

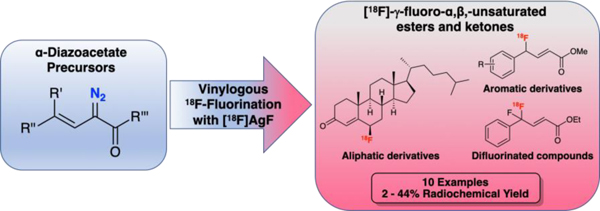
Abstract
This communication reports a method for the vinylogous radiofluorination of α-diazoacetates to generate γ-[18F]fluoro-α,β-unsaturated esters and ketones in moderate to good radiochemical yields. The method uses no-carrier-added [18F]AgF and is compatible with aromatic and non-aromatic substrates and a number of different functional groups. The labeling method is showcased in the synthesis of a fluorinated 5-cholesten-3-one derivative as well as a difluorinated product pertinent to drug discovery.
Keywords: Fluorine-18, late-stage fluorination, PET radiochemistry, diazonium chemistry, positron emission tomography
Graphical Abstract
Positron emission tomography (PET) is a functional imaging technique that is used for clinical diagnostic imaging as well as research applications in academic medical centers and pharmaceutical companies.1,2 Fluorine-18 (18F) is one of the most commonly used PET radionuclides because of its useful half-life (110 min) and favorable imaging properties. Reflecting this, the development of new methods for accessing novel radiotracer motifs using fluorine-18 is an exciting area of research that has led to development of numerous new methods for the fluorination of a diverse array of substrates in recent years.3 New C–18F bond-forming reactions need to be compatible with the unique challenges of working with 18F, such as short reaction times (usually ≤20 min), automated synthesis and purification, and cGMP compliant dose-on-demand production. In this context, transition metal-mediated nucleophilic radiofluorination methods have emerged as particularly practical approaches for forming new C–18F bonds.4
Incorporation of 18F at an sp3 carbon is one of the most widely used labelling strategies. This is typically achieved via nucleophilic displacement of an appropriate leaving group with [18F]fluoride, such as in the production of 2-[18F]fluoro- 2-deoxy-D-glucose ([18F]FDG) by the reaction of mannose triflate with K[18F]F, one of the most commonly used labelling reactions in PET radiochemistry.5 However, this approach is not compatible with all substrates and new methods for generating C(sp3)–F bonds are essential to simplify production of diverse libraries of PET radiotracers for preclinical and clinical evaluation.
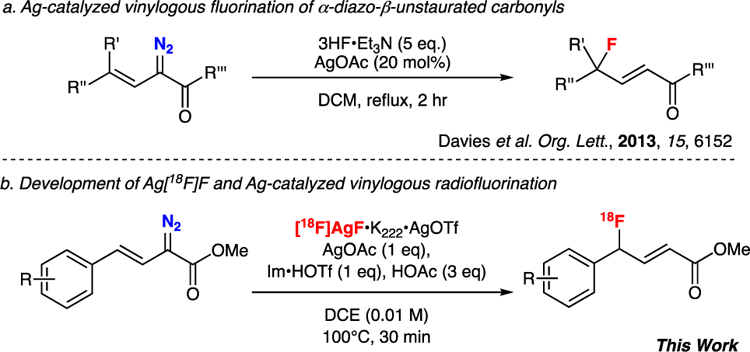
In recent years we and others have reported new reactions for generating both C(sp2)–F and C(sp3)–F bonds that have proven challenging using traditional nucleophilic substitution reactions with [18F]fluoride.6 Key to developing such reactions has been our discovery that different forms of [18F]fluoride (beyond traditional [18F]KF) are easily accessible by simple adjustment of the solution used to elute [18F]fluoride from the quaternary methylammonium (QMA) cartridge employed to reprocess [18F]fluoride obtained from cyclotron-irradiated [18O]H2O.6 For example, we recently reported a new method for generating [18F]AgF6a and utilized it in the C(sp3)–H radiofluorination of a series of 8-methyl quinoline derivatives.6d This work demonstrated that new fluorine-18 radiochemistry could be accessed using [18F]AgF, and we were interested to explore whether we could use [18F]AgF in the development of other novel radiofluorination methodology. In particular, vinylogous fluorination is quite challenging, and we were particularly interested in the Ag-catalyzed vinylogous fluorination of α-diazoacetates recently described by Davies and co-workers (Scheme 1a).7 This paper describes our efforts towards translating Ag-mediated vinylogous fluorination to radio-fluorination using [18F]AgF (Scheme 1b). The transformation was particularly attractive as (1) gamma (γ) functionalization of carbonyl compounds generally remains challenging,8 relying primarily on SN2’ reactions with α-substituted-β,γ-unsaturated esters, reaction of activated enol ethers with electrophiles, or Wittig-type reactions with prefunctionalised reagents, all of which are challenging transformations to accomplish with [18F]fluoride; (2) while diazo compounds have been demonstrated as useful precursors for radiofluorination9 and related methods for allylic 18F-fluorination have been reported,10 both remain unexploited as methods for accessing candidate PET radiotracers and radioligands; (3) the resultant γ-fluoro-α,β,-unsaturated esters are useful synthons for further reaction and (4) difluorinated moieties are attracting attention as bioisosteres and for the synthesis of PET imaging agents.11,12
Scheme 1.
Strategies for (radio)fluorination of α-diazoacetates and motivation for this work
Initial examination of the reaction, as reported by Davies,7 identified two parameters which could potentially make such a reaction challenging to adapt for use with fluorine-18. Firstly, the reaction requires the use of an excess of fluoride (15 equivalents) with respect to the diazonium reagent. In PET radiochemistry, [18F]fluoride is produced in pmol to nmol amounts and, under no-carrier-added conditions, is always the limiting reagent, often by several orders of magnitude. Secondly, the reaction requires a proton source as the reaction involves the (formal) addition of HF and in most cases, [18F]fluoride is used in under basic conditions.
