated for 30 min at 37 °C. Medium was aspirated, and 250 μL of trichloroacetic acid (TCA, 7.5% w/v) was added to each well. cAMP content in TCA extracts was determined by EIA (Cayman Chemical) following the manufacturer’s instructions. The amount of cAMP was normalized to the amount of protein per sample as determined using a dye-binding protein assay (Bio-Rad).
Real-time quantitative reverse transcription polymerase chain reaction
After 48-h incubation with the lentivirus, medium was aspirated, cells were washed once in 1× PBS, and buffer RLT was added to disrupt cell membranes. Cells were collected and RNA was isolated using RNeasy columns with a QIAcube robot (Qiagen). RNA was quantified using the NanoDrop 2000/2000c (Thermo Fisher). Reverse transcription was performed on 400 ng of RNA samples. All steps followed the SuperScript® III First-Strand Synthesis System for RT-PCR protocol (Thermo Fisher). The steps included a denaturation step at 65 °C for 5 min in the RNA/primer mixture, containing 50 ng/μL of random hexamers. Complementary DNA (cDNA) synthesis mixture was added to the RNA/primer mixture and incubated for 10 min at 25 °C, followed by 50 min at 50 °C. The reaction was terminated at 85 °C for 5 min, then chilled on ice. Finally, 1 μL of RNase H was added to each tube followed by incubation for 20 min at 37 °C. cDNA samples were either used immediately for quantitative reverse transcription polymerase chain reaction (qRT-PCR) experiments or stored at − 20 °C.
TaqMan® Gene Expression Assays (Thermo Fisher) were used for qRT-PCR reactions. The ratio of components for each reaction was as follows: 1 μL 20× TaqMan® Gene Expression Assay, 10 μL 2× TaqMan® Gene Expression Mater Mix, 4 μL cDNA (1 ng to 100 ng), and 5 μL of RNase-free water. The thermal cycler settings were as follows: step 1, 50 °C for 2 min; step 2, 95 °C for 10 min; step 3, 95 °C for 15 s; and step 4, 60 °C for 1 min. Steps 3 and 4 were repeated for a total of 40 cycles. The housekeeping genes GAPDH, β-actin, and CDKN1A were used to normalize for differences in RNA samples. PCR data were analyzed using the 2[−ΔΔCt] method, where Ct is the cycle threshold.
PDE activity assays
PDE assays were performed according to the methods of Bauer and Schwabe (1980). Following pretreatments, cells were washed, scraped from the dishes, and lysed by sonication. Cell lysates were pre-incubated for 5 min with inhibitors or vehicle. Assay mix containing [3H] cAMP was added followed by incubation for 25 min at 37 °C with shaking. The reaction was stopped by adding 0.2 N HCl, and samples were placed on ice for at least 10 min. Crotalus atrox snake venom–derived 5′-nucleotidase was added, and samples were incubated for 20 min at 37 °C. The assay mixture was then added to QAE-Sephadex A25 columns, and nucleosides were eluted with 30 mM ammonium formate. [3H] adenosine formation was quantified by liquid scintillation counting and taken as total PDE activity. In some experiments, roflumilast (1 μM) or 3-isobutyl-1-methylxanthine (IBMX) (1 mM) was included during the PDE assays to assess PDE4 and total PDE activities, respectively.
Statistics
Statistical comparison tests (one-way and two-way analyses of variance) were performed with GraphPad Prism 7.0. Tukey’s multiple comparison test was performed where appropriate.
Results
To determine whether prolonged exposure of HFL-1 cells to PGE2 causes desensitization of subsequent PGE2 responses, cells were pretreated with 1 nM, 10 nM, or 100 nM PGE2 for 24 h. Cells were then washed to remove the pretreatment drug, and cAMP responses to various concentrations of PGE2 were measured using the cADDis assay. The change of fluorescence caused by each concentration ofPGE2 was measured for 30 min, and the data was fit to a one-site decay model (Fig. 1a, b). The plateau of each response was then normalized to the maximal response elicited by forskolin plus IBMX and plotted to generate a concentration-response curve to PGE2 for each pretreatment condition. PGE2 stimulated cAMP production in vehicle-treated cells with a log EC50 value of −7.40 ±0.16 (Fig. 1c, Table 1). By contrast, PGE2 responses in cells pretreated with 1 nM PGE2 required moderately higher concentrations (log EC50 = − 7.10 ± 0.25) and cells pretreated with 10 nM PGE2 displayed PGE2 responses that required even higher concentrations (log EC50 = − 6.79 ± 0.20, Table 1). Pretreatment with 100 nM PGE2 induced the largest rightward shift of the PGE2 concentration-response curve to − 6.09 ± 0.15 (Fig. 1c, Table 1). None of the pretreatment conditions produced a significant reduction in Emax. Therefore, pre-exposure to PGE2 causes reductions in the potency of PGE2 for increasing cAMP levels.
Fig. 1.
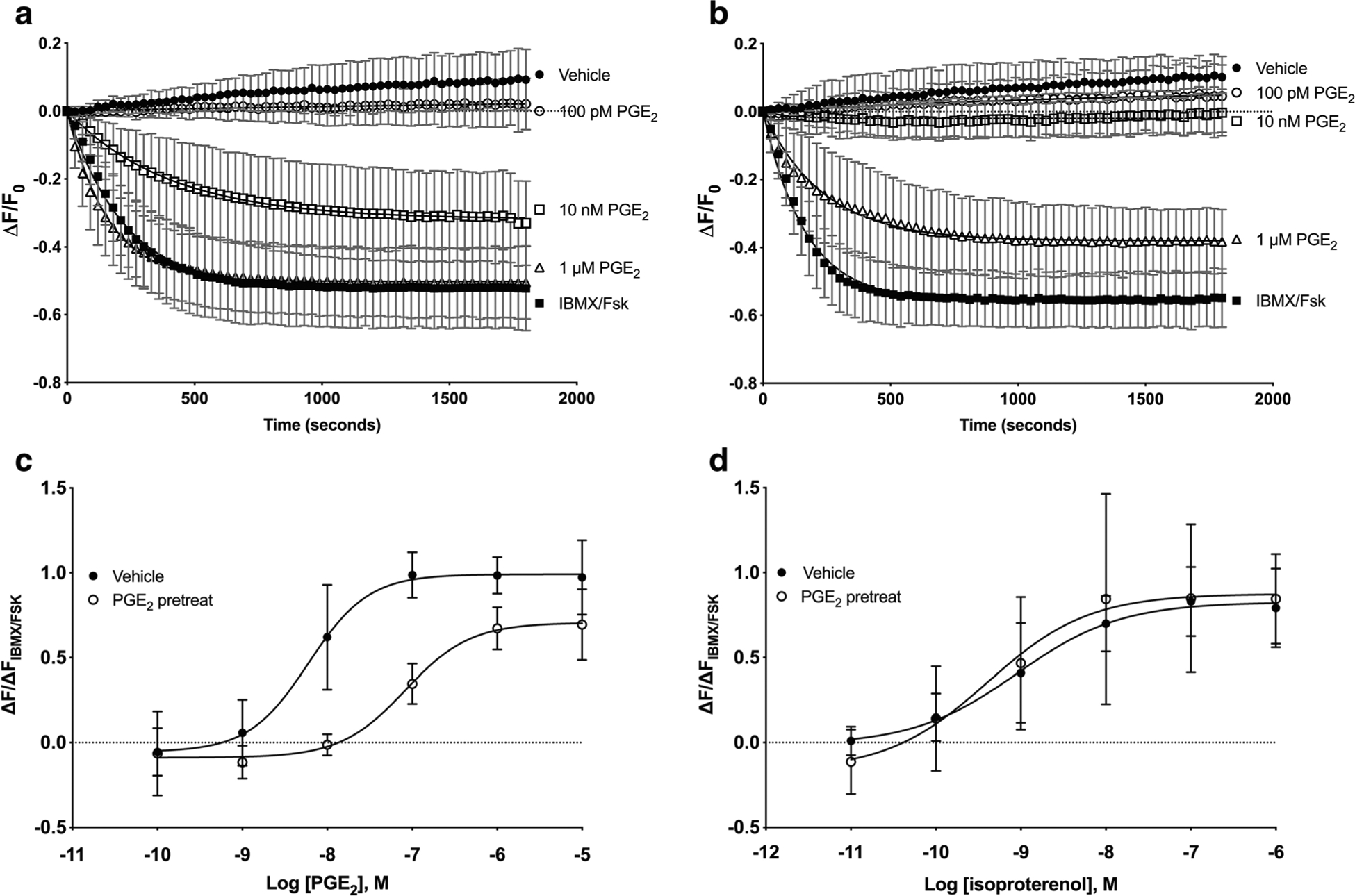
Twenty-four-hour pretreatment with PGE2 induces desensitization of cAMP responses to PGE2 but not isoproterenol. HFL-1 cells were treated with vehicle or 100 nM PGE2 for 24 h and washed, then acute responses to PGE2 were measured using the cADDis cAMP sensor. Fluorescent decay curves are shown for three concentrations of PGE2 in vehicle-pretreated (a) or PGE2-pretreated (b) cells. The plateau from each decay curve was normalized to the maximal response to 200 mM IBMX plus 1 μM forskolin and plotted as a concentration-response curve. Concentration-response curves to PGE2 (c) or isoproterenol (d) are shown in both vehicle and 100 nM PGE2-pretreated cells. Data are mean ± SEM of n = 3–10. c Significant (p < 0.0001) and d not significant (p = 0.070) by 2-way ANOVA
Table 1.
