

Abstract
Site selective amide reductions of the cyclic undecapeptide, cyclosporine A, have been developed using the combination of a heteroleptic borane catalyst and a silane reductant. Tertiary silane Me2EtSiH provides two unique cyclosporine A derivatives, one of which can be readily diversified in subsequent reactions. The secondary silane Et2SiH2 enables divergent reactivity that uses a free hydroxyl group to direct the reduction. The transient O-silyl hemiaminal intermediate of this reduction can additionally be trapped by reducing to the amine or by reductive cyanation.
Graphical Abstract
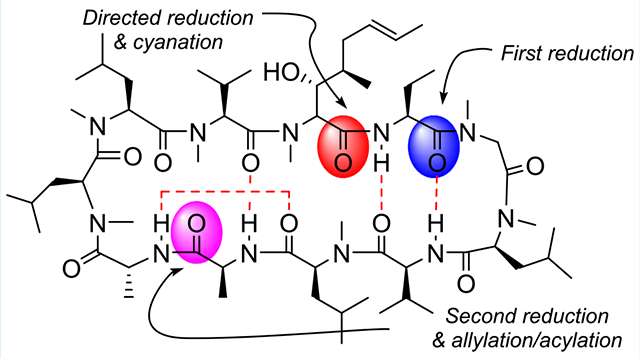
Achieving specificity over which chemically distinct but functionally identical groups are modified in a complex molecule (site selectivity) represents a significant challenge in homogeneous catalysis.1,2 Nowhere is this challenge more acute than in the realm of late-stage functionalization, a strategy in medicinal chemistry where complex natural products or drug candidates are site-selectively modified to optimize their physical, biological or pharmaceutical properties.3–6 The targeted diversification of an already bioactive structure removes the bottleneck imposed by de novo synthesis. While orthogonal reactivity can be exploited to achieve chemoselectivity, the challenge is magnified when a substrate bears multiple copies of the same reactive functional group. In these cases strategic use of catalysts and reagents can provide uniquely enabling methods.
Approaches for site-selective amide reduction in polyamides is not well studied.7,8 Although many ways to reduce amides exist,9,10 including stoichiometric metal hydrides (i.e., LiAlH4), these reactive reagents are not prone to tunable selectivity. Metal catalyzed hydrogenation of amides11,12 is in principle possible, but usually requires extreme conditions (temperature and pressure), which are inhospitable to selectivity. Arguably, the mildest subclass of amide reductions includes those employing silane reductants. Various metal catalysts13 (Ir, Rh, Pt, Zn, Ni) are effective and display varying degrees of chemoselectivity. Metal-free variations exist and use boranes14–17 and boronic acids18 as catalysts. We have recently shown that heteroleptic perfluoroaryl/alkyl