Our initial investigations focused on the radiofluorination of model α-diazoacetate substrate 1N2 using the previously-described preparation of [18F]AgF, with kryptofix-222 (K222) as a phase transfer catalyst (Table 1).6d Early experiments focused on the reaction of Ag[18F]F•K222•AgOTf with 1N2 at 40 °C in dichloromethane (DCM) in the dark, using a protocol similar to that described by Davies.7 Under these conditions, radiochemical yields (RCY)13 of [18F]1F were found to be ≤0.5% (Table 1, entry 1), which was not unexpected considering the lack of a proton source in this system. Screening of protic additives in the reaction identified imidazolium triflate (Im•HOTf) as an additive that led to improved RCYs (Table 1, entry 2). RCYs were further increased to 23 ± 11 % (n = 7) upon addition of 3 equivalents of acetic acid (AcOH, Table 1, entry 3). Further optimization of the reaction revealed that higher temperatures (which necessitated changing the reaction solvent from DCM to dichloroethane (DCE)) also led to improved yields. At 40 °C in DCE in the presence of Im•HOTf and AcOH, [18F]1F was produced in 12 ± 6 % RCY (n = 4) (Table 1, entry 4), which was lower than the observed RCY in DCM at that temperature. However, increasing the temperature of the reaction to 100 °C increased the RCY of [18F]1F to 40 ± 12 % (n = 26) (Table 1, entry 5). Further increases in temperature under these conditions led to erosion of the observed RCY. Interestingly, RCYs for the transformation when [18F]KF•K222•KOTf was used as a source of [18F]fluoride are similar to those observed when using [18F]AgF•K222•AgOTf (Table 1, entries 5 and 6). Since the transformation using [18F]AgF•K222•AgOTf requires 1 equivalent of AgOAc for the reaction to proceed, it is perhaps not surprising that the reaction proceeds equally well with [18F]AgF and [18F]KF. Omitting AgOAc from the reaction greatly suppressed RCY, regardless of whether [18F]AgF or [18F]KF was used as the [18F]fluoride source (Table 1, entries 7 and 8). Ultimately however, in order to maintain a common counter ion, we elected to move forward with [18F]AgF•K222•AgOTf for further development of this methodology. Other variables, including the identity and loading of a variety of silver and imidazolium salts, and weak acids as well as the concentration of 1N2 and AgOAc were optimized for their effect on the reaction (see Supporting Information). Ultimately, the optimal conditions for the conversion of 1N2 to [18F]1F were as follows: 10 µmol of 1N2, 10 µmol AgOAc, 10 µmol of Im•HOTf, 30 µmol AcOH, [18F]AgF•K222•AgOTf in DCE (100 µL, 100–1000 µCi) in a total volume of 1 mL DCE, heated to 100 °C for 30 minutes. These optimal conditions generated [18F]1F RCY of 40 ± 12 % (n = 26) (Table 1, entry 5). Analysis of a typical radiosynthesis employing 12 MBq of [18F]fluoride provided 151 MBq of [18F]1F (46% RCY) with a molar activity of 18 GBq / mmol (typical for a lower activity synthesis run in our laboratory6d).
Table 1.
Optimization of radiofluorination of α-diazoacetates using [18F]AgF
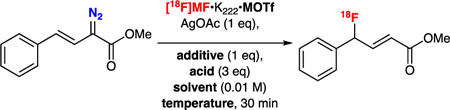 | ||||||
|---|---|---|---|---|---|---|
| Entry | M | Additive | Acid | Solvent | Temp | RCY (%)13 |
| 1 | Ag | - | - | DCM | 40 | 0.5±0.1 (n=3) |
| 2 | Ag | Im•HOTf | - | DCM | 40 | 8±3 (n=3) |
| 3 | Ag | Im•HOTf | HOAc | DCM | 40 | 23±11 (n=7) |
| 4 | Ag | Im•HOTf | HOAc | DCE | 40 | 12±6 (n=4) |
| 5 | Ag | Im•HOTf | HOAc | DCE | 100 | 40±12 (n=26) |
| 6 | K | Im•HOTf | HOAc | DCE | 100 | 40±3 (n=3) |
| 7a | Ag | Im•HOTf | HOAc | DCE | 100 | 3±2 (n=3) |
| 8a | K | Im•HOTf | HOAc | DCE | 100 | 1±0 (n=3) |
Conditons: 10 µmol of 1N2, 10 µmol AgOAc, 10 µmol of additive, 30 µmol of acid, [18F]MF•K222•MOTf in solvent (100 µL, 100–1000 µCi) in a total volume of 1 mL solvent heated to the indicated temperature for 30 min. RCY are non-solated yield estimated by radioTLC and are reported as mean ± standard deviation for n runs;
Reactions run without any AgOAc.
Following optimization of the reaction conditions using simple unsubstituted arene substrate 1N2, the substrate scope of the reaction was explored using a series of substituted arene precursors without further optimization of the reaction conditions (Figure 1). Electron-deficient ([18F]2F) and electron-neutral arenes ([18F]3F – [18F]6F) were well tolerated in the reaction. Contrastingly, electron rich arenes, such as anisoles ([18F]7F) and N,N-dimethylanilines ([18F]8F)proved poor substrates presumably due to instability of the 18F-fluorinated product, which is implied by the in situ analysis of such substrates in the original reaction.7,14 While most substrates tested contained para-substituents, the reaction was also tolerant of ortho- and meta-substituents on the phenyl ring. RCYs of the para- ([18F]4F), meta- ([18F]5F) and ortho-bromo ([18F]6F) substituted products were comparable, with the slightly lower RCYs of the ortho- and meta-substituted products likely attributable to steric effects. Using a fluorovinyl-α-diazo precursor (9N2), it was found that the difluorinated product [18F]9F could be obtained in 45 ± 5 % (n = 4) RCY, suggesting that this transformation may be useful for the synthesis of [18F]CF2 groups which is of interest as the CF2 motif is being explored for PET imaging and as a bioisostere in drug design.11,12 Finally, it was demonstrated that this method was suitable for radiolabeling non-aromatic precursors, whereby 4-diazo-functionalized 5-cholesten-3-one was successfully radiofluorinated using Ag[18F]F•K222•AgOTf to generate [18F]10F in modest radiochemical yield (15 ± 8 %, n = 4). The latter compound is of interest for our program radiolabeling steroid derivatives for applications, such as use of PET to quantify cholesterol metabolism.6e,15
Figure 1.