Twenty-four-hour pretreatment with PGE2 desensitizes PGE2 responses in a concentration-dependent manner
| Pretreatment condition | Log EC50 |
|---|---|
| Vehicle | − 7.40 ±0.16 |
| 1 nM PGE2 | − 7.10 ± 0.25 |
| 10 nM PGE2 | − 6.79 ± 0.20 |
| 100 nM PGE2 | − 6.09 ±0.15 |
HFL-1 cells were treated with vehicle and 1 nM, 10 nM, or 100 nM PGE2 for 24 h and washed, then acute cAMP responses to PGE2 were measured using the cADDis assay. Various concentrations of PGE2 ranging from 0.1 to 10 μM were added and responses measured as the change in fluorescence normalized to the change in fluorescence of maximal response stimulated by 1 μM forskolin plus 200 μM IBMX. The log EC50 was then calculated for PGE2 in each pretreatment condition. Data are mean ± SEM of n = 3–10
We assessed basal cAMP levels in cells pretreated with either vehicle or 100 nM PGE2 for 24 h. Because biosensors such as cADDis can only display changes in cAMP levels, we lysed cells after treatment and measured cAMP levels using an EIA. Basal cAMP level in vehicle-treated cells was 57.7 ±15.4 fmol/mg protein while basal cAMP level in PGE2-pretreated cells was 57.9 ± 13.8 fmol/mg protein (not significant p = 0.815 by Student’s t test, data not shown). Thus, PGE2 pretreatment does not alter basal levels of cAMP in the bulk cytosol.
We investigated whether a 24-h pretreatment with PGE2 alters responses mediated by other receptors. cAMP levels stimulated by the β-adrenoceptor (β-AR) agonist isoproterenol were measured in cells pretreated with 100 nM PGE2 or vehicle for 24 h. In contrast to the 22-fold rightward shift in the PGE2 concentration-response curve in PGE2-pretreated cells, responses mediated by isoproterenol were not right-shifted by PGE2 pretreatment, with log EC50 values of − 9.05 ± 0.33 and − 9.39 ± 0.60 for vehicle and PGE2 pretreatment, respectively (Fig. 1d). Thus, PGE2 pre-exposure does not cause desensitization of β-AR–mediated cAMP signaling.
To establish if decreased responsiveness to PGE2 occurs upon pretreatment with other cAMP-elevating agents, the effects of pretreating cells with either vehicle, 100 nM isoproterenol, or 1 μM forskolin (a direct activator of AC) were assessed. Log EC50 values for PGE2 stimulation were − 7.78 ± 0.52 and − 8.44 ± 0.69 for vehicle and isoproterenol pretreatment, respectively (Fig. 2a). cAMP responses to isoproterenol displayed log EC50 values of − 9.05 ± 0.33 and −8.65 ± 0.15 for vehicle and isoproterenol, respectively (Fig. 2b). Thus, pretreatment with isoproterenol induces desensitization of β-AR responses but does not decrease responses to PGE2. Pretreatment of HFL-1 cells with 1 μM forskolin for 24 h also did not cause desensitization of PGE2 responses, with log EC50 values of − 7.78 ± 0.52 and − 9.36 ±3.54 for vehicle and forskolin pretreatment, respectively (Fig. 3). The small increase in PGE2 potency observed in these studies may be due to residual forskolin from the pretreatment phase that could not be washed out.
Fig. 2.
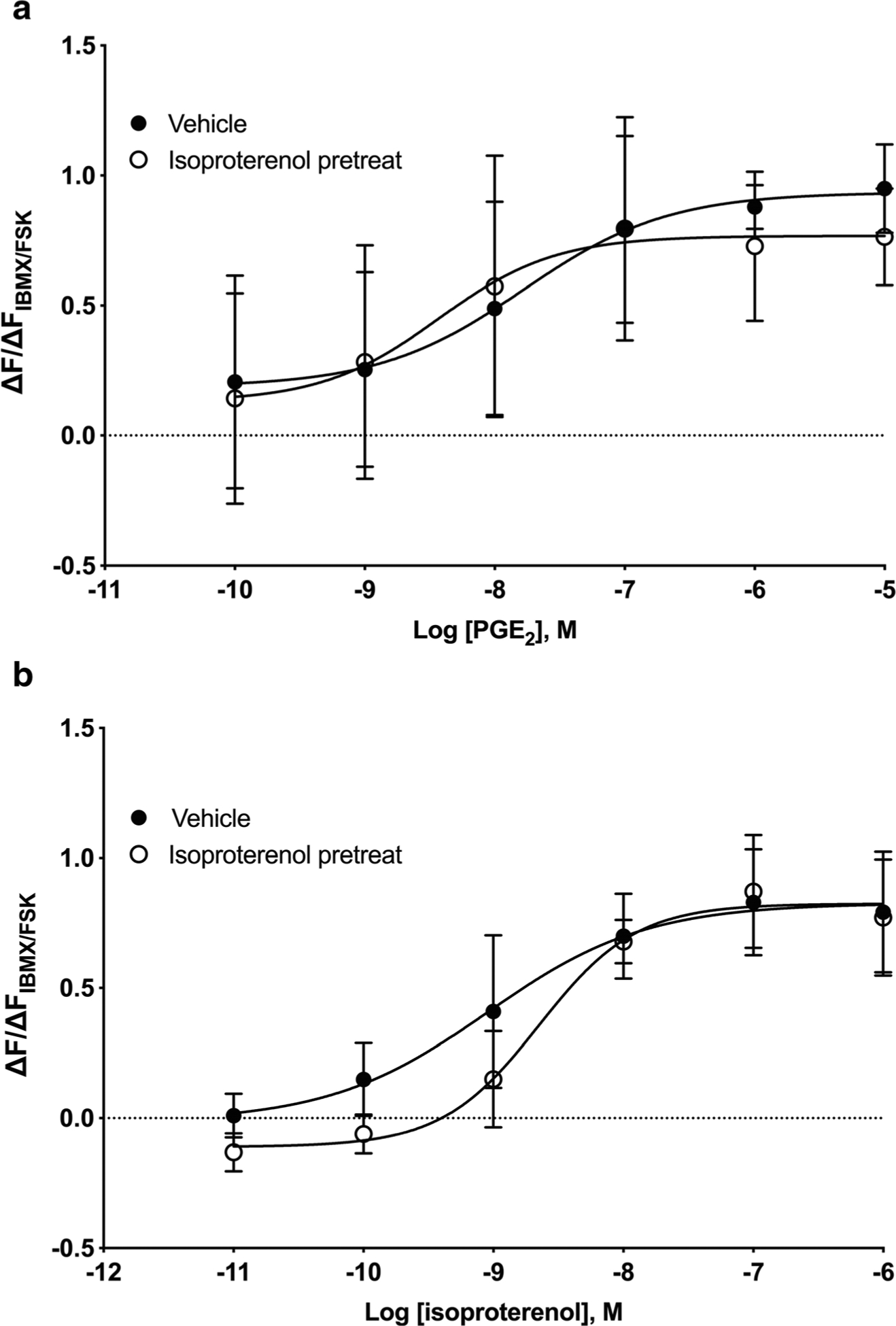
Twenty-four-hour pretreatment with isoproterenol does not induce desensitization of PGE2 cAMP responses. HFL-1 cells were treated with 100 nM isoproterenol for 24 h then washed, and acute responses to PGE2 (a) or isoproterenol (b) were measured. cAMP levels were measured as a function of change in fluorescence normalized to the change in fluorescence of maximal response stimulated by 1 μM forskolin plus 200 μM IBMX using the cADDis assay. Data are mean ± SEM of n = 5. a Not significant (p = 0.612) and b significant (p = 0.0318) by 2-way ANOVA
Fig. 3.