borane catalysts, such as B2-Cat, are highly selective for amide reductions relative to their homoleptic counterparts (i.e., B(C6F5)3). Given the successful application of heteroleptic borane catalysts on substrates with the complexity of di- and tripeptides,19 we questioned whether they may also enable site-selective reductions in complex polyamide substrates, and that these reductions would then enable additional structural modifications.
The cyclic undecapeptide natural product, cyclosporine A (CsA, Figure 1), was used to investigate this question, as it provides an excellent test bed for developing site selective amide chemistries.7,8 CsA is a potent immunosuppressant used in the clinic to treat graft rejection, rheumatoid arthritis, and psoriasis. Its immunosuppressant activity stems from its ability to inhibit calcineurin (T cell activator) through formation of the ternary complex cyclophilin·CsA·calcineurin.20 Since the protein cyclophilin is also a target for treating viral infections (hepatitis and HIV) and neural protection, considerable effort has gone into discovering nonimmunosuppresant analogs of CsA,21–23 i.e., those that on binding cyclophilin do not inhibit calcineurin.
Figure 1.
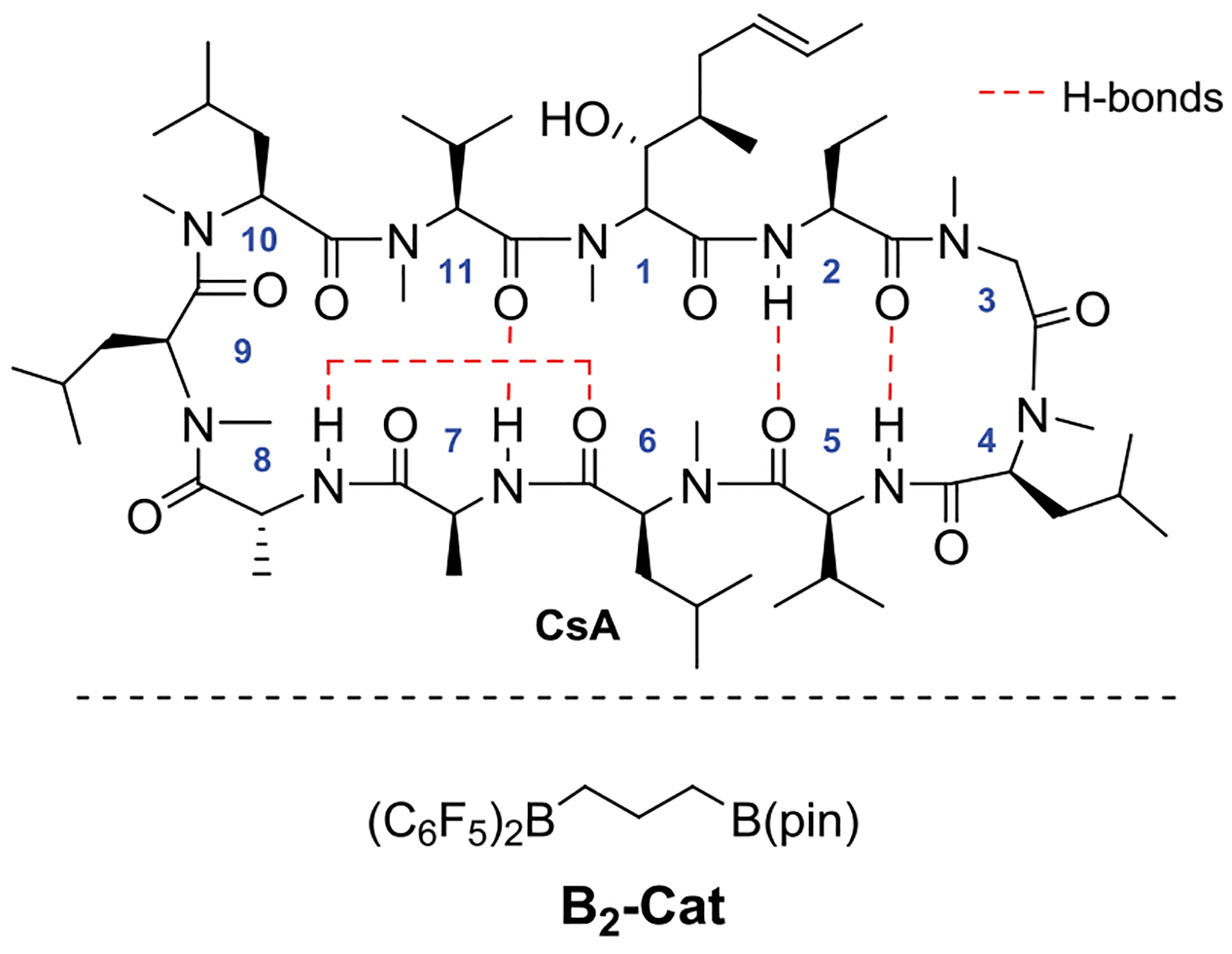
Structure of cyclosporine A and borane catalyst.
The conversion of an amide to an amine alters a peptide’s metabolism, membrane permeability, and bioavailability, all of which can be useful in optimizing a drug’s properties.24,25 Site selective reductions of CsA therefore enable interesting but largely inaccessible structural spaces to be populated. Positionally selective reduction of CsA’s secondary amides is particularly intriguing, as the resulting secondary amines provide new handles for further diversification. Catalysts able to reduce secondary amides, however, are rare.17,26–28
We began our studies using the heteroleptic catalyst, B2-Cat, which is conveniently prepared in situ by reacting Piers borane29,30 ((C6F5)2BH) with allyl-B(pin). B2-Cat in combination with a tertiary silane such as Me2EtSiH in toluene can reduce tertiary amides in the presence of secondary amides, alcohols, heterocycles, etc. Dehydrogenative silylation of the free hydroxyl group of CsA was fast as assessed by the rapid effervescence of the solution. Since the subsequent amide reduction was relatively slow, the actual substrate for the reaction is the OH-silylated CsA-OSiR3. A brief solvent screen indicated that CHCl3 and trifluorotoluene gave higher conversions over 24 h and were thus used for further studies (Scheme 1).
