

Abstract
Methamphetamine (METH) is a highly addictive psychostimulant. Cannabidiol (CBD) is an exogenous cannabinoid without psychostimulating activity, which has potential therapeutic effects on opioid addiction. However, it is unclear whether CBD has therapeutic effects on METH-induced motivational effects. The present study examines whether CBD has a protective effect on METH-induced conditioned place preference (CPP) in rats by regulating the Sigma1R and AKT-GSK3β-CREB signaling pathway. Seventy rats were equally and randomly divided into seven groups. The rat CPP model was established via the intraperitoneal injection (IP) of 2 mg/kg of METH. Next, the intraperitoneal injection of 10, 20, 40, and 80 mg/kg CBD was performed 1 h prior to the injection of saline or METH. The protein expression levels of Sigma1R, AKT, p-AKT, GSK-3β, p-GSK-3β, CREB, and p-CREB in the rats’ prefrontal cortex, nucleus accumbens, and hippocampus and ventral tegmental were detected using western blot analysis. CBD was found to inhibit METH-induced CPP in a dose-dependent fashion. The expression levels of Sigma1R, p-AKT, p-GSK3β, and p-CREB increased significantly in the METH-induced CPP model. Treatment involving different doses of CBD caused differential inhibitory responses in the cellular protein abundance of Sigma1R, p-AKT, p-GSK3β, and p-CREB across various brain regions. The present study found that METH can induce CPP in rats. When a pretreatment of CBD is applied, the CBD can weaken CPP in METH-induced rats by regulating the SigmaR1/AKT/GSK-3β/CREB signaling pathway. The results of this study indicate that CBD has a potential therapeutic effect on METH-induced rewarding effects.
Keywords: cannabidiol, methamphetamine, conditioned place preference, Sigma1R, AKT, GSK-3β, CREB
Introduction
Methamphetamine (METH), a white, odorless, crystalline powder commonly referred to as crystal METH, is a highly addictive synthetic psychostimulant. It is one of the most popular synthetic drugs abused recreationally worldwide. Long-term consumption can produce notable toxic effects on the central nervous system (CNS) and cause serious physical and mental damage to abusers [1–4].
Cannabis (marijuana) is a widely used drug for medical or recreational purposes. It contains Δ9-tetrahydrocannabinol (THC), cannabidiol (CBD), cannabichromene (CBC), cannabigerol (CBG), cannabinol (CBN), cannabidivarin (CBDV), and other compounds. Although these components have similar chemical structures, they exert distinct pharmacological effects [5–7]. CBD is the second largest active component of cannabis after THC. It appears to be nonpsychoactive and can interact with numerous drug targets of the CNS. This makes it attractive for wide use in treating neurodegenerative diseases, such as brain trauma, stroke [8], and epilepsy [9, 10]. In addition, CBD has the therapeutic potential toward drug-associated rewarding effects [11–13], but there is limited research on the neuroprotective effects caused by CBD on METH-induced motivational effects.
The sigma receptor (σR) is a novel class of receptors ubiquitously expressed in dopaminergic regions such as striatum, substantia nigra, and ventral tegmental area (VTA) [14]. The σR consists of Sigma1R and Sigma2R. Sigma1R is a small transmembrane protein mainly concentrated in the endoplasmic reticulum (ER) and mitochondria-associated ER membranes (MAM), as well as the cell membrane and nuclear membrane [15]. Sigma1R is involved in the pathophysiology of a panel of neurological and psychiatric diseases, including schizophrenia, depression, stroke, Alzheimer’s disease, amyotrophic lateral sclerosis, and Parkinson’s disease [16–18]. Additionally, Sigma1R can be used as a potential target for drug abuse treatment. The receptor can regulate the behavioral effects induced by cocaine, METH, ethanol, and nicotine [17, 19]. METH exposure activates Sigma1R, disrupts intracellular calcium homeostasis, and induces a large amount of dopamine (DA) release in the nucleus accumbens (NAc), which leads to significant neurotoxic effects [20]. As such, blocking the Sigma1R can inhibit DA release and the METH-induced production of reactive oxygen species [21].
AKT/PKB (protein kinase B), a serine/threonine kinase, mediates cell proliferation, protein synthesis, transcription, and apoptosis [22]. The phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway plays an important role in regulating the survival of dopaminergic neurons [23]. Studies have shown that METH can induce autophagy in human neuroblastoma SH-SY5Y cell lines via the PI3K/AKT/mTOR signaling pathway [24]. This suggests that the PI3K/AKT signaling pathway has a regulatory effect on drug-induced toxicity.
The cyclic adenosine monophosphate (cAMP) reactive element-binding protein (CREB) is a nuclear transcription factor commonly expressed in all organs and is necessary for physiological processes, such as cell proliferation, growth, survival, and differentiation. CREB contributes to functions such as learning, memory, synaptic transmission, neuron differentiation, survival, and axonal growth [25]. Studies have shown that in addition to the formation of memory and learning, the cAMP/PKA/CREB signaling pathway is involved in the regulation of drug tolerance and addiction mechanisms [26, 27]. As a downstream target protein of the PI3K/AKT signaling pathway, glycogen synthase kinase-3 (GSK-3β) is inhibited by AKT through the induced phosphorylation of serine (Ser)9 at GSK-3β’s N terminal [28]. This phosphorylation and subsequent inactivation further activate CREB and NF-κB (the nuclear factor kappa-light-chain-enhancer of activated B cells) [29, 30]. For example, previous research demonstrates that arsenic causes the adrenal pheochromocytoma (PC12) cells in rats to undergo apoptosis through the AKT/GSK-3β/NFAT signaling pathway [31]. Rifampin can also attenuate rotenone-induced apoptosis in SH-SY5Y cells via the PI3K/AKT/GSK-3β/CREB signaling pathway [32], as observed by the regulation of anxiety-like behaviors in rats undergoing alcohol dependence withdrawal [33]. However, it is still unclear whether the PI3K/AKT/GSK-3β/CREB pathway contributes to the mediation of METH-induced rewarding effects.
The PI3K/AKT/GSK-β/CREB signaling pathway has a wide range of regulatory effects on the CNS and thus has gained significant attention in the research community [34]. Several studies report that drug abuse such as morphine, cocaine, and METH relate closely to the midbrain DA reward system. The prefrontal cortex (PFC), striatum, NAc, hippocampus, VTA, and amygdala are key reward system projection areas involved in drug reward [33, 35, 36]. The present study used this information as a basis for examining the activity of the Sigma1R/PI3K/AKT/GSK-β/CREB signaling pathway in the PFC, NAc, hippocampus, and VTA of METH-exposed rats, some of which are received CBD treatment.