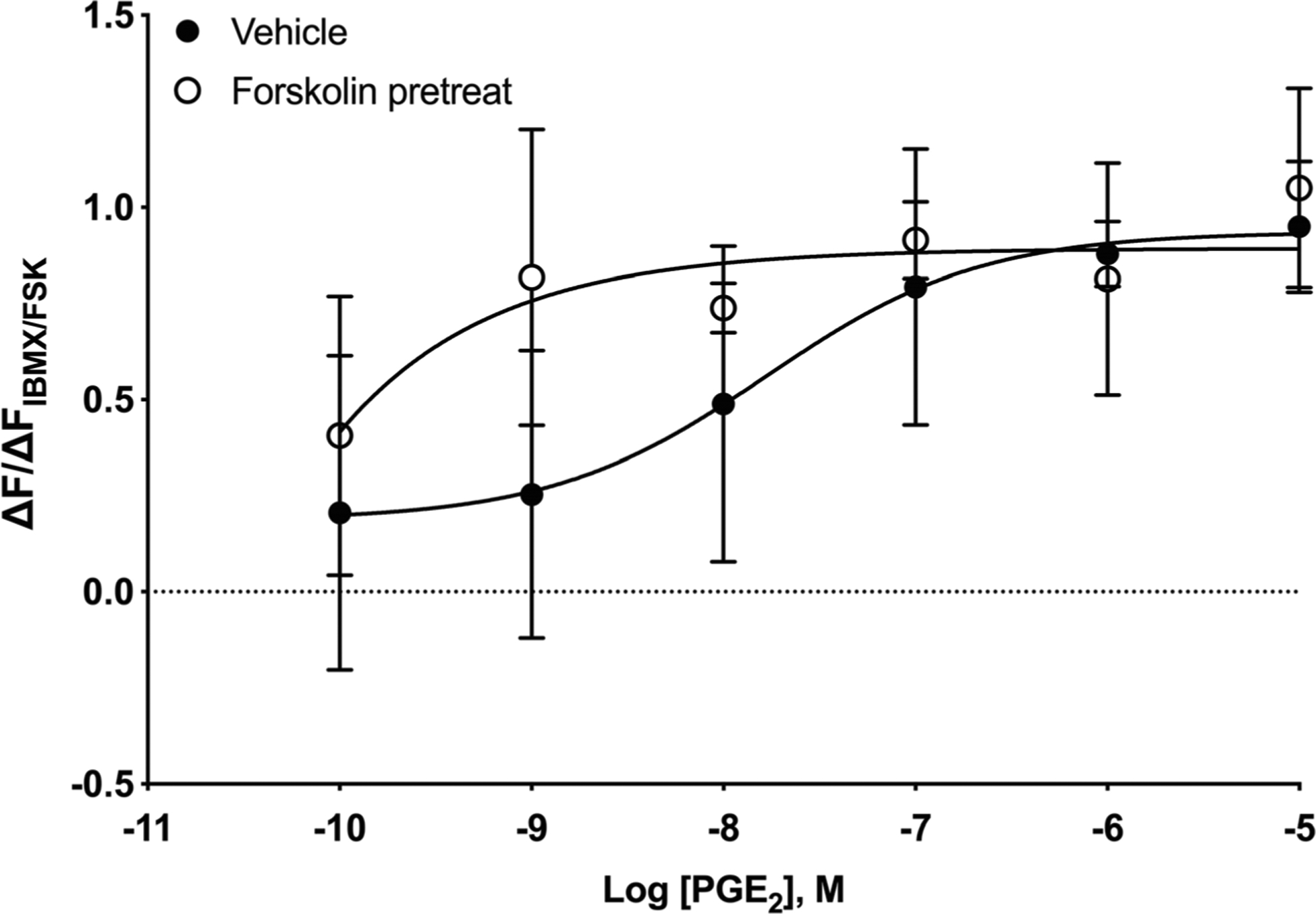
Twenty-four-hour pretreatment with forskolin does not induce desensitization of PGE2 cAMP responses. HFL-1 cells were treated with 1 μM forskolin for 24 h then washed, and acute responses to PGE2 were measured. cAMP levels were measured as a function of change in fluorescence normalized to the change in fluorescence of maximal response stimulated by 1 μM forskolin plus 200 μM IBMX using the cADDis assay. Data are mean ± SEM of n = 5. Not significant (p = 0.287) by 2-way ANOVA
PGE2 can increase cAMP by activation of EP2 or EP4 receptors. To assess which of these two EP receptor types mediates PGE2-stimulated cAMP responses, we examined the effect of PF-0441894 (EP2 receptor–specific antagonist) or GW-627368X (EP4 receptor–specific antagonist) on PGE2-stimulated cAMP responses. PF-0441894 (100 nM) shifted the PGE2 concentration-response curve by nearly 10-fold the right, while GW-627368X (100 nM) did not cause a rightward shift (Table 2). We also examined the effects of both PF-0441894 and GW-627368X on PGE2-stimulated cAMP responses in cells that had been pretreated with PGE2 for 24 After a 24-h pretreatment with either PGE2 or vehicle, cells were washed and equilibrated for 30 min, and then each antagonist was added. Ten minutes after addition of antagonist, various concentrations of PGE2 were added and cAMP responses were measured for 30 min. Unlike our results Figs. 1, 2, and 3, PGE2 pretreatment caused a reduction PGE2 maximal response in addition to a rightward shift (vehicle 1.06 ± 0.10, PGE2-pretreated 0.60 ± 0.09). Just as vehicle-pretreated cells, PF-0441894 shifted PGE concentration-response curves to the right (Fig. 4a, Table 2 but GW-627368X did not (Fig. 4b, Table 2). Based on published Ki values, 100 nM PF-0441894 occupies 95% of EP receptors while 100 nM GW-627368X occupies just 1.2% EP2 receptors (Wilson et al. 2006; af Forselles et al. 2011 These results suggest that EP2 receptors, but not EP4 receptors, mediate the PGE2-induced increase in cAMP and that pretreatment with PGE2 for 24 h does not alter this receptor response profile.
Table 2.
EP2 receptors mediate PGE2 responses both before and after desensitization
| Pretreatment condition | Antagonist | Log EC50 |
|---|---|---|
| Vehicle | None | − 7.39 ±0.26 |
| Vehicle | 100 nM PF-0441894 | − 6.44 ± 0.40 |
| Vehicle | 100 nM GW-627368X | − 7.92 ±0.24 |
| 100 nM PGE2 pretreatment | None | − 6.65 ± 0.27 |
| 100 nM PGE2 pretreatment | 100 nM PF-0441894 | − 5.68 ± 1.27 |
| 100 nMPGE2 pretreatment | 100 nMGW-627368X | − 6.55 ± 0.37 |
HFL-1 cells were treated with vehicle or 100 nM PGE2 for 24 h, washed, then incubated for 10 min with either 100 nM PF-0441894 (EP2 receptor antagonist) or 100 nM GW-627368X (EP4 receptor antagonist). cAMP responses to various concentrations of PGE2 were measured using the cADDis assay. For each concentration of PGE2, the change in fluorescence was normalized to the change in fluorescence of maximal response stimulated by 1 μM forskolin plus 200 μM IBMX. The log EC50 was then calculated for PGE2 in each pretreatment condition. Data are mean ± SEM of n = 3–10. Plots of these data are shown in Fig. 4
Fig. 4.