Scheme 1.
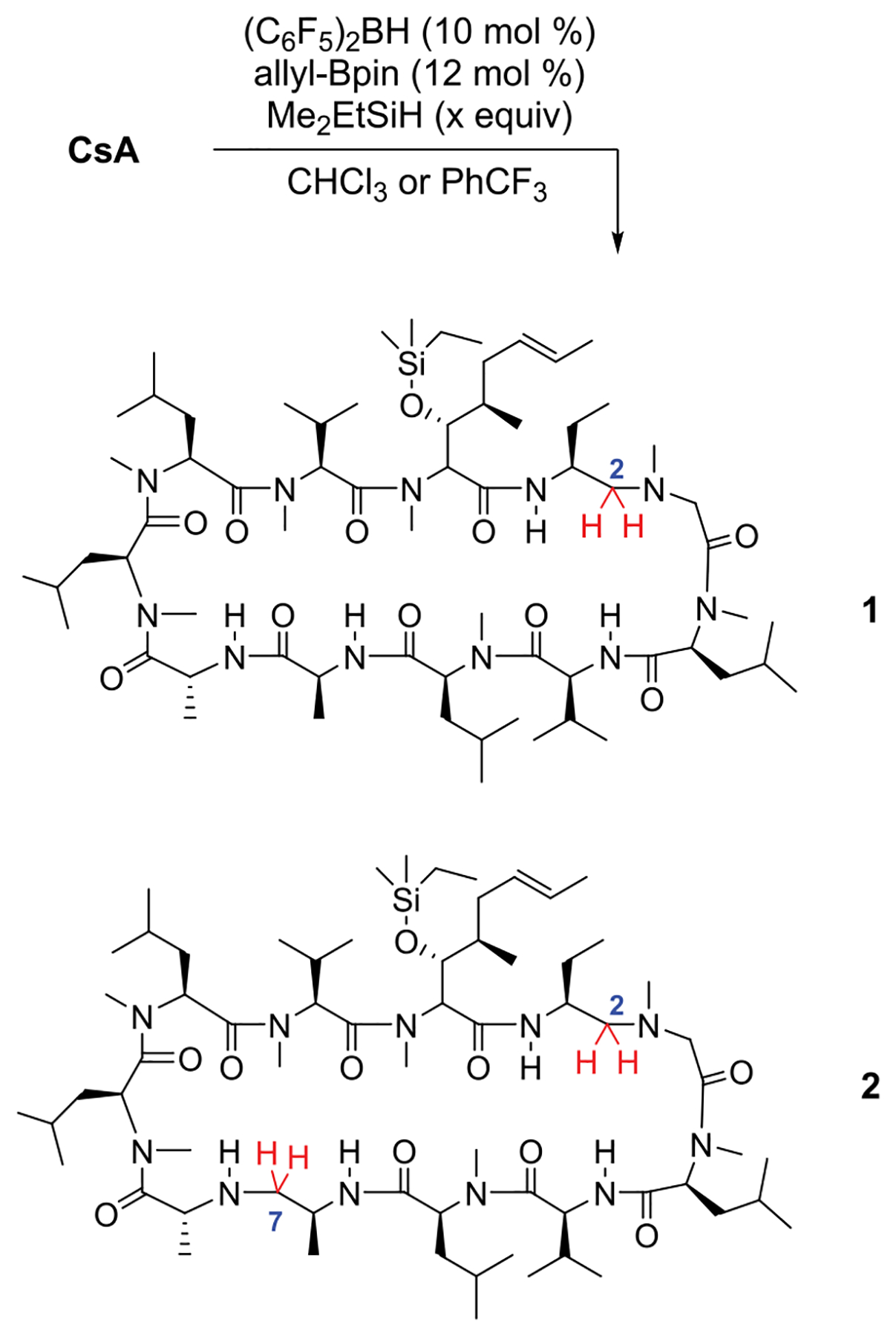
Reduction of 1 with Me2EtSiH
Analysis of the reaction mixture by LC-MS revealed two major products, a single and a double amide reduction. The same products were observed across the solvents tested. The single (1, 37%) and double reduction (2, 16%) products were isolable by flash column chromatography. The structure of the double reduction (2) product was ascertained by a suite of 1H and 13C NMR experiments. Briefly, the amide region of the 13C NMR spectrum confirmed the loss of two amides; the amide region of the 1H NMR spectrum revealed that one secondary amide had been reduced. Selectively irradiating the remaining amide NH resonances via 1D-TOCSY experiments localized the reduction to Ala7 or Ala8, and 2D spectra (HSQC, HMBC, and COSY) identified the site to be Ala7. The site of tertiary amide reduction could then be traced to Abu2 by analysis of the 2D spectra (HSQC, HMBC, COSY).
In contrast to 2, the NMR spectra of 1 were significantly broadened. Since resubjecting 1 to reaction conditions smoothly converted it to 2, the site of reduction was logically either Abu2 or Ala7. This was disambiguated by adapting a peptide sequencing mass spectrometry experiment.
The characterization of peptides via MS/MS is a powerful proteomics technique, but applying these methods to cyclic n-mer peptides is problematic, as the first (ring-opening) fragmentation can occur at any of the (O)C–N bonds to give “n” distinct primary ions, all of which have the same molecular weight. We sought to distinguish between Abu2 or Ala7 being the first site reduced by analyzing the fragmentation of each of the 10 possible ions created by ring opening of a monoreduced CsA. To illustrate, consider the putative linear peptides that result from a ring-opening cleavage at the 5/6 junction in the mass spectrometer of the two isomeric monoreduced possibilities (1a and 1b, Scheme 2). Although the two parent ions (1a and 1b) have the same m/z, sequential