The conditional place preference (CPP) paradigm has been commonly used as a preclinical behavioral model to assess the rewarding and aversive effects of addictive drugs [37]. In this study, the question of whether METH-induced CPP in rats is associated with alternations to the Sigma1R and AKT/GSK-3β/CREB signaling pathways will be examined.
Materials and Methods
Animals
Seventy male Sprague–Dawley (SD) rats weighing between 180 and 220 g were purchased from the Laboratory Animal Center of Kunming Medical University (Kunming, China). The rats were housed in temperature-controlled rooms (23 ± 1°C) with 45–55% humidity and a 12-h alternating light–dark cycle. A standard diet (Beijing Ke-ao Xie-li Feed Co., LTD, SPF Rat Feed) and water were administered ad libitum. All rats were acclimated to the facility for 3 days before being used in the experiments. The rats were equally and randomly divided into seven groups: the control group (saline, 10 ml/kg, IP), METH group (METH, 2 mg/kg, IP), CBD group (CBD, 40 mg/kg, IP), CBD (10 mg/kg) + METH group, CBD (20 mg/kg) + METH group, CBD (40 mg/kg) + METH group, and CBD (80 mg/kg) + METH group. The rats received METH (2 mg/kg, IP) or saline (10 ml/kg, IP) injections following 1 h treatments of CBD (10, 20, 40, 80 mg/kg, IP) or saline. The body weights of the rats were measured weekly for dose adjustment.
All procedures were conducted in accordance with the National Institutes of Health (NIH)‘s Guidelines for the Care and Use of Laboratory Animals, and the experiments were approved by the Experimental Animal Ethics Committee of Kunming Medical University.
Reagents and chemicals
METH (methamphetamine hydrochloride) was purchased from the National Institutes for Food and Drug Control (Cat. #: 171212-200603, Beijing, China). The METH was dissolved in saline and administered at a dose of 2 mg/kg via intraperitoneal injection (IP). CBD at 99 percent purity was sourced from Hebei Fanzhangtang Commercial and Trading Co., Ltd. (Cat. #: 13956-29-1, Heibei, China). The CBD was dissolved in a vehicle solution of 5% dimethyl sulfoxide (DMSO) and 5% polysorbate 80 (TWEEN® 80) in saline and then administered at doses of 10, 20, 40, and 80 mg/kg via IP. DMSO was purchased from Sigma-Aldrich (St. Louis, MO, USA). Several antibodies were used, of which anti-rabbit SigmaR1 (D4J2E) (Cat. #: 61994S), anti-rabbit AKT (pan) (C67E7) (Cat. #: 4691S), anti-rabbit phospho-AKT (Ser473) (Cat. #: S473(D9E)), anti-rabbit phospho-GSK-3β (S9) (Cat. #: 9323S), anti-rabbit CREB (48H2) (Cat. #: 9197S), and anti-rabbit phospho-CREB (ser133) (87G3) (Cat. #: 9198S) were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-β-actin (Cat. #: A1978) was purchased from Sigma-Aldrich (St. Louis, MO, USA), anti-rabbit GSK-3β was purchased from Calbiochem (Cat. #: PK1111, USA), and anti-mouse (Cat. #: 7076S) and anti-rabbit (Cat. #: 7074S) secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA).
Apparatus
The CPP apparatus (Shanghai Xinruan Information Technology Co., Ltd., model #XR-XT401, Shanghai, China) consisted of three compartments. One compartment had a grid floor and black walls (L × W × H: 35 × 35 × 35 cm). Another same-sized compartment had mesh floor and white walls. The middle compartment had flat, gray floor, and gray walls (L × W × H: 15 × 35 × 35 cm) and was connected to the other two compartments via removable doors. The black and white compartments each contained a ceiling lamp. The general activity within the compartments was monitored via video, and the time the rats spent in each compartment was recorded using XR-XT401 software.
Conditioned place preference procedure
The conditioned place preference (CPP) was performed as previously described [37, 38]. The CPP apparatus consisted of two equal-sized compartments joined by a small central compartment accessed through guillotine doors. One compartment has white walls with a grid floor, while the other compartment has black walls with the mesh floor. The rats did not display any preference for either grid or mesh floor. The CPP procedure used in this experiment consisted of three phases, the performance of which is shown in Fig. 1a. The preconditioning phase lasted from days 1 to 3. To identify any pre-existing bias toward the individual compartments, each rat was placed in the middle chamber and then allowed to move freely between the black and white chambers for 30 min each day for three consecutive days before drug administration. The time spent in each compartment on the third day was used as the preconditioning data. The most visited compartment was designated as the preferred compartment and the other as the non-preferred compartment. Each animal received individual CPP training.
Figure 1.
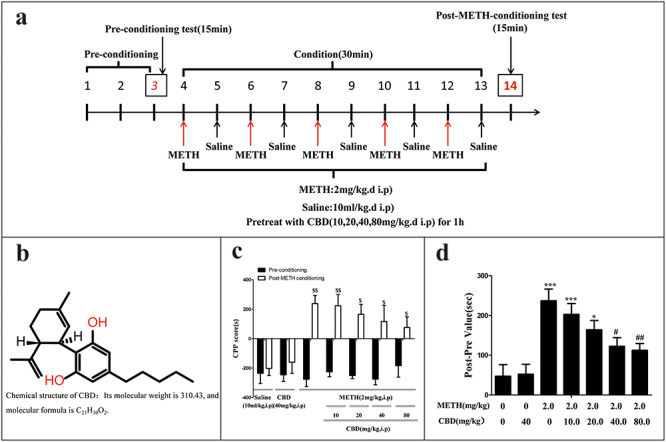
Effect of CBD on METH-induced CPP in rats. CPP experiment procedure; rats received METH (2 mg/kg, IP) or saline (10 ml/kg, IP) injections following 1 h pretreatments of CBD (10, 20, 40, 80 mg/kg, IP) or saline (10 ml/kg, IP) treatments (a). Chemical structure of CBD (b). CPP score was calculated by subtracting the time spent in the saline-paired compartment from the time spent in the drug-paired compartment [39] ($P < 0.05, $$P < 0.01 vs. preconditioning, N = 10 per group) (c). Place preference data were expressed as the proportion of time spent in the drug-paired conditioning compartment [40] (d). Data were presented as mean ± SD. (*P < 0.05, **P < 0.01 vs. saline-paired control; #P < 0.05, ##P < 0.01 vs. saline + METH group, N = 10 animals per group).