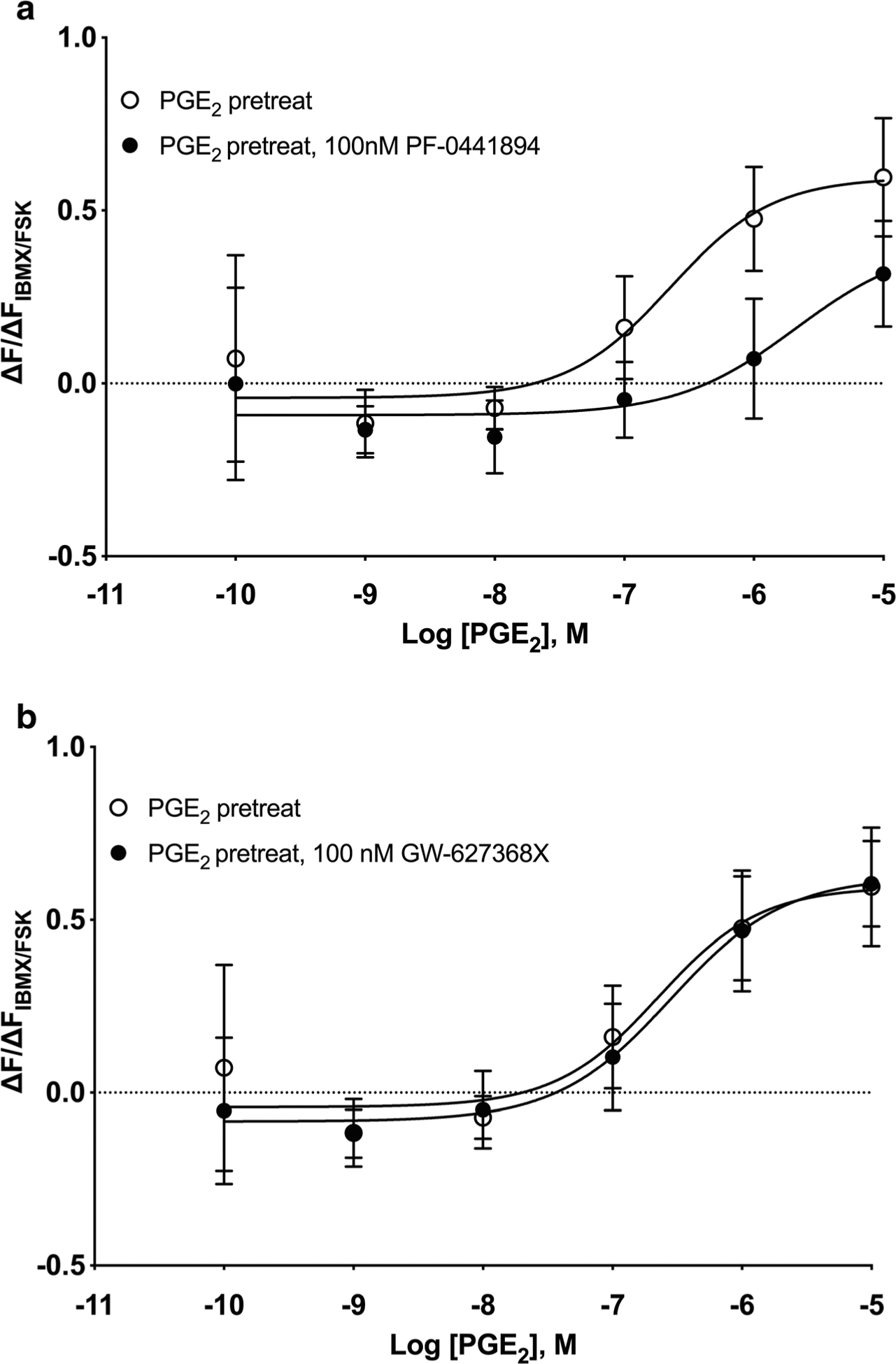
PGE2-stimulated cAMP responses in HFL-1 cells are mediated by EP2 not EP4 receptors. HFL-1 cells were pretreated with 100 nM PGE2 for 24 h. a 10 min prior to measuring cAMP responses to PGE2 with addition of either vehicle or 100 nM PF-0441894 (EP2-selective antagonist). b 10 min prior to measuring cAMP responses to PGE2 with addition of either vehicle or 100 nM GW-627368X (EP4-selective antagonist). cAMP levels were measured as a function of change in fluorescence normalized to the change in fluorescence of maximal response stimulated by 1 μM forskolin plus 200 μM IBMX using the cADDis assay. Data are mean ± SEM of n = 6. a Significant (p < 0.0001) and b not significant (p = 0.421) by 2-way ANOVA
To understand which EP receptors mediate PGE2-induced desensitization, the receptor subtype–selective agonists ONO-AE1–259 (EP2) and ONO-329 (EP4) were tested for their abilities to induce desensitization. Both ONO-AE1–259 and ONO-AE1–329 increased cAMP levels acutely in HFL-1 cells, but ONO-AE1–259 did so with a log EC50 value of −8.29 (similar to the potency of PGE2, log EC50 value of −8.48) while ONO-AE1–329 was far less potent (log EC50 value of − 7.49, Fig. 5a). Cells were pretreated with vehicle, 100 nM PGE2, 100 nM ONO-AE1–259, or 100 nM ONO-AE1–329 for 24 h and washed, and then cAMP responses to PGE2 were measured. ONO-AE1–259 at 100 nM is predicted to occupy 97% of EP2 receptors but just 4.1% of EP4 receptors based on published Ki values (Suzawa et al. 2000; Ganesh 2014). ONO-AE1–329 at 100 nM is predicted to occupy 4.7% of EP2 receptors but 91% of EP4 receptors (Suzawa et al. 2000). ONO-AE1–259 pretreatment shifted PGE2 responses rightward by 4.0 ± 1.2-fold (n = 4) compared to vehicle pretreatment, with log EC50 values of − 7.51 ± 0.18 and − 8.00 ± 0.09, respectively (Fig. 5b). This degree of desensitization was nearly equivalent to the 8.0 ± 2.2-fold shift induced by pretreatment with 100 nM PGE2 in the same experiments. ONO-AE1–329 pretreatment did not induce a change compared to vehicle pretreatment, with PGE2 log EC50 values of − 8.09 ± 0.17 and − 8.00 ± 0.09, respectively. Together, these data indicate that EP2 receptors are responsible for desensitization of PGE2 responses in HFL-1 cells. Our findings are supported by other studies that report the relative abundance of mRNA levels among Gs-coupled GPCRs in HFL-1 cells. Using qRT-PCR and cAMP measures with PDE inhibitors, Roberts et al. (2018) determined the relative abundance of receptors to be IP receptors > EP2 receptors >>> EP4 receptors. RNA-Seq studies of HFL-1 cells reported in the Gene Expression Omnibus show the relative abundance of mRNA to be EP2 receptors > IP receptors = EP4 receptors (GEO accession GSE73555). Thus, while the expression level of IP receptors is debatable, it is clear that EP2 receptors are expressed at much higher levels than EP4 receptors in HFL-1 cells.
Fig. 5.
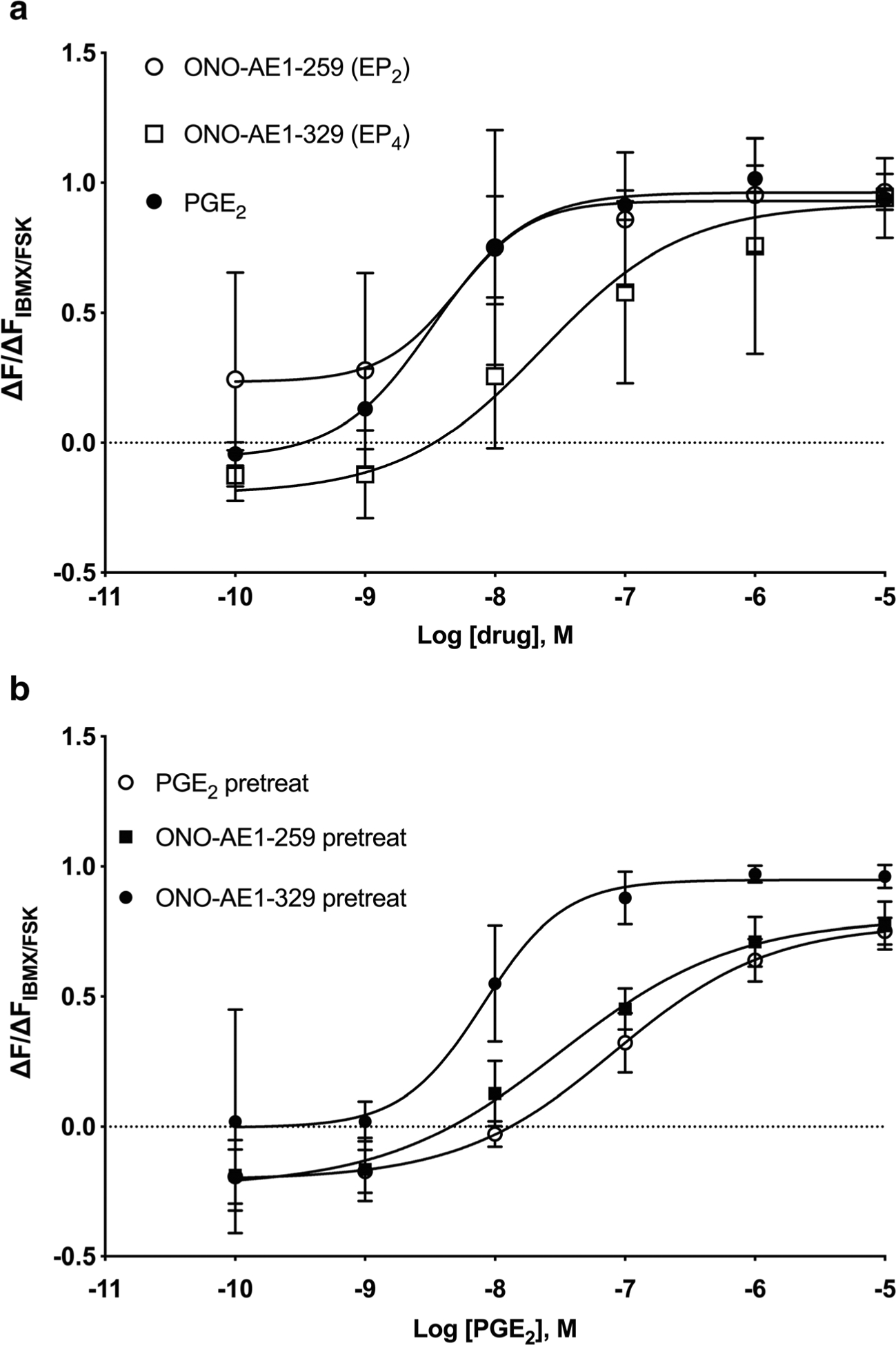
PGE2 responses are desensitized following the 24-h pretreatment with an EP2R-selective agonist but not by an EP4R-selective agonist. a Acute cAMP responses in HFL-1 cells to various concentrations of PGE2, ONO-AE1–259 (EP2 agonist), or ONO-AE1–329 were measured. b HFL-1 cells were treated with 100 nM PGE2, 100 nM ONO AE1–259 (EP2 agonist), or 100 nM ONO-AE1–329 (EP4 agonist) for 24 h then washed, and acute responses to PGE2 were measured. cAMP levels were measured as a function of change in fluorescence normalized to the change in fluorescence of maximal response stimulated by 1 μM forskolin plus 200 μM IBMX using the cADDis assay. Data are mean ± SEM of n = 4. ONO-AE1–259 is not significant (p = 0.264), while ONO-AE1–329 is significant (p < 0.0001) from PGE2 by 2-way ANOVA
cAMP levels are also regulated by phosphodiesterase activity, which hydrolyzes cAMP to AMP (Kohyama et al. 2002b), so increases in PDE activity provide another possible explanation for the desensitization we observed. HFL-1 cells primarily express PDE3B, PDE4A, PDE4B, PDE4C, PDE4D, PDE7A, PDE7B, and PDE8A (GEO accession GSE73555) (Kohyama et al. 2002b). qRT-PCR experiments were performed to determine whether the expression of any of these PDE isoforms is altered by PGE2 pretreatment. Cells were pretreated with 1 μM forskolin, 100 nM isoproterenol, or 100 nM PGE2 for 24 h, and changes in PDE subtype expression were quantified by qRT-PCR. Significant upregulation of PDE3A, PDE3B, PDE4B, and PDE4D occurred following pretreatment with each of these cAMP-elevating agents (Fig. 6). Of these isoforms, PDE3A was selectively upregulated by PGE2 pretreatment (7.3-fold), with forskolin and isoproterenol having no effect on PDE3A. PGE2 and forskolin pretreatment upregulated PDE4B expression (2.6- and 3.9-fold, respectively), but isoproterenol had no effect (1.3-fold). PDE4D was upregulated significantly by all three drugs, but more by PGE2 (22.3-fold) than forskolin or isoproterenol (7.9- and 5.0-fold, respectively). While a previous report suggests that a 12-h pretreatment with 1 μM PGE2 can downregulate expression of both EP2R and EP4R when exogenously expressed in Chinese hamster ovary cells (Nishigaki et al. 1996), we did not observe any change in EP2R mRNA levels following a 24-h pretreatment with 100 nM PGE2 (Fig. 6a).
