

SUMMARY
Reactive stromal cells are an integral part of tumor microenvironment (TME) and interact with cancer cells to regulate their growth. Although targeting stromal cells could be a viable therapy to regulate the communication between TME and cancer cells, identification of stromal targets that make cancer cells vulnerable has remained challenging and elusive. Here, we identify a previously unrecognized mechanism whereby metabolism of reactive stromal cells is reprogrammed through an upregulated glutamine anabolic pathway. This dysfunctional stromal metabolism confers atypical metabolic flexibility and adaptive mechanisms in stromal cells, allowing them to harness carbon and nitrogen from noncanonical sources to synthesize glutamine in nutrient-deprived conditions existing in TME. Using an orthotopic mouse model for ovarian carcinoma, we find that co-targeting glutamine synthetase in stroma and glutaminase in cancer cells reduces tumor weight, nodules, and metastasis. We present a synthetic lethal approach to target tumor stroma and cancer cells simultaneously for desirable therapeutic outcomes.
In Brief
Yang et al. reveal that cancer-associated fibroblasts boost glutamine production by harnessing carbon and nitrogen from atypical nutrient sources to maintain cancer cell growth when glutamine is scarce. Co-targeting stromal glutamine synthetase and cancer cell glutaminase disrupts this metabolic crosstalk, inducing tumor regression in an ovarian carcinoma mouse model.
Graphical Abstarct
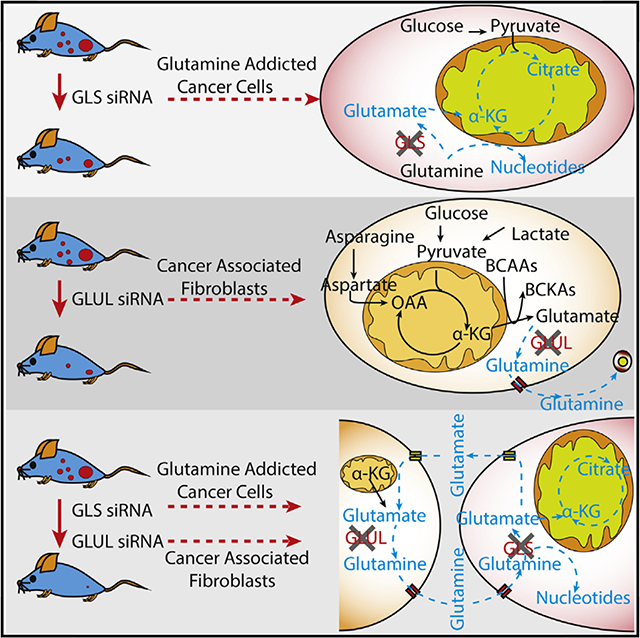
INTRODUCTION
The transformation of healthy epithelial cells into aggressive tumors is a gradual process that is characterized by cancer hallmarks such as drug resistance, dysregulated energy metabolism, and metastatic proclivity (Hanahan and Weinberg, 2011; Kroemer and Pouyssegur, 2008). As cancer cells grow, they recruit stromal cells, forming a complex tumor microenvironment (TME). These reactive stromal cells co-evolve and continually interact with cancer cells, becoming an integral part of their physiology, and are indispensable for their survival (Icard et al., 2014). Increasing evidence suggests that reactive stroma is not an innocent bystander (Chu et al., 2007; Yeung et al., 2013), but rather mediates a network of paracrine signals conferring resistance to cancer cells in nutrient-deprived conditions observed in TME (Valencia et al., 2014). Targeting reactive stromal cells is emerging as an attractive and viable therapy to regulate the channels of communication between stromal and cancer cells (Goveia et al., 2014; Hansen et al., 2016). To target non-autonomous mechanisms of cancer cell aberrations, the mechanistic underpinnings of reactive stroma vis a vis quiescent or normal stroma is required. TME consists of several non- cancerous cells such as fibroblasts, endothelial cells, pericytes, and immune cells embedded within extracellular matrix proteins and vasculature. These interactions present numerous opportunities for targeting TME for effective therapy. Since cancer-associated fibroblasts (CAFs) are the most populous cells in the TME and are genetically stable relative to cancer cells, therapies targeting them would theoretically be more effective (Hansen et al., 2016).
Glutamine (Gln) has pleiotropic roles in tumorigenesis including initiation of neoplasia, progression of disease, and metastasis. Several studies have shown that tumors utilize multiple sources of Gln, such as macropinocytosis of extracellular fluid, endocytosis of exosome, and de novo synthesis of Gln, in order to meet their requirements for Gln (Bott et al., 2015; Commisso et al., 2013; Zhao et al., 2016). The de novo Gln synthesis is mediated by the intracellular Gln synthetase (GS), which catalyzes condensation of glutamate and ammonia to synthesize Gln. In tumors, multiple oncogenic signaling pathways have been shown to also affect the expression of GS. For instance, the phosphorylation of FOXO transcription factor regulates GS expression (van der Vos et al., 2012). In cancer cells, the upregulation of GS expression increases the level of intracellular Gln, promotes synthesis of asparagine and nucleotides, and enhances uptake of essential amino acids from extracellular regions of TME (Bott et al., 2015). Hence, a positive correlation of GS activity with cell survival and proliferation is observed in Myc-driven cancers. Interestingly, metabolic heterogeneity in different components of the tumor enables nutrient crosstalk. In Gln-independent breast luminal cancer cells, higher expression level of GS enables Gln synthesis and provides Gln for the growth of Gln-dependent breast basal cancer cells (Kung et al., 2011). Recently, it was reported that heterogeneity exists in GS expression among glioblastoma (GBM) cells of the same tumor; this characteristic allowed GS-deficient GBM cells to acquire Gln from GS-positive GBM cells and astrocytes for de novo nucleotide synthesis (Tardito et al., 2015). These observations strongly imply that investigation into how the TME protects cancer cells from Gln deprivation is necessary for developing novel therapy that combinatorially targets TME and cancer cells. Such therapeutic methods will effectively achieve systematic regression in tumor growth, progression, and metastasis.
The objective of this study was to examine if altered reactive stromal metabolism is the driver for regulating cancer growth in its harsh microenvironment and if targeting this aberration could create metabolic vulnerability in cancer cells by disrupting the metabolic crosstalk between stromal and cancer cells. Our study revealed the fundamental metabolic differences between CAFs and normal ovarian fibroblasts (NOFs), which support tumor growth and progression. Comparing the gene expression profiles of fibroblastic stromal components from a series of advanced stage, high-grade serous ovarian adenocarcinomas to NOFs revealed a strikingly higher Gln anabolic pathway in CAFs compared to NOFs. We also demonstrated, for the first time, that CAFs had remarkably higher metabolic flexibility compared to NOFs. Dysregulated CAF metabolism induced adaptive mechanisms for harnessing carbon and nitrogen from atypical sources to synthesize Gln in environments where Gln is scarce. Significantly, co-targeting highly expressed stromal GS with highly expressed cancer cell glutaminase (GLS) in an orthotopic intra-ovarian mouse model revealed that the metabolic interdependence of CAF and ovarian cancer (OVCA) cells conferred a synthetic lethality in tumor and stromal compartments. Our work underscores the reliance of cancer cells on stromal CAFs, presenting an opportunity to target tumor stroma and cancer cells simultaneously to improve therapeutic outcomes.