fragment loss from these ions predicts a set of b-series ions that are unique to the two possible reduction sites.31 Using a sensitivity threshold of 6σ, 247 fragments were found for 1 in the MS/MS experiment. These ions were then matched to those predicted for 1a and 1b. As indicated by the blue (observed) and red (not observed) coding in Scheme 2, the fidelity of the predicted/observed ions of deoxy-Ala7 is a significantly poorer match than that predicted for deoxy-Abu2. The analyses of the other nine ring opening sites were similarly consistent with 1 being deoxy-Abu2 as described in the Supporting Information.
Scheme 2.
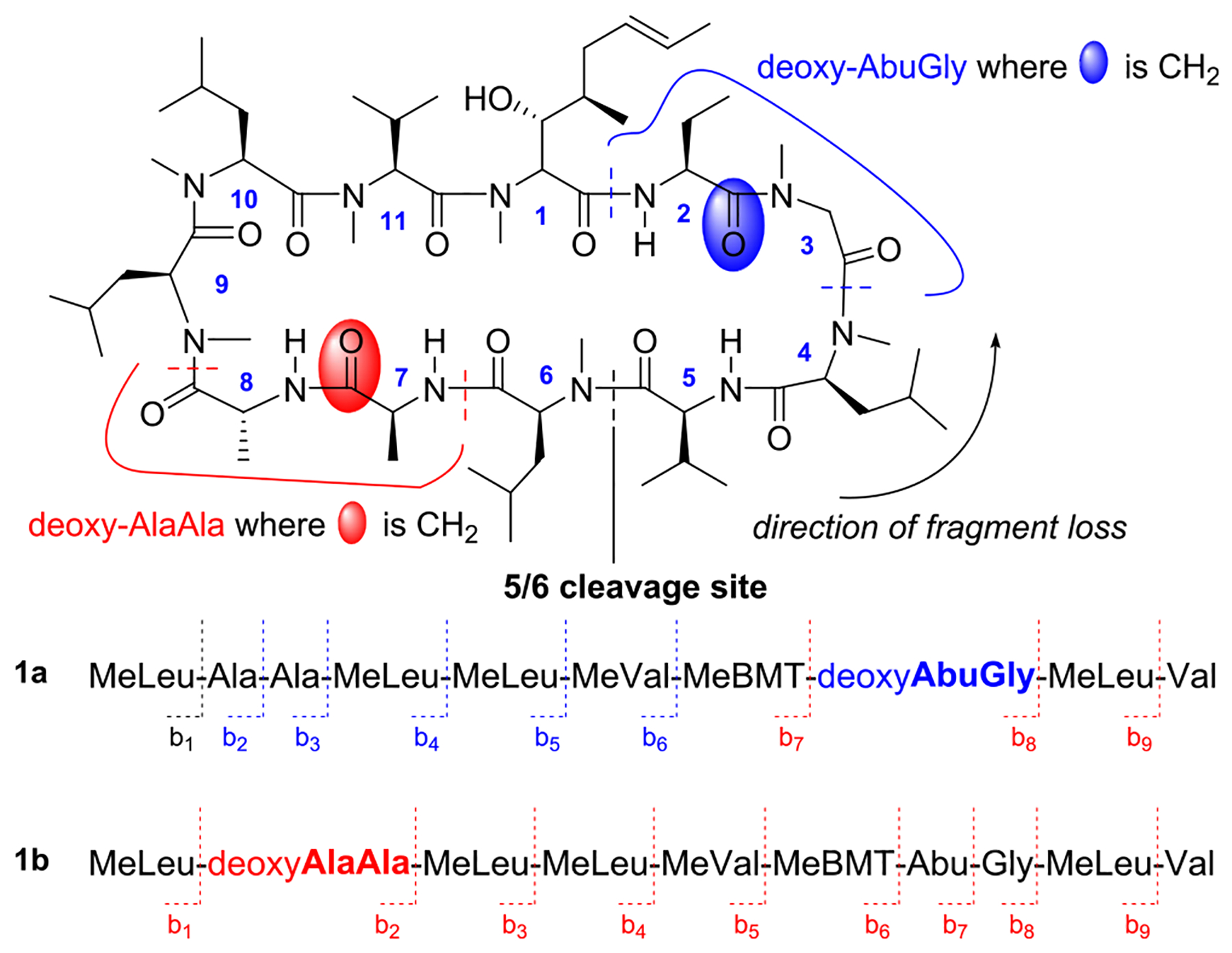
Sample MS2 Analysis of 1
Having successfully identified 1 and 2 we sought to optimize the reaction conditions. As shown in Table 1, the best conditions (entry 7) utilized 15 equiv of silane and were robust to scale up (×8, ~500 mg CsA). The reaction was quite sensitive to the sterics of the silane, as the bulkier Et3SiH (5 or 15 equiv) stops after dehydrocoupling of the free OH.
Table 1.
| entry | solvent | x equiv of Me2EtSiH | yield (%) 1 | yield (%) 2 |
|---|---|---|---|---|
| 1 | PhCF3 | 5 | 24 | 13 |
| 2 | PhCF3 | 9 | 38 | 16 |
| 3 | CHCl3 | 5 | 37 | 16 |
| 4b | CHCl3 | 5 | 31 | 17 |
| 5 | CHCl3 | 9 | 39 | 26 |
| 6c | CHCl3 | 9 | 40 | 26 |
| 7 | CHCl3 | 15 | 35 | 44 |
All reactions were performed with 10 mol % B2-Cat. on 0.05 mmol scale. Isolated yields are given.
Reaction performed at 50 °C.
Reaction performed on 0.4 mmol scale
B2-Cat’s selectivity for first reducing a tertiary (Abu2) over a secondary site in 1 agrees with results on simple substrates. We surmise that B2-Cat selects Abu2 over the tertiary valine and isoleucine sites on steric grounds; however, its preference over the normally more reactive Gly3 is unprecedented. For example, Gly3 is the site reduced by Beller in his Rh catalyzed hydrosilylation protocol7 and it is also the site activated by [Me3O][BF4]32 en route to acyclic CsA derivatives (after MeOH treatment). The preferred reduction sites of B2-Cat are therefore not those that are most sterically accessible, and they additionally differ from previous derivation efforts.21