The conditioning phase lasted for 10 days (days 4–13), consisting of five drug sessions and five saline sessions. The saline- and drug-paired compartments were separated by a closed door. On day 4, day 6, day 8, day 10, and day 12, the rats were pretreated for 1 h with CBD or saline and then confined to the drug-paired compartment for 30 min where they were administered 2 mg/kg of METH via IP. On day 5, day 7, day 9, day 11, and day 13, the rats were administered with saline (10 ml/kg, IP) and confined to the saline-paired compartment for 30 min after the 1 h pretreatment of CBD (10, 20, 40, 80 mg/kg, IP) or saline. The floor and walls of each compartment were wiped daily with a 70% ethanol solution after each trial.
The post-conditioning phase lasted 14 days. On day 14, the post-conditioning test was performed for 15 min. The amount of time spent in the non-preferred (drug-paired) compartment was recorded as the post-conditioning data. Preference (the CPP score) was calculated by subtracting the time spent in the saline-paired compartment from the time spent in the drug-paired compartment [39], and the CPP behaviors were expressed as post–pre values [40]. These were calculated as [(post-value) − (pre-value)] to reflect the difference in time spent in drug conditioning in the post- and preconditioning stages, respectively.
Western blot analysis
The rats’ brain tissue was dissected and put on ice. The PFC, hippocampal area (HPC), NAc, and ventral tegmental area (VTA) were isolated from the brain tissue and stored at −80°C until used in a western blot analysis. Brain tissue protein lysates were homogenized in an enhanced radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime, China) containing 1% phenylmethanesulfonyl fluoride (Beyotime, China) and a protease inhibitor cocktail and then centrifuged at 14 000g for 15 min at 4°C. The supernatant was collected, and the proteins were measured using the enhanced bicinchoninic acid (BCA) protein assay kit (Beyotime, China). Equal amounts of proteins (25 μg) were separated via 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to 0.45 μm polyvinylidene fluoride (PVDF) membranes (Millipore, MA, USA). The membranes were blocked for 2 h with 5% bovine serum albumin (BSA) (Sigma-Aldrich, USA) or 5% nonfat milk at 37°C, and the membrane was incubated with primary antibodies at a dilution of 1:1000 in the blocking buffer overnight at 4°C. This was followed by incubation with a secondary antibody (1:5000 dilution with 5% defatted milk) at room temperature for 1 h, at which point the membranes were washed three times with 0.1% Tween® 20 (TBST) for 10 min. The blots were then developed using an enhanced Chemiluminescence Plus detection kit (Cat. #: WBKLS0500, Millipore, USA) and visualized using the Bio-Rad imaging system (Bio-Rad, USA). The protein band was quantified using ImageJ software and expressed as the densitometric quantization of the protein band’s intensities, normalized against the corresponding β-actin band’s intensities. This experiment was repeated at a minimum in triplicate, and representative western blot images were collected for presentation.
Statistical analysis
Statistical analyses were performed using SPSS 19.0 (IBM SPSS, Chicago, USA) and GraphPad Prism 7.04 (GraphPad Software, USA). All data were expressed as mean ± standard deviation (SD) from at least three independent experiments. Comparisons between groups were made via a one-way analysis of variance (ANOVA), followed by a Tukey’s test for comparing individual groups. P-values of <0.05 were considered statistically significant.
Results
Effects of CBD on the development of METH-induced CPP in rats
CPP, a model widely used to assess drug-seeking behaviors, was used in the present study to assess rats’ preference for visual and tactile cues associated with METH after being administered with varying doses of CBD [37]. As shown in Fig. 1c, saline-paired controls and 40 mg/kg of CBD did not produce motivational effects before or after conditioning, which set the baseline measurement for the CPP assay. METH administration at 2 mg/kg significantly increased METH-induced CPP compared to the controls (P < 0.01). As shown in Fig. 1d, 1 h pretreatments of 10, 20, 40, and 80 mg/kg of CBD administered via IP gradually reduced METH-induced motivational effects in a dose-dependent manner, compared to the METH-only controls (P < 0.05). The METH-induced effects decreased as the dosage of CBD increased. Per these results, it may be concluded that 2 mg/kg of METH is sufficient to induce CPP, and the coadministration of CBD dose dependently attenuates the development of METH-induced CPP.
CBD downregulates the expression of Sigma1R in METH-induced CPP rats
As shown in Fig. 2, the western blot assay indicates that the protein expression levels of Sigma1R in the METH-treated group were significantly elevated in the PFC (P < 0.001), NAc (P < 0.001), HPC (P < 0.01), and VTA (P < 0.001) compared to the saline-paired controls. However, when the rats were pretreated with CBD for 1 h prior to METH administration, Sigma1R expression generally decreased across the four examined brain regions as CBD concentrations increased. These results suggest that CBD can broadly attenuate METH-induced Sigma1R protein expression in the PFC, NAc, HPC, and VTA.
Figure 2.
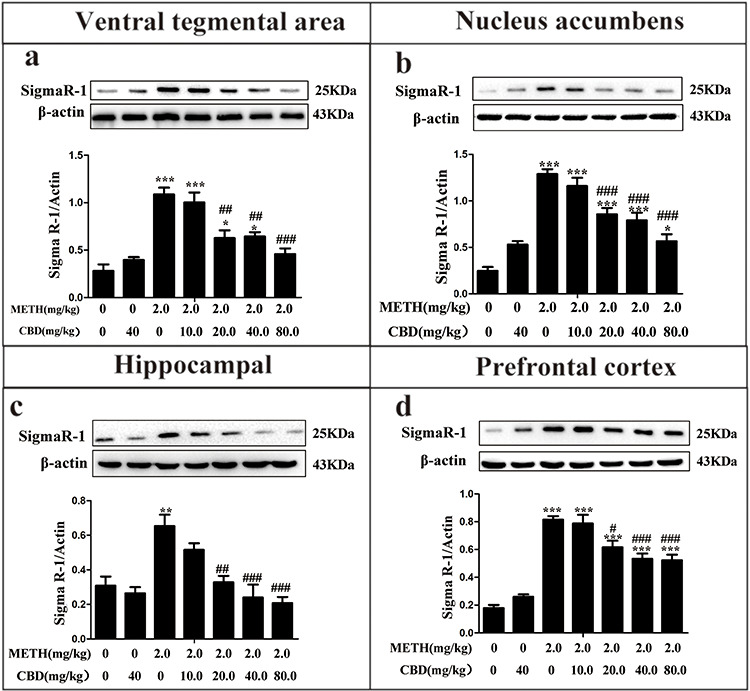
Effect of CBD on Sigma1R in the VTA, NAc, HPC, and PFC of METH-induced CPP rats. Rats were pretreated with CBD (10, 20, 40, 80 mg/kg, IP) for 1 h before the administration of METH (2 mg/kg, IP). Sigma1R expression was detected via western blot analysis in the VTA (a), NAc (b), HPC (c), and PFC (d). Data were expressed as Sigma1R/β-actin and presented as mean ± SD from three independent experiments. (**P < 0.01, ***P < 0.001 vs. control; ##P < 0.01, ###P < 0.001 vs. METH group, N = 3 independent biological replicates per group). Representative images are shown.