RESULTS
Upregulated Gln Anabolic Pathway in CAFs Compared to NOFs
To analyze metabolic reprogramming in reactive stroma, we analyzed expression of genes encoding enzymes in central carbon metabolism of the fibroblastic stromal components microdissected from a series of advanced stage, high-grade serous ovarian adenocarcinomas and compared that to NOFs (Leung et al., 2014). Interestingly, we found that CAFs had significantly higher expression of Gln pathway genes, especially GLUL, responsible for Gln synthesis and genes encoding amino acid transferases (glutamic-oxaloacetic transaminase ½, GOT½; branch chain amino acid transaminase 1, BCAT1), which facilitate intracellular glutamate synthesis (Figures 1A, 1B, and S1A, available online; GEO: GSE40595). Immunohistochemical (IHC) staining found that CAFs have higher GS, GOT2, and BCAT1 expression than NOFs (Figures S1B and S1C). Further, CAFs had increased expression of genes encoding glycolysis, tricarboxylic acid (TCA) cycle, and electron transport chain (ETC) (Figures 1A and S1D-S1F), which increased their metabolic activity. Consistent with these expression results, transcription factors known to target these enzymes were also indeed upregulated (Figure 1C; Table S1).
Figure 1. Gln Anabolic Pathway Is Upregulated in CAFs Compared to NOFs.

(A) Differential expression of genes encoding metabolic enzymes in cancer-associated fibroblasts (CAFs; n = 33) relative to normal ovarian fibroblasts (NOFs; n = 8). CAFs were derived from fibroblastic stromal components microdissected from a series of advanced-stage, high-grade serous ovarian adenocarcinomas; NOFs were derived from normal ovaries obtained from patients.
(B) Expression level of branch-chain aminotransferase 1 (BCAT1), glutamic-oxaloacetic transaminase 1 (GOT1), GOT2, and GS (GLUL) In CAFs and NOFs.
(C) Network of transcription factors regulating Gln anabolic and glycolysis pathway genes in CAFs relative to NOFs.
(D) GLUL expression levels in paired tumor epithelial and stromal compartments. Lines connecting tumor and stromal data points signify tumor and stromal samples derived from the same patient (Wisconsin test).
(E) Average pathway expression of Gln anabolism levels in paired tumor epithelial and stromal compartments (Wisconsin test).
(F) Representative IHC staining image comparing GS protein expression between stromal and tumor compartments.
(G) Proliferation after 72 hr of patient-derived NOF1, NOF2, and CAF1–5 under Gln deprivation relative to nutrient-rich media.
(H) Gln secretion rate of three patient-derived CAF1–3 and patient-derived NOF2/NOF1 under Gln deprivation conditions.
(I) Gln uptake rate of high-grade OVCA cell lines, HeyA8 and SKOV3, in Gln-replete medium.
Error bars indicate mean ± SEM of n ≥ 3 independent experiments.*p < 0.05, **p < 0.01, ***p < 0.001.
To confirm that the increased gene expression of metabolic enzymes is associated with the stromal compartment, we compared expressions of microdissected paired fibroblastic stromal and epithelial components. We found that stromal CAFs had significantly higher expression of GLUL and higher average expression of Gln anabolic pathway genes (Figures 1D, 1E, and S2A). However, tumor stroma had similar expression of glycolysis genes as tumor epithelial cells (Figure S1G). The GS staining in OVCA patient-derived tumor tissues further substantiated that the fibroblastic stromal component had higher expression of GS compared to its epithelial compartment (Figure 1F). The increased GS expression in reactive stromal CAFs implied that CAFs could survive under Gln deprivation conditions. Indeed, patient-derived CAFs were Gln independent for proliferation, while NOFs were Gln addicted (Figure 1G). CAFs and NOFs in complete medium were found to have similar growth rates in complete medium (Figure S1H). Previously, we showed that high-grade OVCA cells were Gln dependent for cell proliferation and metastasis (Yang et al., 2014). This hypernutrient dependency of cancer cells depletes nutrients from TME and suggests that Gln anabolic metabolism of stromal CAFs may play a role in maintaining growth of these cancer cells by secreting Gln. To verify Gln secretory capacity of reactive stroma, we measured the concentration of Gln in Gln-free spent media and found that patient-derived CAFs secreted Gln at a rate of around 25 pmol/K cells/hr, while NOFs barely secreted any detectable Gln (Figure 1H). The secretion rate of Gln substantiates a higher expression of GLUL in CAFs compared to NOFs (Figures 1A and 1B). Within complete medium containing 2 mM Gln, OVCA cell lines, HeyA8 and SKOV3, uptake around 100 pmol/K cells/hr Gln for sustaining cell growth (Figure 11). The physiological ratio of CAFs to cancer is reported to be between 1 and 10 (Hu et al., 2015), thereby suggesting that CAFs in TME are well equipped to maintain the Gln uptake flux of Gln-addicted cancer cells.
Upregulated Asparagine and Aspartate Flux through GOT in CAFs Promote Gln Synthesis
To obtain the mechanistic underpinnings of the deregulated CAF metabolism, we performed 13C-labeled metabolic flux analysis (13C-MFA) using U-13C6 glucose (Figure S3A) to estimate metabolic fluxes in CAFs and NOFs. We found that NOFs were sensitive to Gln-depletion stress, and there was significant reduction in glutamate, α-ketoglutarate (α-KG), malate, aspartate, and citrate pools in NOFs, whereas CAFs suffered only slight reduction in the metabolite pools (Figure 2A). The consumption and secretion profiles of CAFs revealed an increase in the uptake of asparagine and branched-chain amino acids (BCAAs) from the medium and reduction in glutamate secretion (Figure S3B). Further, metabolic tracing revealed that the relative abundance of TCA cycle metabolites, such as M2 citrate, and M2 glutamate, directly derived from U-13C6 glucose, was significantly enhanced in CAFs under Gln deprivation as compared to complete media (Figures 2B-2D). In the case of NOFs, Gln deprivation dramatically decreased glucose’s conversion to TCA cycle metabolites and glutamate (indicated by heavy isotopologues of metabolites) (Figures 2B-2D). By quantifying intracellular fluxes, we found that in NOFs under Gln deprivation, TCA cycle fluxes were affected significantly, and conversion of α-KG to succinate, fumarate, malate, and oxaloacetate (OAA) was reduced by 45% as compared to Gln-replete condition (Figure 2E; Table S2). Further, there is no Gln synthesis in NOFs. In contrast, in CAFs under Gln deprivation, the increased contribution of glucose-derived carbon to citrate and α-KG, along with an increase in aspartate and asparagine intake, contributes to Gln synthesis (Figure 2E; Table S3). On comparing fluxes normalized to pyruvate transport flux in Gln-deprived CAF to those in Gln-deprived NOF, we observed that all TCA cycle fluxes are 2.5–4 times higher (Figure 2F; Table S4). The above results establish a major metabolic reprogramming of reactive stroma, evident through upregulated Gln metabolism in CAFs compared to quiescent NOFs.
Figure 2. Upregulated Asparagine and Aspartate Flux through GOT in CAFs Promote Gln Synthesis.

(A) Intracellular concentrations of metabolites in CAFs and NOFs under Gln deprivation relative to nutrient-rich medium.
(B-D) Fractional enrichments of glutamate (B), citrate (C), and malate (D) in CAF1 and NOF2 cultured with U-13C6 glucose under Gln-replete condition or Gln starvation.