The secondary amine created in 2 resides at the edge of the calcineurin binding domain but could also modulate its binding to cyclophilin A.21–23 Despite being a difficult nitrogen to alkylate in CsA,21 the amine form in 2 is readily allylated, propargylated, benzylated, and benzoylated in moderate yields (Scheme 3). The allyl and propargyl groups provide excellent handles for further derivations.33–35 Traces of overalkylation were observable by LC-MS. In situ protection of the alcohol under the reduction conditions conveniently prevents O-alkylation in the follow-up derivation steps.
Scheme 3.
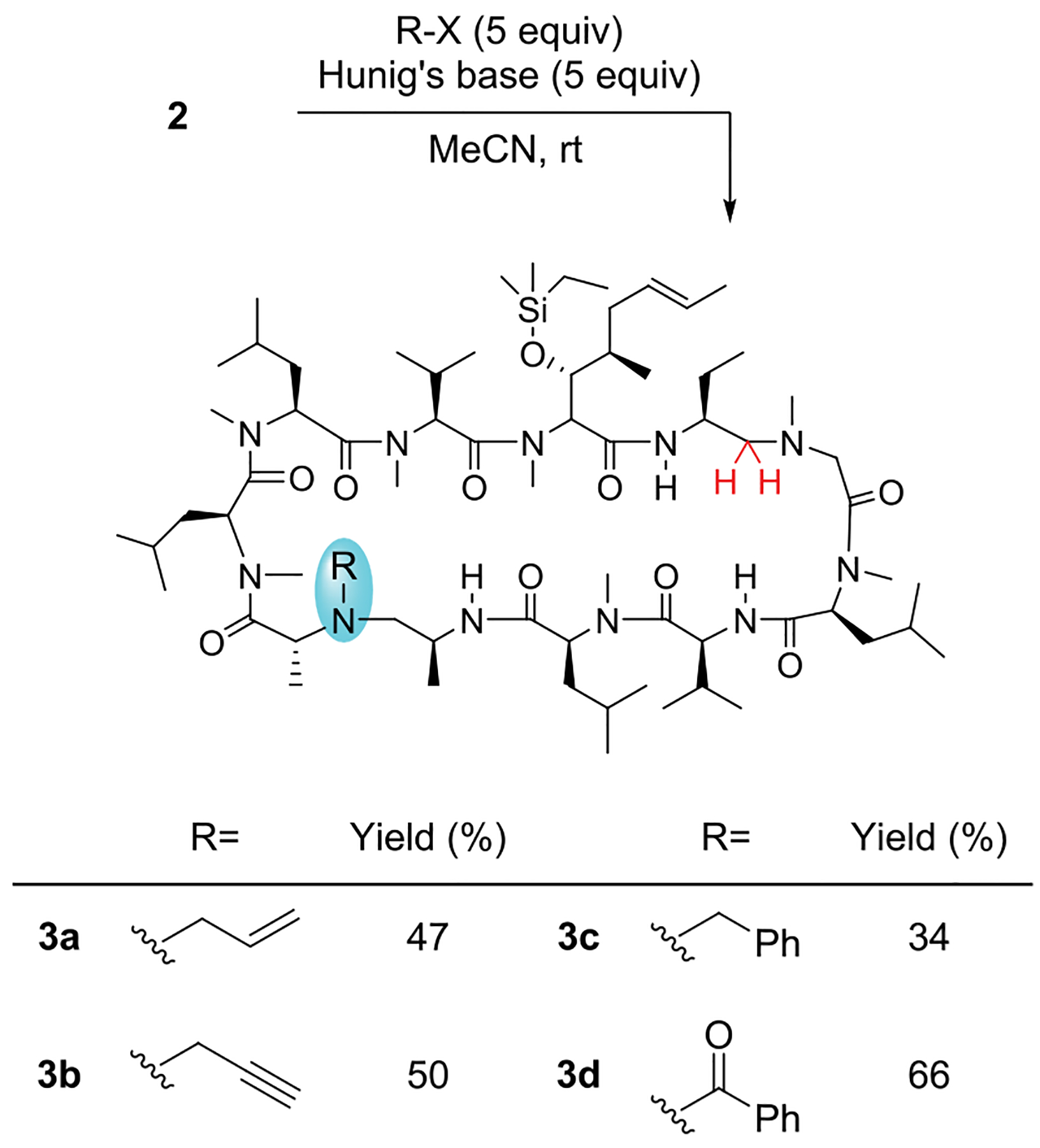
Derivations of 3
We next envisioned that the free hydroxyl group in CsA could be used to direct reactivity to proximal amide sites, as we previously demonstrated with natamycin.2 While preparing this manuscript, the Kanai group applied this strategy using (C6F5)3B and PhMeSiH2.8 Our own conditions (Scheme 4) similarly provided the directed reduction product 3 (at MeBmT1) in 62% yield. HRMS analysis of the product indicated that the residual silyl group is the bis silyl ether that would result from ring opening the intermediate O-silyl hemiaminal. As shown in Scheme 4, the targeted proximal amide generates the six-membered ring intermediate A. Reaching back to Val11 was also considered feasible, but this would require a seven-membered ring intermediate. Since NMR spectra of 3 were sharp, the site of reduction could be identified through selective irradiation of the amide proton resonances in 1D-TOCSY experiments. The reaction also performed well on a 0.5 gram scale giving a 50% isolated yield of chromatographically purified 3.
Scheme 4.
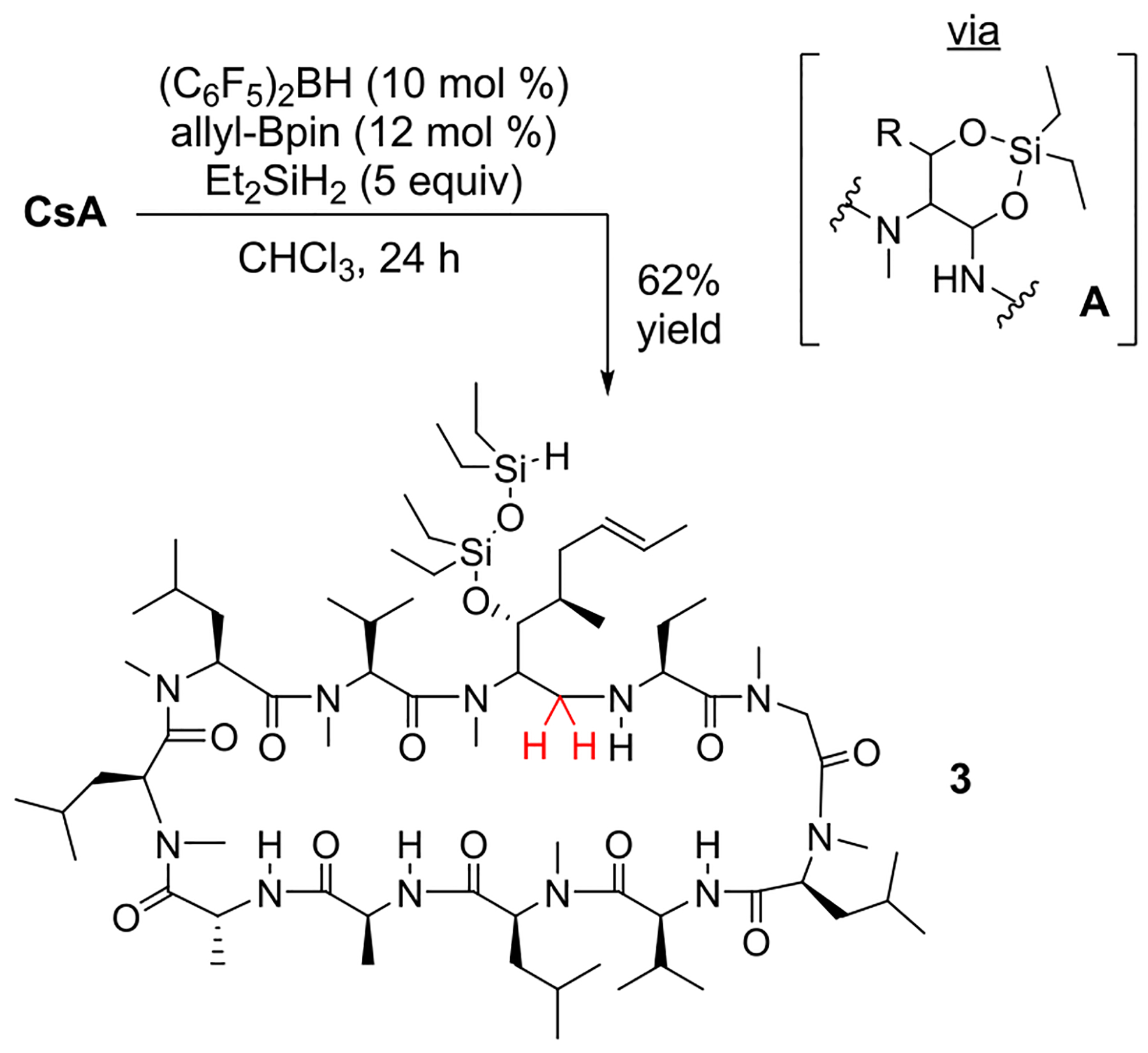
Directed Reduction of 1
O-Silyl hemiaminals such as A are additionally prone to selective chemistry.36–39 Although we have not directly observed this intermediate by in situ 13C or 1H NMR spectroscopy, we hoped that it could be intercepted productively. To this end, CsA was reacted first with stoichiometric Et2SiH2 to putatively generate A. After 1 h, 5 equiv of Bu3Sn-CN were added and the residual B2-Cat was able to catalyze the opening/cyanation to yield the α-amino nitrile 4 in 43% yield as a single diastereomer (Scheme 5). α-Amino nitriles can also be diversely transformed to other potentially interesting structures.36,40
Scheme 5.