CBD downregulates the expression of p-AKT and p-AKT/AKT in METH-induced CPP rats
As shown in Fig. 3, METH significantly augmented the protein expression levels of AKT and p-AKT in the PFC (P < 0.01) and NAc (P < 0.001), and p-AKT levels were elevated across all four brain regions compared to those in the saline-paired controls (P < 0.001). The relative expression levels of p-AKT showed a general trend of downregulation in the PFC, NAc, and HPC after pretreatment with different CBD doses compared to those in the METH-only group (P < 0.05). AKT protein expression did not exhibit a significant dose-dependent change in the four brain regions compared to the METH-only group (P > 0.05). However, METH notably enhanced p-AKT/AKT ratios in the PFC (P < 0.001), NAc (P < 0.01), HPC (P < 0.01), and VTA (P < 0.001) compared to the saline-paired control group. Pretreatment with different CBD doses before METH administration generally attenuated p-AKT/AKT ratios dose dependently in PFC, HPC, and VTA, compared to the METH-only group (P < 0.05). Both 40 and 80 mg/kg of CBD reduced p-AKT/AKT ratios to a similar extent in NAc. These results suggest that CBD can broadly attenuate METH-induced AKT protein expression and relative p-AKT/AKT expression in the PFC, NAc, HPC, and VTA.
Figure 3.
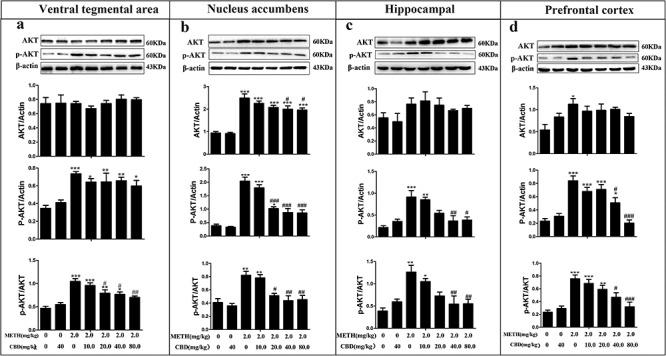
CBD decreases the levels of p-AKT and p-AKT/AKT in the VTA, NAc, HPC, and PFC in METH-induced CPP rats. The expression levels of AKT, p-AKT, and p-AKT/AKT in the VTA (a), NAc (b), HPC (c), and PFC (d) were analyzed via western blotting. Data were expressed as AKT/β-actin, p-AKT/β-actin, and p-AKT/AKT and then presented as mean ± SD from three independent experiments. (**P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. METH group, N = 3 independent biological replicates per group). Representative images are shown.
CBD downregulates the expression of p-GSK-3β and p-GSK-3β/GSK-3β in METH-induced CPP rats
As shown in Fig. 4, METH significantly increased the protein expression of p-GSK-3β and p-GSK-3β/GSK-3β in the PFC (P < 0.001), NAc (P < 0.01), HPC (P < 0.01), and VTA (P < 0.001) compared to those in the saline-paired controls. However, GSK-3β protein expressions were not found to change in any of the examined brain tissues (P > 0.05). Pretreating with CBD for 1 h, followed by METH administration, generally reduced the expression levels of p-GSK-3β and p-GSK-3β/GSK-3β in a dose-dependent manner compared to those in the METH-only group across the four brain regions. These results suggest that CBD can broadly attenuate METH-induced p-GSK-3β protein expression and relative p-GSK-3β/GSK-3β expression in the PFC, NAc, HPC, and VTA.
Figure 4.
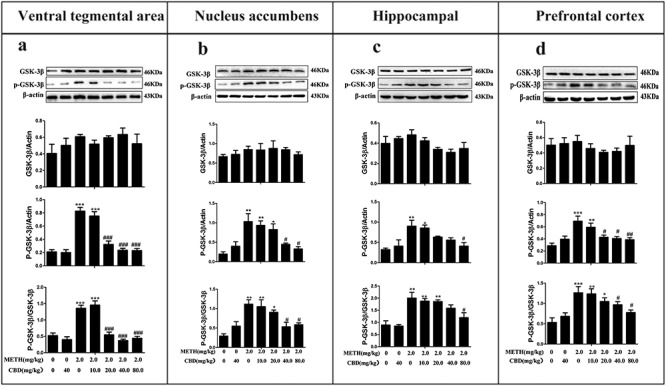
CBD decreases levels of p-GSK-3β and p-GSK-3β/GSK-3β in the VTA, NAc, HPC, and PFC of METH-induced CPP rats. The expression levels of GSK-3β, p-GSK-3β, and p-GSK-3β/GSK-3β in the VTA (a), NAc (b), HPC (c), and PFC (d) were analyzed by western blotting. Data were expressed as GSK-3β/β-actin, p-GSK-3β/β-actin, and p-GSK-3β/GSK-3β and then presented as mean ± SD. (*P < 0.05, **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. METH group, N = 3 independent biological replicates per group). Representative images are shown.
CBD downregulates the expression of p-CREB and p-CREB/CREB in METH-induced CPP rats
As shown in Fig. 5, METH significantly increased the protein expression levels of p-CREB and p-CREB/CREB in the PFC (P < 0.01), NAc (P < 0.01), HPC (P < 0.05), and VTA (P < 0.01) compared to the saline-paired controls. However, CREB expressions remained unaltered in all examined brain tissues (P > 0.05). Pretreating with different CBD doses for 1 h, followed by METH administration, was observed to decrease the expression levels of p-CREB and p-CREB/CREB dose dependently in the four brain regions compared to the METH-only group. These results suggest that CBD can broadly attenuate METH-induced p-CREB protein expression and relative p-GSK-3β/GSK-3β expression in the PFC, NAc, HPC, and VTA.