(E) Intracellular fluxes in Gln-deprived NOF/CAF (NOF/CAF 0Q) relative to NOF/CAF in nutrient-rich medium (NOF/CAF1Q) quantified using U-13C6 glucose tracer experiments and 13C metabolic flux analysis. Line thickness is proportional to flux values in CAF 1Q conditions. Color key represents increased (red) or decreased (green) fluxes in conditions compared to control. Blue arrows represent fluxes that are reversed in the condition compared to control, and gray arrows represent no significant change in fluxes between conditions.
(F) Intracellular fluxes in Gln-deprived CAFs (CAF 0Q) relative to Gln-deprived NOFs (NOF 0Q) quantified using U-13C6 glucose tracer experiments and 13C metabolic flux analysis (13C-MFA). Line thickness is proportional to flux values (relative to pyruvate flux to TCA) in NOF 0Q conditions. Confidence intervals for 13C-MFA fluxes estimated using Monte-Carlo sampling are reported in Tables S2-S4.
Error bars indicate mean ± SEM of n ≥ 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
CAFs, but Not NOFs, Maintain Gln-Addicted Cancer Cell Growth under Gln Deprivation Condition
To substantiate the role of CAFs in maintaining OVCA cell growth under Gln deprivation, we measured the proliferation rate of GFP-labeled high-grade OVCA cell lines, HeyA8 and SKOV3, in direct co-cultures with patient-derived CAFs or NOFs. Remarkably, CAFs rescued the proliferation rate of cancer cells under Gln deprivation while NOFs had no effect (Figures 3A-3C). However, CAFs and NOFs have a similar impact on the growth rate of OVCA in complete medium (Figures S3C and S3D). To exclude the possibility that soluble factors could rescue proliferation and confirm that rescue is through CAF-secreted Gln, we performed co-cultures with L-asparaginase, which hydrolyzes extracellular Gln to glutamate and ammonia, thereby blocking uptake of CAF-secreted Gln by the cancer cells. As expected, CAF-mediated rescue of cancer cell proliferation was attenuated, thereby confirming the role of CAF-secreted Gln (Figure 3C). To understand the effects of CAF-derived Gln in cancer cells, we measured gene expression of cancer cells in monocultures and co-cultures with CAFs using lllumina microarrays (GEO: GSE87773). Gene set enrichment analysis (GSEA; Subramanian et al., 2005) of transcriptional changes revealed that expression of cell-cycle genes was higher in cancer cells co-cultured with CAFs as compared to monocultures, whereas those of apoptosis were lower (Figure 3D). Similarly, it was found that CAFs can significantly enhance the gene expression of the cell cycle and antiapoptosis in HeyA8, compared to NOFs (Figure S3E). CAFs also enhanced the expression level of unsaturated fatty acid synthesis, an important pathway of lipid bilayer synthesis for cell division, in cancer cells (Figure S3F). Comparing HeyA8 gene expression in Gln-replete and Gln-deprived medium, we observed higher enrichment scores of cell-cycle, unsaturated fatty acid synthesis genes (Figures S3G and S3H). Furthermore, we used the conditioned medium (CM) derived from NOFs or CAFs to treat cancer cells, and the results corroborated our findings that CM from CAFs rescued the proliferation, while CM from NOFs could not rescue it (Figure S3I). Furthermore, the addition of L-asparaginase into the CM abrogated the rescue effect from CAFs. Both CAFs and NOFs cannot support cancer cell survival under glucose deprivation, since CAFs and NOFs were themselves dependent on glucose for proliferation (Figures S4A and S4B). To exclude the possibility that Gln is directly catabolized from cell autophagy-induced protein degradation, we used the autophagy inhibitor, chloroquine. We found that chloroquine does not inhibit the rescue effect of CAFs on cancer cell growth under Gln-deprivation conditions (Figures S4C and S4D).
Figure 3. CAFs, but Not NOFs, Could Sustain Gln-Addicted Cancer Cell Growth under Gln Deprivation.

(A) Fluorescence microscopy images comparing growth of GFP-labeled HeyA8 and SKOV3 cells in contact co-cultures with CAFs or NOFs under Gln deprivation.
(B) Relative proliferation rates of GFP-labeled SKOV3 co-cultured with CAFs or NOFs under Gln deprivation quantified from fluorescence intensities. GFP fluorescence values under Gln-free condition are normalized with one in Gln-replete medium.
(C) Relative proliferation rates of HeyA8 co-cultured with CAFs or NOFs at different seeding ratios under Gln deprivation and treated with L-asparaginase. Proliferation rates are normalized to HeyA8 co-cultured with NOFs In Gln-replete media.
(D) Gene set enrichment analysis of cell cycle and apoptosis genes in HeyA8 co-cultured with CAFs with respect to monocultured HeyA8 in Gln-deprived medium. Gene expressions are measured 48 hr after culturing in respective media. (E) Gln secretion rates of CAF1 treated with MSO, a GS inhibitor, relative to untreated control.
(F) Relative proliferation rates of Gln-deprived HeyA8 treated with MSO cultured with and without CAFs normalized to HeyA8 mono-cultured in Gln-replete medium.
(G) Fluorescence microscopy images and quantified growth rates of HeyA8 co-cultured with CAF transfected with three independent Glul siRNA in Gln-rich and Gln-deprived media.
(H) GS protein expression in HeyA8 cells treated with shGLUL and their proliferation rates when co-cultured with CAFs in Gln-rich and Gln-deprived media. Errors bars indicate mean ± SEM of n ≥ 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
To associate GS, a central enzyme regulating Gln anabolism, with CAF-mediated rescue of cancer cells under Gln-deprived conditions, we added MSO, a GS inhibitor in co-cultures. Upon inhibition of the GS activity via MSO, cancer cells in Gln-deprived media could not survive without Gln secreted by CAFs (Figures 3E and 3F). Similarly, inhibiting GS expression and Gln secretion by small interfering RNA (siRNA)-GLUL in CAFs significantly impacted rescue of cancer cell growth under Gln deprivation (Figures 3G, S4E, and S4F). To ascertain that CAF-mediated rescue is conferred via CAF-secreted Gln and not due to a possible CAF-induced overexpression of GLUL in cancer cells, we silenced GLUL in HeyA8 cells using short hairpin RNA (shRNA)-GLUL. As expected, knocking down GLUL in cancer cells had no effect on CAF-mediated rescue of cancer cell growth under Gln deprivation (Figure 3H). These data provide evidence that stromal CAF-secreted Gln maintains cancer cell growth under nutrient-stressed TME.