Fig. 6.
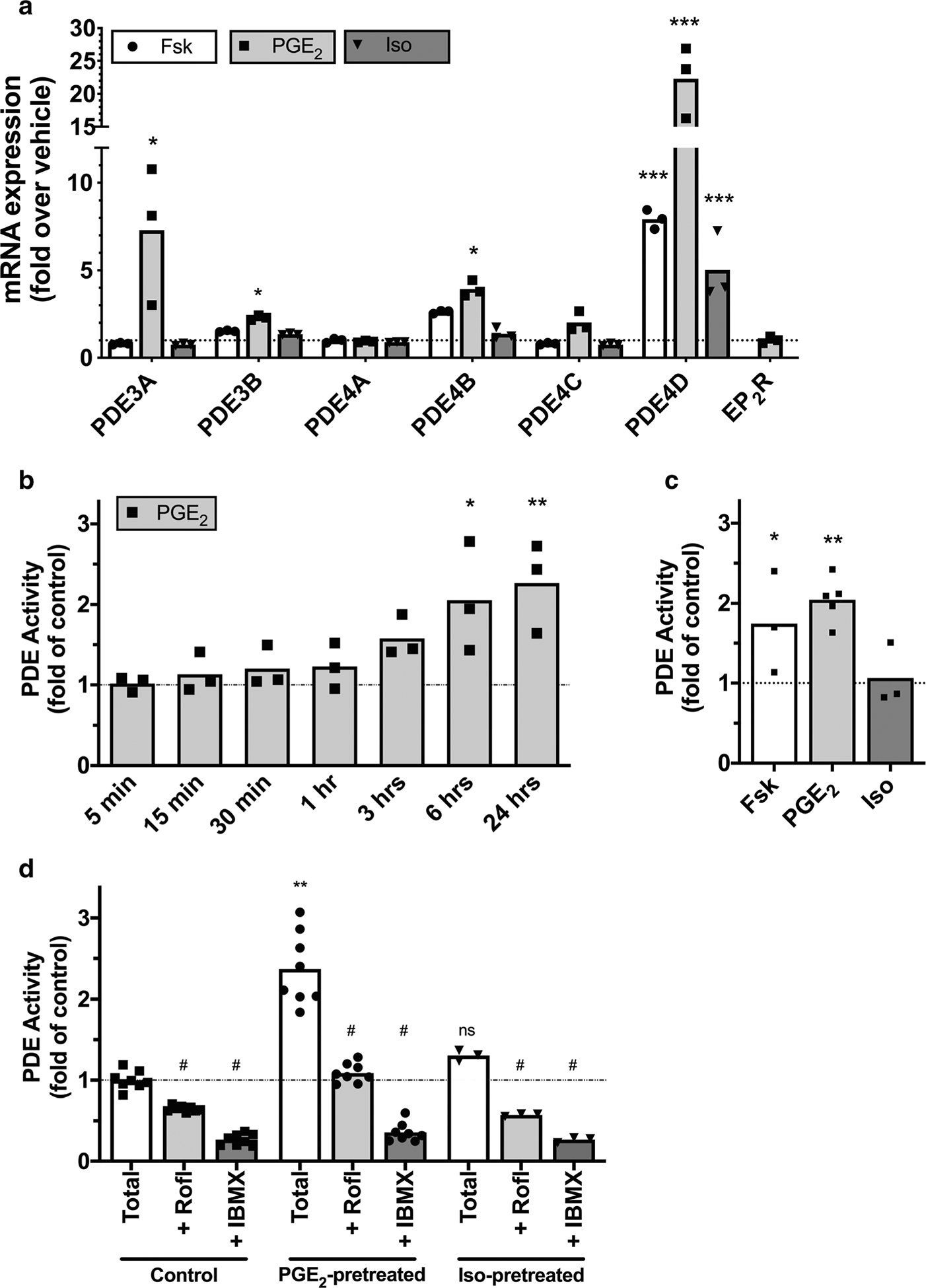
PDE isoform expression and activity is increased by PGE2 but not isoproterenol pretreatment. a HFL-1 cells were incubated with vehicle, 1 μM forskolin, 100 nM PGE2, or 100 nM isoproterenol for 24 h, then the expression of PDE isoform mRNAwas assessed by quantitative RT-PCR. Expression of EP2 receptors was assessed in the same way following PGE2 pretreatment. b Cells were incubated for the indicated times with vehicle or 100 nM PGE2, then PDE enzyme activity in cell lysates was assayed. c Cells were incubated for 24 h with vehicle, 30 μM forskolin, 100 nM PGE2, or 1 μM isoproterenol, then PDE enzyme activity in cell lysates was assayed. d Cells were incubated for 24 h with vehicle, 100 nM PGE2, or 1 μM isoproterenol, then PDE enzyme activity in cell lysates was assayed in the presence of either vehicle (total), roflumilast (1 μM), or IBMX (1 mM). Data are expressed as the fold change over vehicle-treated cells (dotted line). Bars show means from n = 3–8 with individual data points plotted in symbols. *p < 0.05, **p < 0.01, and ***p < 0.0001 by two-way ANOVA (a, d) or one-way ANOVA (b, c) as compared to vehicle. #p < 0.05 by two-way ANOVA as compared to total activity in the same condition
PDE activity assays performed in cell lysates from HFL-1 cells pretreated with 100 nM PGE2 confirm that the observed increase in PDE mRNA translates to increased cAMP hydrolyzing activity. We treated cells for various time points from 5 min to 24 h and found significant increases in bulk PDE activity after 6 h or more of PGE2 treatment as compared to vehicle (Fig. 6d). We then treated cells with either vehicle, 30 μM forskolin, 100 nM PGE2 or 1 μM isoproterenol for 24 h and examined bulk PDE activity. Forskolin and PGE2, but not isoproterenol, significantly increased PDE activity (Fig. 6c). Inclusion of the PDE4 family inhibitor roflumilast (1 μM) or the broad-spectrum PDE inhibitor IBMX (1 mM) significantly reduced PDE activity in vehicle (control) and in PGE2- and isoproterenol-pretreated cells (Fig. 6d). Therefore, PGE2 pretreatment significantly upregulates PDE activity while isoproterenol does not. Much of the upregulated PDE activity appears sensitive to a PDE4 inhibitor.