Figure 5.
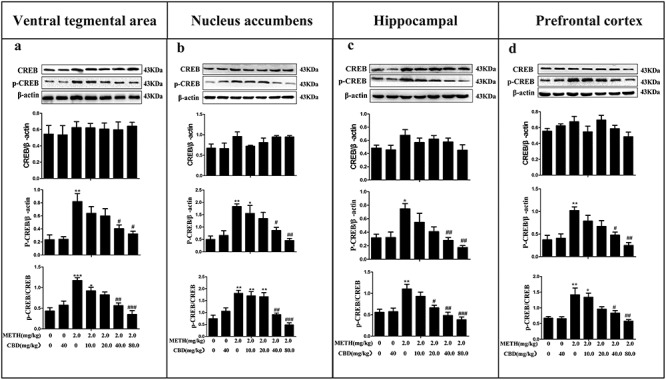
CBD reduces the levels of p-CREB and p-CREB/CREB in the VTA, NAc, HPC, and PFC in METH-induced CPP rats. The expression levels of CREB, p-CREB, and p-CREB/CREB in the VTA (a), NAc (b), HPC (c), and PFC (d) were analyzed via western blotting. Data were expressed as CREB/β-actin, p-CREB/β-actin, and p-CREB/CREB and then presented as mean ± SD. (*P < 0.05, **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. METH group, N = 3 independent biological replicates per group). Representative images are shown.
Discussion
In this study, CPP was employed to evaluate the addiction behaviors of METH-induced rats, as well as to identify the molecular mechanism by which early interventions using CBD can modulate METH-induced CPP. It has been consistently shown that 2 mg/kg/day of METH is able to induce CPP in rats [38, 41, 42]. We have previously reported that 2 mg/kg of METH elicits a stronger effect on METH-induced behavioral sensitization in a preclinical animal model mediated by the DA system compared to the METH doses of 1 and 4 mg/kg [43]. Here, 2 mg/kg of METH was observed to induce CPP formation significantly while pretreating with CBD at different doses attenuated METH-induced CPP in a dose-dependent manner (Fig. 1). In the experiment, CBD doses of 40 and 80 mg/kg were observed to significantly inhibit METH-induced CPP. These results are consistent with those of prior studies that recommend CBD’s use as a potential pharmacological intervention to attenuate drug-induced rewarding effects [44].
The relationship between the METH-induced neurotoxicity and METH-induced motivational effects has not been vigorously tested. The incentive-sensitization theory of addiction is a generally accepted theory to explain METH-induced behavioral sensitization. Addictive drugs, e.g. METH act on the brain’s reward system to induce the excessive attribution of incentive salience to drug-associated stimuli, producing compulsive motivation and the ingraining of drug-seeking habits [42, 45]. It has been reported that METH increases the synaptic release of DA and inhibits the dopamine transporter (DAT) reuptake of DA, thereby activating DA receptors and dopaminergic signaling in the brain’s reward pathway to elicit reward-motivated behavior [39, 46]. Dopamine D3 receptor (D3R) is required in METH addiction and associates with METH-induced hyperactivity and behavioral sensitization in preclinical animal models [39, 47–49]. Excess DA release, metabolized by monoamine oxidase, is associated with induction of oxidative stress, degeneration of dopaminergic neurons, and mitochondrial dysfunction resulting in severe METH-induced neurotoxicity [20, 50–52].
Previous studies have shown that 80 mg/kg of CBD can significantly reduce patient motivation to self-medicate with METH [44]. However, doses of 20 and 40 mg/kg of CBD in these studies had no such effect [44]. Doses of 20 mg/kg of CBD were observed to reduce the cocaine-induced rewarding effects in mice [53], while 10 mg/kg of CBD effectively blocked the memory reconstruction induced by cocaine [54] and attenuated cocaine- and METH-induced CPP [13]. CBD at 5 and 10 mg/kg significantly reduced the rewarding effects and drug-seeking behaviors induced by heroin [12, 55]. Further, CBD at 10 mg/kg has been shown to inhibit morphine-induced CPP in mice [56].
The present research demonstrates that CBD has potential therapeutic use for mitigating the rewarding effects induced by METH. However, the CBD dosage levels identified as effective in previous studies differ from those in the present study [12, 55]. This discrepancy could be explained by the choices of drugs across the studies, the varying pharmacological effects of CBD on those drugs, the use of different animal models for experimentation, and the varied drug delivery methods and experimental protocols used [38, 41–43].
The results of this study suggest that METH can induce strong Sigma1R expression in the PFC, NAc, hippocampus, and VTA (Fig. 2). Our data is consistent with previous research results that demonstrate that Sigma1R expression levels increase with chronic and repeated METH administration [57]. The expression levels of Sigma1R in the substantia nigra and the VTA were also observed to rise significantly in the self-administering METH rat model, suggesting that Sigma1R plays a vital role in METH-induced CPP and behavioral sensitization [58].
It was observed that the expression levels of σRs vary across the different brain regions, such as the PFC, amygdala, hippocampus, substantia nigra, hypothalamus, and cerebellum. Neuropsychosomatic diseases induced by repeated METH use may be associated with increased σR expression levels in specific brain regions [59]. Previous studies have shown that σRs are involved in drug-associated rewarding effects [20, 60]. Sigma1R antagonists, therefore, should effectively prevent METH-induced behavioral sensitization [61]. Preconditioning with selective Sigma1R antagonists has been shown to significantly reduce cocaine-induced hyperactivity, behavioral sensitization, and CPP, as well as METH-induced hyperactivity [62]. In addition, Sigma1R selective antagonists reduce the cytotoxic effects induced by cocaine, while nonselective antagonists reduce METH-induced neuronal degeneration [19, 63, 64]. Many studies have shown that Sigma1R antagonists exhibit robust neuroprotective effects against neuronal injury induced by METH [65–68]. A prior study indicated that pretreatment of CBD promotes morphine-induced supraspinal antinociception, reduces glutamate N-methyl-D-aspartate acid receptor (NMDA)-mediated convulsive syndrome, and stroke damage via Sigma1R [16]. In the same study, CBD was able to exhibit Sigma1R antagonist-like effects to disrupt the association between Sigma1R and NR1 subunit of NMDA receptor (NMDAR) in vitro [16]. In the present study, pretreating with 20, 40, and 80 mg/kg of CBD was observed to attenuate METH-induced Sigma1R expression in the PFC, NAc, HPC, and VTR. These results demonstrate that CBD can be used as an antagonist of Sigma1R to reverse CPP in METH-induced rats.
