

Abstract
Acute graft-vs-host disease (GVHD) is a frequent complication of hematopoietic transplantation, yet patient risk stratification remains difficult and prognostic biomarkers to guide early clinical interventions are lacking. We developed an approach to evaluate the potential of human T cells from hematopoietic grafts to produce GVHD. Nonconditioned NBSGW mice transplanted with titrated doses of human bone marrow developed GVHD that was characterized by widespread lymphocyte infiltration and organ pathology. Interestingly, GVHD was not an inevitable outcome in our system and was influenced by transplant dose, inflammatory status of the host and type of graft. Mice that went on to develop GVHD showed signs of rapid proliferation in the human T cell population during the first 1–3 weeks post-transplant and had elevated human IFNγ in plasma that correlated negatively with the expansion of the human hematopoietic compartment. Furthermore, these early T cell activation metrics were predictive of GVHD onset 3–6 weeks before phenotypic pathology. These results reveal an early window of susceptibility for pathological T cell activation following hematopoietic transplantation that is not simply determined by transient inflammation resulting from conditioning-associated damage and show that T cell parameters during this window can serve as prognostic biomarkers for risk of later GVHD development.
Introduction
Graft-vs-host disease (GVHD) is a major complication following allogeneic hematopoietic stem cell transplantation (HSCT) that is associated with non-relapse mortality (NRM), delayed immune reconstitution, elevated risk of infections and overall poor quality of life1–2. The central feature of GVHD is the reactivity of donor T cells against host tissues, primarily manifesting as liver, gut and mucous membrane pathology3. Despite the risk of GVHD after allogeneic HSCT, the probability of any given patient developing GVHD is difficult to predict, since it is dependent on a variety of factors including the number of major and minor MHC mismatches between donor and recipient, type of pre-transplant conditioning regimen (myeloablative vs reduced intensity vs non-myeloablative), type of graft source used for the transplant (bone marrow, G-CSF mobilized blood or umbilical cord blood) and the nature of the post-transplant GVHD prophylaxis (traditional calcineurin regimens vs post-transplant cytoxin/ATG)1–2. Without a clear consensus on optimal treatment regimens, murine model systems have been integral to elucidating the biology of GVHD development and pathology.
Studies in murine models of GVHD have revealed a general mechanism of immunological events prior to disease pathology. In brief, host hematopoietic and non-hematopoietic antigen presenting cells (APCs) that are not cleared by the conditioning regimen become activated in the presence of damage-associated-molecular-patterns (DAMPs) released during the conditioning3–7. The resulting inflammatory host APCs then activate donor naïve T cells transplanted within the graft against host antigens, which leads to TH1, TH17 and CD8+ cytotoxic responses against host organs3–4,8.
While murine models have provided a critical mechanistic understanding of processes leading up to GVHD, these models are limited in their capacity to directly evaluate the impact and functional differences associated with the various clinically used graft tissues. Xenotransplantation models using immunodeficient mice provide an opportunity to address these gaps in our understanding of the post-transplant behavior of different sources of human graft tissues. However, possibly as a result of inefficient competition for murine hematopoietic factors, pre-transplant irradiation is required for human cells to successfully engraft most strains of immunodeficient mice9–11. Since irradiation causes damage that promotes GVHD by activating host responses, we sought an approach that would allow us to investigate the inherent propensity of human T cells to cause pathology in a host environment lacking radiation-associated damage. To do this, we utilized a recently derived immunodeficient mouse strain (NBSGW) that is highly permissive for transplanted human immune cells in the absence of conditioning12. This novel model revealed distinct differences among the various human hematopoietic graft tissues in the cellular dose required to cause GVHD and identified early T cell activation metrics that are predictive of subsequent GVHD pathology irrespective of the source of hematopoietic tissue used.
Methods
Isolation of Primary Human Cells.
Human bone marrow cells and G-CSF mobilized blood were collected from the remnants left-over in de-identified bags and filters used for clinical HSCT procedures. De-identified human umbilical cord blood samples were acquired from University of Colorado’s ClinImmune Labs cord blood bank or the Medical College of Wisconsin’s tissue bank. Venous peripheral blood was drawn from healthy consenting donors. Graft sources were isolated by ficoll density-gradient centrifugation (1100xg for 15min with 0 brake) to remove red blood cells, neutrophils and other non-leukocytes. Where indicated, StemCell Technologies RosetteSep T cell enrichment cocktail was used (catalog #15061) to remove lineage positive cells (excluding T cells), leaving untouched CD34+ HSPCs and T cells; T cells were depleted using StemCell Technologies RosetteSep T cell depletion kit (catalogue # 15661). HSPCs were isolated using StemCell Technologies EasySep Human CD34 positive selection kit II (catalog #17856) which first depletes lineage positive cells, followed by magnetic labeling of CD34+ cells and isolation using a magnetic column. Cells were counted, washed and resuspended in PBS prior to injection into mice.
Transplantation of human cells into NBSGW Mice.
The immunodeficient mouse strain NBSGW (NOD.Cg-KitW−41JTyr+PrkdcscidIL2rgtm1Wjl/ThomJ) was purchased from Jackson Laboratories and bred and maintained in specific pathogen free facilities using aseptic housing. Equal numbers of male and female mice between 6–10 weeks of age were used for all experiments. Human cells were suspended in 150μL of PBS and injected retro-orbitally. Mice were weighed and monitored weekly for visible signs of GVHD with scoring carried out as follows: 0 = no signs of GVHD, 1 = no weight loss but visible signs of GVHD (hunching, lethargy, ruffled fur), 2 = 0–5% weight loss, 3 = 5–10% weight loss, 4 = >10% weight loss. Mice that lost 10% or more of their maximum body weight were euthanized. Blood was drawn at the indicated time points, and after 3 months all mice were euthanized and the spleen, one lobe of the liver and the left leg were collected for further processing. For some experiments, LPS was co-injected with human cells at 1ng/mL or I.P. injected at 50ng/g (Invitrogen).
Flow Cytometry.
Single cell suspensions were blocked in 10% human serum and stained for flow cytometric analysis using fluorophore-conjugated antibodies purchased from Biolegend: CD3 (OKT3), CD4 (SK3), CD8 (SK1), CD10 (HI10A), CD19 (HIB19), CD28 (C28.2), CD33 (HIM3–4), CD34 (8G12), CD38 (HIT2), mCD45.1 (A20), CD45RA (HI100), CD45RO (UCHL1), CD56 (HCD56), CD66b (610F5), CD123 (6H6), IFNγ (4S.B3), TNFα (MAb11), OX40 (Ber-ACT35), ICOS (C398.4A), CTLA4 (BNI3), PD1 (EH12.2H7) and PanHLA (W6/32). Stained samples were resuspended in PBS and analyzed on a BD LSRII flow cytometer equipped with 3 lasers (9 filter channels) and quantified using Precision Count Beads from BioLegend. Data analysis was performed using FlowJo V10.5.
Cell Culture & ELISAs.
Cultures of BM-HSPCs were performed using StemCell Technologies StemSpan media (catalog #09650) supplemented with StemSpan CD34+ Expansion Supplement (catalog #02691) for 7 days at 37°C and 5% CO2. BM-HSPCs were stained prior to culturing with ThermoFisher Scientific’s CellTrace Violet (catalog #C34557). Cell culture supernatants or mouse plasma was diluted either 1:5 or 1:25 in ELISA Assay Buffer respectively before performing an IFNγ-specific ELISA using the antibodies clones MD-1 (coating), biotinylated 4S.B3 (detection) and a streptavidin-HRP antibody (BioLegend).
Statistics.
Graphs and statistical tests were completed using GraphPad Prism 6. Means are shown for data plotted on linear axes, while geometric means are used when the data are presented on a logarithmic axis. Trendlines on plots of logarithmic data show results of non-linear regression analyses. Correlations were determined using either a parametric Pearson’s coefficient (for data following a gaussian distribution) or a nonparametric Spearman coefficient and denoted as a correlation coefficient (r). Unpaired, non-parametric t-tests were used to assess significance of aggregated data shown on columnar scatter plots. Receiver operator characteristic (ROC) analyses were done by Graphpad Prism based on mice having no GVHD as the control condition. For the T cell blasting analysis, mice with less than 10 collected T cell events were excluded from the analysis to avoid skewing the result.
Study Approval.
All work involving human cells and tissues was performed in accordance with IRB protocol 2018–0304 (JG), 2016–0298 (PH), and 2017–0870 (JG). All animal work performed in accordance with UW-Madison IACUC approved protocol number M005199.
Results
Development of GVHD in nonconditioned NBSGW mice depends on experimentally manipulatable variables
Immunodeficient NBSGW mice have been shown to accept purified human hematopoietic stem and progenitor cells (HSPC) without prior conditioning and no evidence of GVHD development12–13. However, clinical HSCT protocols are typically performed using unmanipulated graft sources that contain a mixture of cell types. To investigate the ability of clinical human grafts to mediate GVHD in a nonconditioned environment, we injected total mononuclear bone marrow cells (BM-MNC) retro-orbitally into NBSGW mice at various doses and monitored them for 13 weeks (Fig 1A). There was a dose-dependent increase in GVHD mortality (>10% loss of max body weight) and GVHD score (based on weight loss, hunching, squinting, lethargy) with a dose of 1E7 BM-MNC yielding an approximate 50% penetrance of lethal GVHD (Fig 1B–C). Interestingly, analysis of the BM of mice with or without GVHD at the time of euthanasia revealed no statistical difference in the total number of human T cells in the murine bone marrow or spleen (Fig 1D–E), but did show a distinct difference in the number of other human immune cell types (Fig 1F). Moreover, the number of other human immune cells (i.e. human cells excluding T cells) in the murine bone marrow showed a significant negative correlation with GVHD severity (Fig 1G). Thus, despite not inducing damage-associated ligands in this model, GVHD still occurred and was not associated with a significant increase in the absolute numbers of human T cells, but instead was negatively associated with the expansion of non-T cell human immune lineages.