We used PDE3- and PDE4-specific inhibitors to examine if these isozymes are involved in mediating desensitization of PGE2 cAMP responses. HFL-1 cells were pretreated for 24 h with 100 nM PGE2, washed, and then incubated with 10 μM rolipram (a PDE4 family inhibitor), 10 μM cilostazol (a PDE3 family inhibitor), or 10 μM IBMX for 10 min prior to measuring cAMP levels in response to various concentrations of PGE2. The PDE4-selective inhibitor rolipram was able to reverse the desensitization caused by PGE2 pretreatment by shifting the concentration-response curve leftward by 2.8-fold (Fig. 7a). The PGE2 log EC50 value in the PGE2-pretreated condition was − 6.85 ± 0.25 while the log EC50 value in the presence of 10 μM rolipram was − 7.30 ± 0.11. The addition of the PDE3-selective inhibitor cilostazol at 10 μM had no effect, with the PGE2 log EC50 value of − 7.03 ± 0.39 when 10 μM cilostazol was present (Fig. 7b). IBMX, a nonselective inhibitor of all PDE isozymes except PDE8, induced a small leftward shift that was not statistically significant. The PGE2
Fig. 7.
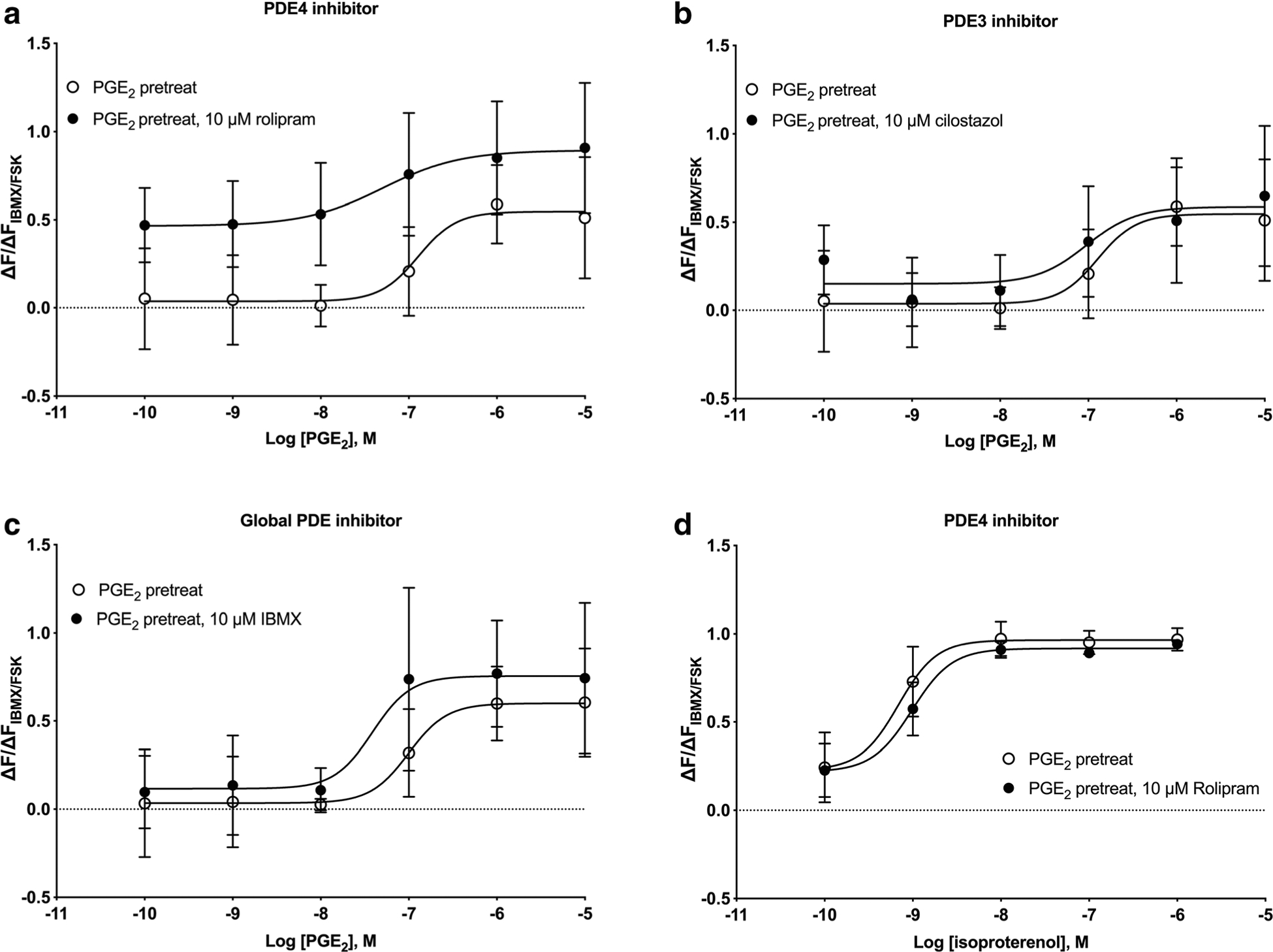
Inhibition of PDE4, but not PDE3, re-sensitizes PGE2-mediated cAMP responses. HFL-1 cells were pretreated with 100 nM PGE2 for 24 h, prior to measuring cAMP responses to either PGE2 (a–c) or isoproterenol (d). Either vehicle, 10 μM rolipram (PDE4 inhibitor, panels a and d), 10 μM cilostazol (PDE3 inhibitor, panel b), or 10 μM IBMX (broad PDE isoform inhibitor, panel c) was added 10 min prior to addition of PGE2 or isoproterenol. cAMP production was measured as a function of change in fluorescence normalized to the change in fluorescence of maximal response stimulated by 1 μM forskolin and 200 μM IBMX using the cADDis assay. Data are mean ± SEM of n = 3. a Significant (p < 0.0001), b not significant (p = 0.286), c not significant (p = 0.114), and d not significant (p = 0.259) by 2-way ANOVA
log EC50 value in the PGE2-pretreated condition was − 7.00 ± 0.22 while the log EC50 value in the presence of 10 μM IBMX was − 7.47 ± 0.62 (Fig. 7c). A lower concentration of IBMX was used in these studies since higher concentrations stimulated increases in cAMP that nearly saturated the cADDis biosensor. Rolipram had no effect on isoproterenol responses in cells pretreated with PGE2, implying that the increased PDE activity does not regulate β-AR signaling (Fig. 7d). Taken together, these results are consistent with the idea that PGE2 pretreatment induces expression of PDE4 isozyme(s) that regulates cAMP signals stimulated by PGE2 but not those stimulated by β-AR.
Discussion
Evidence that PGE2 produces antifibrotic effects is relatively clear in the literature (Kolodsick et al. 2003; White et al. 2005; Bozyk and Moore 2011). PGE2 is produced as part of the normal injury response, to promote lung homeostasis and to inhibit fibrotic processes and help promote alveolar epithelial cell regeneration and thus restore airway barrier function (Wilborn et al. 1995; Bozyk and Moore 2011). The signaling pathways that PGE2 activates remain attractive targets for therapies in PF. For example, PGE2 activates EP receptors that are coupled to the Gs-AC-cAMP pathway that promote an antifibrotic response (Bozyk and Moore 2011). However, some studies demonstrate diminished COX-2 expression in lung fibroblasts cultured from patients with PF, which would lead to reduced PGE2 synthesis (Wilborn et al. 1995). Despite this, PGE2 levels remain elevated in fibrotic lungs, so the reason for its limited antifibrotic action in PF is not explained. It is possible that PGE2 loses its antifibrotic action following long-term exposure due to attenuated cAMP signaling in lung fibroblasts (Michalski et al. 2012). Because PF is characterized by ongoing airway injury and presumably chronic elevation of PGE2 (Fastres et al. 2017), the current study examined how EP receptor signaling is affected by prolonged agonist exposure. Our findings demonstrate that a 24-h exposure of lung fibroblasts to PGE2 elicits desensitization of EP2 receptor responses. Thus, even if PGE2 could be restored to normal levels or administered as a therapeutic agent, this desensitization of EP2 receptors would likely inhibit its antifibrotic effects.
The data here show that PGE2-induced desensitization of PGE-stimulated cAMP accumulation occurs primarily through activation of EP2 receptors and not EP4 receptors. Studies with both PGE2 receptor subtype–selective antagonists and agonists demonstrated that desensitization of PGE2-stimulated cAMP levels requires activation of EP2 receptors and provided no evidence for contributions of EP4 receptors. Whether EP4 receptors are not expressed or rather are not involved in the desensitization response is not clear from our studies. Transcripts for EP4 receptors are low but detectible by qRT-PCR and RNA sequencing. PGE2 itself also activates EP3 and EP4 receptors, which can have unwanted profibrotic effects (Bozyk and Moore 2011). More importantly, defining the EP receptor subtype responsible for desensitization allows the future exploration of the molecular signaling components co-localized with EP2 receptors in the subcellular compartment in which it resides. This includes PDE isoforms that might be upregulated by prolonged PGE2 exposure. Identification of these downstream components co-localized with EP2 receptors would increase understanding of how these receptors signal and perhaps provide additional therapeutic approaches that would overcome the loss of signaling caused by chronic agonist exposure.