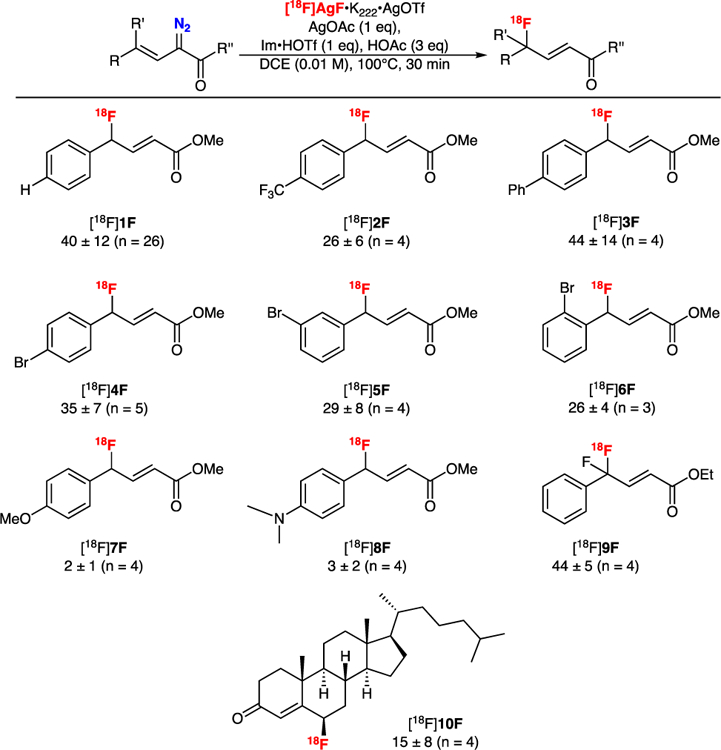
Substrate scope of AgOAc-mediated radiofluorination of α-diazoacetates using Ag[18F]F. Conditions: 10 µmol of 1N2 – 8N2, 10 µmol AgOAc, 10 µmol of Im•HOTf, 30 µmol AcOH, Ag[18F]F•K222•AgOTf in DCE (100 µL, 100–1000 µCi) in a total volume of 1 mL DCE heated to 100 °C for 30 minutes. Non-isolated RCYs were estimated by radioTLC and product identities were confirmed by radioHPLC.
In conclusion, we have developed a simple method for the vinylogous radiofluorination of substituted α-diazoacetates in moderate to good radiochemical yield using [18F]AgF. This method provides access to γ-fluoro-α,β,-unsaturated esters which are useful synthons for further elaboration. These labeled substrates are challenging to access via traditional radiochemistry, and this method has potential for labeling drug molecules and preparing new PET radiotracers. In the latter case, we are in the process of using the new methodology to label steroids such as [18F]10F for pre-clinical PET imaging and anticipated translation into clinical trials.
General Considerations
Reagents were purchased from Sigma Aldrich, Alfa Aesar, Oakwood, Fisher Scientific, EMD Millipore Corporation and Acros Organics. 1H NMR spectra were obtained on a Varian 400 MHz MR NMR (399.7 MHz for 1H, 100.5 MHz for 13C and 376.1 MHz for 19F) and Varian 500 MHz VNMRS (499.5 MHz for 1H, 125.6 MHz for 13C) spectrometers. Chemical shifts are reported in parts per million (ppm) and referenced to tetramethylsilane as in internal standard (1H: δ = 0.00) or residual solvent peak (CDCl3: 1H: δ = 7.26 ppm, 13C: δ = 77.16 ppm). 19F NMR spectra are referenced to an external standard trichlorofluoromethane (CFCl3: δ = 0.00 ppm for 19F). NMR spectra were recorded at room temperature. The abbreviations for 1H and 19F multiplicities are reported as follows: singlet (s), doublet (d), triplet (t), quartet (q), doublet of doublets (dd), doublet of doublets of doublets (ddd), doublet of triplets (dt), triplet of doublets (td), and multiplet (m). Broad signals are indicated by “br”. Coupling constants (J) are reported in hertz (Hz). High-resolution mass spectra were recorded on a Micromass AutoSpec Ultima Magnetic Sector mass spectrometer. Thin layer chromatography (TLC) was performed on Merck KGaA. pre-coated TLC Silica gel 60 F254 plates. Flash column chromatography was conducted using a Biotage Isolera Prime system with SNAP KP-Sil column cartridges (10 g or 25 g).
Reagents were purchased from Sigma Aldrich, Alfa Aesar, Oakwood, Fisher Scientific, EMD Millipore Corporation and Acros Organics. Ultrapure water was obtained from a Millipore MilliQ Gradient A10 system. Sterile vials were purchased from Hollister-Stier.
QMA-light Sep-Paks were purchased form Waters Corporation, and were preconditioned with 10 ml ethanol, followed by 10 mL of water, followed by 10 mL of 0.5 M aqueous potassium triflate solution, followed by 10 mL of water before use.
Glass backed thin layer chromatography (TLC) plates coated with silica gel 60F254 were used for TLC- and radio-TLC analysis and were purchased from EMD-Millipore. TLC plates were visualized with KMnO4 or anisaldehyde stain. Radio-TLC analysis was performed using a Bioscan AR 2000 Radio-TLC scanner (Ekert and Ziegler).
Activity in vials was counted using a CRC-15 (Capintec) detector, calibrated for fluorine-18.
High performance liquid chromatography (HPLC) was performed using a Shimadzu LC-2010A HT system equipped with a Bioscan B-FC-1000 radiation detector in series. A 0.2 min offset was applied to all traces below to account for the detectors being in series. The following set of HPLC conditions were used, as specified in the Supporting Information.