Figure 1. Human cell dose and inflammatory activation determine GVHD penetrance in nonconditioned NBSGW mice.
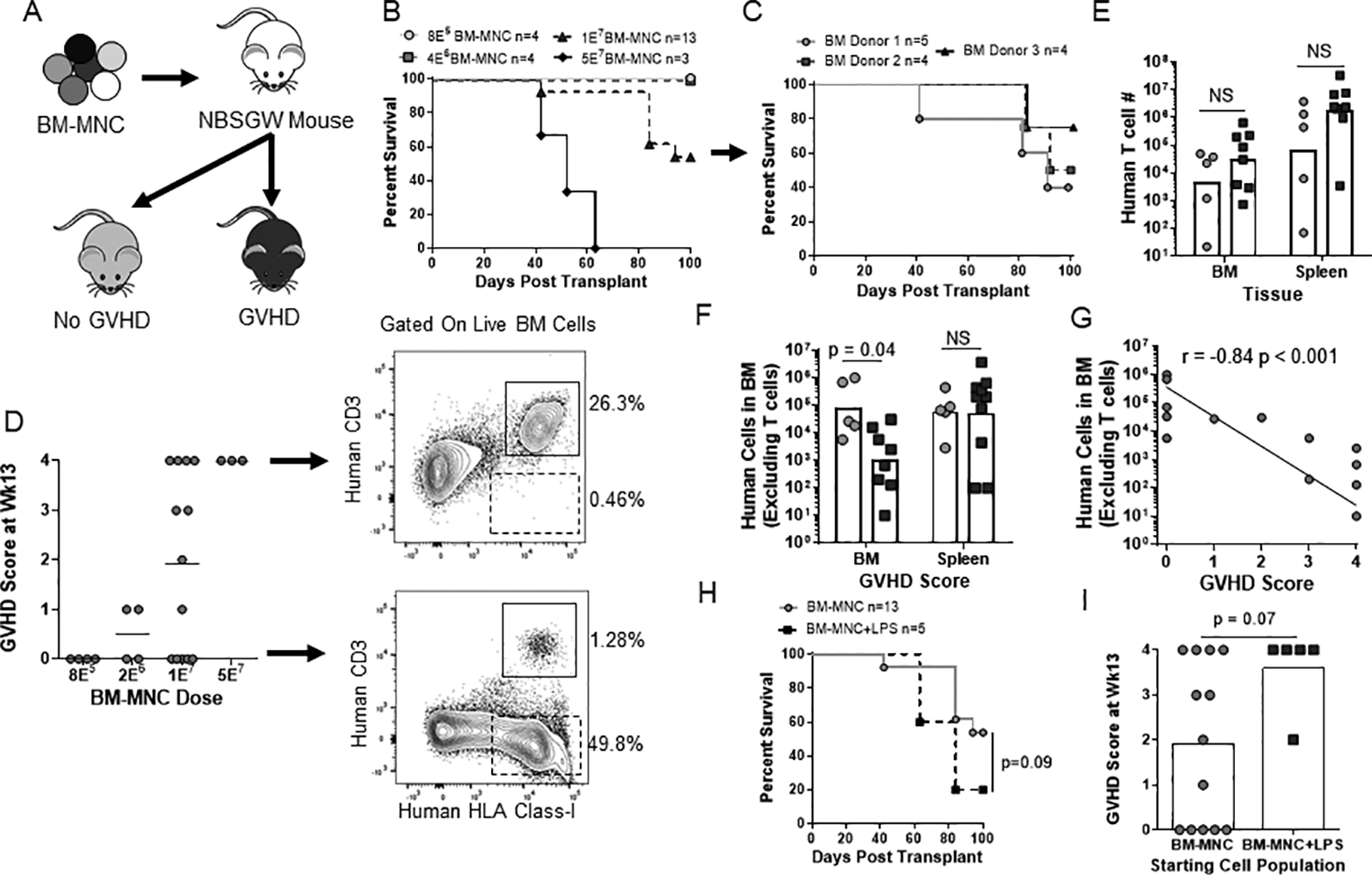
(A) Schematic of experimental layout. (B-C) Kaplan-Meier survival frequency of nonconditioned NBSGW injected with BM-MNCs over a 100-day period. (B) BM-MNC dose is indicated in the figure with the 1E7 BM-MNC dose compiled from three independent BM samples shown in (C). (D) Final GVHD score of each mouse is shown with arrows indicating representative flow plots of the murine bone marrow from a mouse with a final GVHD score of 0 or 4. (E-G) At the time of euthanasia, the number of human T cells and non-T cells in the murine bone marrow (femur+tibia) and spleen from the 1E7 BM-MNC dose was quantified and separated by the presence/absence of GVHD (E-F) or correlated with GVHD score (G). (H-I) Nonconditioned NBSGW mice were either injected with BM-MNC or BM-MNC+LPS (sub-lethal dose of LPS at day +0 and +3) with survival (H) and final GVHD score (H) shown. Data are aggregate of three (B-G) or two (H-I) independent experiments.
Importantly, however, we found that GVHD was not an inevitable outcome of human xenotransplantation in this system. The percentage of mice developing GVHD was dependent on the dose of human bone marrow cells administered (Fig 1B). Mice that received up to 4E6 human BM cells had human chimerism that included T cells but did not develop detectable GVHD within 3 months (Fig 1B and 1D). Moreover, we found that mice that received 1E7 human BM cells had a 50% chance of developing lethal GVHD that was consistent across multiple human BM donors (Fig 1C). Additionally, administration of a sub-lethal dose of LPS at day +0 and +3 post-transplant produced a trend toward increased incidence of GVHD in mice that received 1E7 human BM cells (Fig 1H–I). These results demonstrated that the induction of GVHD is not an intrinsic feature of xenotransplantation and instead is related to human cell dose and probably also to the presence of inflammatory factors after transplantation.
GVHD pathology in the NBSGW mice was characterized by widespread lymphocytic infiltration of the liver, kidney, lung and salivary gland while the skin and gastrointestinal tract remained clear (Fig 2A). Liver tissue collected at the time of euthanasia and scored for lymphocytic infiltration by blinded assessment of H&E stained sections revealed a dose-dependent increase in liver pathology (Fig 2A–B). To further show GVHD mice developed liver damage, we assayed the plasma of GVHD and non-GVHD mice for the liver enzymes ALT, ALP and AST and compared them to a murine reference range14. Most mice with GVHD had elevated levels of AST and some also showed elevated ALT, while levels of ALP, blood urea nitrogen and albumin were mostly unaffected (Fig 2C, data not shown). Lastly, all mice that developed GVHD had marked decreases in body weight and was the primary phenotypic indicator of GVHD pathology used in this study (Fig 2D–E).
Figure 2. GVHD is characterized by widespread organ infiltration and damage.