The mechanism by which EP2 receptors desensitize in lung fibroblasts also has not been studied directly. Since EP2 receptors do not internalize due to their shorter C-terminus (Desai et al. 2000), we hypothesized that chronic exposure to PGE2 might drive an increase in expression of one or more of the cAMP-hydrolyzing PDE isoforms. Data here show that PGE2 pretreatment upregulates the expression of PDE3A, PDE3B, PDE4B, and PDE4D mRNA. The changes in expression of PDE3A and PDE4B were highly specific to PGE2 pretreatment, as exposure to the β-AR agonist isoproterenol had no effects on these isoforms. The PDE4 inhibitor rolipram but not the PDE3 inhibitor cilostazol was able to partially reverse desensitization to PGE2, suggesting that EP2 receptor cAMP signals are regulated by a PDE4 isoform in a way that β-AR signaling is not. This mode of regulation has been described in other cells (Bogard et al. 2012; Agarwal et al. 2017), but further studies are needed to confirm the precise PDE isoforms that are active in lung fibroblasts. Previous studies have also implicated PDE4 isoform upregulation following PGE2 treatment. Michalski and co-workers (2012) pretreated primary human lung fibroblasts and found PGE2 pretreatment attenuated cAMP and chemotactic responses in a manner consistent with upregulation of PDE4. They also found this response was altered in cells isolated from patients with chronic obstructive pulmonary disease, furthering the therapeutic potential of PDE inhibition. We attempted to knockdown the expression of specific PDE isoforms using small interfering RNA (siRNA) approaches but were unable to confirm protein knockdown due to either failure of the siRNA or limitations of available antibodies to detect PDE proteins (data not shown). Therefore, more work is needed to reveal which specific PDE isoforms are responsible for the desensitization to PGE2 that we observed.
Another important remaining question is how PGE2 pretreatment leads to the increased expression of a specific PDE isoform. Pretreatment with the β-AR agonist isoproterenol had no effect on subsequent PGE2 stimulation of cAMP, implying that this effect is limited to PGE and perhaps a subset of other cAMP-elevating agents. Pretreatment with forskolin, which stimulates AC directly to increase cAMP levels throughout the cell, also induced little desensitization. Importantly, isoproterenol and forskolin also induced upregulation of fewer PDE genes, providing a possible explanation for the greater decrease in cAMP accumulation with PGE2 pretreatment than for pretreatment with other cAMP-elevating agents. These results imply that either cAMP signaling alone is insufficient for inducing desensitization of EP receptor–stimulated cAMP accumulation or that cAMP signals are highly compartmentalized. cAMP signaling in a very specific subcellular compartment may be required for the desensitization mechanism to be activated, and/or EP2 receptors may localize to different compartments than β-AR. In fact, EP2 receptors exist in discrete membrane microdomains in various cell types where they can couple to specific AC isoforms, specifically AC2 (Johnstone et al. 2018a). β-AR exists primarily in a different compartment with different AC isozymes and PDE isoforms. Fibroblasts may interpret the localized EP2 receptor signal differently and respond by upregulating a specific PDE isoform that selectively regulates cAMP signaling in that domain. This response would leave signaling via β-ARs in their own microdomain unaffected, explaining the results in Figs. 1b and 7d. This hypothesis requires further study but is consistent with that of previous studies and would explain the specific desensitization of PGE2 responses observed here. The concept that a single PDE isoform can selectively regulate cAMP signaling in a specific compartment has been previously demonstrated (Johnstone et al. 2018b).
Several limitations to our study should be noted. First, we observed significant variability in the EC50 values for PGE2 after pretreatment with vehicle or PGE2 across different experiments. This results in some studies having large variability in the EC50 and/or Emax values. This may also explain some of the quantitative differences in the PGE2 responses seen across different figures. Some of this variability may be due to the difficulty in washing out PGE2 (or isoproterenol or forskolin in other experiments) after pretreatment before subsequent responses to PGE2 were measured. Our experimental protocol required limited wash steps because more extensive washing negatively affected cell attachment and viability. Residual drug from the pretreatments would be expected to artificially increase potency of subsequent drug additions, as is particularly noted in Fig. 3 when cells were pretreated with forskolin. The variability could also come from differences in expression of the cADDis sensor. Even though we normalize responses to a maximal stimulus, differences in the biosensor levels could alter the observed sensitivity. Nonetheless, the desensitization induced by pretreatment with PGE2 was highly reproducible in a qualitative sense and consistently different statistically using 2-way ANOVA. Second, our data do not explain why PGE2-induced desensitization was greater than that with pretreatment with the selective EP2 receptor agonist ONO-AE1–259 (22-fold vs. 4-fold, respectively), given that our data taken together suggest that the desensitization is mediated selectively by EP2 receptor activation. This observation leaves open the possibility that other receptors activated by PGE2 but not by ONO-AE1–259 contribute to the induction of desensitization (perhaps EP1 and/or EP3 receptors coupling to Gq). The possibility that EP1 and/or EP3 receptors are involved is diminished by the fact that transcripts for these receptors are very low in HFL-1 cells (GEO accession GSE73555). While several groups have reported that EP2 receptors do not internalize upon agonist exposure, we did not directly test this in our studies. Finally, our studies examined responses in a widely utilized cultured human fetal lung fibroblast cell line, and studies in primary pulmonary fibroblasts from humans as well as in vivo studies in animal models of lung fibrosis may shed new light on the unresolved issues from the present studies.
Given that cAMP has potent antifibrotic effects, any therapy addressing PF should consider cAMP-elevating agents to combat fibrosis. While PGE2 has long been an attractive candidate, its nonspecific activation of all EP receptors could activate unwanted signaling cascades. For this reason, EP2-specific agonists such as ONO-AE-259 should be considered due to their ability to only activate EP2-associated signaling pathways. However, EP2-specific agonists should likely be combined with PDE4-specific inhibitors so that cAMP elevation is promoted while simultaneously inhibiting EP2-mediated desensitization, thus achieving sustained high levels of cAMP that may be sufficient for reducing fibrotic activity. Selective and combinatorial inhibition of PDE isoforms is gaining traction in a wide array of pulmonary diseases (Zuo et al. 2019).
In conclusion, this study demonstrates that long-term exposure of lung fibroblasts to PGE2 induces agonist-specific desensitization of cAMP signaling without altering signaling by β-ARs. Selective agonists and antagonists demonstrate that the EP2 receptor subtype is responsible for both the cAMP signal stimulated by PGE2 and the subsequent desensitization. EP2 receptors do not undergo GRK-mediated phosphorylation and β-arrestin–mediated internalization, making this an unlikely mechanism for the observed desensitization. Increased PDE activity appears responsible for the desensitization, since PDE3A and PDE4D expression levels were selectively upregulated and bulk PDE activity was increased PGE2 pretreatment. The PDE4 inhibitor rolipram reversed the effect of PGE2 pretreatment. These findings provide potential new insights into PF pathology and point to new therapeutic approaches for treating PF.
Funding information
This work was supported by the National Institutes of Health grants GM107094 (RO), HL058506, and HL136209 (RP).