During the present study’s examination of the PI3K/AKT/GSK-3β/CREB signaling pathway, it was found that METH can induce the phosphorylation of AKT (Ser473), GSK-3β (Ser9), and CREB (Ser133) in four brain regions. The expression levels of AKT, GSK-3β, and CREB did not alter significantly in general, but the p-AKT/AKT, p-GSK-3β/GSK-3β, and p-CREB/CREB ratios exhibited notable increases. This indicates that METH can activate the AKT/GSK-3β/CREB signaling pathway and promote the phosphorylation of AKT, GSK-3β, and CREB (Figs 3–5).
Xu et al. reported that METH promotes the hyperphosphorylation of an Alzheimer’s disease (AD)-related pathological protein p-tau mediated by the downregulation of the insulin signaling pathway and activation of GSK-3β in Neuro2A cells [69]. Treatment with the GSK-3β inhibitor TWS119 inhibited METH-induced hyperphosphorylation of tau [69]. Here, we consistently showed that CBD can broadly attenuate METH-induced p-GSK-3β protein expression and relative p-GSK-3β/GSK-3β expression in the PFC, NAc, HPC, and VTA, which could contribute to the weakening of the METH-induced CPP.
The present study demonstrates that pretreating with different doses of CBD causes respective reductions to the relative expressions of p-AKT/AKT, p-GSK3β/GSK3β, and p-CREB/CREB in the brain’s regions. Per these results, CBD at 40 and 80 mg/kg significantly reduces p-AKT/AKT, p-GSK-3β/GSK-3β, and p-CREB/CREB ratios in the PFC. These data were statistically significant compared to the group that did not receive CBD treatment. This indicates that METH addiction can activate the phosphorylation of PI3K/AKT/GSK-3β/CREB signaling pathway-related proteins and that the early use of CBD can inhibit METH addiction.
Conclusion
Similar to the results of previous research, the present study found that METH can induce CPP in rats. When a pretreatment of CBD was applied, however, the CBD weakened the METH-induced CPP by regulating the Sigma1R/AKT/GSK-3β/CREB signaling pathway. CBD at doses of 40 and 80 mg/kg had the strongest effects. Further investigations should seek to confirm the functional specificities of the Sigma1R/AKT/GSK-3β/CREB signaling pathway as related to METH-induced rewarding effects.
Conflict of Interest Statement
The authors declare no conflicts of interest.
Funding
This work was supported by the National Nature Science Foundation of China (No. 81960340, 81660310, and 81560303), the Yunnan Applied Basic Research Projects-Joint Special Project (No. 2017FE467(-018) and 2018FE001(-182)).
Acknowledgements
We acknowledge G.Y., L.L., R.Z., J.L., C.K.L., J.H., Y.L., and B.S. who participated in the literature search, experimental validation, data mining, bioinformatics analysis, and interpretation and writing the manuscript and creating figures and tables. X.Z. and D.Z. supervised the study design and critically read and edited the manuscript. All the authors approved the final version for publication.
References
- 1. Shaerzadeh F, Streit WJ, Heysieattalab S et al. Methamphetamine neurotoxicity, microglia, and neuroinflammation. J Neuroinflammation 2018;15:341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Robinson JD, Howard CD, Pastuzyn ED et al. Methamphetamine-induced neurotoxicity disrupts pharmacologically evoked dopamine transients in the dorsomedial and dorsolateral striatum. Neurotox Res 2014;26:152–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Friend DM, Keefe KA. Glial reactivity in resistance to methamphetamine-induced neurotoxicity. J Neurochem 2013;125:566–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xu X, Huang E, Tai Y et al. Nupr1 modulates methamphetamine-induced dopaminergic neuronal apoptosis and autophagy through CHOP-Trib3-mediated endoplasmic reticulum stress signaling pathway. Front Mol Neurosci 2017;10:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Russo EB. Taming THC: potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br J Pharmacol 2011;163:1344–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hirst RA, Lambert DG, Notcutt WG. Pharmacology and potential therapeutic uses of cannabis. Br J Anaesth 1998;81:77–84. [DOI] [PubMed] [Google Scholar]
- 7. Elsohly MA, Slade D. Chemical constituents of marijuana: the complex mixture of natural cannabinoids. Life Sci 2005;78:539–48. [DOI] [PubMed] [Google Scholar]
- 8. Fernandez-Ruiz J, Moro MA, Martinez-Orgado J. Cannabinoids in neurodegenerative disorders and stroke/brain trauma: from preclinical models to clinical applications. Neurotherapeutics 2015;12:793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Samanta D. Cannabidiol: a review of clinical efficacy and safety in epilepsy. Pediatr Neurol 2019;96:24–9. [DOI] [PubMed] [Google Scholar]
- 10. Millar SA, Stone NL, Bellman ZD et al. A systematic review of cannabidiol dosing in clinical populations. Br J Clin Pharmacol 2019; 85:1888–1900. doi: 10.1111/bcp.14038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Prud'homme M, Cata R, Jutras-Aswad D. Cannabidiol as an intervention for addictive behaviors: a systematic review of the evidence. J Subst Abuse: Res Treat 2015;9:33–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ren Y, Whittard J, Higuera-Matas A et al. Cannabidiol, a nonpsychotropic component of cannabis, inhibits cue-induced heroin seeking and normalizes discrete mesolimbic neuronal disturbances. J Neurosci Off J Soc Neurosci 2009;29:14764–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parker LA, Burton P, Sorge RE et al. Effect of low doses of delta9-tetrahydrocannabinol and cannabidiol on the extinction of cocaine-induced and amphetamine-induced conditioned place preference learning in rats. Psychopharmacology 2004;175:360–6. [DOI] [PubMed] [Google Scholar]
- 14. Sambo DO, Lin M, Owens A et al. The sigma-1 receptor modulates methamphetamine dysregulation of dopamine neurotransmission. Nat Commun 2017;8:2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007;131:596–610. [DOI] [PubMed] [Google Scholar]