Higher Metabolic Flexibility in CAFs Compared to NOFs Induces Adaptive Mechanisms for Harnessing Carbon and Nitrogen from Atypical Sources toward Gln Synthesis
To dissect the contribution of dominant substrates toward carbons of glutamate and Gln and to identify the potential metabolic vulnerability in reactive stroma, we cultured CAFs with 13C-labeled substrates to estimate their contribution toward Gln synthesis (Figure 4A). Interestingly, from the U-13C6 Glc isotope-labeling experiment, non-glucose substrate contribution, measured through MO levels, was much higher in glutamate, indicating dominant contribution from other carbon sources in Gln-deprived CAFs (Figures 2C and 2D). Culturing CAFs under Gln deprivation with 1 mM U-13C5 glutamate revealed that CAFs incorporated over 60% extracellular glutamate for Gln synthesis (Figure 4B). Surprisingly, supplementing lactate in culture media not only increased intracellular levels of TCA cycle metabolites, glutamate, and Gln, but also enhanced Gln secretion (Figures 4C and S4G). Although glucose contributed to around 50% of citrate and 30% of intracellular glutamate and Gln in CAFs, adding 11 mM lactate dramatically decreased the glucose contribution to TCA cycle metabolites, glutamate, and Gln, displacing glucose as the major carbon source for Gln precursors (Figures 4D-4F). Using U-13C3 lactate revealed high lactate contribution toward M2 Gln and M3 alanine in CAFs (Figures 4F and S4H-S4J). The addition of lactate in extracellular medium partially rescues the growth arrest in CAFs induced by glucose deprivation, further confirming that lactate can significantly affect stromal growth (Figure 4G). Even with labeled lactate in the media, at least 30% citrate is found to be unlabeled, indicating that other sources provide carbon for Gln’s backbone in CAFs (Figure 4D). Apart from glucose and lactate, acetate, BCAAs, and fatty acids are anaplerotic sources of acetyl-CoA. Both labeled acetate and leucine were found not to provide significant acetyl-CoA for Gln synthesis (Figure 4H). To reveal if fatty acid oxidation (FAO) can account for unlabeled glutamate, we cultured CAFs in U-13C6 glucose with 10 μM etomoxir, an FAO inhibitor. The fractional enrichment of extracellular M2 Gln and intracellular M2 glutamate significantly increased, thereby suggesting that FAO inhibition increased glucose’s contribution to glutamate synthesis and Gln secretion (Figures 4H and S4K). Our results demonstrate that fatty acids, glucose, and lactate were the major sources of acetyl-CoA and TCA metabolites.
Figure 4. Higher Metabolic Flexibility in CAFs Compared to NOFs Induced Adaptive Mechanisms for Harnessing Carbon from Atypical Substrates for Gln Synthesis.

(A) Schematic describing the fate of different stable isotope-labeled nutrient sources in CAFs used for synthesizing Gln.
(B) Contribution of U-13C5 glutamate (1 mM) to Gln secretion in spent medium after incubating with labeled glutamate for 24 hr.
(C) Relative extracellular Gln concentration in conditioned media of CAFs treated with lactate or ammonium in Gln-deprived media.
(D and E) U-13C6 glucose or U-13C3 lactate incorporation into intracellular citrate (D) and glutamate (E) after 24 hr of incubation with labeled substrates in CAF and NOF.
(F) U-13C6 glucose or U-13C3 lactate incorporation into extracellular Gln.
(G) Effect of lactate addition on growth rate of CAFs with Gln and glucose deprivation.
(H) U-13C6 glucose incorporation into extracellular Gln with and without fatty acid oxidation (FAO) inhibitor, etomoxir, to reveal FAO contribution to extracellular Gln. U-13C2 acetate and U-13C6 leucine contribution to extracellular Gln secreted by CAFs.
(I) Gene expression of glucose, lactate, and amino acid transporters in CAFs and NOFs.
(J) Contribution of aspartate and asparagine to intracellular malate, citrate, and glutamate using 1 mM 1,4-13C2 aspartate and 1 mM U-13C4 asparagine after 24 hr incubation.
(K) Schematic demonstrating different nutrient sources contributing to acetyl-CoA, OAA, glutamate, and Gln in CAFs.
(L) Uptake of 13C-labeled nutrients in CAFs compared to NOFs after 1 and 3 hr of incubation. M3 Lac is from U-13C3 lactate, M2 Asp is from [1,4] 13C2 aspartate, M4 Asn is from U-13C4 asparagine, and M5 Glu is from U-13C5 glutamate.
Error bars indicate mean ± SEM of n ≥ 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
OAA is an important anaplerotic point of entry in the TCA cycle and is derived either from pyruvate via pyruvate carboxylase (PC), or from asparate or asparagine through GOT. The relatively low M5 citrate with U-13C6 glucose labeling ruled out PC activity in CAFs (Figure 4D). We then analyzed the aspartate and asparagine transporter expression level in CAFs and compared it to NOFs. Interestingly, CAFs had significantly higher expression of aspartate transporter SLC1A3 and asparagine transporter SLC38A2, suggesting their significance in supplying OAA with a higher-expression GOT½ in CAFs (Figure 4I). Next, we used 1 mM [1,4]-13C2 aspartate or U-13C4 asparagine to trace their contribution in the TCA cycle (Figure S4L). Interestingly, aspartate provided 40% of M2 citrate, 60% of M2 malate in CAFs, as well as 40% of M1 glutamate (Figure 4J). Using U-13C4 asparagine, we observed that asparagine can also contribute to around 40% aspartate, which further contributes to more than 20% of citrate and malate and 15% of glutamate pools (Figures 4J and S4M). Therefore, asparagine can also be a major source of OAA for the TCA cycle through aspartate in CAFs. Collectively, glucose only accounted for 19% of acetyl-CoA synthesis while lactate contributed to the majority (48%) (Figure 4K). Aspartate and asparagine contributed to 69% and 30% OAA, respectively (Figure 4K). Approximately 50% of intracellular glutamate in CAFs was derived from the intracellular Gln anabolic pathway, while the remaining half was from the microenvironment (Figure 4K). The hyperactive Gln anabolism of CAFs was maintained by high lactate, aspartate, asparagine, and glutamate intake mediated by upregulated transporter expression levels in CAFs compared to NOFs (Figures 4I and 4L).
To identify the substrates contributing toward nitrogen supply for Gln synthesis in CAFs, we used 1 mM 15N-labeled BCAA, aspartate, alanine, serine, and ammonia. Analysis of intracellular glutamate and CAF-secreted Gln showed that BCAAs (leucine, 20%; isoleucine, 20%; valine, 6%) contributed significantly to amine (Figures 5A-5C). We found that aspartate is another major nitrogen donor because it can contribute to 40% of amine in the glutamate pool while consuming α-KG and producing OAA via GOT. Alanine is deaminated by glutamate pyruvate transaminase (GPT) and the amino group is transferred to glutamate and subsequently to Gln. Although only 10% of the glutamate pool was derived from labeled alanine, it is interesting to note that 30% of Gln nitrogen is from alanine. Serine contribution of nitrogen was almost negligible to glutamate and Gln (Figures 5B and 5C). Since GS exclusively utilizes free ammonia to generate Gln from glutamate, we saw 5% ammonia incorporation into glutamate and 80% incorporation into Gln, and its addition increased Gln secretion (Figures 4C, 5B, and 5C). In NOFs, the contribution to intracellular glutamate from BCAA, alanine, and ammonia was less than 10% and negligible from all other nitrogen sources (Figure 5D). This highlights the disparate nitrogen metabolism in CAFs and NOFs that allowed for efficient acquisition of nitrogen in CAFs for intracellular glutamate synthesis (Figure 5E). Removal of BCAAs, or aspartate and asparagine, from Gln-free medium was detrimental to not only Gln synthesis but also to the growth of CAFs (Figures 5F and S4N). To provide further support for the idea that targeting the stromal Gln anabolic pathway disrupts the cancer growth, we deprived asparagine and aspartate from co-culture medium and observed a marked decrease in cancer cell proliferation (Figure 5G). The addition of either asparagine or aspartate can completely reverse the proliferation (Figure 5G). Treatment with exogenous malate rescued the growth arrest, suggesting that aspartate and asparagine are imperative in maintaining Gln synthesis in CAFs (Figure 5G). Treating co-cultures with 20 mM gabapentin, an inhibitor of BCAT1, abolished the positive effect of CAFs on cancer cells under Gln deprivation (Figures 5H and S40). The addition of 250 μM aminooxyacetic acid (AOA), an inhibitor of amino transferase, had a similar effect as gabapentin (Figures 5H and S40). Flence, targeting incorporation of any of these nitrogen donors can reverse the supportive effects of CAFs on cancer cells.