Abbreviations
- AC
Adenylyl cyclase
- β-AR
Beta-adrenoceptor
- cAMP
Cyclic 3′,5′-adenosine monophosphate
- Epac
Exchange protein activated by cAMP
- EP2R
Prostaglandin E2 receptor
- GPCR
G protein–coupled receptor
- PDE
Phosphodiesterase PF Pulmonary fibrosis
- PGE2
Prostaglandin
- E2 PKA
Protein kinase A
- qRT-PCR
Quantitative reverse transcription polymerase chain reaction
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- af Forselles KJ, Root J, Clarke T, Davey D, Aughton K, Dack K, Pullen N (2011) In vitro and in vivo characterization of PF-04418948, a novel, potent and selective prostaglandin EP2 receptor antagonist. Br J Pharmacol 164:1847–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SR, Miyashiro K, Latt H, Ostrom RS, Harvey RD (2017) Compartmentalized cAMP responses to prostaglandin EP2 receptor activation in human airway smooth muscle cells. Br J Pharmacol 174:2784–2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer AC, Schwabe U (1980) An improved assay of cyclic 3′,5′-nucleotide phosphodiesterases with QAE-Sephadex columns. Naunyn Schmiedeberg’s Arch Pharmacol 311:193–198 [DOI] [PubMed] [Google Scholar]
- Bogard AS, Adris P, Ostrom RS (2012) Adenylyl cyclase 2 selectively couples to E prostanoid type 2 receptors, whereas adenylyl cyclase 3 is not receptor-regulated in airway smooth muscle. J Pharmacol Exp Ther 342:586–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozyk PD, Moore BB (2011) Prostaglandin E2 and the pathogenesis of pulmonary fibrosis. Am J Respir Cell Mol Biol 45:445–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dackor RT, Cheng J, Voltz JW, Card JW, Ferguson CD, Garrett RC, Bradbury JA, DeGraff LM, Lih FB, Tomer KB, Flake GP, Travlos GS, Ramsey RW Jr, Edin ML, Morgan DL, Zeldin DC (2011) Prostaglandin E2 protects murine lungs from bleomycin-induced pulmonary fibrosis and lung dysfunction. Am J Phys Lung Cell Mol Phys 301:L645–L655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai S, Ashby B (2001) Agonist-induced internalization and mitogen-activated protein kinase activation of the human prostaglandin EP4 receptor. FEBS Lett 501:156–160 [DOI] [PubMed] [Google Scholar]
- Desai S, April H, Nwaneshiudu C, Ashby B (2000) Comparison of agonist-induced internalization of the human EP2 and EP4 prostaglandin receptors: role of the carboxyl terminus in EP4 receptor sequestration. Mol Pharmacol 58:1279–1286 [DOI] [PubMed] [Google Scholar]
- Deshpande DA, Theriot BS, Penn RB, Walker JK (2008) Beta-arrestins specifically constrain beta2-adrenergic receptor signaling and function in airway smooth muscle. FASEB J 22:2134–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fastres A, Felice F, Roels E, Moermans C, Corhay JL, Bureau F, Louis R, Clercx C, Guiot J (2017) The lung microbiome in idiopathic pulmonary fibrosis: a promising approach for targeted therapies. Int J Mol Sci:18 10.3390/ijms18122735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh T (2014) Prostanoid receptor EP2 as a therapeutic target. J Med Chem 57:4454–4465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel PA, Murray F, Yokoyama U, Romano S, Yun H, Brown L, Snead A, Lu D, Aroonsakool N (2012) cAMP and Epac in the regulation of tissue fibrosis. Br J Pharmacol 166:447–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone TB, Agarwal SR, Harvey RD, Ostrom RS (2018a) cAMP signaling compartmentation: adenylyl cyclases as anchors of dynamic signaling complexes. Mol Pharmacol 93:270–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone TB, Smith KH, Koziol-White CJ, Li F, Kazarian AG, Corpuz ML, Shumyatcher M, Ehlert FJ, Himes BE, Panettieri RA Jr, Ostrom RS (2018b) PDE8 is expressed in human airway smooth muscle and selectively regulates cAMP signaling by beta2-adrenergic receptors and adenylyl cyclase 6. Am J Respir Cell Mol Biol 58:530–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohyama T, Liu X, Kim HJ, Kobayashi T, Ertl RF, Wen FQ, Takizawa H, Rennard SI (2002a) Prostacyclin analogs inhibit fibroblast migration. Am J Phys Lung Cell Mol Phys 283:L428–L432 [DOI] [PubMed] [Google Scholar]
- Kohyama T, Liu X, Wen FQ, Zhu YK, Wang H, Kim HJ, Takizawa H,Cieslinski LB, Barnette MS, Rennard SI (2002b) PDE4 inhibitors attenuate fibroblast chemotaxis and contraction of native collagen gels. Am J Respir Cell Mol Biol 26:694–701 [DOI] [PubMed] [Google Scholar]
- Kolodsick JE, Peters-Golden M, Larios J, Toews GB, Thannickal VJ, Moore BB (2003) Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol 29:537–544 [DOI] [PubMed] [Google Scholar]
- Liu X, Ostrom RS, Insel PA (2004) cAMP-elevating agents and adenylyl cyclase overexpression promote an antifibrotic phenotype in pulmonary fibroblasts. Am J Phys Cell Physiol 286:C1089–C1099 [DOI] [PubMed] [Google Scholar]
- Liu X, Sun SQ, Ostrom RS (2005) Fibrotic lung fibroblasts show blunted inhibition by cAMP due to deficient cAMP response element-binding protein phosphorylation. J Pharmacol Exp Ther 315:678–687 [DOI] [PubMed] [Google Scholar]
- Liu X, Thangavel M, Sun SQ, Kaminsky J, Mahautmr P, Stitham J, Hwa J, Ostrom RS (2008) Adenylyl cyclase type 6 overexpression selectively enhances beta-adrenergic and prostacyclin receptor-mediated inhibition of cardiac fibroblast function because of colocalization in lipid rafts. Naunyn Schmiedeberg’s Arch Pharmacol 377:359–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li F, Sun SQ, Thangavel M, Kaminsky J, Balazs L, Ostrom RS (2010) Fibroblast-specific expression of AC6 enhances beta-adrenergic and prostacyclin signaling and blunts bleomycin-induced pulmonary fibrosis. Am J Phys Lung Cell Mol Phys 298: L819–L829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski J, Kanaji N, Liu X, Nogel S, Wang X, Basma H, Nakanishi M, Sato T, Gunji Y, Fahrid M, Nelson A, Muller KC, Holz O, Magnussen H, Rabe KF, Toews ML, Rennard SI (2012) Attenuation of inhibitory prostaglandin E2 signaling in human lung fibroblasts is mediated by phosphodiesterase 4. Am J Respir Cell Mol Biol 47:729–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Sheng W, Michkov A, Sriarm K, Sun R, Dvorkin-Gheva A, Insel PA, Janssen LJ (2019) Prostaglandin E2 inhibits profibrotic function of human pulmonary fibroblasts by disrupting Ca2+ signaling. Am J Phys Lung Cell Mol Physiol 316:L810–L821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishigaki N, Negishi M, Ichikawa A (1996) Two Gs-coupled prostaglandin E receptor subtypes, EP2 and EP4, differ in desensitization and sensitivity to the metabolic inactivation of the agonist. Mol Pharmacol 50:1031–1037 [PubMed] [Google Scholar]
- Ostrom RS (2014) A two-pronged weapon in the fight against fibrosis. Focus on “inhibition of Wnt/beta-catenin signaling promotes epithelial differentiation of mesenchymal stem cells and repairs bleomycin-induced lung injury”. Am J Phys Cell Physiol 307: C232–C233 [DOI] [PubMed] [Google Scholar]
- Penn RB, Pascual RM, Kim YM, Mundell SJ, Krymskaya VP, Panettieri RA Jr, Benovic JL (2001) Arrestin specificity for G protein-coupled receptors in human airway smooth muscle. J Biol Chem 276:32648–32656 [DOI] [PubMed] [Google Scholar]
- Roberts MJ, Broome RE, Kent TC, Charlton SJ, Rosethorne EM (2018) The inhibition of human lung fibroblast proliferation and differentiation by Gs-coupled receptors is not predicted by the magnitude of cAMP response. Respir Res 19:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzawa T, Miyaura C, Inada M, Maruyama T, Sugimoto Y, Ushikubi F, Ichikawa A, Narumiya S, Suda T (2000) The role of prostaglandin E receptor subtypes (EP1, EP2, EP3, and EP4) in bone resorption: an analysis using specific agonists for the respective EPs. Endocrinology 141:1554–1559 [DOI] [PubMed] [Google Scholar]
- White ES, Atrasz RG, Dickie EG, Aronoff DM, Stambolic V, Mak TW, Moore BB, Peters-Golden M (2005) Prostaglandin E(2) inhibits fibroblast migration by E-prostanoid 2 receptor-mediated increase in PTEN activity. Am J Respir Cell Mol Biol 32:135–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M (1995) Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Invest 95:1861–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RJ, Giblin GM, Roomans S, Rhodes SA, Cartwright KA, Shield VJ, Brown J, Wise A, Chowdhury J, Pritchard S, Coote J, Noel LS, Kenakin T, Burns-Kurtis CL, Morrison V, Gray DW, Giles H (2006) GW627368X ((N-{2-[4-(4,9-diethoxy-1-oxo-1,3-dihydro-2H-benzo [f]isoindol-2-yl)phenyl]acety)l} benzene sulphonamide): a novel, potent and selective prostanoid EP4 receptor antagonist. Br J Pharmacol 148:326–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi DT, Hahn TJ, Beeker TG, Kleeman CR, Muallem S (1988) Relationship of cAMP and calcium messenger systems in prostaglandin-stimulated UMR-106 cells. J Biol Chem 263: 10745–10753 [PubMed] [Google Scholar]
- Zmajkovicova K, Menyhart K, Bauer Y, Studer R, Renault B, Schnoebelen M, Bolinger M, Nayler O, Gatfield J (2019) The antifibrotic activity of prostacyclin receptor agonism is mediated through inhibition of YAP/TAZ. Am J Respir Cell Mol Biol 60: 578–591 [DOI] [PubMed] [Google Scholar]
- Zuo H, Cattani-Cavalieri I, Musheshe N, Nikolaev VO, Schmidt M (2019) Phosphodiesterases as therapeutic targets for respiratory diseases. Pharmacol Ther 197:225–242 [DOI] [PubMed] [Google Scholar]