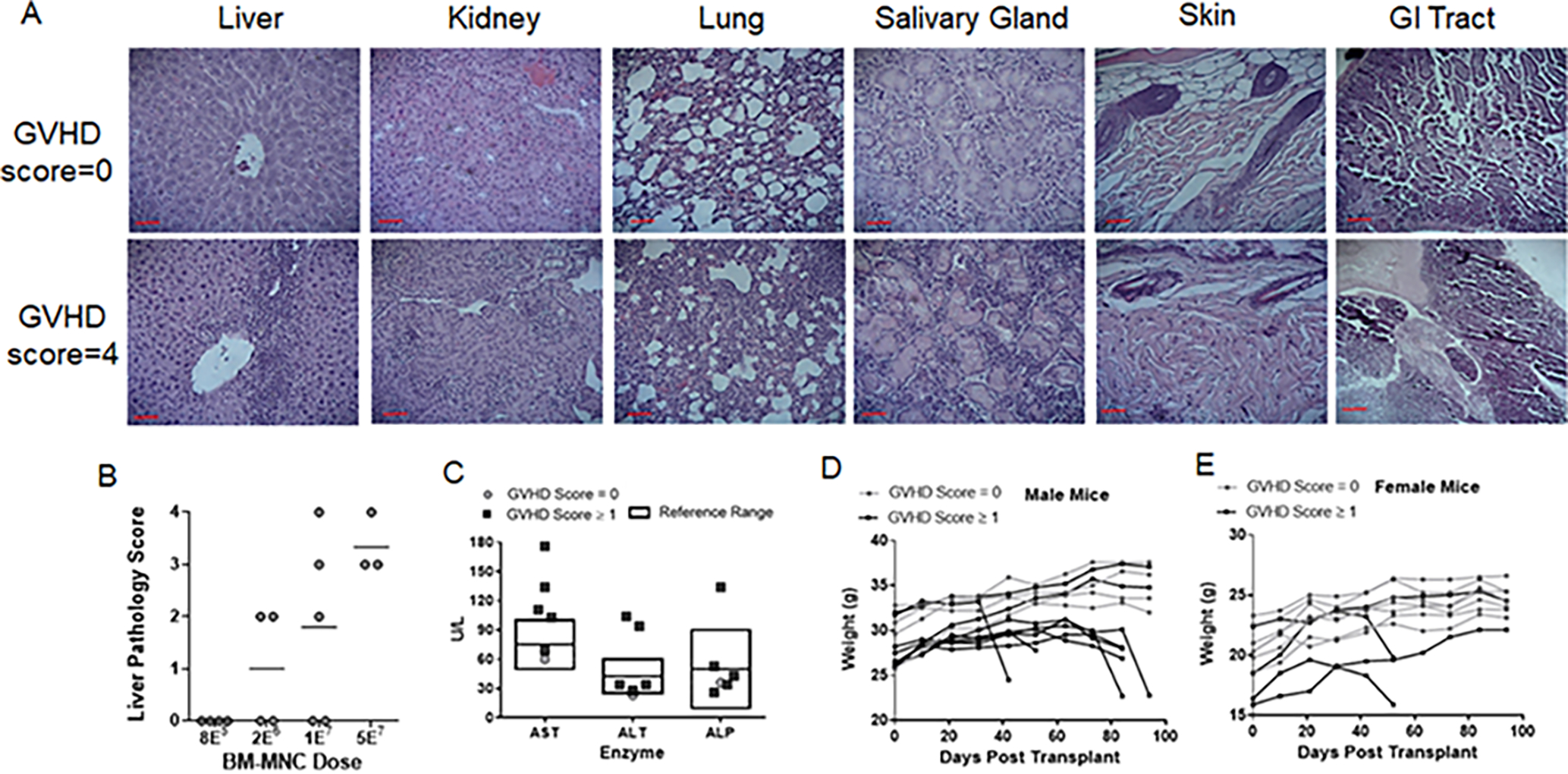
(A) Light microscopy of representative H&E stained histological sections taken at 40X magnification of the indicated tissues from either healthy (GVHD score=0) or severely GVHD mice (GVHD score=4). Scale bars represent 100μm. (B) Blinded histological pathology analysis of H&E liver sections. (C) Quantification of the liver enzymes ALT, ALP and AST in plasma collected from mice with a GVHD score between 3–4 (dark squares) or with a score of 0 (light circles). Boxed areas show reference ranges from Otto et. al.14. (D-E) Weight of each mouse transplanted with BM-MNC over the course of the experiment(s).
T cells are required and sufficient to induce GVHD
Studies in murine models indicate that donor T cells but not donor APCs are required for the induction of GVHD5. We investigated the role of donor T cells and donor APCs in our model by transplanting total BM-MNCs compared with either T cell depleted BM or isolated BM-T cells (lineage negative CD34+ HSPCs were included as an internal control, see materials and methods). Mice receiving T cell depleted grafts showed no evidence of GVHD, while mice given isolated BM-T cells developed GVHD at the same frequency as BM-MNC when controlling for injection dose (Fig 3A–D). Mice given BM-MNC and isolated BM-T cells also had equivalent number of T cells and human non-T cells derived from the co-transplanted HSPCs in their bone marrow and spleen at the time of euthanasia (Fig 3E–F). These data show that similar to what has been observed about the induction of GVHD in murine models, transplantation of human T cells, but not co-transplantation of human APCs, is required for GVHD in this model.
Figure 3. Isolated human T cells are necessary and sufficient to cause GVHD in NBSGW mice.
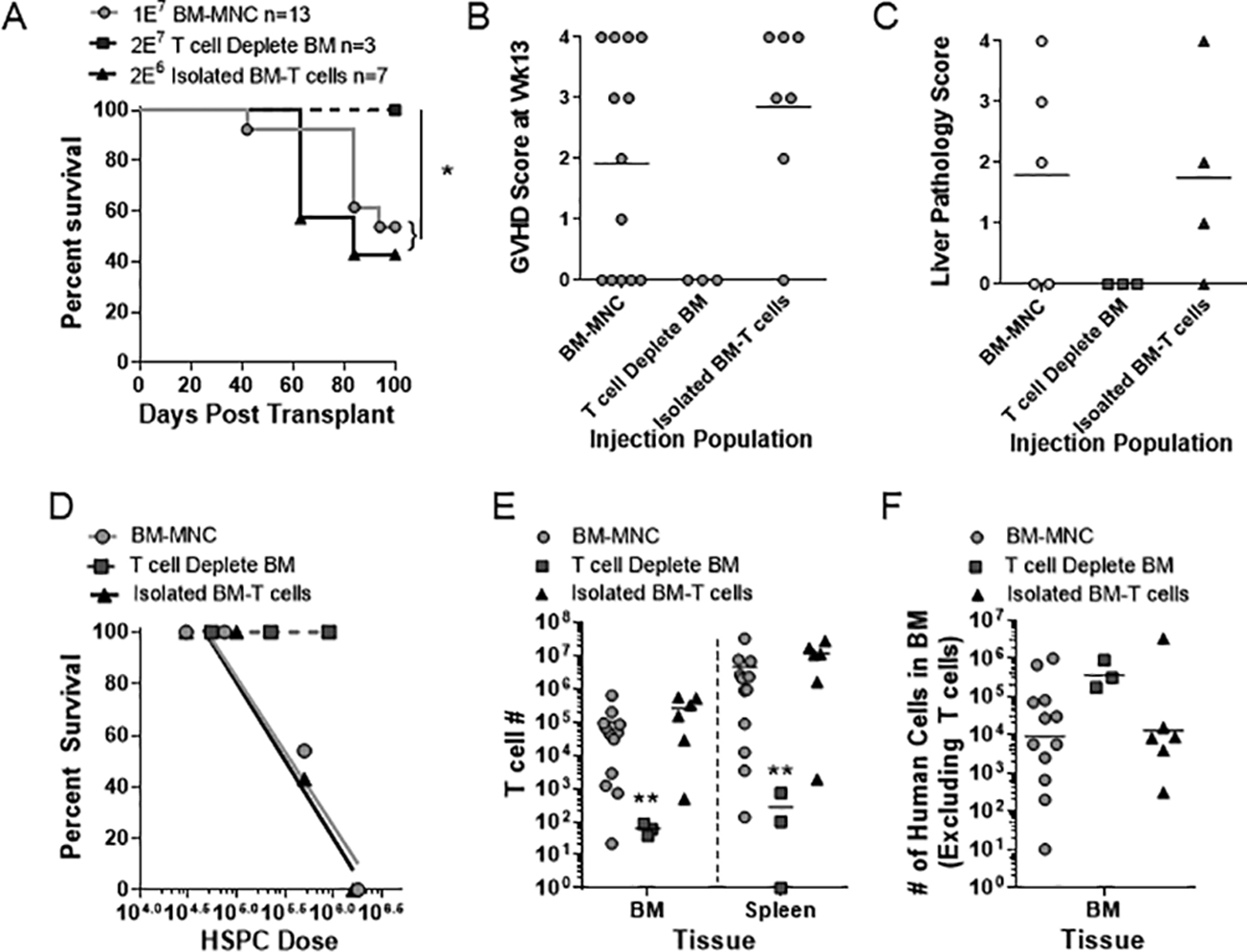
Nonconditioned NBSGW mice were retro-orbitally injected with either human BM-MNC (red, 1E7), T cell depleted BM-MNC (light blue, 2E7) or isolated CD3+ BM-T cells (black, 2E6). (A-D) Kaplan Meier survival frequencies (A), final GVHD score (B), liver pathology scoring (C) and LD50 values (calculated from 3–4 different doses and normalized to CD34+ HSPC injection numbers) (D) are shown. Upon euthanasia, CD3+ T cell numbers (E) or the number of human non-T cells (F) were quantified in the murine bone marrow and spleen for each condition. (A-F) Data are aggregate of three independent experiments. * p < 0.05; ** p < 0.01.
Cellular T cell activation metrics and IFNγ are associated with GVHD pathology
To investigate the changes in the human T cell population over time, mice transplanted with either BM-MNC or isolated BM-T cells were bled at regular intervals and the human T cells in the blood were analyzed and quantified by flow cytometry. Mice that eventually developed GVHD had significantly higher number of human T cells in the blood by 3 weeks’ post-transplant (Fig 4A, Suppl Fig 1A). The difference in blood T cell numbers remained significant throughout the remainder of the experiment (Fig 4A), but differences in T cell numbers were less apparent in the bone marrow and spleen of mice given BM-MNC when compared between one BM-MNC dose (Fig 1E) or multiple doses (Fig 4B). However, mice given isolated BM-T cells that went on to develop GVHD showed significantly elevated T cell numbers in bone marrow, spleen, as well as peripheral blood throughout the experiment (Suppl Fig 1A–B). T cell expression of ICOS, CTLA-4 and PD-1 were upregulated compared to pre-transplant controls, but did not differ between mice that ultimately did or did not develop GVHD (Fig 4C, Suppl Fig 2A). Furthermore, almost all T cells transitioned to CD45RO+ by 6 weeks’ post-transplant in both GVHD and non-GVHD mice suggesting that T cells gained antigenic experience regardless of whether GVHD pathology developed (Fig 4G, Suppl Fig 1C, Suppl Fig 2B). Thus, GVHD may not simply be an outcome of antigenic stimulation but may instead depend on the nature of the T cell activation process.