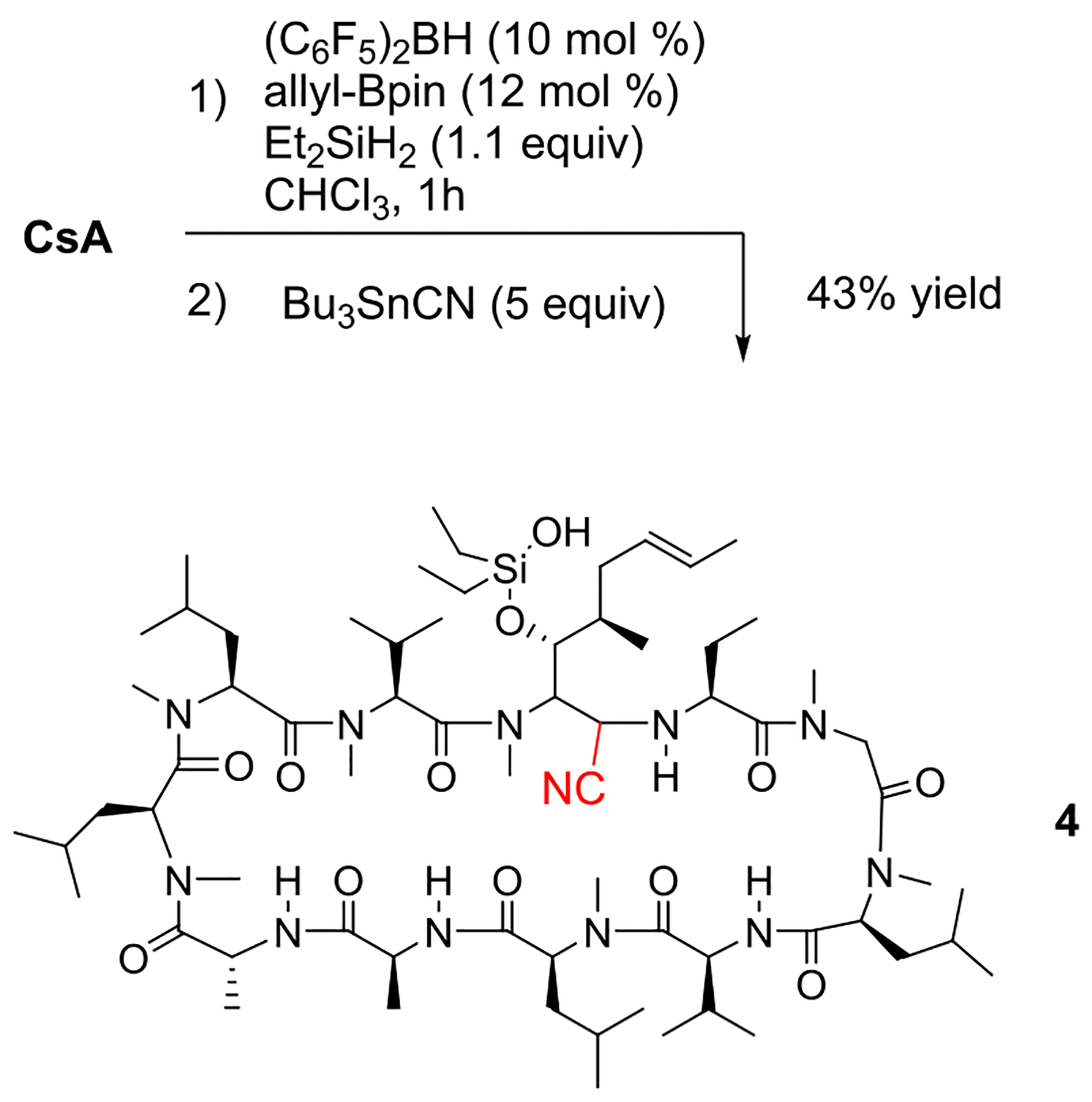
Reductive Cyanation of CsA
The development of amide reduction catalysts able to function under mild conditions has enabled the development of diverse site selective reactions on a complex polyamide structure. Despite being the subject of extensive synthetic studies for its diverse medicinal effects, the observed selectivities with tertiary silanes are novel. Even though the origins of the innate site selectivity are not yet clear, the B2-Cat system reduces amide sites not targeted in other systems and doubles the number of known amide reduced CsA derivatives. Reduction of secondary amide Ala7 additionally reveals a secondary amine that is prone to diversity generating reactivity. Furthermore, the B2-Cat catalyzed amide reductions can be directed using secondary silanes, in this case using the MeBmT1 hydroxyl to select for MeBmT1 reduction over other sites.8 We also report that the O-silyl hemiaminal intermediate (A) can be intercepted to form new C–C bonds, and products that can be further diversified. These studies provide a framework for using nonmetal catalysts to site selectively diversify complex polyamides with important biological properties.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM130693. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. S.J.L. thanks the Army Research Office for support. Equipment grants were also enabling, and we thank the National Science Foundation (CHE-1726291), and UNC’s School of Medicine Office of Research. We thank UNC’s Department of Chemistry Mass Spectrometry Core Laboratory, especially Dr. Brandie Ehrmann, for her assistance with the mass spectrometry analysis.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:10.1021/acs.orglett.9b01245.