- 16. Rodriguez-Munoz M, Onetti Y, Cortes-Montero E et al. Cannabidiol enhances morphine antinociception, diminishes NMDA-mediated seizures and reduces stroke damage via the sigma 1 receptor. Mol Brain 2018;11:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kourrich S, Su TP, Fujimoto M et al. The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci 2012;35:762–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang K, Wang C, Sun T. The roles of intracellular chaperone proteins, sigma receptors, in Parkinson's disease (PD) and major depressive disorder (MDD). Front Pharmacol 2019;10:528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Robson MJ, Noorbakhsh B, Seminerio MJ et al. Sigma-1 receptors: potential targets for the treatment of substance abuse. Curr Pharm Des 2012;18:902–19. [DOI] [PubMed] [Google Scholar]
- 20. Hedges DM, Obray JD, Yorgason JT et al. Methamphetamine induces dopamine release in the nucleus accumbens through a sigma receptor-mediated pathway. Neuropsychopharmacology 2018;43:1405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaushal N, Elliott M, Robson MJ et al. AC927, a sigma receptor ligand, blocks methamphetamine-induced release of dopamine and generation of reactive oxygen species in NG108-15 cells. Mol Pharmacol 2012;81:299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu Y, Huang Y, Ding J et al. Targeting Akt by SC66 triggers GSK-3beta mediated apoptosis in colon cancer therapy. Cancer Cell Int 2019;19:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakaso K, Tajima N, Horikoshi Y et al. The estrogen receptor beta-PI3K/Akt pathway mediates the cytoprotective effects of tocotrienol in a cellular Parkinson's disease model. Biochim Biophys Acta 2014;1842:1303–12. [DOI] [PubMed] [Google Scholar]
- 24. Yang G, Zeng X, Li J et al. Protective effect of gastrodin against methamphetamine-induced autophagy in human dopaminergic neuroblastoma SH-SY5Y cells via the AKT/mTOR signaling pathway. Neurosci Lett 2019;707:134287. [DOI] [PubMed] [Google Scholar]
- 25. Sun H, Wu H, Liu J et al. Prenatal stress impairs spatial learning and memory associated with lower mRNA level of the CAMKII and CREB in the adult female rat hippocampus. Neurochem Res 2017;42:1496–503. [DOI] [PubMed] [Google Scholar]
- 26. Vandesquille M, Baudonnat M, Decorte L et al. Working memory deficits and related disinhibition of the cAMP/PKA/CREB are alleviated by prefrontal alpha4beta2*-nAChRs stimulation in aged mice. Neurobiol Aging 2013;34:1599–609. [DOI] [PubMed] [Google Scholar]
- 27. Nestler EJ. Historical review: molecular and cellular mechanisms of opiate and cocaine addiction. Trends Pharmacol Sci 2004;25:210–8. [DOI] [PubMed] [Google Scholar]
- 28. Lei G, Xia Y, Johnson KM. The role of Akt-GSK-3beta signaling and synaptic strength in phencyclidine-induced neurodegeneration. Neuropsychopharmacology 2008;33:1343–53. [DOI] [PubMed] [Google Scholar]
- 29. Teng L, Meng Q, Lu J et al. Liquiritin modulates ERK and AKT/GSK3betadependent pathways to protect against glutamateinduced cell damage in differentiated PC12 cells. Mol Med Rep 2014;10:818–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kwon SH, Ma SX, Lee SY et al. Sulfuretin inhibits 6-hydroxydopamine-induced neuronal cell death via reactive oxygen species-dependent mechanisms in human neuroblastoma SH-SY5Y cells. Neurochem Int 2014;74:53–64. [DOI] [PubMed] [Google Scholar]
- 31. Tan Z, Kang T, Zhang X et al. Nerve growth factor prevents arsenic-induced toxicity in PC12 cells through the AKT/GSK-3beta/NFAT pathway. J Cell Physiol 2019;234:4726–38. [DOI] [PubMed] [Google Scholar]
- 32. Wu X, Liang Y, Jing X et al. Rifampicin prevents SH-SY5Y cells from rotenone-induced apoptosis via the PI3K/Akt/GSK-3beta/CREB signaling pathway. Neurochem Res 2018;43:886–93. [DOI] [PubMed] [Google Scholar]
- 33. Qiao X, Gai H, Su R et al. PI3K-AKT-GSK3beta-CREB signaling pathway regulates anxiety-like behavior in rats following alcohol withdrawal. J Affect Disord 2018;235:96–104. [DOI] [PubMed] [Google Scholar]
- 34. Wu Y, Shang Y, Sun S et al. Erythropoietin prevents PC12 cells from 1-methyl-4-phenylpyridinium ion-induced apoptosis via the Akt/GSK-3beta/caspase-3 mediated signaling pathway. Apoptosis 2007;12:1365–75. [DOI] [PubMed] [Google Scholar]
- 35. Karimi-Haghighi S, Dargahi L, Haghparast A. Cannabidiol modulates the expression of neuroinflammatory factors in stress- and drug-induced reinstatement of methamphetamine in extinguished rats. Addict Biol 2020;25:e12740. doi: 10.1111/adb.12740. [DOI] [PubMed] [Google Scholar]
- 36. Ferguson D, Shao N, Heller E et al. SIRT1-FOXO3a regulate cocaine actions in the nucleus accumbens. J Neurosci Off J Soc Neurosci 2015;35:3100–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Havlickova T, Charalambous C, Lapka M et al. Ghrelin receptor antagonism of methamphetamine-induced conditioned place preference and intravenous self-administration in rats. Int J Mol Sci 2018;19:pii: E2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu W, Peng QX, Lin XL et al. Effect of rhynchophylline on the expression of p-CREB and sc-Fos in triatum and hippocampal CA1 area of methamphetamine-induced conditioned place preference rats. Fitoterapia 2014;92:16–22. [DOI] [PubMed] [Google Scholar]
- 39. Sun L, Song R, Chen Y et al. A selective D3 receptor antagonist YQA14 attenuates methamphetamine-induced behavioral sensitization and conditioned place preference in mice. Acta Pharmacol Sin 2016;37:157–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fu K, Lin H, Miyamoto Y et al. Pseudoginsenoside-F11 inhibits methamphetamine-induced behaviors by regulating dopaminergic and GABAergic neurons in the nucleus accumbens. Psychopharmacology 2016;233:831–40. [DOI] [PubMed] [Google Scholar]
- 41. Bayat AH, Haghparast A. Effect of insulin deficiency on the rewarding properties of methamphetamine in streptozotocin-induced diabetic rats. Pharmacol Biochem Behav 2015;128:8–13. [DOI] [PubMed] [Google Scholar]