Figure 4. Correlation of early T cell activation with subsequent GVHD.
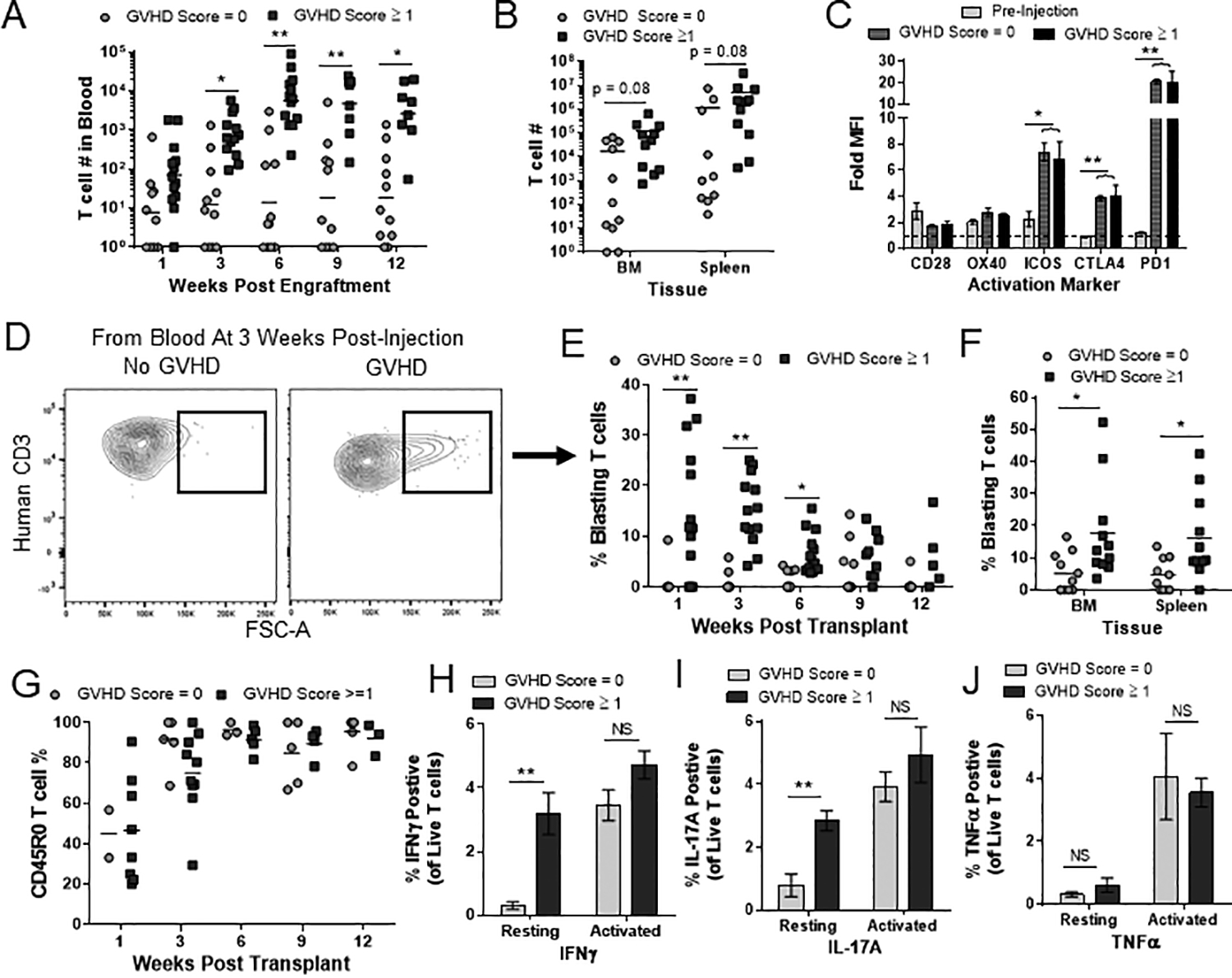
(A-B) Nonconditioned NBSGW mice transplanted with BM-MNC were separated based on their final GVHD score, irrespective of cell dose and quantified for T cell burden in ~150μL of blood (time points indicated in graph) (A), murine bone marrow (tibia+femur) and spleen (B). (C) T cells from pre-injection BM, non-GVHD and GVHD mice were analyzed for the activation markers indicated and expressed as median fluorescent intensity (MFI) over an isotype control (Fold MFI). (D-F) Representative flow plots of blasting T cells from a GVHD and no GVHD mouse (D). The percentage of blasting T cells is shown for blood T cells (E) and for T cells in the murine BM and spleen (F). (G) Frequency of CD45RO+ antigen-experienced cells in the blood was analyzed at the indicated time points. (H-J) T cells collected from the murine bone marrow of non-GVHD and GVHD mice were cultured overnight before adding either brefeldin A (resting) or PMA/Ionomycin (activated) for 6 hrs. The percentage of IFNγ (H), IL-17A (I) and TNFα (J) positive T cells is shown from 4 mice across two different experiments. (A-G) A minimum of 10 T cell events were required to be included in these analyses. Each dot represents one individual mouse. Data are aggregate of three independent experiments. Bars represent the mean and SEM. (NS = not significant; * p < 0.05; ** p < 0.01.
To investigate this further, we examined the frequency of blasting T cells (an increase in cell size that is classically associated with rapid lymphocyte proliferation) in the blood of BM-MNC and isolated BM-T cell transplanted mice over the course of the 13-week experiment. Interestingly, there was a significant difference in the percentage of blasting T cells between GVHD and non-GVHD mice as early as 1-week post-transplant and was maintained in the bone marrow and spleen (Fig 4D–F, Suppl Fig 1D–E). To assess whether blasting frequency is correlated with T cell functional status, we took splenic cells from BM-MNC mice collected at the time of euthanasia and incubated them in vitro in the presence or absence of PMA/ionomycin before assessing their intracellular IFNγ, IL-17A and TNFα production. There was no difference in the number of T cells producing either IFNγ, IL-17A or TNFα after PMA/ionomycin treatment (Fig 4H–J, Suppl Fig 2C). However, in the absence of PMA/ionomycin treatment, mice that had a higher frequency of blasting T cells and went on to develop GVHD showed a significantly elevated frequency of T cells that stained positively for IFNγ and IL17A (Fig 4H–I). In contrast, TNFα production by non-stimulated T cells did not differ significantly (Fig 4J). These results suggested that T cells in the non-GVHD mice did not have an inherent defect in their activation potential but instead had not been activated in vivo to produce IFNγ or IL-17A, whereas T cells from mice that went on to develop GVHD were likely secreting IFNγ and IL-17A at early time points in vivo.
Therefore, we next investigated the role of IFNγ within our model. Plasma taken from regular blood draws was analyzed for IFNγ by ELISA. We observed a significant difference between the GVHD and non-GVHD mice as early as 6 weeks post-transplant in the BM-MNC condition and 3 weeks in the isolated BM-T cells condition (Fig 5A–B). As noted earlier (Fig 1F–G), mice with GVHD had reduced numbers of human non-T cell lineages in their bone marrow. To investigate if IFNγ might be involved in this effect, we collected the supernatant from murine bone marrow harvests and analyzed them for IFNγ by ELISA. Samples from mice with GVHD showed significantly higher amounts of IFNγ that were negatively correlated with the presence of human non-T cell lineages in the murine bone marrow (Fig 5C–D).
Figure 5. GVHD associated IFNγ secretion is correlated with suppression of the hematopoietic compartment.