Experimental methods and characterization data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Kawabata T In Site-Selective Catalysis; Kawabata T, Ed.; Topics in Current Chemistry; Springer International Publishing: Cham, 2016; Vol. 372. [Google Scholar]
- (2).Bender TA; Payne PR; Gagné MR Late-Stage Chemoselective Functional-Group Manipulation of Bioactive Natural Products with Super-Electrophilic Silylium Ions. Nat. Chem 2018, 10, 85–90. [DOI] [PubMed] [Google Scholar]
- (3).Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW The Medicinal Chemist’s Toolbox for Late Stage Functionalization of Drug-like Molecules. Chem. Soc. Rev 2016, 45, 546–576. [DOI] [PubMed] [Google Scholar]
- (4).Wencel-Delord J; Glorius F C–H Bond Activation Enables the Rapid Construction and Late-Stage Diversification of Functional Molecules. Nat. Chem 2013, 5, 369–375. [DOI] [PubMed] [Google Scholar]
- (5).Cohen DT; Zhang C; Fadzen CM; Mijalis AJ; Hie L; Johnson KD; Shriver Z; Plante O; Miller SJ; Buchwald SL; et al. A Chemoselective Strategy for Late-Stage Functionalization of Complex Small Molecules with Polypeptides and Proteins. Nat. Chem 2019, 11, 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Abid Masood M; Farrant E; Morao I; Bazin M; Perez M; Bunnage ME; Fancy SA; Peakman T Lead Diversification. Application to Existing Drug Molecules: Mifepristone 1 and Antalarmin 8. Bioorg. Med. Chem. Lett 2012, 22, 723–728. [DOI] [PubMed] [Google Scholar]
- (7).Das S; Li Y; Bornschein C; Pisiewicz S; Kiersch K; Michalik D; Gallou F; Junge K; Beller M Selective Rhodium-Catalyzed Reduction of Tertiary Amides in Amino Acid Esters and Peptides. Angew. Chem., Int. Ed 2015, 54, 12389–12393. [DOI] [PubMed] [Google Scholar]
- (8).Ni J; Oguro T; Sawazaki T; Sohma Y; Kanai M Hydroxy Group Directed Catalytic Hydrosilylation of Amides. Org. Lett 2018, 20, 7371–7374. [DOI] [PubMed] [Google Scholar]
- (9).Volkov A; Tinnis F; Slagbrand T; Trillo P; Adolfsson H Chemoselective Reduction of Carboxamides. Chem. Soc. Rev 2016, 45, 6685–6697. [DOI] [PubMed] [Google Scholar]
- (10).Chardon A; Morisset E; Rouden J; Blanchet J Recent Advances in Amide Reductions. Synthesis 2018, 50, 984–997. [Google Scholar]
- (11).Werkmeister S; Junge K; Beller M Catalytic Hydrogenation of Carboxylic Acid Esters, Amides, and Nitriles with Homogeneous Catalysts. Org. Process Res. Dev 2014, 18, 289–302. [Google Scholar]
- (12).Coetzee J; Dodds DL; Klankermayer J; Brosinski S; Leitner W; Slawin AMZ; Cole-Hamilton DJ Homogeneous Catalytic Hydrogenation of Amides to Amines. Chem. - Eur. J 2013, 19, 11039–11050. [DOI] [PubMed] [Google Scholar]
- (13).Li B; Sortais J-B; Darcel C Amine Synthesis via Transition Metal Homogeneous Catalysed Hydrosilylation. RSC Adv. 2016, 6, 57603–57625. [Google Scholar]
- (14).Blondiaux E; Cantat T Efficient Metal-Free Hydrosilylation of Tertiary, Secondary and Primary Amides to Amines. Chem. Commun 2014, 50, 9349–9352. [DOI] [PubMed] [Google Scholar]
- (15).Tan M; Zhang Y An Efficient Metal-Free Reduction Using Diphenylsilane with (Tris-Perfluorophenyl)Borane as Catalyst. Tetrahedron Lett. 2009, 50, 4912–4915. [Google Scholar]
- (16).Huang PQ; Lang QW; Wang YR Mild Metal-Free Hydrosilylation of Secondary Amides to Amines. J. Org. Chem 2016, 81, 4235–4243. [DOI] [PubMed] [Google Scholar]
- (17).Chadwick RC; Kardelis V; Lim P; Adronov A Metal-Free Reduction of Secondary and Tertiary N -Phenyl Amides by Tris(Pentafluorophenyl)Boron-Catalyzed Hydrosilylation. J. Org. Chem 2014, 79, 7728–7733. [DOI] [PubMed] [Google Scholar]
- (18).Li Y; Molina de La Torre JA; Grabow K; Bentrup U; Junge K; Zhou S; Brückner A; Beller M Selective Reduction of Amides to Amines by Boronic Acid Catalyzed Hydrosilylation. Angew. Chem., Int. Ed 2013, 52, 11577–11580. [DOI] [PubMed] [Google Scholar]
- (19).Peruzzi MT; Mei QQ; Lee SJ; Gagné MR Chemoselective Amide Reductions by Heteroleptic Fluoroaryl Boron Lewis Acids. Chem. Commun 2018, 54, 5855–5858. [DOI] [PubMed] [Google Scholar]
- (20).Kallen J; Mikol V; Quesniaux VFJ; Walkinshaw MD; Schneider-Scherzer E; Schörgendorfer K; Weber G; Fliri HG Cyclosporins: Recent Developments in Biosynthesis, Pharmacology and Biology, and Clinical Applications. Biotechnology; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2008; pp 535–591. [Google Scholar]