Figure 5. Higher Metabolic Flexibility in CAFs Compared to NOFs Induced Adaptive Mechanisms for Harnessing Nitrogen from Atypical Substrates for Gln Synthesis.

(A) Schematic showing transfer of amine group for glutamate and Gln synthesis.
(B-D) Contribution of 15N-labeled substrates (1 mM NH4+, 1 mM 15N serine, 1 mM 15N alanine, 1 mM 15N aspartate, and 1 mM 15N leucine/isoleucine/valine) to intracellular glutamate in CAF1 (B), extracellular Gln in CAF1 (C), and intracellular glutamate in N0F2 (D).
(E) Absolute intracellular concentrations of labeled glutamate derived from 15N-labeled BCAAs, serine, aspartate, alanine, and ammonia in CAFs and NOFs.
(F) Gln secretion rates in CAFs under Gln deprivation combined with depletion of BCAAs or aspartate/asparagine from media, relative to CAFs in only Gln-deprived medium.
(G) Proliferation of Gln-deprived HeyA8 co-cultured with CAF1 supplemented with combinations of 1 mM asparagine, 1 mM dimethyl-aspartate, and 1 mM dimethyl-malate relative to HeyA8 co-cultured with CAF1 in Gln-free medium.
(H) Proliferation of HeyA8 co-cultured with CAF1 treated with 20 mM gabapentin (branch-chain aminotransferase, BCAT inhibitor) and 250 μM AOA (aminotransferase inhibitor) relative to mono-cultured HeyA8 in Gln-free medium.
Error bars indicate mean ± SEM of n ≥ 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
Crosstalk between Stromal-Epithelial Cells Augments Dysregulated Metabolism in CAFs and Supports Nucleotides and TCA Cycle Metabolite Levels in Cancer Cells
We next asked if neighboring cancer cells influenced the utilization of the carbon substrates by CAFs in order to support their Gln anabolic characteristics. We found that cancer cells increased accumulation of lactate and pyruvate, along with TCA cycle metabolites like citrate and α-KG in CAFs when they were co-cultured, as compared to monocultures under Gln deprivation (Figure 6A). This was confirmed through stable isotope tracing kinetic flux experiments with U-13C6 glucose. CAFs co-cultured with cancer cells had higher fractional labeling of Gln and glutamate as compared to CAFs in monoculture (Figures 6B, 6C, and S5A-S5C). Cancer cells enhanced TCA cycle activity in CAFs by increasing their glucose contribution to the TCA cycle to maintain glutamate and citrate levels for Gln synthesis (Figures S5D and S5E). This effect is independent of GLUL expression in cancer cells, for both control and HeyA8-shGLUL showed similar fractional labeling (Figures 6B, 6C, and S5A). To prove that CAFs can consume extracellular lactate for Gln synthesis, we knocked down MCT1, which is responsible for uptake of lactate (Figure S5F). We found that MCT1 knockdown (KD) decreased Gln secretion rate and suppressed the rescue effect of CAF on HeyA8 when Gln was deprived from the culture medium (Figures 6D, S5G, and S5H).
Figure 6. Metabolic Symbiosis between Stromal and Epithelial Cells Enhanced Dysregulated Gln Metabolism in CAFs and Maintained Nucleotide and TCA Cycle Metabolite Levels in Cancer Cells.

(A) Intracellular concentrations of lactate, pyruvate, alanine, citrate, α-KG, and glutamate in Gln-deprived CAF1 when transwell co-cultured with HeyA8 cells relative to monocultured CAF1.
(B and C) Dynamic isotope labeling of M2 (B), M4 (C), extracellular Gln secreted by CAF1 when contact co-cultured with or without HeyA8 and HeyA8 GLUL KD cells.
(D) Fluorescence images representing growth rate of HeyA8 co-cultured with CAF transfected with three independent MCT1 siRNA.
(E) Gln concentration in spent medium of HeyA8 and HeyA8 GLUL KD co-cultured with CAF1.
(F and G) GSEA analysis of purine (F) and pyrimidine (G) synthesis genes of HeyA8 transwell co-cultured with CAFs with respect to monocultured HeyA8 cells.
(H) Intracellular concentration of Gln in HeyA8 transwell co-cultured with CAF1 relative to monocultured HeyA8.
(I) Intracellular concentrations of glutamate, pyruvate, lactate and TCA cycle metabolites in HeyA8 when transwell co-cultured with CAF1 relative to monocultured HeyA8.
(J) Proliferation rates of Gln-deprived HeyA8 supplemented with α-KG or nucleotides precursors (GITUAC indicates the mixture of 500 μM guanine, inosine, thymine, uracil, adenine, and cytosine; GAIUMP indicates the mixture of 500 μM GMP, AMP, IMP, and UMP), relative to HeyA8 in Gln-replete medium.
(K) Contribution of extracellular glutamate and CAF intracellular glutamate synthesis to intracellular glutamate in CAF, estimated using a linear regression model using MID measurements at 24 and 48 hr.
(L) Contribution of cancer cell-secreted glutamate to extracellular glutamate in medium estimated using a linear regression model using MID measurements at 24 and 48 hr.
(M) Contribution of CAF-secreted extracellular Gln to intracellular glutamate synthesis in cancer cell estimated using a linear regression model using MID measurements at 24 and 48 hr.
(N) Schematic showing the mutual transfer of metabolites between CAFs and cancer cells. Cancer cells secrete glutamate (M4 and M5) and lactate (M3) when cultured with U-13C6 glucose. The secreted lactate and glutamate is absorbed and metabolized by CAFs to synthesize lighter Gln isotopologues, which is secreted by CAFs and subsequently absorbed by cancer cells for glutaminolysis and nucleotide synthesis. Circles with colors indicate 13C labeled; blank circles indicate 12C.
Error bars indicate mean ± SEM of n ≥ 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001. #p < 0.05, ##p < 0.01, ###p < 0.001.
To test whether metabolic changes in cancer cells were induced by CAF-secreted Gln, we performed metabolic isotope tracing studies of cancer cells when they were co-cultured with CAFs or were cultured alone. As expected, extracellular Gln secreted by CAFs was found to be rapidly depleted in the co-culture media in presence of cancer cells (HeyA8) due to high Gln consumption by these cancer cells (Figure 6E). The metabolic effects of CAF-secreted Gln on cancer cells were also recapitulated at the transcriptional levels using GSEA. This analysis revealed that purine and pyrimidine biosynthesis pathway genes in cancer cells were highly enriched (Figures 6F and 6G). Concurrently, gene expression of lysosomal pathways responsible for protein degradation under nutrient stress was reduced (Figure S6A). Gap junction genes that regulate exogenous uptake of amino acids were also upregulated (Figure S6B). The reduced expression of lysosome and endocytosis genes was indicative of a switch in nutrient acquisition of cancer cells to extracellular sources under Gln deprivation that are potentially provided by CAFs (Figures S6A-S6C). These transcriptional changes were recapitulated in Gln-deprived cancer cells by supplementing Gln, thereby suggesting that CAF-derived Gln is sufficient to rescue OVCA cells in nutrient-stressed conditions (Figures S6D-S6F).