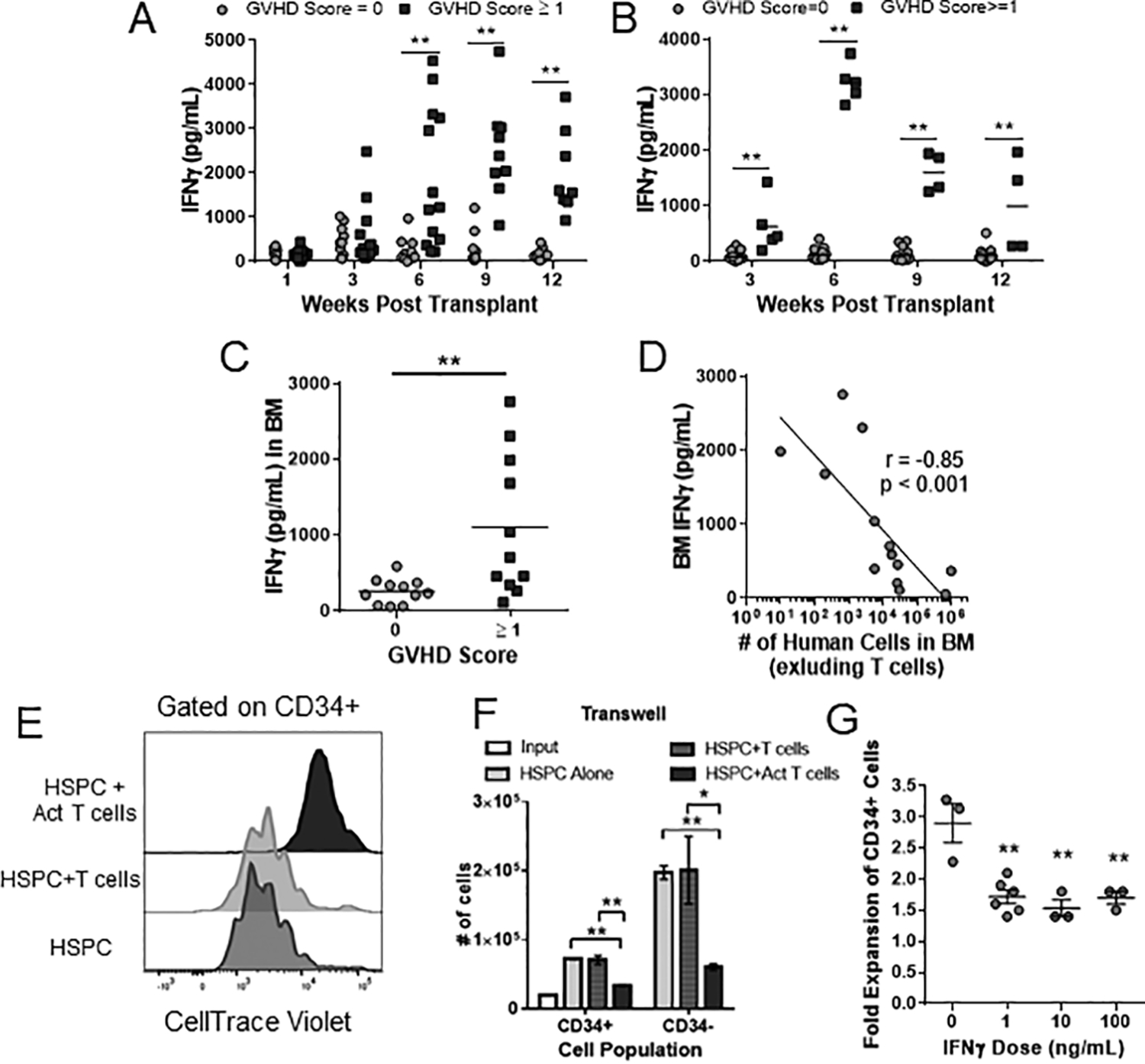
NBSGW mice were separated by GVHD score as in Fig 4 and the levels of human IFNγ in blood plasma samples was quantified at the indicated time points from mice injected with BM-MNC (A) or isolated BM-T cells (B). (C) Diluted supernatant from murine BM at time of euthanasia was collected and analyzed for human IFNγ by ELISA. (D) IFNγ concentration from murine BM was correlated with the number of human non-T cells in the murine BM at the time of euthanasia. (E-G) Isolated CD34+ BM-HSPCs were labeled with cell-division indicator dye (CTV) and culture for 7 days in the presence or absence of autologous T cells in medium containing a hematopoietic expansion cocktail. For “Act T cells”, T cell were activated with αCD3 and αCD28 antibodies. (E) Representative CTV histograms of indicated cultures from a transwell experiment that separated BM-HSPCs from autologous T cells. (F) Quantification of total HSPC-derived cell number that either maintained for lost the expression of CD34. Bars represent the mean and SEM of three replicate wells from one of two independent experiments. (G) Isolated BM-HSPCs were cultured for 7 days with the indicated concentration of recombinant human IFNγ and fold expansion of CD34+ cells quantified by flow cytometry. * p < 0.05; ** p < 0.01
It is well established in murine model systems that chronic IFNγ exposure can prevent HSPC proliferation15–17. Therefore, we investigated if IFNγ had a similar effect on HSPCs isolated from our human BM samples. Isolated human HSPCs were cultured for 7 days alone or in a transwell culture with autologous BM-T cells or CD3/CD28 activated BM-T cells with their proliferation monitored with a CTV dye and overall cell number quantitated. While there were no differences between HSPCs cultured alone or in the presence of autologous T cells, there was a significant reduction in proliferation and cell number of HSPCs cultured in the presence of activated T cells (Fig 5E–F). We also confirmed that IFNγ was produced only in the condition with activated T cells and that the HSPCs had detectable levels of IFNγR1 (CD119) (Suppl Fig 3). Lastly, isolated HSPCs were given recombinant IFNγ at varying doses and showed a significant reduction in cell number in the presence of as little as 1ng/mL of IFNγ (Fig 5G).
Different GVHD dose-response curves for different human hematopoietic graft tissues
Currently, there is no clear consensus on the optimal source of tissue for allogeneic HSCT. The most commonly used tissues are G-CSF mobilized peripheral blood (MB), bone marrow (BM) and umbilical cord blood (CB) (in that order) but their use also varies among clinics1–2,18. Within the clinic, CB transplantation has been shown to have the lowest rates of acute GVHD while MB in the context of allogeneic transplants have the highest rates of acute GHVD1–2. Despite these observations, there have been few studies that have directly investigated the propensity of each graft tissue to mediate GVHD in a controlled laboratory setting. To directly compare the propensity of these three types of tissue to produce GVHD in our model, we transplanted nonconditioned NBSGW mice with titrated cell doses and monitored them for signs of GVHD over time. Similar to what has been observed clinically, CB had the lowest propensity to produce GVHD requiring approximately 7.3E7 CB-MNC to produce lethal GVHD in 50% of the mice (Fig 6B, E). Surprisingly however, bone marrow (BM) produced GVHD at lower cell doses than MB, with calculated LD50 values of 1E7 and 2E7 MNC respectively that is not explained by differences in the cellular composition of the grafts (Fig 6A,C,E, Suppl Fig 4). To further investigate, we compared transplantation of peripheral blood (PB) from healthy control subjects to transplantation of mobilized peripheral blood from G-CSF treated donors (MB). Strikingly, MB had a 10X higher LD50 than PB (Fig 6C–E). Hence, G-CSF treatment of donors may result in reduced propensity of the graft tissue to produce GVHD, compared to normal peripheral blood19. Notably, however, for the mice that did get GVHD, the time to death was shortest for mice that received MB tissue (Fig 6F). Thus, this model suggests that MB has a comparatively low propensity to produce GVHD, but when pathology does occur it may be more severe than that produced by other types of tissue.
Figure 6. Distinct GVHD-potential of different graft tissue sources.
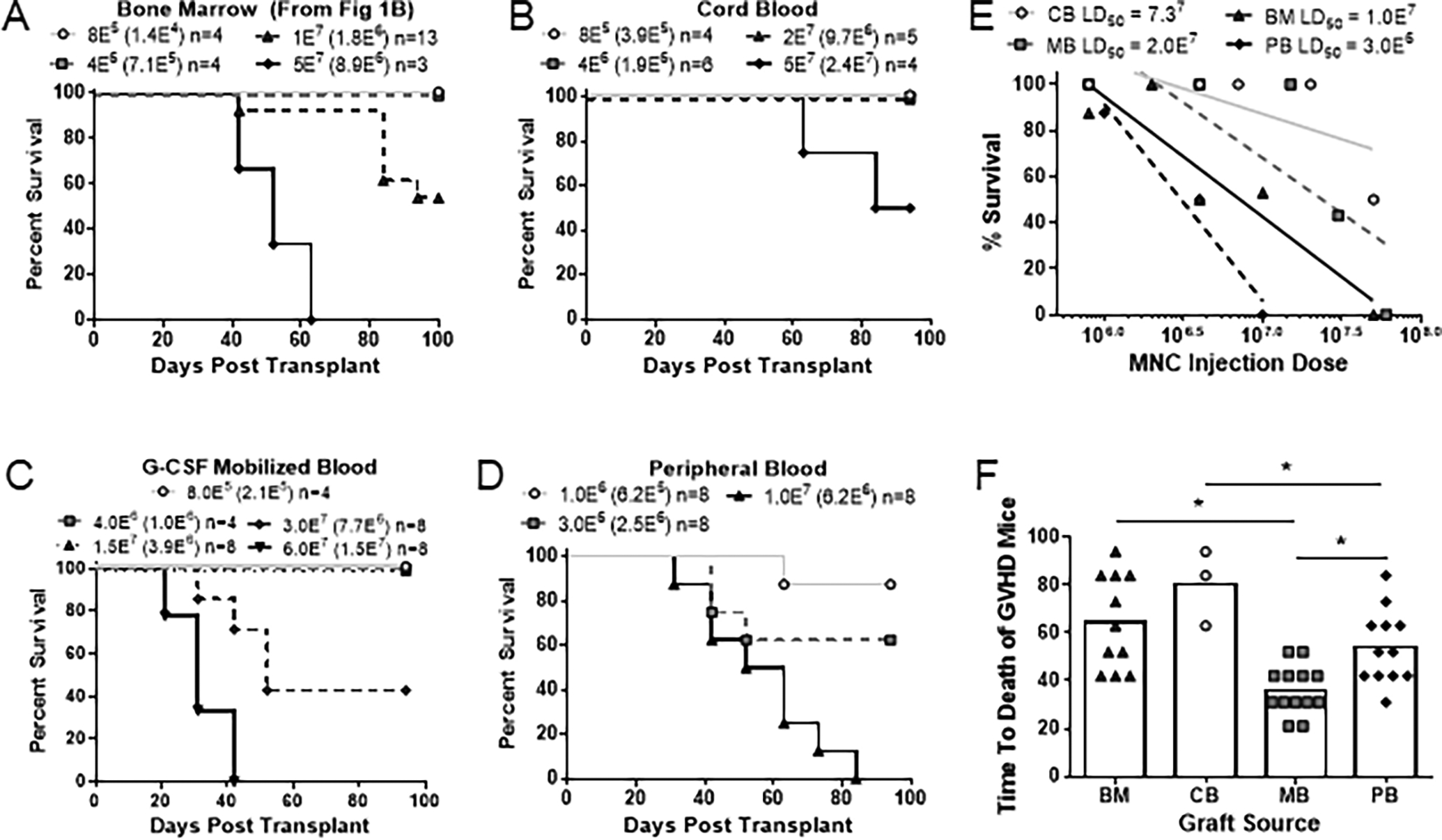
(A-D) Kaplan-Meier survival frequencies over a 100-day period of mice transplanted with the following tissues: (A) human bone marrow (note that this is the same plot as Figure 1B, and is repeated here to facilitate comparison with other tissue grafts), (B) umbilical cord blood, (C) G-CSF mobilized blood, or (D) peripheral blood. Symbol legends show total number of mononuclear cells transplanted, with the number of T cells within each dose shown in parentheses. (E) Calculated LD50 values for each HSCT graft source. (F) Graph showing the average time to death of each graft source irrespective of dosage. (G) Relative frequency of T cells, B cells, myeloid cells and HSPCs in the mononuclear fraction of each respective graft source. (H) Relative frequency of CD4+, CD8+, double negative, naïve and memory T cells within the T cell population of each respective graft source. (G-H) Data aggregated from 3–13 different graft donors.