Chemistry
Synthesis of α-diazoacetate substrates (1N2 – 10N2); General Procedure for Diazo Transfer
α-diazoacetate substrates (1N2 – 10N2) were prepared through adaptation of literature methods reported by Davies.7 Briefly, the appropriate ester (1 eq., see precursors S1 – S10 in Supporting Information) and p-acetamidobenzenesulfonyl azide (1.1 eq.) were taken up in dry MeCN and the suspension cooled to 0 °C. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU, 1.1 eq) was taken up in dry MeCN, and added to the suspension dropwise over 10 minutes. The total volume of MeCN used gave a final concentration of the ester of 0.25 M. Two thirds of the total volume of MeCN was used to dissolve the ester/ketone, and the remaining third used for the DBU. The suspension was stirred at 0 °C for a further 30 mins, and at RT for a further 30–60 minutes, until the reaction was found to be complete by TLC. The deep orange solution was concentrated, and the residue was taken up into DCM, and dry-loaded onto silica gel, before being passed through a short (5 cm) silica plug, eluting with ethyl acetate in hexanes (20% v/v). The product was further purified by automated silica gel chromatography using a hexane-ethyl acetate gradient to yield the corresponding diazo ester.
The isolated diazo products are unstable at RT, and are best stored at −20 °C in the dark. Prolonged storage under vacuum (i.e. to remove residual solvents) was found to accelerate decomposition. If stored for long periods (>1–2 months), diazo precursors should be re-purified before use. The 13C NMR signal for the diazo carbon is not observed, due to the long T1 for such carbons.
Methyl (E)-2-diazo-4-phenylbut-3-enoate (1N2)
Synthesised according to the general procedure to yield 1N2 (766 mg, 83%) as an orange-red oil that solidified upon storage at −20 °C. δH (399.7 MHz, CDCl3): 7.36–7.29 (4 H, m, ArH), 7.22–7.17 (1 H, m, ArH), 6.48 (1 H, d, J = 16.3 Hz, ArCH=CH), 6.19 (1 H, d, J = 16.3 Hz, ArCH=CH), 3.85 (3 H, s, COOCH3); δC (100.5 MHz, CDCl3): 165.7, 136.9, 128.8, 127.2, 126.0, 123.1, 111.3, 52.5; HRMS (ESI+) calc. for C11H11O2N2 [M+H]+ 203.0815, found 203.0813.
Methyl (E)-2-diazo-4-(4’-(trifluoromethyl)phenyl)but-3-enoate (2N2)
Synthesised according to the general procedure to yield 2N2 (184 mg, 83%) as an orange solid. δH (399.7 MHz, CDCl3): 7.55 (2 H, app. d, J = 8.3, ArH), 7.43 (2 H, app. d, J = 8.3, ArH), 6.61 (1 H, J = 16.3 Hz, ArCH=CH), 6.22 (1 H, d, J = 16.3 Hz, ArCH=CH), 3.87 (3 H, s, COOCH3); δC (100.5 MHz, CDCl3): 165.1, 140.2 (q, J = 1.5 Hz), 128.6 (q, J = 32.5 Hz), 125.8, 125.6 (q, J = 3.8 Hz), 124.2 (q, J = 271.8 Hz), 121.2, 114.4, 52.5; δF (376.1 MHz, CDCl3): −62.46 (3F, s, CF3); HRMS (ESI+) calc. for C12H10O2F3N2 [M+H]+ 271.0689, found 271.0687.
Methyl (E)-2-diazo-4-([1’,1’’-biphenyl]-4’-yl)but-3-enoate (3N2)
Synthesised according to the general procedure to yield 3N2 (107 mg, 65%) as an orange solid. δH (399.7 MHz, CDCl3): 7.61–7.55 (4 H, m, ArH), 7.46–7.42 (4 H, m, ArH), 7.36–7.32 (1 H, m, ArH), 6.52 (1 H, d, J = 16.3 Hz, ArCH=CH), 6.24 (1 H, d, J = 16.3 Hz, ArCH=CH), 3.87 (3 H, s, COOCH3); δC (100.5 MHz, CDCl3): 165.7, 140.7, 140.0, 135.9, 128.9, 127.5, 127.4, 127.0, 126.4, 122.7, 111.4, 52.0; HRMS (ESI+) calc. for C17H15O2 [M+H]+ 279.1128, found 279.1127.
Methyl (E)-2-diazo-4-(4’-bromophenyl)but-3-enoate (4N2)
Synthesised according to the general procedure to yield 4N2 (134 mg, 61%) as an orange-red oil that solidified upon storage at −20 °C. δH (399.7 MHz, CDCl3): 7.44–7.41 (2 H, m, ArH), 7.22–7.20 (2 H, m, ArH), 6.48 (1 H, d, J = 16.3 Hz, ArCH=CH), 6.13 (1 H, d, J = 16.3 Hz, ArCH=CH), 3.85 (3 H, s, COOCH3); δC (100.5 MHz, CDCl3): 165.4, 135.7, 131.8, 127.3, 121.6, 120.7, 112.3, 52.4; HRMS (EI+) calc. for C11H10O2N2Br [M]+ 279.9847, found 298.9847.
Methyl (E)-2-diazo-4-(3’-bromophenyl)but-3-enoate (5N2)
Synthesised according to the general procedure to yield 5N2 (206 mg, 75%) as an orange-red oil which solidified upon standing. δH (399.7 MHz, CDCl3): 7.49–7.47 (1 H, m, ArH), 7.31–7.29 (1 H, m, ArH), 7.25–7.23 (1 H, m, ArH), 7.17–7.13 (1 H, m, ArH), 6.41 (1 H, d, J = 16.3 Hz, ArCH=CH), 6.11 (1 H, d, J = 16.3 Hz, ArCH=CH), 3.85 (3 H, s, COOCH3); δC (100.5 MHz, CDCl3): 165.3, 139.0, 130.2, 129.9, 128.7, 124.5, 123.0, 121.4, 113.2, 52.5; HRMS (ESI+) calc. for C11H11O2N2Br [M+H]+ 280.9920, found 280.9918.