To ascertain the utilization of CAF-secreted Gln by cancer cells, we measured intracellular Gln level in cancer cells with or without co-culture with CAFs and observed a dramatic increase of intracellular Gln concentration in cancer cells in co-cultures with CAFs (Figure 6H). Furthermore, we found that TCA cycle metabolite levels in cancer cells were increased (Figures 6I and S6G). Another important role of Gln in OVCA is to maintain nucleotide synthesis, and we hypothesized that CAF-derived Gln is essential for its maintenance in Gln-deprived cancer cells. Gln enters the TCA cycle after being metabolized to α-KG, and its amine group is incorporated into purine and pyrimidine precursors (Lunt and Vander Heiden, 2011; Son et al., 2013). We supplied Gln-deprived OVCA with either exogenous dimethyl-α-KG, GITUAC (guanine, inosine, thymine, uracil, and adenosine), or GAIUMP (GMP, AMP, IMP, and UMP) to unearth if they could individually rescue OVCA growth. Interestingly, α-KG did not show any rescue when administered alone, proving that the anaplerotic role of Gln into the TCA cycle is not sufficient to support tumor growth. Concurrently, nucleotide precursors could only partially rescue proliferation. However, complete proliferation rescue in cancer cells was observed upon addition of GMP, AMP, IMP, and UMP along with α-KG (Figure 6J).
Paradoxically, we saw high enrichment of M4 and M5 CAF-secreted Gln under Gln deprivation in co-cultures and not in monocultures (Figures 6C and S5A). We postulated that cancer cells could be secreting glutamate, which, when used by CAFs, led to secretion of high-mass isotopologue Gln. Indeed, we found that the glutamate secreted by cancer cells is highly enriched as M4 and M5 glutamate in Gln-free medium (Figures S6H and S6I). This was also supported by the observation of negligible secretion of M4 and M5 glutamate by CAFs in monocultures (Figures S6H and S6I). To further understand the role of cancer cells in modulating CAFs’ Gln synthesis, we estimated the relative contributions of cancer cell-derived glutamate and endogenous glutamate synthesized to glutamate pools within CAFs. Using a one-parameter regression-based balance model (see Experimental Procedures), we found that both endogenous and exogenous glutamate contributions were important, since extracellular glutamate contributed around 40% of total glutamate, and the remaining was contributed by endogenous glutamate synthesis (Figure 6K). On estimating the contribution of cancer cell-synthesized glutamate toward glutamate in extracellular medium, we found it to be approximately 65% after 48 hr of co-cultures (Figure 6L). These results suggest that glutamate derived from cancer cells contributes to at least 25% of glutamate in CAFs for Gln synthesis. We then investigated whether cancer cells could modulate CAF utilization of nitrogen sources and found that there was an increase in M1 glutamate derived from 15N BCAAs (Figure S6J) in CAFs when they are in co-culture compared to monoculture. However, we did not find any increase in other nitrogen donors toward glutamate in CAFs co-cultured with cancer cells (Figure S6K). We next evaluated if CM from cancer cells could have similar effects of co-culture in CAFs. We found that CM significantly increased CAF Gln secretion rate (Figure S6L). These results collectively demonstrate that cancer cells enhanced the capacity of CAFs to use different nutrient sources for synthesizing Gln in order to support cancer cell survival in stressed microenvironments. To clearly demonstrate the influence of CAF-secreted Gln on cancer cell metabolism, we formulated a similar balance model as described above for intracellular glutamate in HeyA8 cells co-cultured with CAFs (see Experimental Procedures) (Figures S6M and S6N). Notably, we found that more than 60% of intracellular glutamate in cancer cells is derived from CAF-secreted Gln (Figure 6M). These results revealed a novel metabolic crosstalk between reactive stromal and cancer cells, where stromal cells provided Gln for cancer cells and cancer cells secreted glutamate and lactate for Gln synthesis in stromal cells (Figure 6N).
Orthotopic OVCA Mouse Model Highlights Stromal GLUL and OVCA GLS as Potential Therapeutic Targets
On the basis of our extensive in vitro co-culture experiments, we next examined whether targeting the reactive stroma (GLUL) and OVCA cells (GLS) simultaneously could result in enhanced therapeutic effect. We used our well-characterized chitosan nanoparticle delivery system for these experiments (Krzeszinski etal., 2014). For this purpose, we devised a targeted therapy in a well-characterized orthotopic mouse model of ovarian carcinoma. Following surgical implantation of SKOV3 cells directly into the left ovary, mice (n = 10 per group) were randomized into one of four groups: (1) control siRNA-CH, (2) human GLS (hGLS) siRNA-CH, (3) murine Glul (mGlut) siRNA-CH, and (4) hss+ mGlul siRNA-CH. We did, in fact, find that the combinational therapy to target the tumor stromal and tumor epithelial compartment significantly improves the therapeutic outcomes of tumor-bearing mice (Figures 7A-7E). By using hGLS siRNA to repress GLS expression in SKOV3 tumor cells, or mGlul siRNA to repress GS expression in the mouse stromal cells, we found that single treatment can decrease tumor weight, number of tumor nodules, and the percent of positive Ki67-staining cells (Figures 7B-7D and S7A-S7C). Most importantly, the combination therapy significantly improves treatment efficiency, as seen from dramatic decrease in tumor weight, and number of tumor nodules (Figures 7B-7D). Furthermore, we also observed a significant decline in metastases to other organs, including mesentery, pelvis, omentum, peritoneum, peri-spleen, and peri-hepatic and diaphragmatic tissue (Figures 7E and S7D). The efficacy of mGlul siRNA was verified by using IHC staining to measure GS intensity. Reduced staining in tissue treated with mGlul siRNA and combo siRNA signified decrease of Glul expression when the mGlul siRNA was injected into mice (Figure 7F). Above results were validated using two independent mGlul siRNA (Figure S7A). In order to verify whether Glul siRNA is a direct bind to Glul mRNA, we cloned the Glul reporter plasmid (wild-type, WT) and not-binding mutant Glul reporter plasmid (TACT-Mut) (Figures S7E and S7F). ID8 cells were transfected with WT-Glul and Mut-Glul luciferase reporter plasmid, plus the control PRL-CMV plasmid and 100 pmol of each control siRNA and Glul siRNA. As shown in Figure S7FI, the transfected Glul siRNA effectively inhibited WT-Glul luciferase reporter gene expression. Flowever, results of Glul siRNA on mutation Glul reporter cannot affect the luciferase reporter gene expression, indicating the Glul siRNA sequence-specific activity (Figures S7G and S7H). These experiments demonstrate that Glul siRNA directly interact with the Glul gene. In order to illustrate that sufficient glucose is present in tumor interstitial fluid for Gln synthesis, we directly isolated interstitial fluid from mouse tumors and measured the concentration of glucose and lactate. Interestingly, we found that interstitial fluid maintained high concentrations of glucose and lactate, which were 3.5–6 mM for glucose and 20–30 mM for lactate (Figures 7G and 7H). These concentration ranges were in support of the hypothesis that the interstitial fluid could sufficiently supply carbon for Gln synthesis. Our results, therefore, highlight the synthetic lethality of targeting stromal GS and tumor GLS and expose vulnerabilities in the metabolic interaction between stromal CAFs and cancer cells for developing clinically relevant therapy.