T cell activation metrics predict GVHD across distinct graft tissues
We next assessed whether the T cell activation metrics we identified as early events that precede the onset of GVHD pathology in mice that received BM transplants (Fig 4–5) show a similar predictive value for other types of graft tissue. Regardless of the type of human tissue transplanted, mice that went on to develop GVHD had an elevated frequency of blasting T cells at week 1 (Fig 7A), a higher burden of T cells in the blood at week 3 (Fig 7B) and an increased concentration of IFNγ in their plasma at week 6 (Fig 7C). We analyzed each parameter using a receiver operator characteristic (ROC) curve that tests the efficacy of each metric to predict an outcome. This analysis revealed that each of the three parameters was significantly predictive of GVHD pathology between 1- and 6-weeks post-transplant across all four graft sources tested, with percent blasting T cells in blood showing the most accuracy at the earliest time point (Fig 7D–F). These results establish that a proliferative burst by circulating T cells as early as one week post-transplant is a reliable predictor for subsequent development of GVHD pathology, regardless of the type of human tissue transplanted and even in the absence of radiation-induced damage in the host.
Figure 7. T cell activation metrics are predictive for GVHD across graft sources.
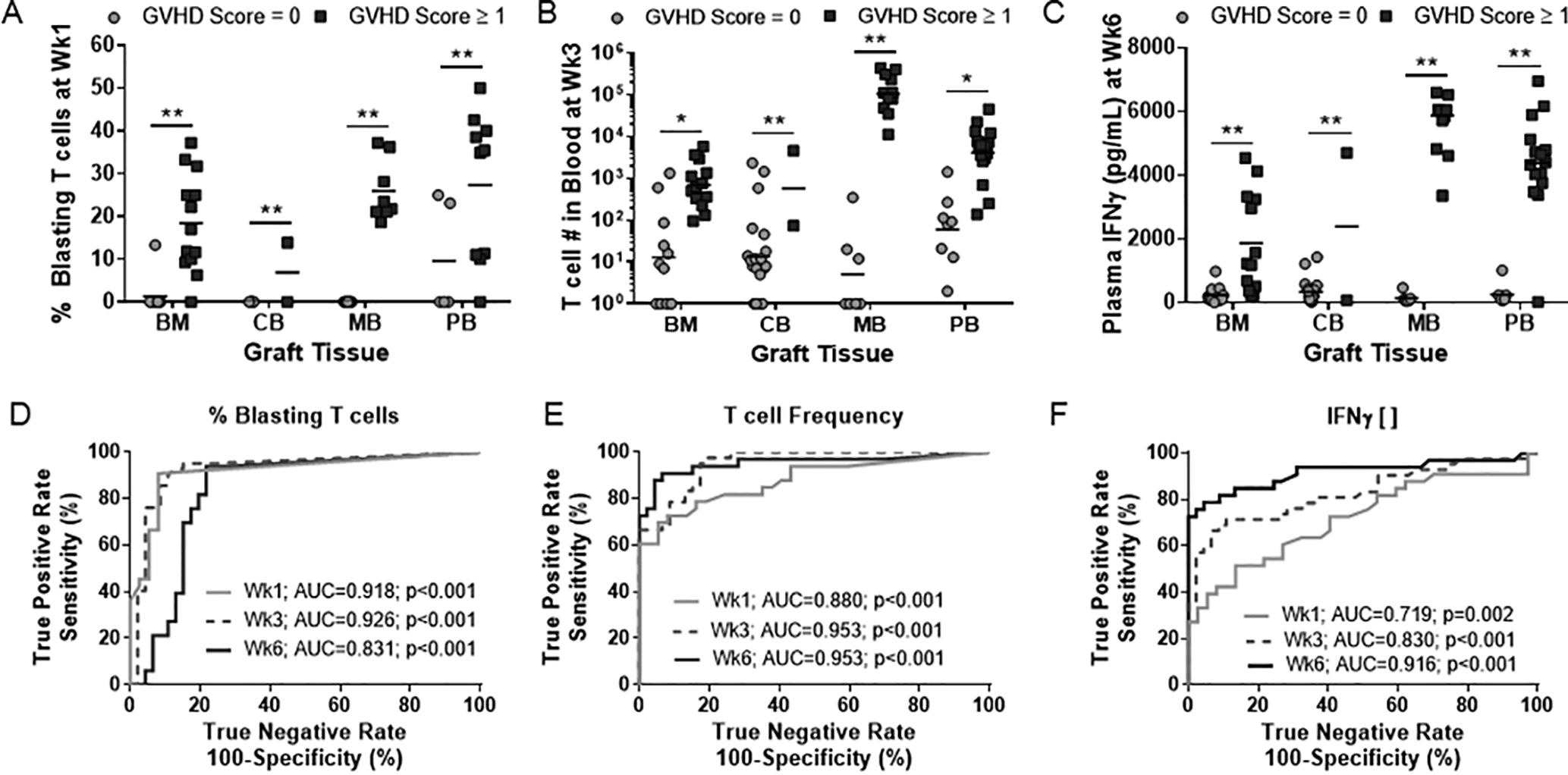
T cell activation markers that were significantly different between non-GVHD and GVHD NBSGW mice (Fig 4 & 5) were validated in BM, CB, MB and PB graft sources (A-C). (D-F) Receiver operator characteristic (ROC) analysis was performed on % blasting T cells (D), T cell burden in peripheral blood (E) and plasma IFNγ concentration (F) to test their efficacy as predictive biomarkers of GVHD.
Discussion
In this study, we have established a nonconditioned immunodeficient mouse model to evaluate the GVHD propensity of clinically used human graft sources and defined several early T cell activation metrics that are predictive of GVHD pathology irrespective of graft source. While using immunodeficient mice provides a powerful approach to study in vivo human immunological events, there are important limitations to consider with xenogeneic transplant models20. Cytokines that are critical for lymphocyte homeostasis such as IL-2 and IL-7 and cytokines that are required for HSPC function such as SCF and SDF-1 are all fully cross-reactive between mice and humans12. However, the full extent of cross-reactive cytokines between mice and humans is not yet known, and many key cytokines (such as IL-3, IL-6, IL-15, M-CSF, GM-CSF and IFNγ) are not highly cross-reactive12,21. Additionally, the level of stimulation received by human T cells from murine MHC and co-stimulatory molecules remains unclear22. Our data show that while pathological activation of human T cells is not an inevitable outcome following xenotransplantation, the murine environment is capable of activating human T cells to produce GVHD pathology regardless of whether human APCs are co-transplanted (Fig 1–3).
Another unique aspect of this study is the use of nonconditioned mice. While harsh myeloablative conditioning (MAC) regimens have classically been used to eliminate malignant cells, conditioning regimens with lower toxicity are now used on older patients and those with specific comorbidities23. The trend of using reduced toxicity conditioning regimens has continued with the introduction of a bead-based conditioning protocol, that has recently entered clinical trials, that allows for engraftment without causing any damage to the host24–26. Since these types of conditioning protocols may become increasingly common in HSCT protocols in the future, it is imperative to understand the potential of HSCT grafts to mediate GVHD in a non-inflammatory environment.
Acute GVHD in the clinic is characterized by donor T cell-driven pathologies of the gastrointestinal tract, liver and skin3. We confirmed that our model recapitulates the infiltration of lymphocytes into the liver and that the resulting damage is measurable by the release of the liver enzymes AST/ALT (Fig 2B–C). Interestingly, we did not detect any infiltration or damage in the murine gastrointestinal tract or skin (Fig 2A). While gastrointestinal GVHD is extremely common in clinical cases, it is also an organ that is predominantly damaged by conditioning regimens27–28. Thus, the absence of a conditioning regimen in our model may prevent the induction of gastrointestinal GVHD, although additional experiments are required to confirm this.