Methyl (E)-2-diazo-4-(2’-bromophenyl)but-3-enoate (6N2)
Synthesised according to the general procedure to yield 6N2 (150 mg, 68%) as an orange-red oil. δH (399.7 MHz, CDCl3): 7.54–7.51 (2 H, m, ArH), 7.28–7.23 (1 H, m, ArH), 7.07–7.03 (1 H, m, ArH), 6.55 (1 H, d, J = 16.3 Hz, ArCH=CH), 6.47 (1 H, d, J = 16.3 Hz, ArCH=CH), 3.85 (3 H, s, COOCH3); δC (126 MHz, CDCl3):165.4, 136.6, 133.2, 128.4, 127.8, 126.6, 123.3, 121.5, 114.7, 52.6; HRMS (ESI+) calc. for C11H11O2N2Br [M+H]+ 280.9920, found 280.9921.
Methyl (E)-2-diazo-4-(4’-methoxyphenyl)but-3-enoate (7N2)
Synthesised according to the general procedure to yield 7N2 (59 mg, 49%) as an orange-red oil that solidified upon storage at −20 °C. δH (399.7 MHz, CDCl3): 7.30–7.28 (2 H, m, ArH), 6.87–6.85 (2 H, m, ArH), 6.30 (1 H, d, J = 16.3 Hz, ArCH=CH), 6.14 (1 H, d, J = 16.3 Hz, ArCH=CH), 3.85 (3 H, s, COOCH3), 3.81 (3 H, s, ArOCH3); δC (100.5 MHz, CDCl3): 165.9, 158.9, 129.7, 127.0, 122.8, 114.3, 108.7, 55.3, 52.3; HRMS (ESI+) calc. for C12H13O3N2 [M+H]+ 233.0921, found 233.0914.
Methyl (E)-2-diazo-4-(4’-(dimethylamino)-phenyl)but-3-enoate (8N2)
Synthesised according to the general procedure to yield 8N2 (44 mg, 85%) as an orange-red oil which solidified upon standing. δH (399.7 MHz, CDCl3): 7.27–7.24 (2 H, m, ArH), 6.69–6.67 (2 H, m, ArH), 6.19 (1 H, d, J = 16.3 Hz, ArCH=CH), 6.11 (1 H, d, J = 16.3. Hz, ArCH=CH), 3.84 (3 H, s, COOCH3), 2.96 (6H, s, ArN(CH3)2; δC (100.5 MHz, CDCl3): 149.9, 127.0, 125.5, 123.7, 112.6, 105.9, 52.4, 40.6 (C=O not observed); HRMS (ESI+) calc. for C13H15O2N3 [M+H]+ 246.1237, found 246.1236.
Ethyl (Z)-2-diazo-4-fluoro-4-phenylbut-3-enoate (9N2)
Synthesised according to the general procedure to yield 9N2 (102 mg, 93%) as a pale orange solid. δH (399.7 MHz, CDCl3): 7.48–7.46 (2 H, m, ArH), 7.38–7.34 (2 H, m, ArH), 7.29–7.25 (1 H, m, ArH), 5.71 (1 H, d, J = 37.2, CFCHCN2), 4.30 (2 H, q, J = 7.1 Hz, CH2CH3), 1.32 (3 H, t, J = 7.1 Hz, CH2CH3); δC (100.5 MHz, CDCl3): 165.7, 131.8 (d, J = 26.4 Hz), 128.7 (d, J = 2.4 Hz), 128.6, 123.5 (d, J = 7.3 Hz), 89.9 (d, J = 12.0 Hz), 61.7, 14.6 (note, CF resonance as well as CN2 resonance not observed); δF (376.1 MHz, CDCl3): −117.77 (1F, br s, CFCHCN2). HRMS (ESI+) calc. for C12H12FO2 [M-N2+H]+ 207.0816, found 207.0815.
5-Cholesten-4-diazo-3-one (10N2)
5-Cholesten-3-one (100 mg, 0.25 mmol, 1 eq.) and p-acetamidobenzenesulfonyl azide (70 mg, 0.29 mmol, 1.1 eq.) were taken up in dry MeCN (4 mL) and the suspension cooled to 0 °C. DBU (43 µL, 0.29 mmol, 1.1 eq) was taken up in dry MeCN (1 mL), and added to the suspension dropwise over 10 minutes. The suspension was stirred at 0 °C for a further 30 mins, and at RT for a further 90 minutes, until the reaction was found to be complete by TLC. The deep orange solution was concentrated, and the residue was taken up into DCM, and dry-loaded onto silica gel, before being passed through a short (5 cm) silica plug, eluting with ethyl acetate in hexanes (20% v/v). The product was further purified by automated silica gel chromatography using a hexane-ethyl acetate gradient to give 10N2 (13.7 mg, 13%) as a yellow oil. δH (399.7 MHz, CDCl3): 5.18 (1 H, dd, J = 4.9, 2.8 Hz), 2.47–2.44 (2 H, m), 2.23 (1 H, dt, J = 18.0, 5.2 Hz), 2.05 (1 H, dt, J = 12.6, 3.3 Hz), 2.00–1.95 (1 H, m), 1.90–1.82 (1 H, m), 1.79–0.96 (23 H, m), 0.92 (3 H, d, J = 6.5 Hz), 0.87 (6 H, dd, J = 6.6, 1.7 Hz), 0.72 (3 H, s); δC (100.5 MHz, CDCl3): 192.90, 129.42, 114.87, 56.6, 56.0, 48.3, 42.4, 39.5, 39.5, 36.1, 35.8, 35.4, 33.4, 32.4, 31.5, 31.3, 28.2, 28.0, 24.2, 23.8, 22.8, 22.6, 21.2, 19.4, 18.7, 11.9; HRMS (ESI+) calc. for C27H43O [M-N2+H]+ 383.3308, found 383.3308.