Figure 7. Targeted Therapy in Orthotopic OVCA Mouse Model Highlights Stromal GLUL and OVCA GLS as Potential Therapeutic Targets.

(A) Using a well-characterized chitosan nanoparticle delivery system, mice were treated with control siRNA-CH, human GLS (hGLS) siRNA-CH, murine Glul (mGlul) siRNA-CH, and combinations of hGLS + mGlul siRNA-CH. n > 10 for each group.
(B) Weight of tumors extracted from mice subjected to control siRNA, hGLS siRNA, mGlul siRNA, and combinations of hGLS and mGlul siRNA.
(C) Number of tumor nodules in mice subjected to control siRNA, hGLS siRNA, mGlul siRNA, and combinations of hGLS and mGlul siRNA.
(D) Ki67 staining to quantify proliferative tumor cells in mice tumors subjected to control siRNA, hGLS siRNA, mGlul siRNA, and combination of hGLS and mGlul siRNA.
(E) Metastasis of ovarian tumor to different organ sites in mice treated with control siRNA, hGLS siRNA, mGlul siRNA, and combination of hGLS and mGlul siRNA.
(F) IHC staining indicating expression of GS in stromal cells of mice treated with control siRNA, mGlul siRNA, and combination of hGLS and mGlul siRNA.
(G) Glucose concentration in tumor interstitial fluid.
(H) Lactate concentration in tumor interstitial fluid.
Error bars indicate mean ± SEM of n ≥ 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
DISCUSSION
The TME, specifically CAFs, has been found to play a multifaceted role in tumor initiation and development. Failure of traditional therapy is due, in part, to our limited understanding of how the TME can facilitate the rapid progression or recurrence of tumors. Targeting tumor stroma for therapeutic purpose is a burgeoning idea that has gained traction in the recent past. In fact, metabolic interactions of cancer cells with TME have been associated as one of the emerging hallmarks of cancer (Pavlova and Thompson, 2016). Under stress conditions, cancer cells have been reported to enhance enzymatic activity for increasing anabolic metabolism (Tardito et al., 2015), lysosome degradation to recycle intracellular waste (Dibble and Manning, 2013; Perera et al., 2015), or intake of lysophospholipids and albumin from microenvironment (Commisso et al., 2013; Kam-phorst et al., 2013). We recently identified that TME-secreted exosomes can provide metabolites to cancer cells under nutrient stress (Zhao et al., 2016). However, the identification of stromal pathways that can be exploited to make cancer cells vulnerable has remained elusive. In order to decode such stromal targets, we adopted an orthogonal approach where differential metabolic transformations in reactive stromal versus normal stroma were identified. Remarkably, we found the Gln anabolic pathway to be distinctly upregulated in CAFs as compared to NOFs. Through comprehensive metabolic isotope tracing and flux analysis, we identified that CAFs have extraordinary metabolic flexibility that helps them adapt to harness carbon and nitrogen from atypical carbon and nitrogen sources for Gln synthesis under Gln deprivation. Contrary to the conventional wisdom, CAFs used more lactate than glucose for acetyl-CoA synthesis. Similarly, aspartate and asparagine were major sources for OAA in CAFs, instead of pyruvate through PC. Our results were in line with the metabolic hallmark, where cancer cell-secreted metabolites were posited to induce adaptations in stroma by altering the microenvironment. There is mounting evidence that indicates acidification of the extracellular space by cancer cells, but not stromal cells, influenced the phenotype, intracellular signaling, and metabolic programming of other cells in their vicinity (Goetze etal., 2011; Pavlova and Thompson, 2016). Furthermore, we found that BCAA and aspartate were the major substrates contributing to the nitrogen supply for Gln synthesis in CAFs. This was also confirmed by increased expression of BOAT and GOT genes.
In line with our hypothesis, disrupting Gln anabolism in CAFs via pharmacological inhibitors or nutrient deprivation proved deleterious to OVCA growth. In addition to inherent reprogramming in CAFs, cancer cells also remarkably influenced CAF metabolism when in co-culture. We found that due to the metabolic pressure applied by cancer cells, CAFs increased their incorporation of glucose/lactate-derived carbon into TCA metabolites and BCAA-derived nitrogen to glutamate. Our data suggested that cancer cells influenced CAF metabolism to maximize Gln synthesis, and in a symbiotic manner the CAF-derived Gln influenced the cancer cell metabolism and growth. Interestingly, our analysis of microarray data of the cancer cell transcriptome showed that co-culturing with CAFs reversed the transcriptional programming induced by nutrient stress. There are potential therapeutic opportunities resulting from targeting stromal metabolic pathways. Our study demonstrated that Gln anabolism in CAFs is a potential target for exploiting systemic metabolic vulnerabilities in ovarian tumors. Previously, researchers have focused on discovering drugs that block intracellular Gln catabolism in cancer cells, which led to the development of inhibitors such as BPTES, CB839, and other such potent drugs (Gross et al., 2014; Shukla et al., 2012). However, recently, environmental influences on non-small-cell lung cancer have been reported (Davidson et al., 2016; Hensley et al., 2016). These results highlight limitations in efficacy of targeting tumors with Gln catabolism monotherapy. Our study uncovers a novel target of Gln source in the stroma. As illustrated by our data from orthotopic intra-ovarian mouse model, combination therapy to simultaneously target Gln anabolism in CAFs and Gln catabolism in cancer cells may prove to be a viable synthetic lethal approach to target tumors systemically for achieving desirable therapeutic outcomes.
EXPERIMENTAL PROCEDURES
Cells and Reagents
SKOV3-GFP and HeyA8-GFP cells were kindly provided by Dr. Samuel Mok from MD Anderson. Ovarian CAFs were derived from advanced-stage high-grade serous OVCA samples and NOFs were derived from normal ovaries obtained from patients with benign gynecologic malignancies. Both CAFs and NOFs were kindly provided by Dr. Jinsong Liu and Dr. Samuel Mok from MD Anderson. All tissue samples were collected under the approval of the institution review board (IRB).
Isotope Labeling Analysis Using Gas Chromatography-Mass Spectrometry
Metabolite Extraction
Cells were seeded in six-well plates overnight, and the next day, media were replaced with fresh media containing U-13C6 glucose or other 13C, 15N labeling nutrients. After 24/72 hr, media were removed, and cells were washed with cold PBS and added with 400 μL ice-cold methanol. The same volume of water containing 1 ng norvaline was added before cells were scraped using cell lifter. A total of 800 μL chloroform was added into the tubes, vortexed at 4°C for 30 min, and centrifuged at 7,300 rpm for 10 min at 4°C. The aqueous layer was collected for metabolite analysis.
Derivatization
Aqueous samples were dried using SpeedVac and dissolved in 30 μL of 2% methoxyamine hydrochloride in pyridine (Pierce) before sonicating for 10 min. Samples were kept in 37°C for 2 hr followed by 1 hr at 55°C after addition of 45 μL MBTSTFA+1 % TBDMCS (Pierce). Samples were transferred into vials containing 150 μL insert.