Another aspect of clinical HSCT that our model recapitulates is the inverse correlation between systemic IFNγ concentration and de novo expansion of the human hematopoietic compartment in the bone marrow of our mice (Fig 5). It has become well established from both murine studies and immune reconstitution data in the clinic that chronic IFNγ exposure severely limits the expansion and differentiation of HSPCs15–17,29. Clinically, this often manifests as a drop in neutrophil and platelet counts after the onset of GVHD pathology In our model the total human non-T cell compartment was reduced in GVHD mice (Fig 1F–G), which is due to the adverse effect of GVHD-associated cytokines (e.g. IFNγ) on HSPC function (Fig 5). The inclusion of human HSPCs within the grafts transplanted into our model system also highlights the possibility of de novo T cell development. Importantly, we and others have not detected any de novo T cell development in the murine bone marrow when we transplanted T cell deplete or isolated HSPCs (Fig 3 and data not shown). Furthermore, necropsies of these mice have shown that the murine thymus atrophies shortly after birth rendering it non-functional at the ages used in this study12–13.
A key feature of our model system is the incomplete penetrance of GVHD pathology at intermediate cell doses. This suggests that GVHD is not simply a function of transplanting human T cells into a xenogeneic environment and is instead associated with activation events that do not necessarily occur in all similarly transplanted mice (Fig 1 & 6). While the nature of these activation events is currently not clear, we have identified several variables that influence the probability of an individual mouse to develop GVHD22. We show that cellular dose, graft source and probably also systemic inflammation can all modulate GVHD risk. Since our model is highly permissive to transplantation of human cells without prior conditioning, the baseline inflammation within our model is extremely low. Similar to what is reported in the clinical setting, we observed a trend for systemic inflammation (mediated by sub-lethal doses of LPS) to increase the penetrance of GVHD in our model but found that it is not required for GVHD initiation (Fig 1F–G). In addition to the dose-escalation of GVHD penetrance, this data implies that upon transplant, there is a threshold of activation that must occur within the T cell population for later GVHD pathology to develop which occurs stochastically within each mouse. Thus, both systemic inflammation and increased cellular dose increase the chances of any given mouse of surpassing that activation threshold30. Importantly, the putative activation threshold appears to be independent of T cell acquisition of antigenic experience, as both T cells from GVHD and non-GVHD mice become CD45RO+ and have elevated levels of PD-1, CTLA-4 and ICOS. Thus, the nature of the activation mechanism driving GVHD pathology requires further investigation.
It is well established that clinical transplantation of G-CSF mobilized blood (MB) is associated with higher rates of GVHD than BM transplantation1–2. However, our data suggests that on a per-cell-basis, BM is more GVHD-prone that MB. The most striking difference in GVHD propensity as measured by the calculated LD50 values is the differences between peripheral blood (PB) and MB. Despite both being comprised of cells circulating in the blood, these two graft sources have an approximate 10-fold difference in LD50 values (Fig 6). This supports the possibility that G-CSF treatment prior to transplantation may have suppressive effects, although prior studies have come to contradictory conclusions on this point31–33. These findings highlight the importance of further examination of the biological factors that influence the GVHD propensity of tissues used for HSCT.
While the events leading to T cell activation remain unclear, we have identified three novel biomarkers of early T cell activation that are all predictive of GVHD 3–6 weeks before visible pathology and are independent of graft source (Fig 4 & 7). Due to the number of variables impacting the development of GVHD in clinical settings, there is a significant need to identify prognostic indicators of GVHD risk before the onset of pathology. Studies to date have predominantly focused on plasma biomarkers, with ST2 and REG3α both rigorously validated as indicators of GVHD severity, probability of NRM and steroid-refractory GVHD34–35. Additionally, the recovery of microbial diversity or lack thereof after transplant is a biomarker of GVHD development that is currently gaining traction36. Our data suggest that percent blasting T cells, total T cell burden and blood IFNγ concentration may provide important new biomarkers for GHVD risk. Moreover, these parameters may turn out to provide a valuable way to identify risk of subsequent pathological activation for cellular immunotherapies in which T cell activation is involved, such as CAR-T cell therapies. In conclusion, this study outlines a novel model system to investigate differences in human HSCT graft sources as well as the ability to define specific early T cell activation events that are predictive of GVHD pathology.
Supplementary Material
Key Points.
Xenogeneic transfer of human graft tissue does not lead to inevitable GVHD.
Degree of graft-vs-host response varies among clinical transplantation tissues.
Peripheral blood T cell responses can predict GVHD development.
Acknowledgements
The authors would also like to acknowledge Melissa Graham and Beth Gray at the Comparative Pathology Lab for assistance with histology and Ashley Weichmann at the Small Animal Imaging Facility for assistance with measuring serum analytes.
Grant Support
NJH supported by NIH T32 AI125231 and T32 HL07899. Additional support from NIH R21 AI116007 and R01 AI136500 to JEG, and R21 AI105841 (AWH).
Footnotes
Disclosure of Conflicts of Interest
The authors have no conflicts of interest to disclose.
References
- 1.Anasetti C, Logan BR, Lee SJ, Waller EK, Weisdorf DJ, Wingard JR, Cutler CS, Westervelt P, Woolfrey A, Couban S, Ehninger G, Johnston L, Maziarz RT, Pulsipher MA, Porter DL, Mineishi S, McCarty JM, Khan SP, Anderlini P, Bensinger WI, Leitman SF, Rowley SD, Bredeson C, Carter SL, Horowitz MM, Confer DL, and Blood and Marrow Transplant Clinical Trials Network. 2012. Peripheral-blood stem cells versus bone marrow from unrelated donors. N. Engl. J. Med 367: 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keating AK, Langenhorst J, Wagner JE, Page KM, Veys P, Wynn RF, Stefanski H, Elfeky R, Giller R, Mitchell R, Milano F, O’Brien TA, Dahlberg A, Delaney C, Kurtzberg J, Verneris MR, and Boelens JJ. 2019. The influence of stem cell source on transplant outcomes for pediatric patients with acute myeloid leukemia. Blood Adv 3: 1118–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang L, Chu J, Yu J, and Wei W. 2016. Cellular and molecular mechanisms in graft-versus-host disease. J. Leukoc. Biol 99: 279–287.Koyama, M., and G. R. Hill. 2019. The primacy of gastrointestinal tract antigen-presenting cells in lethal graft-versus-host disease. Blood 134: 2139–2148. [DOI] [PubMed] [Google Scholar]
- 4.Henden AS, and Hill GR. 2015. Cytokines in Graft-versus-Host Disease. J. Immunol 194: 4604–4612. [DOI] [PubMed] [Google Scholar]
- 5.Koyama M, Kuns RD, Olver SD, Raffelt NC, Wilson YA, Don ALJ, Lineburg KE, Cheong M, Robb RJ, Markey KA, Varelias A, Malissen B, Hämmerling GJ, Clouston AD, Engwerda CR, Bhat P, MacDonald KPA, and Hill GR. 2011. Recipient nonhematopoietic antigen-presenting cells are sufficient to induce lethal acute graft-versus-host disease. Nat. Med 18: 135–142. [DOI] [PubMed] [Google Scholar]
- 6.Koyama M, Mukhopadhyay P, Schuster IS, Henden AS, Hülsdünker J, Varelias A, Vetizou M, Kuns RD, Robb RJ, Zhang P, Blazar BB, Thomas R, Begun J, Waddell N, Trinchieri G, Zeiser R, Clouston AD, Degli-Esposti MA, and Hill GR. 2019. MHC Class II Antigen Presentation by the Intestinal Epithelium Initiates Graft-versus-Host Disease and Is Influenced by the Microbiota. Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koehn BH, Saha A, McDonald-Hyman C, Loschi M, Thangavelu G, Ma L, Zaiken M, Dysthe J, Krepps W, Panthera J, Hippen K, Jameson SC, Miller JS, Cooper MA, Farady CJ, Iwawaki T, Ting JP-Y, Serody JS, Murphy WJ, Hill GR, Murray PJ, Bronte V, Munn DH, Zeiser R, and Blazar BR. 2019. Danger-associated extracellular ATP counters MDSC therapeutic efficacy in acute GvHD. Blood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson BE, McNiff J, Yan J, Doyle H, Mamula M, Shlomchik MJ, and Shlomchik WD. 2003. Memory CD4+ T cells do not induce graft-versus-host disease. J. Clin. Invest 112: 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allen TM, Brehm MA, Bridges S, Ferguson S, Kumar P, Mirochnitchenko O, Palucka K, Pelanda R, Sanders-Beer B, Shultz LD, Su L, and PrabhuDas M. 2019. Humanized immune system mouse models: progress, challenges and opportunities. Nat. Immunol 20: 770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McDaniel Mims B, Jones-Hall Y, Dos Santos AP, Furr K, Enriquez J, and Grisham MB. 2019. Induction of acute graft vs. host disease in lymphopenic mice. Pathophysiology. [DOI] [PubMed] [Google Scholar]