Synthesis of fluorinated reference standards; General Procedure14
AgOAc (0.2 eq.) was placed in a flame dried, foil-covered flask, and the flask evacuated and filled with Ar three times. Dry DCM (final concentration 0.075 M) was added to the flask under Ar, and 3HF.Et3N was added to the suspension. The suspension was heated to reflux before the appropriate α-diazoacetate substrates (1 eq, in ca. 1 mL DCM) was added to the refluxing suspension by syringe pump over 1 h. After addition was complete, a further aliquot of DCM (1 mL) was taken up into the syringe, and this added in one portion to the suspension. The reaction mixture was refluxed for a further 3 h. The reaction was cooled, the foil removed, and the reaction quenched with sat. aq. NaHCO3 (10× the volume of 3HF.Et3N used), and the resultant mixture stirred for 30 min. The mixture was diluted with water (10 mL), then the resultant biphasic mixture was separated and the aqueous phase extracted with DCM (3 × 30 mL). The organic extracts were combined, dried (Na2SO4), filtered and concentrated. The residue was taken up into DCM, dry-loaded onto silica gel, and purified by automated silica gel chromatography using a hexane-ethyl acetate gradient to yield the corresponding fluorinated product.
Methyl (E)-4-fluoro-4-phenylbut-2-enoate (1F)
Synthesised according to the general procedure to yield 1F (27 mg, 36%) as a colourless oil. δH (399.7 MHz, CDCl3): 7.43–7.26 (5 H, m, ArH), 7.06 (1 H, ddd, J = 19.2, 15.7, 4.4 Hz, ArCHF-CH=CH), 6.19 (1 H, ddd, J = 15.7, 1.6, 1.6 Hz, ArCHF-CH=CH), 6.02 (1 H, ddd, J = 47.0, 19.2, 1.6 Hz, ArCHF-CH=CH), 3.76 (3 H, s, COOCH3); δC (100.5 MHz, CDCl3): 166.4, 144.5 (d, J = 22.0 Hz), 136.8 (d, J = 20.0 Hz), 129.4 (d, J = 2.4 Hz), 129.0, 126.8 (d, J = 5.7 Hz), 121.1 (d, J = 10.2 Hz), 91.9 (d, J = 175.3 Hz), 52.0; δF (376.1 MHz, CDCl3): −173.50 (1F, dd, J = 47.0, 19.2 Hz, CHFCHCH); HRMS (ESI+) calc. for C11H12O2F [M+H]+ 195.0816, found 195.0811.
Methyl (E)-4-fluoro-4-(4’-(trifluoromethyl)phenyl)but-2-enoate (2F)
Synthesised according to the general procedure to yield 2F (4.7 mg, 16%) as a colourless oil. δH (399.7 MHz, CDCl3): 7.67 (2 H, d, J = 8.0 Hz, ArH), 7.48 (2 H, d, J = 8.0 Hz, ArH), 7.02 (1 H, ddd, J = 18.8, 15.7, 4.6 Hz, ArCHF-CH=CH), 6.20 (1 H, ddd, J = 15.7, 1.7, 1.7, ArCHF-CH=CH), 6.15–6.02 (1 H, m, ArCHF-CH=CH), 3.77 (3 H, s, COOCH3); δC (100.5 MHz, CDCl3): 166.1, 143.3 (d, J = 21.2 Hz), 127.3 (app. d, J = 32.5 Hz), 126.8 (d, J = 6.2), 126.0 (q, J = 3.8 Hz), 122.0 (d, J = 10.5 Hz), 91.1 (d, J = 177.0 Hz) 52.1, note: quartets for C-CF3 and C-CF3 carbons were not observed due to the low mass of material isolated; δF (376.1 MHz, CDCl3): −177.30 (1F, dd, J = 46.7, 18.8 Hz, CHFCHCH), −62.80 (3F, s, CF3); HRMS (ESI+) calc. for C12H11O2F4 [M+H]+ 263.0690, found 263.0696.
Methyl (E)-4-fluoro-4-([1’,1’’-biphenyl]-4’-yl)but-2-enoate (3F)
Synthesised according to the general procedure to yield 3F (15 mg, 30%) as a colourless oil. δH (399.7 MHz, CDCl3): 7.64–7.57 (4 H, m, ArH), 7.47–7.41 (4 H, m, ArH), 7.39–7.35 (1 H, m, ArH) 7.09 (1 H, ddd, J = 19.0, 15.7, 4.4 Hz, ArCHF-H=CH), 6.22 (1 H, ddd, J = 15.7, 1.6, 1.6 Hz, ArCHF-CH=CH), 6.07 (1 H, ddd, J = 47.0, 4.3, 1.6 Hz, ArCHF-CH=CH), 3.77 (3 H, s, COOCH3); δC (100.5 MHz, CDCl3): 166.4, 144.4 (d, J = 22.1 Hz), 142.44, 140.5, 135.7 (d, J = 20.1 Hz), 129.0, 127.8 (d, J = 6.5 Hz), 127.8, 127.3 (d, J = 5.5 Hz), 127.3, 121.3 (d, J = 10.0 Hz), 91.8 (d, J = 175.2 Hz), 52.0; δF (376.1 MHz, CDCl3): −172.96 (1F, dd, J = 47.0, 19.0 Hz, CHFCHCH); HRMS (ESI+) calc. for C17H16O2F [M+Na]+ 293.0948, found 293.0946.
Methyl (E)-4-fluoro-4-(4’-bromophenyl)but-2-enoate (4F)
Synthesised according to the general procedure to yield 4F (15 mg, 30%) as a colourless oil. δH (399.7 MHz, CDCl3): 7.54 (2 H, d, J = 8.3 Hz, ArH), 7.22 (2 H, d, J = 8.3 Hz, ArH), 7.00 (1 H, ddd, J = 19.0, 15.7, 3.9 Hz, ArCHF-CH=CH), 6.19–6.15 (1 H, m, ArCHF-CH=CH), 6.04–5.92 (1 H, m, ArCHF-CH=CH), 3.77 (3 H, s, COOCH3); δC (100.5 MHz, CDCl3): 166.2, 143.8 (d, J = 21.9 Hz), 135.8 (d, J = 20.5 Hz), 132.2, 128.4 (d, J = 5.7 Hz), 123.6 (d, J = 3.0 Hz), 121.6 (d, J = 10.2 Hz), 91.2 (d, J = 176.1 Hz), 52.1; δF (376.1 MHz, CDCl3): −174.13 (1F, dd, J = 46.8, 19.0 Hz, CHFCHCH); HRMS (ESI+) calc. for C11H11O2FBr [M+H]+ 272.9921, found 272.9916.