GC-MS Measurements
Gas chromatography-mass spectrometry (GC-MS) analysis was performed using an Agilent 6890 GC equipped with a 30 m Rtx-5 capillary column for metabolites samples, connected to an Agilent 5975B MS. For metabolite samples, the following heating cycle was used for the GC oven: 100°C for 3 min, followed by a temperature increase of 5°C/min to 300°C for a total run time of 48 min. The abundance of relative metabolites was calculated from the integrated signal of all potentially labeled ions for each metabolite fragment.
LC-MS
Samples (50 (μL) were mixed with 20 μL 0.1 mM sodium tetraborate buffer and 40 μL 20 mM dansyl chloride solution for 30 min at room temperature (RT). A total of 300 μL ice-cold acetonitrile was added, mixed, and incubated for 30 min at 4°C. Samples were centrifuged at 13,000 rpm for 5 min to remove the protein. The supernatant was transferred to other tubes and dried for 2 hr to make sure it was dry. A total of 60 μL buffer A was added (water/aceto-nitrile/formic acid [95:5:0.1]), and samples were centrifuged for 10 min at 13,000 rpm. Supernatant was transferred to liquid chromatography-mass spectrometry (LC-MS) tubes and analyzed by LC-tandem MS (LC-MS/MS) utilizing a TSG Guantum Ultra System.
In Vivo Models and Tissue Processing
Female athymic nude mice were purchased from the Frederick Cancer Research and Development Center, National Cancer Institute and maintained according to guidelines set forth by the American Association for Accreditation of Laboratory Animal Care and the United States Public Flealth Service policy on Fluman Care and Use of Laboratory Animals. All mouse studies were approved and supervised by the MD Anderson Cancer Center Institutional Animal Care and Use Committee.
Statistical Analysis
Comparison of the datasets obtained from the different experiment conditions was performed with the two-tailed Student’s t test. In the bar graphs, data are shown as mean ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001.
More detailed experimental procedures can be found in Supplemental Information.
Supplementary Material
Highlights.
CAFs have an upregulated glutamine anabolic pathway compared to NOFs
CAFs harness atypical carbon and nitrogen sources for glutamine synthesis
Crosstalk between stromal-epithelial cells augments dysregulated metabolism in CAFs
Targeting stromal GS in an orthotopic ovarian cancer model induces tumor regression
ACKNOWLEDGMENTS
This work was made possible in part through support from the St. Louis Ovarian Cancer Awareness Research Grant to D.N. This work was also funded, in part, by the NIH (CA016672, CA109298, P50 CA083639, and UH3 TR000943), the American Cancer Society Research Professor Award, the Blanton-Davis Ovarian Cancer Research Program, and the Frank T. McGraw Memorial Chair in Cancer Research.
Footnotes
ACCESSION NUMBERS
The microarray data for HeyA8 in co-cultures and monocultures under Gln deprivation and complete medium have been deposited in the GEO under ID code GEO: GSE87773.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and four tables and can be found with this article online at http://dx.doi.Org/10.1016/j.cmet.2016.10.011.
REFERENCES
- Bott AJ, Peng IC, Fan Y, Faubert B, Zhao L, Li J, Neidler S, Sun Y, Jaber N, Krokowski D, et al. (2015). Oncogenic Myc induces expression of glutamine synthetase through promoter demethylation. Cell Metab. 22,1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu GC, Kimmelman AC, Hezel AF, and DePinho RA (2007). Stromal biology of pancreatic cancer. J. Cell. Biochem 101, 887–907. [DOI] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, et al. (2016). Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab. 23, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble CC, and Manning BD (2013). Signal integration by mTORCI coordinates nutrient input with biosynthetic output. Nat. Cell Biol 15, 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetze K, Walenta S, Ksiazkiewicz M, Kunz-Schughart LA, and Mueller-Klieser W (2011). Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol 39, 453–463. [DOI] [PubMed] [Google Scholar]
- Goveia J, Stapor P, and Carmeliet P (2014). Principles of targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction in disease. EMBO Mol. Med 6, 1105–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross MI, Demo SD, Dennison JB, Chen L, Chemov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, et al. (2014). Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther 13, 890–901. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hansen JM, Coleman RL, and Sood AK (2016). Targeting the tumour microenvironment in ovarian cancer. Eur. J. Cancer 56,131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley CT, Faubert B, Yuan Q, Lev-Cohaln N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, et al. (2016). Metabolic heterogeneity in human lung tumors. Cell 164, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y„ Yan C, Mu L„ Huang K, Li X, Tao D, Wu Y, and Qin J (2015). Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS ONE 10, e0125625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Icard P, Kafara P, Steyaert JM, Schwartz L, and Lincet H (2014). The metabolic cooperation between cells in solid cancer tumors. Biochim. Biophys. Acta 1846, 216–225. [DOI] [PubMed] [Google Scholar]
- Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, Thompson CB, and Rabinowitz JD (2013). Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lyso-phospholipids. Proc. Natl. Acad. Sci. USA 110, 8882–8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, and Pouyssegur J (2008). Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 13, 472–482. [DOI] [PubMed] [Google Scholar]
- Krzeszinski JY, Wei W, Huynh H, Jin Z, Wang X, Chang TC, Xie XJ, He L, Mangala LS, Lopez-Berestein G, et al. (2014). miR-34a blocks osteoporosis and bone metastasis by inhibiting osteoclastogenesis and Tgif2. Nature 512, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kung HN, Marks JR, and Chi JT (2011). Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 7, e1002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung CS, Yeung TL, Yip KP, Pradeep S, Balasubramanian L, Liu J, Wong KK, Mangala LS, Armaiz-Pena GN, Lopez-Berestein G, et al. (2014). Calcium-dependent FAK/CREB/TNNC1 signalling mediates the effect of stromal MFAP5 on ovarian cancer metastatic potential. Nat. Commun 5, 5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY, and Vander Heiden MG (2011). Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol 27, 441–464. [DOI] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, et al. (2015). Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla K, Ferraris DV, Thomas AG, Stathis M, Duvall B, Delahanty G, Alt J, Rais R, Rojas C, Gao P, et al. (2012). Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J. Med. Chem 55, 10551–10563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496,101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, Miletic H, Sakariassen PØ, Weinstock A, Wagner A, et al. (2015). Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol 17, 1556–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia T, Kim JY, Abu-Baker S, Moscat-Pardos J, Ahn CS, Reina-Campos M, Duran A, Castilla EA, Metallo CM, Diaz-Meco MT, and Moscat J (2014). Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell 26, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vos KE, Eliasson P, Proikas-Cezanne T, Vervoort SJ, van Boxtel R, Putker M, van Zutphen IJ, Mauthe M, Zellmer S, Pals C, et al. (2012). Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat. Cell Biol 14, 829–837. [DOI] [PubMed] [Google Scholar]
- Yang L, Moss T, Mangala LS, Marini J, Zhao H, Wahlig S, Armaiz-Pena G, Jiang D, Achreja A, Win J, et al. (2014). Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol 10, 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung TL, Leung CS, Wong KK, Samimi G, Thompson MS, Liu J, Zaid TM, Ghosh S, Birrer MJ, and Mok SC (2013). TGF-β modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 73, 5016–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, Marini JC, Tudawe T, Seviour EG, San Lucas FA, et al. (2016). Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 5, e10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.