- (21).Peel M; Scribner A Semi-Synthesis of Cyclosporins. Biochim. Biophys. Acta, Gen. Subj 2015, 1850, 2121. [DOI] [PubMed] [Google Scholar]
- (22).Sweeney ZK; Fu J; Wiedmann B From Chemical Tools to Clinical Medicines: Nonimmunosuppressive Cyclophilin Inhibitors Derived from the Cyclosporin and Sanglifehrin Scaffolds. J. Med. Chem 2014, 57, 7145–7159. [DOI] [PubMed] [Google Scholar]
- (23).Fu J; Tjandra M; Becker C; Bednarczyk D; Capparelli M; Elling R; Hanna I; Fujimoto R; Furegati M; Karur S; et al. Potent Nonimmunosuppressive Cyclophilin Inhibitors With Improved Pharmaceutical Properties and Decreased Transporter Inhibition. J. Med. Chem 2014, 57, 8503–8516. [DOI] [PubMed] [Google Scholar]
- (24).Patani GA; LaVoie EJ Bioisosterism: A Rational Approach in Drug Design. Chem. Rev 1996, 96, 3147–3176. [DOI] [PubMed] [Google Scholar]
- (25).Wlodawer A; Erickson JW Structure-Based Inhibitors of HIV-1 Protease. Annu. Rev. Biochem 1993, 62, 543–585. [DOI] [PubMed] [Google Scholar]
- (26).Das S; Li Y; Lu LQ; Junge K; Beller M A General and Selective Rhodium-Catalyzed Reduction of Amides, N-Acyl Amino Esters, and Dipeptides Using Phenylsilane. Chem. - Eur. J 2016, 22, 7050–7053. [DOI] [PubMed] [Google Scholar]
- (27).Cheng C; Brookhart M Iridium-Catalyzed Reduction of Secondary Amides to Secondary Amines and Imines by Diethylsilane. J. Am. Chem. Soc 2012, 134, 11304–11307. [DOI] [PubMed] [Google Scholar]
- (28).Simmons BJ; Hoffmann M; Hwang J; Jackl MK; Garg NK Nickel-Catalyzed Reduction of Secondary and Tertiary Amides. Org. Lett 2017, 19, 1910–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Parks DJ; von H Spence RE; Piers WE Bis(Pentafluorophenyl)Borane: Synthesis, Properties, and Hydroboration Chemistry of a Highly Electrophilic Borane Reagent. Angew. Chem., Int. Ed. Engl 1995, 34, 809–811. [Google Scholar]
- (30).Parks DJ; Piers WE; Yap GPA Synthesis, Properties, and Hydroboration Activity of the Highly Electrophilic Borane Bis(Pentafluorophenyl)Borane, HB(C6F5)2. Organometallics 1998, 17, 5492–5503. [Google Scholar]
- (31).Liu W; Ng J; Meluzzi D; Bandeira N; Gutierrez M; Simmons TL; Schultz AW; Linington RG; Moore BS; Gerwick WH; et al. Interpretation of Tandem Mass Spectra Obtained from Cyclic Nonribosomal Peptides. Anal. Chem 2009, 81, 4200–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Riss B; Grandeury A; Gut T; Seeger-Weibel M; Zuercher C; Li J; Gallou F Development of a Practical Process for the Opening of Macrocyclic Cyclosporin a and Amino Acid Deletion. Org. Process Res. Dev 2014, 18, 1763–1770. [Google Scholar]
- (33).Grubbs RH; Chang S Recent Advances in Olefin Metathesis and Its Application in Organic Synthesis. Tetrahedron 1998, 54, 4413–4450. [Google Scholar]
- (34).Ogba OM; Warner NC; O’Leary DJ; Grubbs RH Recent Advances in Ruthenium-Based Olefin Metathesis. Chem. Soc. Rev 2018, 47, 4510–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Thirumurugan P; Matosiuk D; Jozwiak K Click Chemistry for Drug Development and Diverse Chemical–Biology Applications. Chem. Rev 2013, 113, 4905–4979. [DOI] [PubMed] [Google Scholar]
- (36).Fuentes de Arriba ÁL; Lenci E; Sonawane M; Formery O; Dixon DJ Iridium-Catalyzed Reductive Strecker Reaction for Late-Stage Amide and Lactam Cyanation. Angew. Chem., Int. Ed 2017, 56, 3655–3659. [DOI] [PubMed] [Google Scholar]
- (37).Huang YY; Cai C; Yang X; Lv ZC; Schneider U Catalytic Asymmetric Reactions with N,O-Aminals. ACS Catal. 2016, 6, 5747–5763. [Google Scholar]
- (38).Dixon D; Xie L-G Tertiary Amine Synthesis via Reductive Coupling of Amides with Grignard Reagents. Chem. Sci 2017, 8, 7492–7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Xie LG; Dixon DJ Iridium-Catalyzed Reductive Ugi-Type Reactions of Tertiary Amides. Nat. Commun 2018, 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kouznetsov VV; Galvis CEP Strecker Reaction and α-Amino Nitriles: Recent Advances in Their Chemistry, Synthesis, and Biological Properties. Tetrahedron 2018, 74, 773–810. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.