Methyl (E)-4-fluoro-4-(3’-bromophenyl)but-2-enoate (5F)
Synthesised according to the general procedure to yield 5F (15 mg, 30%) as a colourless oil. δH (399.7 MHz, CDCl3): 7.52–7.50 (2 H, m, ArH), 7.29–7.27 (2 H, m, ArH), 7.00 (1 H, ddd, J = 19.0, 15.7, 4.5 Hz, ArCHF-CH=CH), 6.19 (1 H, ddd, J = 15.7, 1.6, 1.6 Hz, ArCHF-CH=CH), 5.98 (1 H, ddd, J = 46.9, 4.4, 1.6 Hz, ArCHF-CH=CH), 3.77 (3 H, s, COOCH3); δC (100.5 MHz, CDCl3): 166.2, 143.6 (d, J = 21.6 Hz), 139.00 (d, J = 20.4 Hz), 132.5, 130.6, 129.7 (d, J = 6.2 Hz), 125.2 (d, J = 5.8 Hz), 121.8 (d, J = 10.4 Hz), 91.0 (d, J = 177.1 Hz), 52.1; δF (376.1 MHz, CDCl3): −175.29 (1F, dd, J = 46.9, 19.0 Hz, CHFCHCH); HRMS (ESI+) calc. for C11H11O2FBr [M+H]+ 272.9921, found 272.9918.
Methyl (E)-4-fluoro-4-(2’-bromophenyl)but-2-enoate (6F)
Synthesised according to the general procedure to yield 6F (16 mg, 55%) as a pale yellow solid. δH (499 MHz, CDCl3): 7.58 (1 H, dt, J = 8.1, 1.2 Hz, ArH), 7.45 (1 H, dd, J = 7.8, 1.8 Hz, ArH), 7.38 (1 H, td, J = 7.6, 1.2 Hz, ArH), 7.26–7.21 (1 H, m, ArH), 7.04 (1 H, ddd, J = 19.0, 15.7, 4.3 Hz, ArCHF-CH=CH), 6.42 (1 H, ddd, J = 45.7, 4.3, 1.8 Hz, ArCHF-CH=CH); 6.22 (1 H, dt, J = 15.7, 1.7 Hz, ArCHF-CH=CH), 3.76 (3 H, s, COOCH3); δC (126 MHz, CDCl3): 166.4, 143. (d, J = 21.9 Hz), 136.5 (d, J = 21.5 Hz), 133.1, 131.7, 130.6 (d, J = 1.9 Hz), 127.9 (d, J = 8.3 Hz), 121.6 (d, J = 10.4 Hz), 90.5 (d, J = 176.5 Hz), 52.1; δF (470 MHz, CDCl3): 1F, dd, J = −180.58, 19.3 Hz, CHFCHCH). HRMS (ESI+) calc. for C11H10BrFO2 [M+H]+ 271.9848, found 271.9853.
Ethyl (E)-4,4-difluoro-4-phenylbut-2-enoate (9F)
Synthesised according to the general procedure to yield 9F (14 mg, 48%) as a colourless oil. δH (399.7 MHz, CDCl3): 7.51–7.43 (5 H, m, ArH), 7.01 (1 H, dt, J = 15.7, 10.5, CF2CHC), 4.30 (2 H, q, J = 7.1 Hz, CH2CH3), 1.32 (3 H, t, J = 7.1 Hz, CH2CH3); δC (100.5 MHz, CDCl3): 165.2, 140.1 (t, J = 30.7 Hz), 135.3 (t, J = 27.1 Hz), 130.6 (t, J = 1.7 Hz), 128.8, 125.4 (t, J = 5.8 Hz), 124.9 (t, J = 8.2 Hz), 118.5 (t, J = 240.4 Hz), 61.4, 14.3; δF (376.1 MHz, CDCl3): −117.77 (1F, br s, CF2CHCH). HRMS (ESI+) calc. for C12H13F2O2 [M+H]+ 227.0878, found 227.0875.
(6R)-4-Cholesten-6-fluoro-3-one (10F)
Synthesised according to the general procedure, replacing AgOAc with AgOTf, and using 10 equivalents of 3HF.Et3N, to yield 10F (14 mg, 30%) as a colourless solid, as a single diastereomer. δH (399.7 MHz, CDCl3): 5.87 (1 H, d, J = 4.9 Hz), 4.99 (1 H, ddd, J = 49.2, 2.5, 2.5 Hz), 2.55 (1 H, ddd, J = 16.8, 15.1, 4.9 Hz), 2.40 (1 H, ddd, J = 16.8, 3.3, 3.3 Hz), 2.25–2.16 (2 H, m), 1.91–1.81 (2 H, m), 1.73 (1 H, J = 14.3, 14.3, 4.4), 1.65–0.94 (22 H, m), 0.92 (3 H, d, J = 6.5 Hz), 0.87 (6 H, dd, J = 6.6, 1.7 Hz), 0.74 (3 H, s); δC (100.5 MHz, CDCl3): 200.2, 162.3 (d, J = 12.3 Hz), 128.4 (d, J = 9.1 Hz), 93.7 (d, J = 166.0 Hz), 56.2, 55.9, 53.4, 42.7, 39.6, 38.0, 37.6, 37.4, 37.0, 36.3, 35.9, 34.4, 30.1, 28.3, 28.2, 24.2, 24.0, 23.0, 22.7, 21.0, 18.8, 18.5 (d, J = 0.9 Hz), 12.1; δF (376.1 MHz, CDCl3): −165.07– −165.58 (1F, m, CCFHCH2). HRMS (ESI+) calc. for C27H44FO [M+H]+ 404.3371, found 404.3375.
Davies et al. report the isolation of a single diastereomer with configuration as shown above.7 Data are in agreement with the reported data.