- 42. Lien WH, Yeh TL, Yang YK et al. Cycloheximide enhances maintenance of methamphetamine-induced conditioned place preference. Chin J Physiol 2004;47:23–30. [PubMed] [Google Scholar]
- 43. Huang J, Yang G, Li Z et al. Involvement of dopamine D3 receptor and dopamine transporter in methamphetamine-induced behavioral sensitization in tree shrews. Brain Behav 2020;10:e01533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hay GL, Baracz SJ, Everett NA et al. Cannabidiol treatment reduces the motivation to self-administer methamphetamine and methamphetamine-primed relapse in rats. J Psychopharmacol 2018;32:1369–78. [DOI] [PubMed] [Google Scholar]
- 45. Camí J, Farré M. Drug addiction. N Engl J Med 2003;349:975–86. [DOI] [PubMed] [Google Scholar]
- 46. Volz TJ, Hanson GR, Fleckenstein AE. The role of the plasmalemmal dopamine and vesicular monoamine transporters in methamphetamine-induced dopaminergic deficits. J Neurochem 2007;101:883–8. [DOI] [PubMed] [Google Scholar]
- 47. Song R, Bi GH, Zhang HY et al. Blockade of D3 receptors by YQA14 inhibits cocaine's rewarding effects and relapse to drug-seeking behavior in rats. Neuropharmacology 2014;77:398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Collins GT, Truong YN, Levant B et al. Behavioral sensitization to cocaine in rats: evidence for temporal differences in dopamine D3 and D2 receptor sensitivity. Psychopharmacology 2011;215:609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jiang L, Zhu R, Bu Q et al. Brain renin-angiotensin system blockade attenuates methamphetamine-induced hyperlocomotion and neurotoxicity. Neurotherapeutics 2018;15:500–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yang X, Wang Y, Li Q et al. The main molecular mechanisms underlying methamphetamine-induced neurotoxicity and implications for pharmacological treatment. Front Mol Neurosci 2018;11:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Davidson C, Gow AJ, Lee TH et al. Methamphetamine neurotoxicity: necrotic and apoptotic mechanisms and relevance to human abuse and treatment. Brain Res Brain Res Rev 2001;36:1–22. [DOI] [PubMed] [Google Scholar]
- 52. Arias-Carrion O, Stamelou M, Murillo-Rodriguez E et al. Dopaminergic reward system: a short integrative review. Int Arch Med 2010;3:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lujan MA, Castro-Zavala A, Alegre-Zurano L et al. Repeated cannabidiol treatment reduces cocaine intake and modulates neural proliferation and CB1R expression in the mouse hippocampus. Neuropharmacology 2018;143:163–75. [DOI] [PubMed] [Google Scholar]
- 54. de Carvalho CR, Takahashi RN. Cannabidiol disrupts the reconsolidation of contextual drug-associated memories in Wistar rats. Addict Biol 2017;22:742–51. [DOI] [PubMed] [Google Scholar]
- 55. Katsidoni V, Anagnostou I, Panagis G. Cannabidiol inhibits the reward-facilitating effect of morphine: involvement of 5-HT1A receptors in the dorsal raphe nucleus. Addict Biol 2013;18:286–96. [DOI] [PubMed] [Google Scholar]
- 56. Markos JR, Harris HM, Gul W et al. Effects of cannabidiol on morphine conditioned place preference in mice. Planta Med 2018;84:221–4. [DOI] [PubMed] [Google Scholar]
- 57. Stefanski R, Justinova Z, Hayashi T et al. Sigma1 receptor upregulation after chronic methamphetamine self-administration in rats: a study with yoked controls. Psychopharmacology 2004;175:68–75. [DOI] [PubMed] [Google Scholar]
- 58. Hayashi T, Justinova Z, Hayashi E et al. Regulation of sigma-1 receptors and endoplasmic reticulum chaperones in the brain of methamphetamine self-administering rats. J Pharmacol Exp Ther 2010;332:1054–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Itzhak Y. Repeated methamphetamine-treatment alters brain sigma receptors. Eur J Pharmacol 1993;230:243–4. [DOI] [PubMed] [Google Scholar]
- 60. Kourrich S, Hayashi T, Chuang JY et al. Dynamic interaction between sigma-1 receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine. Cell 2013;152:236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nguyen EC, McCracken KA, Liu Y et al. Involvement of sigma (sigma) receptors in the acute actions of methamphetamine: receptor binding and behavioral studies. Neuropharmacology 2005;49:638–45. [DOI] [PubMed] [Google Scholar]
- 62. Yasui Y, Su TP. Potential molecular mechanisms on the role of the Sigma-1 receptor in the action of cocaine and methamphetamine. J Drug Alcohol Res 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Matsumoto RR, Nguyen L, Kaushal N et al. Sigma (sigma) receptors as potential therapeutic targets to mitigate psychostimulant effects. Adv Pharmacol 2014;69:323–86. [DOI] [PubMed] [Google Scholar]
- 64. Matsumoto RR, Gilmore DL, Pouw B et al. Novel analogs of the sigma receptor ligand BD1008 attenuate cocaine-induced toxicity in mice. Eur J Pharmacol 2004;492:21–6. [DOI] [PubMed] [Google Scholar]
- 65. Seminerio MJ, Kaushal N, Shaikh J et al. Sigma (sigma) receptor ligand, AC927 (N-phenethylpiperidine oxalate), attenuates methamphetamine-induced hyperthermia and serotonin damage in mice. Pharmacol Biochem Behav 2011;98:12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kaushal N, Seminerio MJ, Robson MJ et al. Pharmacological evaluation of SN79, a sigma (sigma) receptor ligand, against methamphetamine-induced neurotoxicity in vivo. Eur Neuropsychopharmacol 2013;23:960–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kaushal N, Seminerio MJ, Shaikh J et al. CM156, a high affinity sigma ligand, attenuates the stimulant and neurotoxic effects of methamphetamine in mice. Neuropharmacology 2011;61:992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Seminerio MJ, Hansen R, Kaushal N et al. The evaluation of AZ66, an optimized sigma receptor antagonist, against methamphetamine-induced dopaminergic neurotoxicity and memory impairment in mice. Int J Neuropsychopharmacol 2013;16:1033–44. [DOI] [PubMed] [Google Scholar]
- 69. Xu H, Chen X, Wang J et al. Involvement of insulin signalling pathway in methamphetamine-induced hyperphosphorylation of tau. Toxicology 2018;408:88–94. [DOI] [PubMed] [Google Scholar]