- 11.Ito R, Katano I, Kawai K, Yagoto M, Takahashi T, Ka Y, Ogura T, Takahashi R, and Ito M. 2017. A Novel Xenogeneic Graft-Versus-Host Disease Model for Investigating the Pathological Role of Human CD4+ or CD8+ T Cells Using Immunodeficient NOG Mice. Am. J. Transplant 17: 1216–1228. [DOI] [PubMed] [Google Scholar]
- 12.McIntosh BE, Brown ME, Duffin BM, Maufort JP, Vereide DT, Slukvin II, and Thomson JA. 2015. Nonirradiated NOD,B6.SCID Il2rγ−/− Kit(W41/W41) (NBSGW) mice support multilineage engraftment of human hematopoietic cells. Stem Cell Reports 4: 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown ME, Zhou Y, McIntosh BE, Norman IG, Lou HE, Biermann M, Sullivan JA, Kamp TJ, Thomson JA, Anagnostopoulos PV, and Burlingham WJ. 2018. A Humanized Mouse Model Generated Using Surplus Neonatal Tissue. Stem Cell Reports 10: 1175–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Otto GP, Rathkolb B, Oestereicher MA, Lengger CJ, Moerth C, Micklich K, Fuchs H, Gailus-Durner V, Wolf E, and de Angelis MH. 2016. Clinical Chemistry Reference Intervals for C57BL/6J, C57BL/6N, and C3HeB/FeJ Mice (Mus musculus). J Am Assoc Lab Anim Sci 55: 375–386. [PMC free article] [PubMed] [Google Scholar]
- 15.Capitini CM, Herby S, Milliron M, Anver MR, Mackall CL, and Fry TJ. 2009. Bone marrow deficient in IFN-{gamma} signaling selectively reverses GVHD-associated immunosuppression and enhances a tumor-specific GVT effect. Blood 113: 5002–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merli P, Caruana I, De Vito R, Strocchio L, Weber G, Del Bufalo F, Buatois V, Montanari P, Cefalo MG, Pitisci A, Algeri M, Galaverna F, Quintarelli C, Cirillo V, Pagliara D, Ferlin W, Ballabio M, De Min C, and Locatelli F. 2019. Role of IFNγ in immune-mediated graft failure occurring after allogeneic hematopoietic stem cell transplantation. Haematologica. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alvarado LJ, Huntsman HD, Cheng H, Townsley DM, Winkler T, Feng X, Dunbar CE, Young NS, and Larochelle A. 2019. Eltrombopag maintains human hematopoietic stem and progenitor cells under inflammatory conditions mediated by IFN-γ. Blood 133: 2043–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallardo D, de la Cámara R, Nieto JB, Espigado I, Iriondo A, Jiménez-Velasco A, Vallejo C, Martín C, Caballero D, Brunet S, Serrano D, Solano C, Ribera JM, de la Rubia J, and Carreras E. 2009. Is mobilized peripheral blood comparable with bone marrow as a source of hematopoietic stem cells for allogeneic transplantation from HLA-identical sibling donors? A case-control study. Haematologica 94: 1282–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perobelli SM, Mercadante ACT, Galvani RG, Gonçalves-Silva T, Alves APG, Pereira-Neves A, Benchimol M, Nóbrega A, and Bonomo A. 2016. G-CSF-Induced Suppressor IL-10+ Neutrophils Promote Regulatory T Cells That Inhibit Graft-Versus-Host Disease in a Long-Lasting and Specific Way. J. Immunol 197: 3725–3734. [DOI] [PubMed] [Google Scholar]
- 20.Markey KA, MacDonald KPA, and Hill GR. 2014. The biology of graft-versus-host disease: experimental systems instructing clinical practice. Blood 124: 354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mestas J, and Hughes CCW. 2004. Of mice and not men: differences between mouse and human immunology. J. Immunol 172: 2731–2738. [DOI] [PubMed] [Google Scholar]
- 22.van den Broek T, Borghans JAM, and van Wijk F. 2018. The full spectrum of human naive T cells. Nat. Rev. Immunol 18: 363–373. [DOI] [PubMed] [Google Scholar]
- 23.Solomon SR, St Martin A, Shah NN, Fatobene G, Al Malki MM, Ballen KK, Bashey A, Bejanyan N, Bolaños Meade J, Brunstein CG, DeFilipp Z, Champlin RE, Fuchs EJ, Hamadani M, Hematti P, Kanakry CG, McGuirk JP, McNiece IK, Ciurea SO, Pasquini MC, Rocha V, Romee R, Patel SS, Vasu S, Waller EK, Wingard JR, Zhang M-J, and Eapen M. 2019. Myeloablative vs reduced intensity T-cell-replete haploidentical transplantation for hematologic malignancy. Blood Adv 3: 2836–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Czechowicz A, Palchaudhuri R, Scheck A, Hu Y, Hoggatt J, Saez B, Pang WW, Mansour MK, Tate TA, Chan YY, Walck E, Wernig G, Shizuru JA, Winau F, Scadden DT, and Rossi DJ. 2019. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat Commun 10: 617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Z, Czechowicz A, Scheck A, Rossi DJ, and Murphy PM. 2019. Hematopoietic chimerism and donor-specific skin allograft tolerance after non-genotoxic CD117 antibody-drug-conjugate conditioning in MHC-mismatched allotransplantation. Nat Commun 10: 616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pang WW, Czechowicz A, Logan AC, Bhardwaj R, Poyser J, Park CY, Weissman IL, and Shizuru JA. 2019. Anti-CD117 antibody depletes normal and myelodysplastic syndrome human hematopoietic stem cells in xenografted mice. Blood 133: 2069–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piper C, and Drobyski WR. 2019. Inflammatory Cytokine Networks in Gastrointestinal Tract Graft vs. Host Disease. Front Immunol 10: 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piper C, Zhou V, Komorowski R, Szabo A, Vincent B, Serody J, Alegre ML, Edelson BT, Taneja R, and Drobyski WR. 2019. Pathogenic Bhlhe40+ GM-CSF+ CD4+ T Cells Promote Indirect Alloantigen Presentation in the GI Tract during GVHD. Blood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogonek J, Kralj Juric M, Ghimire S, Varanasi PR, Holler E, Greinix H, and Weissinger E. 2016. Immune Reconstitution after Allogeneic Hematopoietic Stem Cell Transplantation. Front Immunol 7: 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Louboutin J-P, Zhu J, Rivera AJ, and Emerson SG. 2002. Preterminal host dendritic cells in irradiated mice prime CD8+ T cell-mediated acute graft-versus-host disease. J. Clin. Invest 109: 1335–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartung T, Döcke WD, Gantner F, Krieger G, Sauer A, Stevens P, Volk HD, and Wendel A. 1995. Effect of granulocyte colony-stimulating factor treatment on ex vivo blood cytokine response in human volunteers. Blood 85: 2482–2489. [PubMed] [Google Scholar]
- 32.Pan L, Delmonte J, Jalonen CK, and Ferrara JL. 1995. Pretreatment of donor mice with granulocyte colony-stimulating factor polarizes donor T lymphocytes toward type-2 cytokine production and reduces severity of experimental graft-versus-host disease. Blood 86: 4422–4429. [PubMed] [Google Scholar]
- 33.Morris ES, MacDonald KPA, Rowe V, Johnson DH, Banovic T, Clouston AD, and Hill GR. 2004. Donor treatment with pegylated G-CSF augments the generation of IL-10-producing regulatory T cells and promotes transplantation tolerance. Blood 103: 3573–3581. [DOI] [PubMed] [Google Scholar]
- 34.Major-Monfried H, Renteria AS, Pawarode A, Reddy P, Ayuk F, Holler E, Efebera YA, Hogan WJ, Wölfl M, Qayed M, Hexner EO, Wudhikarn K, Ordemann R, Young R, Shah J, Hartwell MJ, Chaudhry MS, Aziz M, Etra A, Yanik GA, Kröger N, Weber D, Chen Y-B, Nakamura R, Rösler W, Kitko CL, Harris AC, Pulsipher M, Reshef R, Kowalyk S, Morales G, Torres I, Özbek U, Ferrara JLM, and Levine JE. 2018. MAGIC biomarkers predict long-term outcomes for steroid-resistant acute GVHD. Blood 131: 2846–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srinagesh HK, Özbek U, Kapoor U, Ayuk F, Aziz M, Ben-David K, Choe HK, DeFilipp Z, Etra A, Grupp SA, Hartwell MJ, Hexner EO, Hogan WJ, Karol AB, Kasikis S, Kitko CL, Kowalyk S, Lin J-Y, Major-Monfried H, Mielke S, Merli P, Morales G, Ordemann R, Pulsipher MA, Qayed M, Reddy P, Reshef R, Rösler W, Sandhu KS, Schechter T, Shah J, Sigel K, Weber D, Wölfl M, Wudhikarn K, Young R, Levine JE, and Ferrara JLM. 2019. The MAGIC algorithm probability is a validated response biomarker of treatment of acute graft-versus-host disease. Blood Adv 3: 4034–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Golob JL, Pergam SA, Srinivasan S, Fiedler TL, Liu C, Garcia K, Mielcarek M, Ko D, Aker S, Marquis S, Loeffelholz T, Plantinga A, Wu MC, Celustka K, Morrison A, Woodfield M, and Fredricks DN. 2017. Stool Microbiota at Neutrophil Recovery Is Predictive for Severe Acute Graft vs Host Disease After Hematopoietic Cell Transplantation. Clin. Infect. Dis 65: 1984–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.