Radiochemistry
General Procedure for vinylogous 18F-fluorination
To a 4 mL vial were added AgOAc (1.6 mg, 10 μmol, 1.0 eq.) and imidazolium triflate (2.2 mg,10 μmol, 1.0 eq.), followed by anhydrous dichloroethane (DCE) (700 μL) and acetic acid solution (100 μL of a stock solution in DCE (18 μL HOAc/1 mL DCE), 30 μmol, 3 eq.) The solution was briefly vortexed. The reaction vial was capped with a PTFE/Silicone septum cap and a 100 μL aliquot of [18F]AgF•K222•AgOTf complex in DCE (typically 60–1000 μCi; see Supporting Information) was added to the reaction vial by syringe. Finally the diazo compound (10 μmol in 100 μL DCE) was added by syringe. The reaction was heated in an aluminum block at 100 °C for 30 min with the exclusion of light.16 After 30 min, the reaction was removed from the heat and allowed to cool to room temperature before analysis by radio-TLC and radio-HPLC to confirm RCY and identity, respectively.
Supplementary Material
Funding Information
We acknowledge US DOE/NIBIB (DE-SC0012484 to PJHS) and NIH (R01EB021155 to MSS and PJHS) for financial support.
Footnotes
Supporting Information
Yes
References
- (1).For a general overview of PET, see: Ametamey SM; Honer M; Schubiger PA Chem. Rev, 2008, 108, 1501. [DOI] [PubMed] [Google Scholar]
- (2).For an introduction to the use of PET in drug development, see: Elsinga PH, Waarde AV, Paans AMJ and Dierckx RAJO, Trends on the Role of PET in Drug Development. World Scientific, Singapore, 2012. [Google Scholar]
- (3).(a) For recent reviews of new fluorine-18 radiochemistry, see: Miller PW; Long NJ; Vilar R; Gee AD Angew. Chem. Int. Ed 2008, 47, 8998. [DOI] [PubMed] [Google Scholar]; (b) Cai L; Lu S; Pike VW Eur. J. Org. Chem 2008, 2853. [Google Scholar]; (c) Tredwell M; Gouverneur V Angew. Chem. Int. Ed 2012, 51, 11426. [DOI] [PubMed] [Google Scholar]; (d) Brooks AF; Topczewski JJ; Ichiishi N; Sanford MS; Scott PJH Chem. Sci 2014, 5, 4545. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Jacobson O; Kiesewetter DO; Chen X Bioconjugate Chem 2015, 26, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Preshlock S; Tredwell M; Gouverneur V Chem. Rev 2016, 116, 719. [DOI] [PubMed] [Google Scholar]; (g) Deng X; Rong J; Wang L; Vadev N; Josephson L; Liang SH Angew. Chem. Int. Ed, 2019, 58, 2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Sanford MS; Scott PJH ACS Cent. Sci 2016, 2, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).For a recent example, see: Langstrom B and Blom E, Synthesis of [18F]Fluoromethyl benzene using Benzyl pentafluorobenzenesulfonate. WO 2008/106442 A1, 4 September 2008. [Google Scholar]
- (6).(a) Scott PJH; Brooks AF; Ichiishi N; Sanford MS Preparation of Ag18F and its use in the synthesis of PET radiotracers, U.S. Patent 2016/0317682 A1, November 3, 2016.; (b) Mossine AV; Brooks AF; Ichiishi N; Makaravage KJ; Sanford MS; Scott PJH Sci. Rep 2017, 7, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mossine AV; Brooks AF; Bernard-Gauthier V; Bailey J; Ichiishi N; Schirrmacher R; Sanford MS; Scott PJHJ Labelled Compd. Radiopharm 2018, 61, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lee SJ; Brooks AF; Ichiishi N; Makaravage KJ; Mossine AV; Sanford MS; Scott PJH Chem. Commun 2019, 55, 2976. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lee SJ; Makaravage KJ; Brooks AF; Scott PJH; Sanford MS Angew. Chem. Int. Ed 2019, 58, 3119. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Verhoog S; Brooks AF; Winton WP; Viglianti BL; Sanford MS; Scott PJH Chem. Commun 2019, 55, 6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Qin C; Davies HM L. Org. Lett 2013, 15, 6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Glasspoole BW; Crudden CM; Nat Chem 2011, 3, 912. [DOI] [PubMed] [Google Scholar]
- (9).Gray EE; Nielsen MK; Choquette KA; Kalow JA; Graham TJA; Doyle AG J. Am. Chem. Soc 2016, 138, 10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Hollingworth C; Hazari A; Hopkinson MN; Tredwell M; Benedetto E; Huiban M; Gee A; Brown JM; Gouverneur V Angew. Chem. Int. Ed 2011, 50, 2613. [DOI] [PubMed] [Google Scholar]; (b) Topczewski JJ; Tewson TJ; Nguyen HM J. Am. Chem. Soc, 2011, 133, 19318. [DOI] [PubMed] [Google Scholar]; (c) Benedetto E; Tredwell M; Hollingworth C; Khotavivattana T; Brown JM; Gouverneur V Chem. Sci, 2013, 4, 89. [Google Scholar]
- (11).Carbonnel E; Poisson T; Jubault P; Pannecoucke X; Besset T Front. Chem 2019, 10.3389/fchem.2019.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Frost AB; Brambilla M; Exner RM; Tredwell M Angew. Chem. Int. Ed 2019, 58, 472. [DOI] [PubMed] [Google Scholar]; (b) Tredwell M Synlett, 2019, 30, 1371. [Google Scholar]
- (13). Radiochemical yields (RCY) are non-isolated and were calculated by % integrated area of the 18F product versus 18F- in a radio-TLC trace. Product identities were confirmed by radio-HPLC.
- (14). In addition to low RCYs, we were not able to synthesize reference standards to confirm the identity of [18F]7F and [18F]8F because of stability issues, which is consistent with Davies’ results. For example, they reported an NMR yield for the -OMe product but did not isolate it, suggesting that the compound is unstable.7.
- (15).Winton W; Brooks AF; Wong KK; Scott PJH; Viglianti BL SynOpen, 2019, 3, 55. [Google Scholar]
- (16). Lights in the fumehood where the reactions occurred were turned off as a general precaution. However, ambient laboratory lighting was left on and we do not believe the chemistry is light sensitive.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.